

Abstract
Obesity and insulin resistance are associated with ectopic lipid deposition in multiple tissues, including the heart. Excess lipid may be stored as triglycerides, but are also shunted into non-oxidative pathways that disrupt normal cellular signaling leading to organ dysfunction and in some cases apoptosis, a process termed lipotoxicity. Various pathophysiological mechanisms have been proposed to lead to lipotoxic tissue injury, which might vary by cell type. Specific mechanisms by which lipotoxicity alters cardiac structure and function are incompletely understood, but are beginning to be elucidated. This review will focus on mechanisms that have been proposed to lead to lipotoxic injury in the heart and will review the state of knowledge regarding potential causes and correlates of increased myocardial lipid content in animal models and humans. We will seek to highlight those areas where additional research is warranted.
1. Introduction
It has been suggested that the dramatic increase in the prevalence of obesity and cardiovascular disease worldwide, termed the metabolic syndrome pandemic [1], may result in a decline in the life expectancy of the current generation [2, 3]. Obesity also increases the susceptibility to diabetes, which not only increases atherosclerotic heart disease but also increases the risk of developing heart failure [4]. The metabolic syndrome, which is in essence caused by an imbalance between nutrient uptake and energy expenditure, is associated with ectopic deposition of lipid (steatosis) in non-adipose tissue such as the pancreas, kidneys, blood vessels, liver, skeletal muscle, and heart. Although these organs can initially store some of this surplus as triglycerides, excess lipids are eventually shunted into non-oxidative pathways resulting in the accumulation of toxic lipid species which alter cellular signaling [5], promote mitochondrial dysfunction [6], and increase apoptosis [7]. However, the order, progression and role of each of these cellular changes in the ensuing lipotoxicity have not been clearly defined, depend on lipid composition and differ between cell types [8].
Hypertriglyceridemia and increased circulating free fatty acids (FFA) are correlated with lipotoxicity in many tissues such as the liver and β-cell but not necessarily in the heart [9, 10]. In addition to increased circulating lipids, co-existent hyperglycemia and increased inflammatory cytokines may accelerate progression of cellular dysfunction and death, leading to the concept of glucolipotoxicity [11, 12]. Thus multiple mechanisms may lead to cardiac dysfunction in obesity and diabetes and these have been recently exhaustively reviewed [4, 12–14] and will not be covered in detail here. Instead we will focus specifically on mechanisms by which increased cellular lipid impairs cardiomyocyte structure and function.
Obesity affects cardiac structure and function in various ways [4, 14, 15]. Cardiac triglyceride positively correlates with both body mass index and left-ventricular (LV) mass in subjects with impaired glucose tolerance or obesity and inversely with systolic function [10, 16]. Obesity has been linked to both structural and functional changes of the heart including LV hypertrophy (LVH), contractile dysfunction, apoptosis, fibrosis, lipid accumulation, and metabolic substrate switching and this topic has also been recently reviewed [4, 15].
Disturbances in various cellular pathways such as endoplasmic reticulum (ER) stress [17] and mitochondrial dysfunction [18], both of which may increase apoptosis have been implicated in lipid-induced cardiac dysfunction. Multiple molecular mediators have been proposed to promote these lipotoxic effects, such as reactive oxygen species (ROS) [19–26], nitric oxide (NO) [27–29], ceramide [30–33], phosphatidylinositol-3-kinase [34, 35], ligands of PPAR nuclear receptors [36–38], leptin [39–41], and other adipokines [25, 42]. Evidence for these mechanisms will be reviewed.
2. Lipid accumulation and cardiac dysfunction
This section will review data obtained in cell culture, animal and human studies that link excess lipid delivery and accumulation to cellular apoptosis, contractile and metabolic dysfunction. Cell culture experiments have defined potential mechanisms for lipid induced cell death [43]. Studies in animal models of obesity have demonstrated triglyceride accumulation in the heart and correlated these changes with potential mechanisms such as mitochondrial dysfunction or apoptosis [30]. Recently, the ability to assess lipid content in the human heart has been enhanced by use of 1H-NMR, allowing for the measurement of triglyceride in the hearts of healthy, obese, and diabetic subjects [10, 16]. By examining lipid accumulation in these different contexts and correlating these measures with contractile function, we now have obtained some insight into the relationship between lipid accumulation and cardiac function. These studies also highlight the dynamic nature and complex molecular interplay between lipid metabolites and normal cellular function (Figure 1 and 2).
Fig. 1.
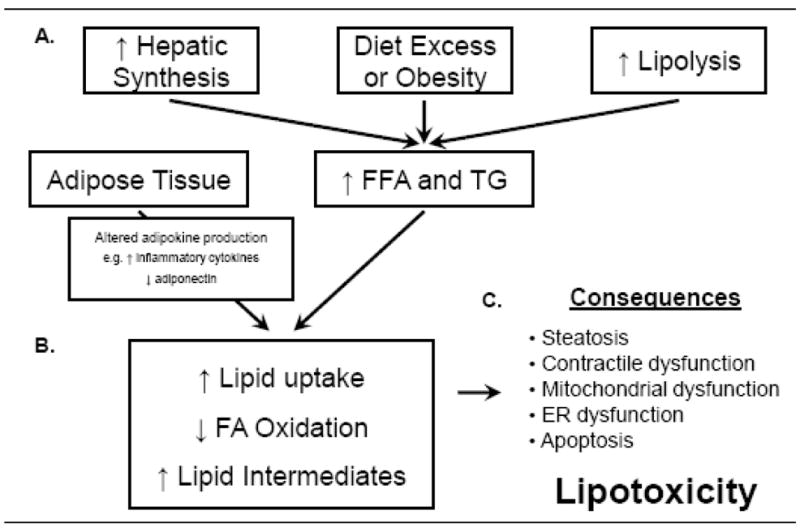
Pathophysiological mechanisms leading to cardiac lipotoxicity. (A) Increased dietary fat intake, hepatic lipogenesis, and lipolysis lead to increased levels of circulating free-fatty acids (FFA) and triglycerides (TG). Obesity and insulin resistance also alter adipokine signaling. (B) Changes in circulating FFA and signaling molecules lead to increased FA uptake, decreased FA oxidation, and increased synthesis of toxic lipid intermediates within the heart. (C) These molecular changes ultimately contribute to cardiac steatosis, contractile dysfunction, mitochondrial dysfunction, endoplasmic reticulum (ER) dysfunction and apoptosis.
Fig. 2.
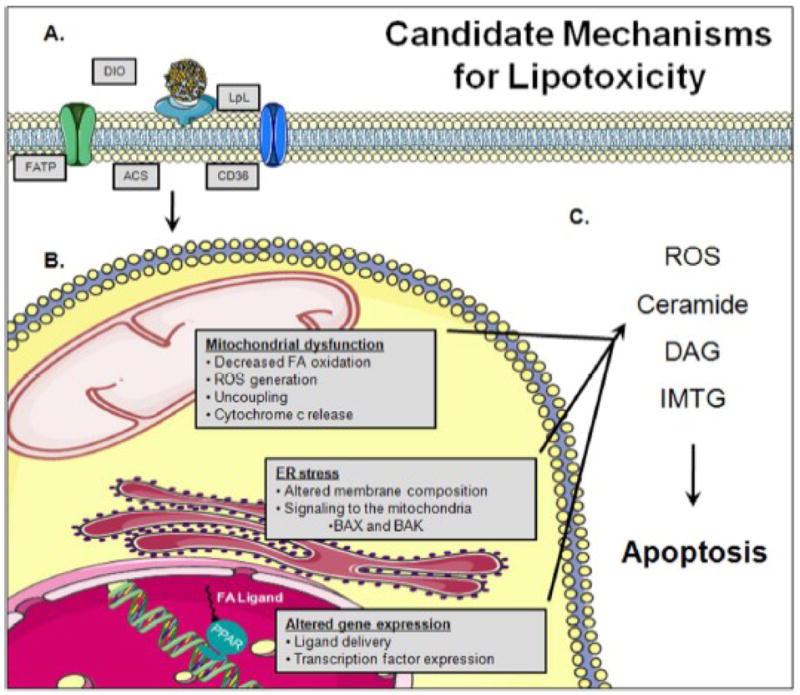
Schematic summary of mechanisms for lipotoxicity. (A) Transgenic models of increased lipid uptake and delivery as well as dietary studies have provided insight into a number of candidate molecular pathways that mediate cardiac lipotoxicity. (B) These include: decreased mitochondrial coupling and oxidative capacity, ER stress, altered membrane composition and function, and altered gene expression through enhanced ligand delivery to transcription factors (e.g. PPARs). However, the specific sequences with which these changes occur and the requirement for each of these pathways is not yet clearly defined. (C) Nevertheless, accumulation of toxic intermediates results in cell death. ACS, acyl-CoA synthase; FATP, fatty acid transport protein; LpL, lipoprotein lipase; DIO, diet-induced obesity; ROS, reactive oxygen species; DAG, diacylglyerol; IMTG, intramyocellular triglycerides; ER, endoplasmic reticulum; FA, fatty acid; PPAR, peroxisome proliferator-activated receptors. Figure was produced using Servier Medical Art (www.servier.com).
2.1. Mechanisms and consequences of lipotoxicity: Insights from cell culture experiments
It has long been known that exposure to saturated fatty acid (SFA) but not to unsaturated fatty acid (UFA) precipitates apoptosis in cell culture [43]. These early studies in fibroblasts provided evidence that lipid accumulation in the ER may lead to the toxic effects of the SFA, and that UFA leads to an increase in cytoplasmic lipid droplets but with maintenance of cell viability. In response to a number of stimuli associated with obesity and increased lipid delivery, there is increased protein flux through the ER. Initially, the influx of unfolded proteins is regulated by increased expression of chaperone proteins, referred to as the unfolded protein response (UPR). When there is a mismatch between the UPR and protein translation, ER stress ensues and may ultimately lead to cell death [44]. ER stress has been suggested as a potential mechanism linking obesity and the development of diabetes by increasing β-cell dysfunction and apoptosis, and has recently been reviewed in detail [45]. When treated with FFA, β-cells in culture exhibit increased levels of known ER stress response proteins in a cytokine-independent manner, suggesting that lipids play a direct role in initiating the ER stress response [46]. Additionally, high fat diet feeding was shown to increase ER stress in liver and adipose, but not muscle [47]. Though the specific mechanism(s) by which obesity leads to ER stress and ultimately cell death remains undefined, ER stress enhances calcium release and signaling to the mitochondria, altering function of the proapoptotic BCL-2 family members, BAX and BAK, resulting in increased apoptosis [48]. The molecular pathways linking ER dysfunction and mitochondria to the ensuing cell death has recently been reviewed [49]. Studies to define the molecular mechanism of lipid-induced cell death in β-cells, found that apoptosis was enhanced when mitochondrial lipid uptake was inhibited by decreasing carnitine palmitoyltransferase 1 (CPT1) activity [50]. Studies by Paumen et al. also found that de novo ceramide synthesis, resulting from increased cytoplasmic FA accumulation, enhances rates of apoptosis. A similar role for ceramide was also found in skeletal muscle cell culture [31]. However, studies in Chinese hamster ovary (CHO) cells suggest that, unlike in β-cells and skeletal muscle cells, a ceramide-independent mechanism involving increased ROS, may be involved [20]. These observations in various cells and tissues raise the possibility that consequences of lipotoxicity in the heart may differ in myocytes [51–53] and non-myocyte cells [11].
Studies examining the role of SFA versus UFA in primary cardiac myocytes found that C16:1 (palmitoleate) or cis-C18:1 (oleate) FA treatment did not alter cell viability, while 24 h of treatment with C16:0 (palmitate) or C18:0 (stearate) precipitated apoptosis as evidenced by DNA-laddering [52]. Similar to studies with β-cells, treatment of primary adult cardiac myocytes with excess SFA leads to ceramide accumulation and cell death [51]. Dyntar et al. further found that increased ceramide levels mediated apoptosis through a mitochondrial-dependent pathway of cytochrome c release [51]. Direct application of ceramide or increased ceramide synthesis by cytokine-mediated activation of sphingolipid metabolism can induce apoptosis in cardiac myocytes [53] and cytochrome c release from mitochondria [54], supporting a mitochondria-dependent role for ceramide-induced apoptosis.
Mitochondrial dysfunction, ceramide synthesis and apoptosis are not completely separable effects. Moreover, the progression of this pathway has been called into question because careful time course analyses following palmitate treatment suggests that in primary neonatal cardiac myocytes cytochrome c release and mitochondrial dysfunction precede ceramide accumulation [55]. Although ceramide treatment is sufficient to induce apoptosis, additional studies of immortalized cardiac cells (H9c2) treated with palmitate found that increased cellular ROS accumulation and ER stress precede apoptosis [17, 19], however the contribution of ROS versus ceramide and the source of the ROS was not determined. These in vitro studies also provide evidence for a mechanism of rapid incorporation of excess lipid into the rough ER membrane, ultimately compromising the structure and integrity of the ER, further enhancing ER stress. These observations support a mechanism of altered membrane composition as a proximal step in the pathogenesis of lipotoxicity [17]. Mitochondria are the primary source of cellular ROS production [6]. When mitochondrial dysfunction was induced by reducing CPT1 activity, the availability of palmitoyl-CoA for ceramide synthesis was increased [50]. Thus a primary defect in mitochondrial function could precipitate ceramide accumulation. For this reason it is difficult to dissect if mitochondrial dysfunction precedes or results from ER stress and/or ceramide accumulation. Additional studies will be required to clarify this issue.
It has recently been shown that circulating factors, such as adipokines may elicit cardiac specific effects. Adiponectin and leptin are adipose-derived signaling molecules that are important regulators of cardiac energy metabolism. Adiponectin treatment is sufficient to increase fatty acid oxidation in the intact neonatal heart [56] and in cardiomyocytes in cell culture [57]. A potential cardiac-specific role for adiponectin is further supported by the finding that adiponectin accumulates in myocardial tissue following ischemic injury [58]. Shibata et al. concluded that this increase results from leakage from the vascular compartment and increased protein stability as opposed to increased local expression. This accumulation may play a cardioprotective role via inhibition of inducible nitric oxide synthase (iNOS) and NADPH-oxidase-expression, leading to decreased oxidative stress [59]. Low levels of plasma adiponectin in diabetics [60], patients with coronary artery disease [61], and in patients following myocardial infarction [62] correlate with increased cardiovascular risk. Obesity in both animals and humans is associated with hypoadiponectinemia [63]. Together these findings suggest that a component of the adverse outcomes of obesity on cardiac function may result from decreased adiponectin signaling.
In contrast, obesity results in hyperleptinemia [64]. In addition to the known roles of leptin on satiety via its actions on the central nervous system, leptin has peripheral actions to increase fatty acid oxidation in adipose, liver [65], skeletal muscle [66], and heart [67]. It has recently been suggested that the leptin resistance that develops in obesity is specific for the metabolic actions of leptin [68]. Isolated skeletal muscle from obese subjects has a blunted response to leptin-dependent fatty acid oxidation [69]. Leptin-mediated hypertrophy in cultured neonatal cardiomyocytes is mediated by an endothelin-1 mediated increase in ROS [70]. By contrast treatment of cultured cardiomyocytes protects cells from H2O2-mediated apoptosis [71]. Thus leptin may mediate complex interactions between cellular redox state and cellular hypertrophy and apoptosis. Whether leptin action in the heart serves to modulate substrate utilization, or oxidative stress in the heart in obesity remains to be clarified and requires further study. Nevertheless, these findings support the notion that some of the cardiac changes in response to lipid excess could be indirectly regulated by signals that emanate from adipose tissue.
These in vitro studies provide evidence that excess lipid delivery can lead to cell death and although the exact mechanism has not been worked out, they provide a strong rationale for studies in animal models that seek to elucidate mechanisms and consequences of myocardial lipid accumulation in vivo.
2.2. Cardiac lipid accumulation in rodent models of obesity and diabetes
Short-term lipid-infusion enhances myocardial lipid accumulation and depresses cardiac function [72]. The role of nutrient excess and tissue lipid accumulation has been studied extensively in vivo in response to high fat diet (HFD) feeding. Multiple aspects of these dietary studies must be taken into consideration such as duration of the dietary intervention, the carbohydrate content and lipid saturation, if the diets are isocaloric or hypercaloric and the animal species studied. These variables mediate disparate effects on the development of obesity and their related comorbidities. For example, 2 wk of HFD feeding of C57BL/J6 mice alters cardiac metabolism by increasing fatty acid oxidation independently of changes in the expression of gene targets of the transcriptional regulator peroxisome proliferator activated receptor-α (PPARα) [73]. This metabolic switch precedes contractile dysfunction which does not develop until after 20 wk of HFD feeding [74]. In contrast, rats fed HFD for 7–8 wk, exhibit mild LVH, triglyceride accumulation, and contractile dysfunction associated with reduced phosphorylation of phospholamban [75] and increased membrane localization of CD36, which was postulated to increase myocardial lipid uptake [76]. These differences in the time to the development of contractile dysfunction in mouse and rat studies likely reflect genetic differences in myocardial susceptibility to lipotoxicity. The caloric content of HFD feeding may also lead to divergent outcomes in response to lipid excess. Specifically, isocaloric HFD attenuates development of diabetes compared to ad libitum HFD feeding, despite an increase in percent body fat compared to animals on a low-fat diet (LFD) [77].
Dietary lipid composition (e.g. saturated versus unsaturated or long-chain versus medium and short chain) also influences the development of lipotoxicity [11, 78]. A number of studies have tested the hypothesis that mono- or polyunsaturated FA may play a protective role in lipotoxicity [79–81]. C57BL/J6 mice fed diets containing fish oil derived ω-3 FA versus soy oil-based diets had lower plasma triglyceride [79], although cardiac function and tissue triglyceride content were not reported. Studies specifically examining the role of diets rich in polyunsaturated FA (PUFA) in the hearts of rats found that a diet high in fish oil ω-3 FA prevented cardiac remodeling and dysfunction following aortic banding-induced pressure overload [80]. Additionally, in mice with pathological cardiac hypertrophy and lipid accumulation resulting from systemic carnitine deficiency, fish oil containing diets reversed LV dysfunction through a proposed mechanism of altered diacylglycerol (DAG) composition and protein kinase c (PKC) redistribution [82]. However, diets rich in ω-6 PUFA, fed to diabetic rats resulted in increased cardiac necrosis [83]. Additional studies are needed to clarify the mechanism for the differences between ω-3 and ω-6 FAs in modulating cardiac remodeling.
Not all HFDs lead to cardiac dysfunction. This seems to be particularly true when isocaloric HFDs are used, which do not precipitate insulin resistance or hyperglycemia. It has also been suggested that certain molecular changes that occur in response to lipid overload may be deleterious under non-stressed conditions but could be protective in the face of additional pathological insults. In a growing number of studies, high-fat feeding has been shown to attenuate some of the defects associated with pressure-overload and ischemic injury. In hypertensive rats fed an isocaloric HFD compared to a LFD, there was reduced LVH and improved contractile function [84, 85]. Additionally, isocaloric HFD feeding for 8 wk following myocardial infarction (MI)-induced heart failure resulted in increased mitochondrial respiration, despite elevated ceramide levels and modest attenuation of contractile dysfunction [86]. Isocaloric HFD feeding for 16 wk post-MI increased myocardial tissue triglyceride accumulation, but did not alter mitochondrial function and increased cardiac function as assessed by fractional shortening. Interestingly, sham-operated animals exhibited decreased mitochondrial function in response to HFD [87]. Epidemiological studies in humans have also suggested the existence of an obesity paradox (described below), but whether or not similar mechanisms account for the potential beneficial effects of high-fat feeding observed in the animal models described above is currently not known.
Rodents with mutations that impair leptin signaling have been extensively studied and have shed important insights into potential mechanisms and consequences of myocardial lipotoxicity. Db/db mice harbor a mutation in the long form of the leptin receptor. In the C57BL6/KsJ background db/db mice develop diabetes by 5 wk of age [18]. Ob/ob mice lack the leptin gene, and on the C57BL/6J background they develop diabetes by ~10–15 wk of age [88]. Db/db mice on the C57BL/KsJ background develop a 2–3 fold increase in myocardial triglyceride accumulation, which is associated with LV contractile dysfunction [89]. Interestingly, myocardial steatosis and increased rates of FA oxidation could be reversed by treatment of mice with a PPARα ligand that normalized serum glucose and lipid concentrations. However, LV contractile dysfunction was not improved. In contrast, perinatal expression of the glucose transporter GLUT4 rescued contractile dysfunction and lowered the increased rates of FA oxidation in db/db mice [90, 91]. It is likely that the inability of PPARα agonist treatment to normalize cardiac function might reflect cellular consequences of lipotoxicity or diabetes, which were not reversible with short-term normalization of systemic metabolism, whereas transgenic GLUT4 upregulation might have prevented cardiac dysfunction because it occurred prior to the onset of obesity and diabetes. The ob/ob mouse model also shows increased myocardial triglyceride content and diastolic dysfunction that is evident as early as 10 wk of age [92]. In a study directly comparing the development of cardiac dysfunction in these two mouse models it was found that metabolic dysfunction and myocardial triglyceride accumulation precedes the onset of contractile dysfunction and hyperglycemia [18]. Taken together, these studies suggest that metabolic dysfunction may precipitate cardiac dysfunction.
Zucker rats have been an extensively studied model of obesity and cardiac lipotoxicity [30]. The Zucker fatty rat (fa/fa) [93–95] has a mutation in the leptin receptor and impaired leptin signaling leads to obesity. In Zucker diabetic fatty rats (ZDF) a defect in β-cells leads to early development of severe diabetes [96]. Because obesity precedes diabetes by variable intervals in these rats, comparisons of these two rat strains have identified cardiac changes that can be attributed to obesity in the presence or absence of diabetes [30, 97–99]. ZDF rats have increased myocardial triglycerides, increased ceramide levels, increased iNOS expression, increased apoptosis, decreased contractile function and decreased PPARα expression [30]. Studies examining non-diabetic fa/fa rats also show higher cardiac triglyceride content compared to lean controls. PPARα expression has been reported to increase or be unchanged in ZDF rats or fa/fa rats [97–99]. However, expression of PPARα targets uniformly increased, implying increased activation by FA ligands. Of interest, many of the transcriptional changes seen in the ZDF rat heart, including increased expression of the β-oxidation genes, MCAD and mCPT1, parallel those found in human failing hearts with intramyocardial lipid accumulation versus failing hearts with no lipid accumulation [98]. Additionally, in response to fasting, obese fa/fa rats show an inability to increase oxidation of exogenous fatty acids and have reduced cardiac function [99]. Oxidation of endogenous FAs was not determined in these experiments. Thus studies in these models provided evidence for a close relationship between cardiac steatosis and cardiac dysfunction in obesity and diabetes.
2.3. Mitochondrial dysfunction and lipotoxicity
Obesity is associated with changes in mitochondrial number and morphology. Despite the observed increases in mitochondrial number in ob/ob and db/db mice [6], recent evidence has suggested that these two models exhibit decreased mitochondrial function [100, 101]. The increase in mitochondrial number occurred without a concomitant increase in the expression of nuclear-encoded genes that encode oxidative phosphorylation subunits. These mitochondria showed reduced oxidative capacity for glucose and although fatty acid utilization was increased, ATP generation was reduced suggesting that the mitochondria were uncoupled. Measurement of reactive oxygen species in db/db mice revealed increases in ROS which was proposed to lead to activation of uncoupling proteins as no increase in uncoupling protein 3 (UCP3) expression was observed [101]. Although changes in substrate utilization occur early in course of obesity in these models [18], mitochondrial dysfunction might represent a later change. This was recently substantiated in a study that examined mice following short-term HFD feeding [73]. Wright et al. found that decreased glucose utilization and increased FA utilization occurred following as little as 2 wk of HFD feeding and these metabolic changes preceded impaired insulin signaling, changes in PPAR gene expression, mitochondrial uncoupling, ROS production or myocardial triglyceride accumulation. Thus altered myocardial substrate utilization represents the earliest change that develops in response to an increase in caloric intake and precedes mitochondrial and contractile dysfunction and cardiac steatosis.
2.4. Myocardial lipid accumulation in humans with obesity and diabetes
Correlations between myocardial lipid accumulation and cardiac dysfunction have been noted in humans for >150 yr [102]. However, only recently has there been renewed interest in the link between lipid accumulation and cardiac dysfunction [15]. Studies of lipid content and substrate utilization, which have long been conducted in rodent models of obesity, are now being extended to humans. Indeed intramyocardial lipid accumulation in the failing heart shares many similarities with that seen in the lipotoxic rat heart [16, 98]. When comparing healthy lean individuals to those with moderate obesity (body mass index (BMI), 28–33 kg/m2), an increase in triglyceride accumulation with increasing body mass appears to precede LVH [103]. To begin to evaluate the relationship between cardiac steatosis and cardiac function, McGavock et al. compared lean normoglycemic individuals, individuals with obesity, impaired glucose tolerance and type 2 diabetes [10]. They found that myocardial lipid accumulation was increased in all groups (obese, IGT and diabetic), preceded the development of diabetes, LVH and systolic dysfunction, but was associated with diastolic dysfunction [104].
The myocardial triglyceride pool is highly dynamic and can increase 3-fold following a 48 h fast in healthy individuals [105], increase 4-fold following a 3 d fast [106], or 55%-2-fold following 3 d of caloric restriction in healthy individuals [106, 107]. However, in healthy individuals no increase in cardiac lipid accumulation was seen following a single high fat meal, despite a 2-fold increase in serum triglyceride content [105] or following 3 d of a high fat/high energy diet [108]. In some of these short-term studies triglyceride accumulation was associated with diastolic dysfunction [106, 107]. Thus in lean subjects fasting or caloric restriction increases cardiac triglyceride, whereas short-term lipid excess does not. In individuals with type 2 diabetes, 3 d of calorie restriction also increased myocardial triglyceride and decreased LV diastolic function [109]. However, after 16 wk of calorie restriction, myocardial triglyceride levels fell and LV diastolic function improved in diabetic subjects in parallel with normalization of glucose tolerance [110]. In contrast, treatment of relatively lean diabetics with metformin and the thiazolidinedione, pioglitazone, improved cardiac function without clearly changing cardiac metabolism or triglyceride content [111]. Taken together, these studies illustrate that the myocardial triglyceride pool in humans is dynamic and can clearly be manipulated by dietary intervention, but do not prove that triglyceride accumulation per se directly influences cardiac function, but might be a biomarker for additional underlying defects.
Obesity has not been linked to increased mortality in all cases. In fact, a number of reports have described a survival benefit for overweight patients, termed the “obesity paradox” (reviewed in [4, 112]). Specifically, in studies examining survival rates in patients post-MI, higher BMI was associated with greater survival [113] and smaller infarct size [114]. Additionally, in hypertensive patients, overweight individuals had lower mortality, stroke risk, and cardiovascular events compared with lean patients [115–117]. These findings bear similarity with rodent studies showing that high-fat diet feeding is protective post-MI or in models of hypertension [84–87]. It is important to note that in some of these studies, both underweight and the most obese individuals had increased mortality [116] while in others the increased mortality in the underweight group was more closely linked to lifestyle (such as alcohol consumption and smoking) than BMI [117]. In either case these reports suggest that there might be some attribute to mild obesity (or high-fat feeding in rodents) that may confer a protective effect against adverse cardiovascular outcomes once they occur. It is important to emphasize though that modest degrees of obesity will increase the risk for developing cardiovascular disease such as heart failure, atherosclerosis, myocardial ischemia and stroke [4]. Therefore from the standpoint of prevention, reducing levels of obesity should reduce the overall burden of cardiovascular disease in terms of prevalence and outcomes. Moreover, given that most of the studies that suggest an “obesity paradox” have been retrospective and cross-sectional, a direct mechanistic link between obesity and improved myocardial outcomes following acute cardiovascular events remains to be elucidated.
The majority of reports describing the effects of obesity on mortality stratified their patient populations based on BMI, as defined by the World Health Organization. Use of BMI to predict the development of cardiovascular disease is commonly used, however it is likely that the distribution of fat might more accurately predict outcome, and consensus regarding the most suitable measure of obesity for epidemiological studies has not yet been obtained [118]. Finally, the epidemiological studies describing the obesity paradox do not directly address the relationship between cardiac steatosis and clinical outcome.
With the development of technologies, such as magnetic resonance spectroscopy (MRS) [119], it is now possible to quantify myocardial triglyceride content as described above [104]. Positron emission tomography (PET) has been used to address changes in cardiac metabolic flux rates and MVO2 in obese and diabetic subjects and in response to changes in circulating FA concentrations or following various therapeutic interventions [4]. For example, examination of patients with type 1 diabetes, found that glucose utilization increased when FFA levels were decreased and fat oxidation increased with increasing FFA levels [120]. Additionally, use of phosphorus 31 (31P) magnetic resonance spectroscopy, has been used to measure high-energy phosphate metabolite levels (phospho-creatine (PCr) and ATP) and found that in the absence of structural and contractile dysfunction, type 2 diabetic patients have a decrease in the PCr/ATP ratio [121]. A recent study of well-controlled type 2 diabetic patients suggested that contractile function could be improved with anti-diabetic drugs in the absence of any change in substrate metabolism or triglyceride content [111]. These studies define new techniques that extend the findings in rodent models to humans by confirming similar patterns of substrate utilization and lipid accumulation. Moreover they suggest that increased triglyceride storage or FFA delivery may reduce cardiac energetics via mechanisms that may be related to FA induced mitochondrial uncoupling or obesity and diabetes-associated reductions in mitochondrial metabolic capacity. These studies indicate a correlation between cardiac function and triglyceride content but do not necessarily prove that changes in triglyceride content directly cause cardiac dysfunction. However, it may be reasonable to conclude that myocardial triglyceride content represents a biomarker for obesity or diabetes-related cardiac dysfunction. In the future it will be important to determine if changes in cardiac lipid or high-energy phosphate content influences or predicts clinical outcome.
3. Transgenic models of lipotoxicity and its reversal
Although diet-induced obesity, or rodents that develop obesity because of impaired leptin signaling develop evidence of lipotoxicity in the heart, analysis of pathophysiological mechanisms are confounded by systemic disturbances, such as hyperglycemia. This section will focus on mouse models that attempt to directly address the role of lipid in the heart in the absence of changes in systemic metabolism in an attempt to elucidate molecular mechanisms leading to lipotoxicity (Fig. 2).
3.1. Mouse models of lipotoxicity via increased lipid uptake and oxidation or reduced turnover
To directly address the role of increased delivery of myocardial fatty acids, a number of groups have generated cardiac-specific models of increased lipid uptake and delivery. One such model is cardiac-specific overexpression of acyl-CoA synthase (ACS), which leads to the accumulation of triglyceride in the heart that is associated with LVH, LV dysfunction and cytochrome c release [122]. The severity of the cardiac phenotypes and mortality rates were directly proportional to the degree of ACS overexpression in the three lines of animals examined. In the highest expressing line, there was a 3.3-fold increase in ceramide levels, increased cardiomyocyte apoptosis, and all animals died by 4-months of age [122]. A second model evaluated the consequence of cardiac-specific overexpression of the fatty acid transport protein (FATP1). These mice had a less severe phenotype, but manifested diastolic dysfunction that was associated with a ~2-fold increase in cardiac FA moieties with no change in triacylglycerols [123]. FATP1 overexpressing mice exhibited increased fatty acid uptake that was accompanied by increased fatty acid oxidation and reduced glucose oxidation.
Overexpression of a membrane anchored lipoprotein lipase (LpL) in cardiomyocytes also induced a lipotoxic cardiomyopathy that was associated with increased myocardial accumulation of various lipid moieties, including ceramide and cholesterol. LpL transgenic animals also exhibited mild LVH, increased PPARα expression, and increased mortality from a dilated cardiomyopathy [124]. These studies are also significant because they underscore that in vivo, lipolysis of triglyceride-rich lipoprotein particles represent an important source of myocardial lipid [125] and that exposure to triglyceride (TG) rich particles recapitulate metabolic abnormalities associated with lipotoxic cardiomyopathy [126]. It is important to note that under physiological circumstances, LpL mediated lipolysis of TG-rich particles represents an important route of substrate delivery to the heart, as evidenced by impaired cardiac contractile function and perivascular fibrosis, despite a compensatory increase in glucose oxidation in mice with cardiomyocyte-restricted deletion of LpL [125]. Germline deletion of adipose tissue triglyceride lipase (ATGL), resulted in dramatic cardiac lipid accumulation, contractile dysfunction and premature death [127]. However, in contrast to models with increased myocardial lipid uptake, ATGL KO mice represent a model with decreased myocardial lipid pool turnover. Taken together, these studies support the hypothesis that increased myocardial lipid delivery, uptake or decreased turnover may impair cardiac contractile function and alter cardiac metabolism.
A number of studies have provided evidence that activation of transcriptional pathways that regulate the expression of FA oxidation enzymes might be sufficient to induce lipotoxicity [128]. Thus activation of PPAR increases the uptake and oxidation of fatty acids by regulating the expression of fatty acid transporters and mitochondrial genes that regulate FA oxidation. Mice with cardiac-specific overexpression of PPARα develop LVH, increased rates of FA oxidation and decreased rates of glucose oxidation [36]. These animals develop increased myocardial lipid triglyceride content, suggesting mismatch between FA uptake and mitochondrial FA oxidative capacity. Consistent with this hypothesis, HFD exacerbated cardiac dysfunction in PPARα overexpressors [33]. Cardiac-specific expression of the related transcription factor PPARγ, also resulted in cardiac dysfunction and increased lipid stores though, the changes in gene expression differed from PPARα overexpressing transgenic mice [129]. Interestingly, cardiac expression of PPARβ/δ preferentially increased glucose utilization without increasing myocardial lipid accumulation and these mice were protected from ischemia [130]. In contrast, PPARβ/δ-null animals developed cardiac dysfunction and myocardial lipid accumulation with reduced survival that likely developed on the basis of reduced mitochondrial FA oxidative capacity [131]. Thus modulation of fuel utilization at the level of mitochondrial oxidation could represent a mechanism for cardiac dysfunction in lipotoxic cardiomyopathies.
3.2. Reversal of lipotoxic phenotypes
Adenoviral overexpression of leptin in liver resulted in hyperleptinemia that rescued the contractile dysfunction observed in the cardiac-specific ACS overexpressing mice, via mechanisms that presumably result from increased myocardial FA oxidation or increased peripheral oxidation of lipids [39]. α-Lipoic acid (α-LA) treatment also normalized LVH, cardiac contractility and triglyceride content in ACS transgenic mice [40]. Treatment with α-LA resulted in a 6-fold increase in the activation of the energy sensing kinase, AMPKα, which is known to increase myocardial FA oxidation. Thus therapeutic strategies that increase myocardial FA oxidation might have utility in treating lipotoxic cardiomyopathies. Strategies that promote triglyceride export from the heart might represent an alternative aproach to combat lipotoxicity. For example, overexpression of apolipoprotein B (apoB) reversed lipotoxic cardiomyopathy in LpL overexpressing transgenic mice and attenuated cardiac dysfunction following high-fat feeding, presumably by increasing export of TG-rich particles from the heart [132, 133].
A direct role for ceramide accumulation in cardiac contractile and metabolic dysfunction was shown in studies examining the cardiac-specific LpL overexpression model of lipotoxicity [32]. Park et al. found that either pharmacologic or genetic inhibition of ceramide synthesis normalized myocardial substrate utilization and improved systolic function. These results underscore the important role of de novo ceramide synthesis in contributing to cardiac dysfunction in lipotoxic cardiomyopathy.
PPARα signaling affects the uptake and utilization of fatty acids. Of interest, ablation of the fatty acid transporter, CD36, was sufficient to rescue the dysfunction observed following PPARα overexpression [134]. CD36 ablation was also shown to protect against age related decreases in cardiac function that are associated with increased intramyocardial lipid accumulation [135]. Thus in addition to its role in the regulation of mitochondrial function, PPAR-mediated regulation of fatty acid uptake pathways are also critical in the development of lipotoxicity in the heart.
Activation of PPARγ receptors in adipose tissue, using the agonist troglitazone, in ZDF rats lowered cardiac triglyceride and ceramide levels, which was associated with prevention of apoptosis and improved cardiac function [30]. It is likely that these effects were secondary to the troglitazone-mediated improvement in glucose homeostasis and insulin resistance. Additionally, troglitazone-treatment of either ZDF rats or ob/ob mice was associated with increased phosphorylation of AMPKα, via a proposed mechanism involving reduced protein phosphatase 2C (PP2C) expression [136]. Palmitate treatment decreases AMPKα phosphorylation in H9c2 cells [136] and red wine polyphenol-treatment of HepG2 hepatocytes has been shown to activate AMPKα [137]. It was recently shown that fa/fa rats fed a diet supplemented with polyphenols had increased cardiac function, potentially through enhanced NO signaling, reduced triglyceride accumulation, and improved glucose metabolism [28]. Together these studies suggest that reversal of triglyceride accumulation and rescue of cardiac contractile dysfunction either via PPAR agonist treatment or polyphenol supplementation may work through a molecular mechanism involving AMPKα.
4. Concluding remarks
Recent advances in imaging technology now make it possible to directly measure human cardiac lipid content and flux providing a novel biomarker that may provide insight into the progression and correlates of lipotoxic heart disease. Although studies in cell culture and animal models have suggested potential mechanisms either associated with or that might link triglyceride accumulation with cardiac dysfunction, more studies are required to determine if these mechanisms also exist in humans. Animal studies that have taken advantage of dietary manipulations, spontaneously occurring mutations, or cardiomyocyte-restricted transgenes, support a causative relationship between lipid accumulation and metabolic and contractile dysfunction. However, differences in the models and the severity of the dysfunction makes the identification of unifying underlying pathophysiological mechanisms a challenge to achieve. The sum of existing evidence suggests that lipid-induced apoptosis, ceramide accumulation, ROS overproduction, ER stress, and mitochondrial dysfunction might play independent and distinct roles in the pathogenesis of lipotoxic cardiomyopathy. However, all of these mechanisms do not necessarily need to be present in a given model. We are hopeful that research efforts in the next few years will further elucidate the pathophysiology of lipotoxic cardiomyopathy and importantly provide additional insights into therapeutic targets and strategies.
Acknowledgments
ARW was supported by NIH Grant 5T32 HL007576 and a post-doctoral fellowship from the American Heart Association Western Affiliates. Work in the Abel laboratory has been supported by the National Institutes of Health: UO1 HL70525, UO1 HL087947 [Animal Models of Diabetes Complications Consortium (AMDCC)], RO1 HL70070 and RO1 HL73167, the American Heart Association, and the Juvenile Diabetes Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol. 2008;28:629–636. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 2.Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J, Hayflick L, Butler RN, Allison DB, Ludwig DS. A potential decline in life expectancy in the United States in the 21st century. N Engl J Med. 2005;352:1138–1145. doi: 10.1056/NEJMsr043743. [DOI] [PubMed] [Google Scholar]
- 3.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world - A growing challenge. N Engl J Med. 2007;356:213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 4.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang R, Barouch LA. Leptin signaling and obesity: Cardiovascular consequences. Circ Res. 2007;101:545–559. doi: 10.1161/CIRCRESAHA.107.156596. [DOI] [PubMed] [Google Scholar]
- 6.Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci. 2008;114:195–210. doi: 10.1042/CS20070166. [DOI] [PubMed] [Google Scholar]
- 7.Unger RH, Orci L. Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta. 2002;1585:202–212. doi: 10.1016/s1388-1981(02)00342-6. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh S, Rodrigues B. Cardiac cell death in early diabetes and its modulation by dietary fatty acids. Biochim Biophys Acta. 2006;1761:1148–1162. doi: 10.1016/j.bbalip.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Slawik M, Vidal-Puig AJ. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res Rev. 2006;5:144–164. doi: 10.1016/j.arr.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 10.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: A 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 11.Garbarino J, Sturley SL. Saturated with fat: new perspectives on lipotoxicity. Curr Opin Clin Nutr Metab Care. 2009;12:110–116. doi: 10.1097/MCO.0b013e32832182ee. [DOI] [PubMed] [Google Scholar]
- 12.Poitout V, Robertson RP. Glucolipotoxicity: Fuel excess and β-cell dysfunction. Endocr Rev. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chess D, Stanley W. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc Res. 2008;79:269–278. doi: 10.1093/cvr/cvn074. [DOI] [PubMed] [Google Scholar]
- 14.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 15.Harmancey R, Wilson CR, Taegtmeyer H. Adaptation and maladaptation of the heart in obesity. Hypertension. 2008;52:181–187. doi: 10.1161/HYPERTENSIONAHA.108.110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szczepaniak LS, Dobbins RL, Metzger GJ, Sartoni-D’Ambrosia G, Arbique D, Vongpatanasin W, Unger R, Victor RG. Myocardial triglycerides and systolic function in humans: In vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn Reson Med. 2003;49:417–423. doi: 10.1002/mrm.10372. [DOI] [PubMed] [Google Scholar]
- 17.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 19.Borradaile NM, Buhman KK, Listenberger LL, Magee CJ, Morimoto ETA, Ory DS, Schaffer JE. A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol Biol Cell. 2006;17:770–778. doi: 10.1091/mbc.E05-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–14895. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 21.Joseph JW, Koshkin V, Saleh MC, Sivitz WI, Zhang C-Y, Lowell BB, Chan CB, Wheeler MB. Free fatty acid-induced β-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004;279:51049–51056. doi: 10.1074/jbc.M409189200. [DOI] [PubMed] [Google Scholar]
- 22.Carlsson C, Hakan Borg LA, Welsh N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology. 1999;140:3422–3428. doi: 10.1210/endo.140.8.6908. [DOI] [PubMed] [Google Scholar]
- 23.Aronis A, Madar Z, Tirosh O. Mechanism underlying oxidative stress-mediated lipotoxicity: Exposure of J774.2 macrophages to triacylglycerols facilitates mitochondrial reactive oxygen species production and cellular necrosis. Free Radic Biol Med. 2005;38:1221–1230. doi: 10.1016/j.freeradbiomed.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 24.Dewald O, Sharma S, Adrogue J, Salazar R, Duerr GD, Crapo JD, Entman ML, Taegtmeyer H. Downregulation of peroxisome proliferator-activated receptor-α gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation. 2005;112:407–415. doi: 10.1161/CIRCULATIONAHA.105.536318. [DOI] [PubMed] [Google Scholar]
- 25.Delaigle A, Senou M, Guiot Y, Many M, Brichard S. Induction of adiponectin in skeletal muscle of type 2 diabetic mice: in vivo and in vitro studies. Diabetologia. 2006;49:1311–1323. doi: 10.1007/s00125-006-0210-y. [DOI] [PubMed] [Google Scholar]
- 26.Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, Ueda S, Sakanashi M, Takasu N. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology. 2007;148:160–165. doi: 10.1210/en.2006-1132. [DOI] [PubMed] [Google Scholar]
- 27.Shimabukuro M, Ohneda M, Lee Y, Unger RH. Role of nitric oxide in obesity-induced beta cell disease. J Clin Invest. 1997;100:290–295. doi: 10.1172/JCI119534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agouni A, Lagrue-Lak-Hal A-H, Mostefai HA, Tesse A, Mulder P, Rouet P, Desmoulin F, Heymes C, Martinez MC, Andriantsitohaina R. Red wine polyphenols prevent metabolic and cardiovascular alterations associated with obesity in Zucker fatty rats (fa/fa) PLoS ONE. 2009;4:e5557. doi: 10.1371/journal.pone.0005557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabkin S, Klassen S. Palmitate-induced NO production has a dual action to reduce cell death through NO and accentuate cell death through peroxynitrite formation. Prostaglandins Leukot Essent Fatty Acids. 2008;78:147–155. doi: 10.1016/j.plefa.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y-T, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc Natl Acad Sci USA. 2000;97:1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turpin SM, Lancaster GI, Darby I, Febbraio MA, Watt MJ. Apoptosis in skeletal muscle myotubes is induced by ceramides and is positively related to insulin resistance. Am J Physiol Endocrinol Metab. 2006;291:E1341–1350. doi: 10.1152/ajpendo.00095.2006. [DOI] [PubMed] [Google Scholar]
- 32.Park T-S, Hu Y, Noh H-L, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang X-C, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49:2101–2112. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARα-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc Natl Acad Sci USA. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardy S, Langelier Y, Prentki M. Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res. 2000;60:6353–6358. [PubMed] [Google Scholar]
- 35.Huang W, Dedousis N, Bhatt BA, O’Doherty RM. Impaired activation of phosphatidylinositol 3-kinase by leptin is a novel mechanism of hepatic leptin resistance in diet-induced obesity. J Biol Chem. 2004;279:21695–21700. doi: 10.1074/jbc.M401546200. [DOI] [PubMed] [Google Scholar]
- 36.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kourtidis A, Srinivasaiah R, Carkner R, Brosnan MJ, Conklin D. Peroxisome proliferator-activated receptor-γ protects ERBB2-positive breast cancer cells from palmitate toxicity. Breast Cancer Res. 2009;11:R16. doi: 10.1186/bcr2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baranowski M, Blachnio-Zabielska A, Zabielski P, Gorski J. Pioglitazone induces lipid accumulation in the rat heart despite concomitant reduction in plasma free fatty acid availability. Arch Biochem Biophys. 2008;477:86–91. doi: 10.1016/j.abb.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 39.Lee Y, Naseem RH, Duplomb L, Park B-H, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci USA. 2004;101:13624–13629. doi: 10.1073/pnas.0405499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee Y, Naseem RH, Park B-H, Garry DJ, Richardson JA, Schaffer JE, Unger RH. α-Lipoic acid prevents lipotoxic cardiomyopathy in acyl CoA-synthase transgenic mice. Biochem Biophys Res Commun. 2006;344:446–452. doi: 10.1016/j.bbrc.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 41.Shimabukuro M, Koyama K, Chen G, Wang M-Y, Trieu F, Lee Y, Newgard CB, Unger RH. Direct antidiabetic effect of leptin through triglyceride depletion of tissues. Proc Natl Acad Sci USA. 1997;94:4637–4641. doi: 10.1073/pnas.94.9.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palanivel R, Vu V, Park M, Fang X, Sweeney G. Differential impact of adipokines derived from primary adipocytes of wild-type versus streptozotocin-induced diabetic rats on glucose and fatty acid metabolism in cardiomyocytes. J Endocrinol. 2008;199:389–397. doi: 10.1677/JOE-08-0336. [DOI] [PubMed] [Google Scholar]
- 43.Gordon GB. Saturated free fatty acid toxicity: II. Lipid accumulation, ultrastructural alterations, and toxicity in mammalian cells in culture. Exp Mol Pathol. 1977;27:262–276. doi: 10.1016/0014-4800(77)90035-1. [DOI] [PubMed] [Google Scholar]
- 44.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 45.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 46.Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: Role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology. 2004;145:5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- 47.Ozcan U, Cao Q, Yilmaz E, Lee A-H, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 48.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 49.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paumen MB, Ishida Y, Muramatsu M, Yamamoto M, Honjo T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. J Biol Chem. 1997;272:3324–3329. doi: 10.1074/jbc.272.6.3324. [DOI] [PubMed] [Google Scholar]
- 51.Dyntar D, Eppenberger-Eberhardt M, Maedler K, Pruschy M, Eppenberger HM, Spinas GA, Donath MY. Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes. 2001;50:2105–2113. doi: 10.2337/diabetes.50.9.2105. [DOI] [PubMed] [Google Scholar]
- 52.de Vries JE, Vork MM, Roemen TH, de Jong YF, Cleutjens JP, van der Vusse GJ, van Bilsen M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res. 1997;38:1384–1394. [PubMed] [Google Scholar]
- 53.Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, Glembotski CC, Quintana PJ, Sabbadini RA. Tumor necrosis factor α-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghafourifar P, Klein SD, Schucht O, Schenk U, Pruschy M, Rocha S, Richter C. Ceramide induces cytochrome c release from isolated mitochondria. Importance of mitochondrial redox state. J Biol Chem. 1999;274:6080–6084. doi: 10.1074/jbc.274.10.6080. [DOI] [PubMed] [Google Scholar]
- 55.Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB. A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2000;279:H2124–2132. doi: 10.1152/ajpheart.2000.279.5.H2124. [DOI] [PubMed] [Google Scholar]
- 56.Onay-Besikci A, Altarejos JY, Lopaschuk GD. gAd-globular head domain of adiponectin increases fatty acid oxidation in newborn rabbit hearts. J Biol Chem. 2004;279:44320–44326. doi: 10.1074/jbc.M400347200. [DOI] [PubMed] [Google Scholar]
- 57.Palanivel R, Fang X, Park M, Eguchi M, Pallan S, De Girolamo S, Liu Y, Wang Y, Xu A, Sweeney G. Globular and full-length forms of adiponectin mediate specific changes in glucose and fatty acid uptake and metabolism in cardiomyocytes. Cardiovasc Res. 2007;75:148–157. doi: 10.1016/j.cardiores.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 58.Shibata R, Sato K, Kumada M, Izumiya Y, Sonoda M, Kihara S, Ouchi N, Walsh K. Adiponectin accumulates in myocardial tissue that has been damaged by ischemia-reperfusion injury via leakage from the vascular compartment. Cardiovasc Res. 2007;74:471–479. doi: 10.1016/j.cardiores.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 59.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115:1408–1416. doi: 10.1161/CIRCULATIONAHA.106.666941. [DOI] [PubMed] [Google Scholar]
- 60.Schulze MB, Shai I, Rimm EB, Li T, Rifai N, Hu FB. Adiponectin and future coronary heart disease events among men with type 2 diabetes. Diabetes. 2005;54:534–539. doi: 10.2337/diabetes.54.2.534. [DOI] [PubMed] [Google Scholar]
- 61.Nakamura Y, Shimada K, Fukuda D, Shimada Y, Ehara S, Hirose M, Kataoka T, Kamimori K, Shimodozono S, Kobayashi Y, Yoshiyama M, Takeuchi K, Yoshikawa J. Implications of plasma concentrations of adiponectin in patients with coronary artery disease. Heart. 2004;90:528–533. doi: 10.1136/hrt.2003.011114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kojima S, Funahashi T, Otsuka F, Maruyoshi H, Yamashita T, Kajiwara I, Shimomura H, Miyao Y, Fujimoto K, Sugiyama S, Sakamoto T, Yoshimura M, Ogawa H. Future adverse cardiac events can be predicted by persistently low plasma adiponectin concentrations in men and marked reductions of adiponectin in women after acute myocardial infarction. Atherosclerosis. 2007;194:204–213. doi: 10.1016/j.atherosclerosis.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 63.Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 64.Unger RH. Hyperleptinemia: Protecting the heart from lipid overload. Hypertension. 2005;45:1031–1034. doi: 10.1161/01.HYP.0000165683.09053.02. [DOI] [PubMed] [Google Scholar]
- 65.Lee Y, Yu X, Gonzales F, Mangelsdorf DJ, Wang M-Y, Richardson C, Witters LA, Unger RH. PPARα is necessary for the lipopenic action of hyperleptinemia on white adipose and liver tissue. Proc Natl Acad Sci USA. 2002;99:11848–11853. doi: 10.1073/pnas.182420899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor α gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the α2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–4327. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Atkinson LL, Fischer MA, Lopaschuk GD. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J Biol Chem. 2002;277:29424–29430. doi: 10.1074/jbc.M203813200. [DOI] [PubMed] [Google Scholar]
- 68.Correia MLG, Kamal R. Role of leptin in the cardiovascular and endocrine complications of metabolic syndrome. Diabetes Obes Metab. 2006;8:603–610. doi: 10.1111/j.1463-1326.2005.00562.x. [DOI] [PubMed] [Google Scholar]
- 69.Steinberg GR, Parolin ML, Heigenhauser GJF, Dyck DJ. Leptin increases FA oxidation in lean but not obese human skeletal muscle: evidence of peripheral leptin resistance. Am J Physiol Endocrinol Metab. 2002;283:E187–192. doi: 10.1152/ajpendo.00542.2001. [DOI] [PubMed] [Google Scholar]
- 70.Xu F-P, Chen M-S, Wang Y-Z, Yi Q, Lin S-B, Chen AF, Luo J-D. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. 2004;110:1269–1275. doi: 10.1161/01.CIR.0000140766.52771.6D. [DOI] [PubMed] [Google Scholar]
- 71.Megumi E, Yuantao L, Eyun-Jung S, Gary S. Leptin protects H9c2 rat cardiomyocytes from H2O2-induced apoptosis. FEBS Journal. 2008;275:3136–3144. doi: 10.1111/j.1742-4658.2008.06465.x. [DOI] [PubMed] [Google Scholar]
- 72.Hexeberg S, Hessevik I, Hexeberg E. Intravenous lipid infusion results in myocardial lipid droplet accumulation combined with reduced myocardial performance in heparinized rabbits. Acta Physiol Scand. 1995;153:159–168. doi: 10.1111/j.1748-1716.1995.tb09847.x. [DOI] [PubMed] [Google Scholar]
- 73.Wright JJ, Kim J, Buchanan J, Boudina S, Sena S, Bakirtzi K, Ilkun O, Theobald HA, Cooksey RC, Kandror KV, Abel ED. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc Res. 2009;82:351–360. doi: 10.1093/cvr/cvp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Park S-Y, Cho Y-R, Kim H-J, Higashimori T, Danton C, Lee M-K, Dey A, Rothermel B, Kim Y-B, Kalinowski A, Russell KS, Kim JK. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54:3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 75.Ouwens DM, Boer C, Fodor M, de Galan P, Heine RJ, Maassen JA, Diamant M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia. 2005;48:1229–1237. doi: 10.1007/s00125-005-1755-x. [DOI] [PubMed] [Google Scholar]
- 76.Ouwens DM, Diamant M, Fodor M, Habets DD, Pelsers MM, El Hasnaoui M, Dang ZC, Evan den Brom C, Vlasblom R, Rietdijk A, Boer C, Coort SL, Glatz JF, Luiken JJ. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia. 2007;50:1938–1948. doi: 10.1007/s00125-007-0735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petro AE, Cotter J, Cooper DA, Peters JC, Surwit SJ, Surwit RS. Fat, carbohydrate, and calories in the development of diabetes and obesity in the C57BL/6J mouse. Metabolism. 2004;53:454–457. doi: 10.1016/j.metabol.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 78.Russo GL. Dietary n-6 and n-3 polyunsaturated fatty acids: From biochemistry to clinical implications in cardiovascular prevention. Biochem Pharmacol. 2009;77:937–946. doi: 10.1016/j.bcp.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 79.Qi K, Fan C, Jiang J, Zhu H, Jiao H, Meng Q, Deckelbaum RJ. Omega-3 fatty acid containing diets decrease plasma triglyceride concentrations in mice by reducing endogenous triglyceride synthesis and enhancing the blood clearance of triglyceride-rich particles. Clin Nutr. 2008;27:424–430. doi: 10.1016/j.clnu.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 80.Duda MK, O’Shea KM, Tintinu A, Xu W, Khairallah RJ, Barrows BR, Chess DJ, Azimzadeh AM, Harris WS, Sharov VG, Sabbah HN, Stanley WC. Fish oil, but not flaxseed oil, decreases inflammation and prevents pressure overload-induced cardiac dysfunction. Cardiovasc Res. 2009;81:319–327. doi: 10.1093/cvr/cvn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aguila MB, Mandarim-de-Lacerda CA. Heart and blood pressure adaptations in Wistar rats fed with different high-fat diets for 18 months. Nutrition. 2003;19:347–352. doi: 10.1016/s0899-9007(02)00934-6. [DOI] [PubMed] [Google Scholar]
- 82.Takahashi R, Okumura K, Asai T, Hirai T, Murakami H, Murakami R, Numaguchi Y, Matsui H, Ito M, Murohara T. Dietary fish oil attenuates cardiac hypertrophy in lipotoxic cardiomyopathy due to systemic carnitine deficiency. Cardiovasc Res. 2005;68:213–223. doi: 10.1016/j.cardiores.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 83.Ghosh S, An D, Pulinilkunnil T, Qi D, Lau HCS, Abrahani A, Innis SM, Rodrigues B. Role of dietary fatty acids and acute hyperglycemia in modulating cardiac cell death. Nutrition. 2004;20:916–923. doi: 10.1016/j.nut.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 84.Okere IC, Chess DJ, McElfresh TA, Johnson J, Ernsberger JRP, Hoit BD, Chandler MP, Stanley WC. High-fat diet prevents cardiac hypertrophy and improves contractile function in the hypertensive dahl salt-sensitive rat. Clin Exp Pharmacol Physiol. 2005;32:825–831. doi: 10.1111/j.1440-1681.2005.04272.x. [DOI] [PubMed] [Google Scholar]
- 85.Okere IC, Young ME, McElfresh TA, Chess DJ, Sharov VG, Sabbah HN, Hoit BD, Ernsberger P, Chandler MP, Stanley WC. Low carbohydrate/High-fat diet attenuates cardiac hypertrophy, remodeling, and altered gene expression in hypertension. Hypertension. 2006;48:1116–1123. doi: 10.1161/01.HYP.0000248430.26229.0f. [DOI] [PubMed] [Google Scholar]
- 86.Rennison JH, McElfresh TA, Okere IC, Vazquez EJ, Patel HV, Foster AB, Patel KK, Chen Q, Hoit BD, Tserng K-Y, Hassan MO, Hoppel CL, Chandler MP. High-fat diet postinfarction enhances mitochondrial function and does not exacerbate left ventricular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H1498–1506. doi: 10.1152/ajpheart.01021.2006. [DOI] [PubMed] [Google Scholar]
- 87.Rennison JH, McElfresh TA, Chen X, Anand VR, Hoit BD, Hoppel CL, Chandler MP. Prolonged exposure to high dietary lipids is not associated with lipotoxicity in heart failure. J Mol Cell Cardiol. 2009;46:883–890. doi: 10.1016/j.yjmcc.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 89.Aasum E, Belke DD, Severson DL, Riemersma RA, Cooper M, Andreassen M, Larsen TS. Cardiac function and metabolism in Type 2 diabetic mice after treatment with BM 17.0744, a novel PPAR-alpha activator. Am J Physiol Heart Circ Physiol. 2002;283:H949–957. doi: 10.1152/ajpheart.00226.2001. [DOI] [PubMed] [Google Scholar]
- 90.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 91.Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002;283:H976–982. doi: 10.1152/ajpheart.00088.2002. [DOI] [PubMed] [Google Scholar]
- 92.Christoffersen C, Bollano E, Lindegaard MLS, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- 93.Zucker LM, Zucker TF. Fatty, a new mutation in the rat. Journal of Heredity. 1961;52:275–278. [Google Scholar]
- 94.Iida M, Murakami T, Ishida K, Mizuno A, Kuwajima M, Shima K. Phenotype-linked amino acid alteration in leptin receptor cDNA from Zucker fatty (fa/fa) rat. Biochem Biophys Res Commun. 1996;222:19–26. doi: 10.1006/bbrc.1996.0691. [DOI] [PubMed] [Google Scholar]
- 95.Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, Caskey CT, Hess JF. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996;13:18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 96.Clark JB, Palmer CJ, Shaw WN. The diabetic Zucker fatty rat. Proc Soc Exp Biol Med. 1983;173:68–75. doi: 10.3181/00379727-173-41611. [DOI] [PubMed] [Google Scholar]
- 97.Pagano C, Calcagno A, Granzotto M, Calabrese F, Thiene G, Federspil G, Vettor R. Heart lipid accumulation in obese non-diabetic rats: Effect of weight loss. Nutr Metab Cardiovasc Dis. 2008;18:189–197. doi: 10.1016/j.numecd.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 98.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 99.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51:2587–2595. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 100.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 101.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 102.Virchow R. Cellular pathology as based upon physiological and pathological histology. John Churchill; London: 1858. A more precise account of fatty metamorphosis; pp. 342–366. [DOI] [PubMed] [Google Scholar]
- 103.Kankaanpää M, Lehto H-R, Pärkkä JP, Komu M, Viljanen A, Ferrannini E, Knuuti J, Nuutila P, Parkkola R, Iozzo P. Myocardial triglyceride content and epicardial fat mass in human obesity: Relationship to left ventricular function and serum free fatty acid levels. J Clin Endocrinol Metab. 2006;91:4689–4695. doi: 10.1210/jc.2006-0584. [DOI] [PubMed] [Google Scholar]
- 104.Rijzewijk LJ, van der Meer RW, Smit JWA, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52:1793–1799. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 105.Reingold JS, McGavock JM, Kaka S, Tillery T, Victor RG, Szczepaniak LS. Determination of triglyceride in the human myocardium by magnetic resonance spectroscopy: reproducibility and sensitivity of the method. Am J Physiol Endocrinol Metab. 2005;289:E935–939. doi: 10.1152/ajpendo.00095.2005. [DOI] [PubMed] [Google Scholar]
- 106.Hammer S, van der Meer RW, Lamb HJ, Schar M, de Roos A, Smit JWA, Romijn JA. Progressive caloric restriction induces dose–dependent changes in myocardial triglyceride content and diastolic function in healthy men. J Clin Endocrinol Metab. 2008;93:497–503. doi: 10.1210/jc.2007-2015. [DOI] [PubMed] [Google Scholar]
- 107.van der Meer RW, Hammer S, Smit JWA, Frolich M, Bax JJ, Diamant M, Rijzewijk LJ, de Roos A, Romijn JA, Lamb HJ. Short-term caloric restriction induces accumulation of myocardial triglycerides and decreases left ventricular diastolic function in healthy subjects. Diabetes. 2007;56:2849–2853. doi: 10.2337/db07-0768. [DOI] [PubMed] [Google Scholar]
- 108.van der Meer RW, Hammer S, Lamb HJ, Frolich M, Diamant M, Rijzewijk LJ, de Roos A, Romijn JA, Smit JWA. Effects of short-term high-fat, high-energy diet on hepatic and myocardial triglyceride content in healthy men. J Clin Endocrinol Metab. 2008;93:2702–2708. doi: 10.1210/jc.2007-2524. [DOI] [PubMed] [Google Scholar]
- 109.Hammer S, van der Meer RW, Lamb HJ, de Boer HH, Bax JJ, de Roos A, Romijn JA, Smit JWA. Short-term flexibility of myocardial triglycerides and diastolic function in patients with type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. 2008;295:E714–718. doi: 10.1152/ajpendo.90413.2008. [DOI] [PubMed] [Google Scholar]
- 110.Hammer S, Snel M, Lamb HJ, Jazet IM, van der Meer RW, Pijl H, Meinders EA, Romijn JA, de Roos A, Smit JWA. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J Am Coll Cardiol. 2008;52:1006–1012. doi: 10.1016/j.jacc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 111.van der Meer RW, Rijzewijk LJ, de Jong HWAM, Lamb HJ, Lubberink M, Romijn JA, Bax JJ, de Roos A, Kamp O, Paulus WJ, Heine RJ, Lammertsma AA, Smit JWA, Diamant M. Pioglitazone improves cardiac function and alters myocardial substrate metabolism without affecting cardiac triglyceride accumulation and high-energy phosphate metabolism in patients with well-controlled type 2 diabetes mellitus. Circulation. 2009;119:2069–2077. doi: 10.1161/CIRCULATIONAHA.108.803916. [DOI] [PubMed] [Google Scholar]
- 112.Lavie CJ, Milani RV, Ventura HO. Obesity and cardiovascular disease: Risk factor, paradox, and impact of weight loss. J Am Coll Cardiol. 2009;53:1925–1932. doi: 10.1016/j.jacc.2008.12.068. [DOI] [PubMed] [Google Scholar]
- 113.Hall JA, French TK, Rasmusson KD, Vesty JC, Roberts CA, Rimmasch HL, Kfoury AG, Renlund DG. The paradox of obesity in patients with heart failure. J Am Acad Nurse Pract. 2005;17:542–546. doi: 10.1111/j.1745-7599.2005.00084.x. [DOI] [PubMed] [Google Scholar]
- 114.Pingitore A, Di Bella G, Lombardi M, Iervasi G, Strata E, Aquaro GD, Positano V, De Marchi D, Rossi G, L’Abbate A, Rovai D. The obesity paradox and myocardial infarct size. J Cardiovasc Med (Hagerstown) 2007;8:713–717. doi: 10.2459/JCM.0b013e328011c984. [DOI] [PubMed] [Google Scholar]
- 115.Uretsky S, Messerli FH, Bangalore S, Champion A, Cooper-DeHoff RM, Zhou Q, Pepine CJ. Obesity paradox in patients with hypertension and coronary artery disease. Am J Med. 2007;120:863–870. doi: 10.1016/j.amjmed.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 116.Wassertheil-Smoller S, Fann C, Allman RM, Black HR, Camel GH, Davis B, Masaki K, Pressel S, Prineas RJ, Stamler J, Vogt TM. S.C.R.G. for the Relation of low body mass to death and stroke in the systolic hypertension in the elderly program. Arch Intern Med. 2000;160:494–500. doi: 10.1001/archinte.160.4.494. [DOI] [PubMed] [Google Scholar]
- 117.Stamler R, Ford CE, Stamler J. Why do lean hypertensives have higher mortality rates than other hypertensives? Findings of the Hypertension Detection and Follow-up Program. Hypertension. 1991;17:553–564. doi: 10.1161/01.hyp.17.4.553. [DOI] [PubMed] [Google Scholar]
- 118.Litwin SE. Which measures of obesity best predict cardiovascular risk? J Am Coll Cardiol. 2008;52:616–619. doi: 10.1016/j.jacc.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 119.Lingvay I, Raskin P, Szczepaniak LS. Cardiomyopathies: The fatty hearts of patients with diabetes. Nat Rev Cardiol. 2009;6:268–269. doi: 10.1038/nrcardio.2009.30. [DOI] [PubMed] [Google Scholar]
- 120.Peterson LR, Herrero P, McGill J, Schechtman KB, Kisrieva-Ware Z, Lesniak D, Gropler RJ. Fatty acids and insulin modulate myocardial substrate metabolism in humans with type 1 diabetes. Diabetes. 2008;57:32–40. doi: 10.2337/db07-1199. [DOI] [PubMed] [Google Scholar]
- 121.Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, Radda GK, Neubauer S, Clarke K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107:3040–3046. doi: 10.1161/01.CIR.0000072789.89096.10. [DOI] [PubMed] [Google Scholar]
- 122.Chiu H-C, Kovacs A, Ford DA, Hsu F-F, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chiu H-C, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 124.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Augustus AS, Buchanan J, Park T-S, Hirata K, Noh H-l, Sun J, Homma S, D’Armiento J, Abel ED, Goldberg IJ. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J Biol Chem. 2006;281:8716–8723. doi: 10.1074/jbc.M509890200. [DOI] [PubMed] [Google Scholar]
- 126.Pillutla P, Hwang YC, Augustus A, Yokoyama M, Yagyu H, Johnston TP, Kaneko M, Ramasamy R, Goldberg IJ. Perfusion of hearts with triglyceride-rich particles reproduces the metabolic abnormalities in lipotoxic cardiomyopathy. Am J Physiol Endocrinol Metab. 2005;288:E1229–1235. doi: 10.1152/ajpendo.00273.2004. [DOI] [PubMed] [Google Scholar]
- 127.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 128.Medina-Gomez G, Gray S, Vidal-Puig A. Adipogenesis and lipotoxicity: role of peroxisome proliferator-activated receptor γ (PPARγ) and PPARγ coactivator-1 (PGC1) Public Health Nutr. 2007;10:1132–1137. doi: 10.1017/S1368980007000614. [DOI] [PubMed] [Google Scholar]
- 129.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of PPARγ leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors PPARβ/δ and PPARα direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 132.Bartels ED, Nielsen JM, Hellgren LI, Ploug T, Nielsen LB. Cardiac expression of microsomal triglyceride transfer protein is increased in obesity and serves to attenuate cardiac triglyceride accumulation. PLoS ONE. 2009;4:e5300. doi: 10.1371/journal.pone.0005300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yokoyama M, Yagyu H, Hu Y, Seo T, Hirata K, Homma S, Goldberg IJ. Apolipoprotein B production reduces lipotoxic cardiomyopathy: Studies in heart-specific lipoprotein lipase transgenic mouse. J Biol Chem. 2004;279:4204–4211. doi: 10.1074/jbc.M311995200. [DOI] [PubMed] [Google Scholar]
- 134.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 135.Koonen DPY, Febbraio M, Bonnet S, Nagendran J, Young ME, Michelakis ED, Dyck JRB. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation. 2007;116:2139–2147. doi: 10.1161/CIRCULATIONAHA.107.712901. [DOI] [PubMed] [Google Scholar]
- 136.Wang M-y, Unger RH. Role of PP2C in cardiac lipid accumulation in obese rodents and its prevention by troglitazone. Am J Physiol Endocrinol Metab. 2005;288:E216–221. doi: 10.1152/ajpendo.00004.2004. [DOI] [PubMed] [Google Scholar]
- 137.Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]