

Abstract
The Ser/Thr kinase family, RSK, has been implicated in numerous types of hormone-dependent and -independent cancers. However, there has been little consideration of RSKs as downstream mediators of steroid hormone non-genomic effects or of their ability to facilitate steroid receptor-mediated gene expression. Steroid hormone signaling can directly stimulate the MEK/ERK/RSK pathway to regulate cellular proliferation and survival in transformed cells. To date, multiple mechanisms of RSK and steroid hormone receptor-mediated proliferation/survival have been elucidated. For example, RSK enhances proliferation of breast and prostate cancer cells via its ability to control the levels of the estrogen receptor co-activator, cyclin D1. While in lung and other tumors RSK may control apoptosis via estrogen-mediated regulation of mitochondrial integrity. Thus the RSKs could be important anti-cancer therapeutic targets in many different transformed tissues. The recent discovery of RSK-specific inhibitors will advance our current understanding of RSK in transformation and drive these studies into animal and clinical models. In this review we explore the mechanisms associated with RSK in tumorigenesis and their relationship to steroid hormone signaling.
Keywords: MAPK pathway, p90 ribosomal S6 kinase, estrogen receptor, androgen receptor, tumorigenesis
Introduction
Steroid hormone-activated receptors regulate gene transcription by directly binding to DNA. They can also drive transcription and other cellular processes via “extra-nuclear” non-genomic effects (reviewed in [1]). These non-genomic effects have been shown to regulate cancer cell proliferation via multiple signaling pathways including the phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinase 1/2 (ERK1/2) (also called p42/p44 mitogen activated protein kinase (MAPK)) pathways [2]. Estrogen exposure rapidly activates the ERK1/2 pathway in tumor cells via stimulation of p21ras and activation of the tyrosine kinase c-Src [2, 3]. Ligand-bound Estrogen Receptor (ER) complexes with c-Src, resulting in phosphorylation of Shc and p190, and promoting interaction with the additional adaptor molecules, MNAR and Cas. Shc phosphorylation engages the upstream components of the ERK1/2 pathway, Grb and SOS, and activates Ras-mediated ERK1/2 signaling [1, 3]. The ribosomal S6 kinase (RSK) family of Ser/Thr kinases are downstream effectors of ERK1/2. Though not well studied, our emerging knowledge of steroid signaling through the RSKs suggests this pathway may be an important contributor to steroid-mediated tumorigenesis (Figure 1).
Figure 1. RSKs are downstream mediators of the ERK1/2 pathway that regulate proliferation in a variety of cancer cell lines and are overexpressed or hyperactivated in many human cancers.
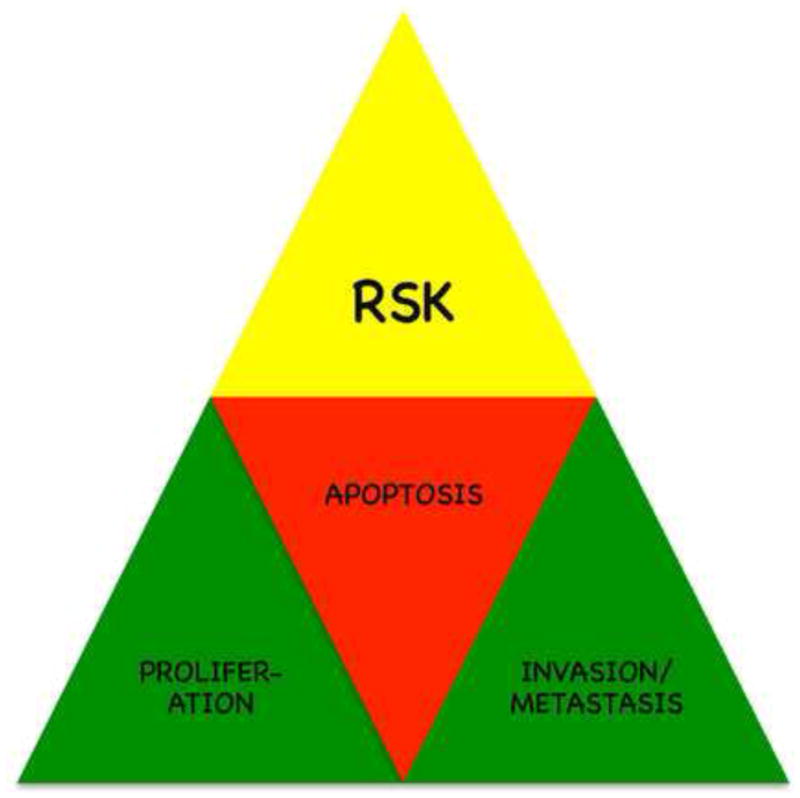
The RSKs can increase proliferation, inhibit apoptosis and promote the invasive phenotype by increasing migration. The participation of RSK in these three major pathways that promote tumorigenesis argues that this kinase family has potential as therapeutic targets.
The ERK1/2 cascade has long been considered a viable source of cancer treatment targets for both hormone-dependent and independent tumors. Consequently, several MAP/ERK kinase (MEK) and Raf inhibitors have been developed and undergone clinical trial evaluation. As yet, targeting these upstream components of the signaling pathway has not been very successful in the clinic [4]. Additionally, inhibiting these “global regulators” produces significant patient side effects. However, the downstream effectors of the ERK1/2 pathway represent an untapped pool of potential therapeutic targets. These targets control a limited set of downstream effectors compared to master regulators like Raf, MEK, and ERK1/2 and are therefore, less likely to mediate severe side effects. Additionally, because they control fewer downstream pathways it is possible they regulate fewer of the feedback loops shown to reduce the efficacy of Raf and MEK inhibitors [5–9]. The RSKs are one such group of downstream mediators of the ERK1/2 pathway. The RSKs are known to regulate proliferation and survival in a variety of cancer cell lines and are found to be overexpressed or hyperactivated in some human cancers [10–15]. Furthermore, RSK has been found to inhibit apoptosis by protecting mitochondrial integrity [16]. Therefore, further investigation of RSK as a potential anti-cancer target would seem warranted.
There are four RSK isoforms, each the product of a different gene. RSK1-4 possess 73–80% amino acid identity and have the same general structure (reviewed in [11]). The RSKs are unusual in that they possess two distinct functional kinase domains (N-terminal; NTKD and C-terminal; CTKD) connected by a linker region. The sequence differences between the RSK isoforms are found in the extreme termini and the linker region. Thus, the specific functions of the individual isoforms may be due to unique sequences in those regions.
In general, RSK is activated by ERK1/2 phosphorylation, which stimulates autophosphorylation resulting in recruitment of 3′-Phosphoinositide Dependent Kinase 1 (PDK1) and subsequent activation of the NTKD. The NTKD is responsible for the phosphorylation of exogenous substrates (Figure 2). The RSK isoforms are activated by the same general mechanism [11]. However, RSK4 is thought to be constitutively active in most tissues [17]. The isoforms have both unique and overlapping functions [10, 11]. RSK1 and RSK2 are the best characterized isoforms. While there are many putative RSK substrates, relatively few have been confirmed using small interfering RNA/short hairpin RNA (siRNA/shRNA) technologies, RSK null-animals or specific inhibitors (Table 1). Confirmed RSK substrate functions can be grouped into three categories—proliferation, survival, and migration (Table 2). The RSKs have diverse functions and substrates in multiple types of human cancer [12, 14, 18–24]. Based on these observations it is reasonable to hypothesize that increased RSK activity mediates transformation. Recently, Cho et al. tested this hypothesis and observed that RSK2 regulates anchorage-independent growth of mouse epidermal JB6 C141 cells and of Ras-transformed NIH 3T3 fibroblasts [21].
Figure 2. RSK activation.

The RSK isoforms are activated by the same general mechanism. That mechanism is summarized in this figure using amino acid numbering corresponding to human RSK2. Color coding identifies the kinase with its associated phosphorylation. A) Inactive ERK1/2 binds to the extreme C-terminus of inactive RSK. In some cell types inactive RSK is also phosphorylated at Tyr-707 and Y529 by FGFR3 or SRC. B) In response to mitogen, ERK1/2 phosphorylates RSK on Ser-369 and Thr-577, activating the CTKD. After phosphorylating its target sites, ERK1/2 disassociates. C) The active CTKD autophosphorylates Ser-386. D) PDK1 is recruited to phospho-Ser-369 and then phosphorylates Ser-227 in the NTKD, completing activation of the NTKD. E) Summary of RSK activation steps leading to NTKD-mediated phosphorylation of exogenous substrates.
Table I.
RSK substrates are grouped under the phosphorylating isoform(s). RSK substrates are considered “validated” if they have been tested using siRNA/shRNA, RSK-specific inhibitors, or RSK null animals/cells. Substrates that have been tested in overexpression systems or in vitro are considered “unconfirmed”. RSK4 is absent from this table because there are no known RSK4 substrates.
| RSK1 substrates | shRNA/siRNA | Specific Inhibitors | Knockout Animals/Cells | In vitro/overexpression | Refs. |
|---|---|---|---|---|---|
| AS160 | X | X | [1] | ||
| Bad | X | X | [2, 3] | ||
| CCTB | X | X | X | [4] | |
| C/EBP | X | X | [5, 6] | ||
| DAPK | X | X | [7] | ||
| EF2K | X | [8, 9] | |||
| eIF4B | X | X | X | [10, 11] | |
| ERα | X (unpublished data) | X | [12] | ||
| ER8 | X | [13] | |||
| Filamin A | X | X | [14] | ||
| IkBa | X | X | [15] | ||
| IKBb | X | [16] | |||
| LKB1 | X | [17] | |||
| MAD1 | X | X | [18] | ||
| Myt1 | X | [19] | |||
| Mi | X | [20] | |||
| NHE1 | X | [9] | |||
| nNos | X | X | [21] | ||
| P27kip | X | X | [22, 23] | ||
| Raptor | X | X | X | [24] | |
| RanBP3 | X | X | [25] | ||
| rpS6 | X | X | X | [26] | |
| Tuberin | X | [27] | |||
| YB-1 | X | X | X | X | [28, 29] |
| RSK2 substrates | |||||
| ATF-4 | X | X | [30] | ||
| Bad | X | X | [31–33] | ||
| CCTB | X | X | X | [4] | |
| c-fos | X | X | [34, 35] | ||
| Emi2 | X | [36] | |||
| ERμ | X (unpublished data) | X | [37] | ||
| Filamin A | X | X | [14] | ||
| MEF2c | X | [38] | |||
| NFAT3 | X | [39] | |||
| NHE1 | X | X | [9, 40] | ||
| Nur-77 | X | X | [41, 42] | ||
| RanBp3 | X | X | [25] | ||
| rpS6 | X | X | X | [26] | |
| STAT-3 | X | X | [43] | ||
| TIF1A | X | [44] | |||
| YB-1 | X | X | X | X | [28] |
| RSK3 substrates | |||||
| Bad | X | [31] | |||
| NHE1 | X | [9] | |||
| rpS6 | X | [45] | |||
| H2B | X | [45] |
References
Geraghty, K.M., Chen, S., Harthill, J.E., Ibrahim, A.F., Toth, R., Morrice, N.A., Vandermoere, F., Moorhead, G.B., Hardie, D.G., and MacKintosh, C. (2007). Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem J 407, 231–241.
Shimamura, A., Ballif, B.A., Richards, S.A., and Blenis, J. (2000). Rsk1 mediates a MEK-MAP kinase cell survival signal. Curr Biol 10, 127–135.
Chaturvedi, D., Cohen, M.S., Taunton, J., and Patel, T.B. (2009). The PKARI{alpha} Subunit of Protein Kinase A Modulates the Activation of p90RSK1 and Its Function. J Biol Chem 284, 23670–23681.
Abe, Y., Yoon, S.O., Kubota, K., Mendoza, M.C., Gygi, S.P., and Blenis, J. (2009). p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J Biol Chem 284, 14939–14948.
Lee, S.J., and Kim, S.G. (2006). Role of p90 ribosomal S6-kinase-1 in oltipraz-induced specific phosphorylation of CCAAT/enhancer binding protein-beta for GSTA2 gene transactivation. Mol Pharmacol 69, 385–396.
Buck, M., and Chojkier, M. (2007). A ribosomal S-6 kinase-mediated signal to C/EBP-beta is critical for the development of liver fibrosis. PLoS One 2, e1372.
Anjum, R., Roux, P.P., Ballif, B.A., Gygi, S.P., and Blenis, J. (2005). The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr Biol 15, 1762–1767.
Wang, X., Li, W., Williams, M., Terada, N., Alessi, D.R., and Proud, C.G. (2001). Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 20, 4370–4379.
Roberts, N.A., Haworth, R.S., and Avkiran, M. (2005). Effects of bisindolylmaleimide PKC inhibitors on p90RSK activity in vitro and in adult ventricular myocytes. Br J Pharmacol 145, 477–489.
Kroczynska, B., Kaur, S., Katsoulidis, E., Majchrzak-Kita, B., Sassano, A., Kozma, S.C., Fish, E.N., and Platanias, L.C. (2009). Interferon-dependent engagement of eukaryotic initiation factor 4B via S6 kinase (S6K)- and ribosomal protein S6K-mediated signals. Mol Cell Biol 29, 2865–2875.
Shahbazian, D., Roux, P.P., Mieulet, V., Cohen, M.S., Raught, B., Taunton, J., Hershey, J.W., Blenis, J., Pende, M., and Sonenberg, N. (2006). The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J 25, 2781–2791.
Joel, P.B., Smith, J., Sturgill, T.W., Fisher, T.L., Blenis, J., and Lannigan, D.A. (1998). pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol 18, 1978–1984.
Wu, J., and Janknecht, R. (2002). Regulation of the ETS transcription factor ER81 by the 90-kDa ribosomal S6 kinase 1 and protein kinase A. J Biol Chem 277, 42669–42679.
Woo, M.S., Ohta, Y., Rabinovitz, I., Stossel, T.P., and Blenis, J. (2004). Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol Cell Biol 24, 3025–3035.
Ghoda, L., Lin, X., and Greene, W.C. (1997). The 90-kDa ribosomal S6 kinase (pp90rsk) phosphorylates the N-terminal regulatory domain of IkappaBalpha and stimulates its degradation in vitro. J Biol Chem 272, 21281–21288.
Xu, S., Bayat, H., Hou, X., and Jiang, B. (2006). Ribosomal S6 kinase-1 modulates interleukin-1beta-induced persistent activation of NF-kappaB through phosphorylation of IkappaBbeta. Am J Physiol Cell Physiol 291, C1336–1345.
Sapkota, G.P., Kieloch, A., Lizcano, J.M., Lain, S., Arthur, J.S., Williams, M.R., Morrice, N., Deak, M., and Alessi, D.R. (2001). Phosphorylation of the protein kinase mutated in Peutz-Jeghers cancer syndrome, LKB1/STK11, at Ser431 by p90(RSK) and cAMP-dependent protein kinase, but not its farnesylation at Cys(433), is essential for LKB1 to suppress cell vrowth. J Biol Chem 276, 19469–19482.
Zhu, J., Blenis, J., and Yuan, J. (2008). Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A 105, 6584–6589.
Palmer, A., Gavin, A.C., and Nebreda, A.R. (1998). A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J 17, 5037–5047.
Wu, M., Hemesath, T.J., Takemoto, C.M., Horstmann, M.A., Wells, A.G., Price, E.R., Fisher, D.Z., and Fisher, D.E. (2000). c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev 14, 301–312.
Song, T., Sugimoto, K., Ihara, H., Mizutani, A., Hatano, N., Kume, K., Kambe, T., Yamaguchi, F., Tokuda, M., and Watanabe, Y. (2007). p90 RSK-1 associates with and inhibits neuronal nitric oxide synthase. Biochem J 401, 391–398.
Fujita, N., Sato, S., and Tsuruo, T. (2003). Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem 278, 49254–49260.
Larrea, M.D., Hong, F., Wander, S.A., da Silva, T.G., Helfman, D., Lannigan, D., Smith, J.A., and Slingerland, J.M. (2009). RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A 106, 9268–9273.
Carriere, A., Cargnello, M., Julien, L.A., Gao, H., Bonneil, E., Thibault, P., and Roux, P.P. (2008). Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol 18, 1269–1277.
Yoon, S.O., Shin, S., Liu, Y., Ballif, B.A., Woo, M.S., Gygi, S.P., and Blenis, J. (2008). Ran-binding protein 3 phosphorylation links the Ras and PI3-kinase pathways to nucleocytoplasmic transport. Mol Cell 29, 362–375.
Roux, P.P., Shahbazian, D., Vu, H., Holz, M.K., Cohen, M.S., Taunton, J., Sonenberg, N., and Blenis, J. (2007). RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem 282, 14056–14064.
Roux, P.P., Ballif, B.A., Anjum, R., Gygi, S.P., and Blenis, J. (2004). Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 101, 13489–13494.
Stratford, A.L., Fry, C.J., Desilets, C., Davies, A.H., Cho, Y.Y., Li, Y., Dong, Z., Berquin, I.M., Roux, P.P., and Dunn, S.E. (2008). Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res 10, R99.
Astanehe, A., Finkbeiner, M.R., Hojabrpour, P., To, K., Fotovati, A., Shadeo, A., Stratford, A.L., Lam, W.L., Berquin, I.M., Duronio, V., et al. (2009). The transcriptional induction of PIK3CA in tumor cells is dependent on the oncoprotein Y-box binding protein-1. Oncogene 28, 2406–2418.
Yang, X., Matsuda, K., Bialek, P., Jacquot, S., Masuoka, H.C., Schinke, T., Li, L., Brancorsini, S., Sassone-Corsi, P., Townes, T.M., et al. (2004). ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 117, 387–398.
Tan, Y., Demeter, M.R., Ruan, H., and Comb, M.J. (2000). BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem 275, 25865–25869.
She, Q.B., Ma, W.Y., Zhong, S., and Dong, Z. (2002). Activation of JNK1, RSK2, and MSK1 is involved in serine 112 phosphorylation of Bad by ultraviolet B radiation. J Biol Chem 277, 24039–24048.
Clark, C.J., McDade, D.M., O’Shaughnessy, C.T., and Morris, B.J. (2007). Contrasting roles of neuronal Msk1 and Rsk2 in Bad phosphorylation and feedback regulation of Erk signalling. J Neurochem 102, 1024–1034.
Chen, R.H., Abate, C., and Blenis, J. (1993). Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci U S A 90, 10952–10956.
David, J.P., Mehic, D., Bakiri, L., Schilling, A.F., Mandic, V., Priemel, M., Idarraga, M.H., Reschke, M.O., Hoffmann, O., Amling, M., et al. (2005). Essential role of RSK2 in c-Fos-dependent osteosarcoma development. J Clin Invest 115, 664–672.
Nishiyama, T., Ohsumi, K., and Kishimoto, T. (2007). Phosphorylation of Erp1 by p90rsk is required for cytostatic factor arrest in Xenopus laevis eggs. Nature 446, 1096–1099.
Clark, D.E., Poteet-Smith, C.E., Smith, J.A., and Lannigan, D.A. (2001). Rsk2 allosterically activates estrogen receptor alpha by docking to the hormone-binding domain. EMBO J 20, 3484–3494.
Wang, Y., Liu, L., and Xia, Z. (2007). Brain-derived neurotrophic factor stimulates the transcriptional and neuroprotective activity of myocyte-enhancer factor 2C through an ERK1/2-RSK2 signaling cascade. J Neurochem 102, 957–966.
Cho, Y.Y., Yao, K., Bode, A.M., Bergen, H.R., 3rd, Madden, B.J., Oh, S.M., Ermakova, S., Kang, B.S., Choi, H.S., Shim, J.H., et al. (2007). RSK2 mediates muscle cell differentiation through regulation of NFAT3. J Biol Chem 282, 8380–8392.
Cuello, F., Snabaitis, A.K., Cohen, M.S., Taunton, J., and Avkiran, M. (2007). Evidence for direct regulation of myocardial Na+/H+ exchanger isoform 1 phosphorylation and activity by 90-kDa ribosomal S6 kinase (RSK): effects of the novel and specific RSK inhibitor fmk on responses to alpha1-adrenergic stimulation. Mol Pharmacol 71, 799–806.
Wang, A., Rud, J., Olson, C.M., Jr., Anguita, J., and Osborne, B.A. (2009). Phosphorylation of Nur77 by the MEK-ERK-RSK cascade induces mitochondrial translocation and apoptosis in T cells. J Immunol 183, 3268–3277.
Wingate, A.D., Campbell, D.G., Peggie, M., and Arthur, J.S. (2006). Nur77 is phosphorylated in cells by RSK in response to mitogenic stimulation. Biochem J 393, 715–724.
Zhang, Y., Cho, Y.Y., Petersen, B.L., Bode, A.M., Zhu, F., and Dong, Z. (2003). Ataxia telangiectasia mutated proteins, MAPKs, and RSK2 are involved in the phosphorylation of STAT3. J Biol Chem 278, 12650–12659.
Zhao, J., Yuan, X., Frodin, M., and Grummt, I. (2003). ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol Cell 11, 405–413.
Zhao, Y., Bjorbaek, C., Weremowicz, S., Morton, C.C., and Moller, D.E. (1995). RSK3 encodes a novel pp90rsk isoform with a unique N-terminal sequence: growth factor-stimulated kinase function and nuclear translocation. Mol Cell Biol 15, 4353–4363.
Table II.
RSK1, RSK2, and RSK3, substrates are grouped according to their cellular functions; proliferation, migration, and survival. Substrates of a given RSK isoform are indicated with an “X”. Many RSK substrates are phosphorylated by multiple isoforms and are therefore labeled with more than one “X”. Some RSK substrates have been identified using reagents that were not isoform-specific. In these cases the “X” is placed in the unknown column because the phosphorylating RSK isoform is unknown. RSK4 is absent from this table because there are no known RSK4 substrates.
| Proliferation | Unknown | RSK1 | RSK2 | RSK3 |
|---|---|---|---|---|
| RanBP3 | X | X | ||
| YB1 | X | X | ||
| Transcription | ||||
| ER8 | X | |||
| H2B | X | |||
| IkBa | X | |||
| IKBb | X | |||
| NFATc4 | X | |||
| NFAT3 | X | |||
| TIF1A | X | |||
| MAD1 | X | |||
| C/EBP | X | |||
| ERa | X | X | ||
| Stat3 | X | |||
| C-fos | X | |||
| MEF2c | X | |||
| Mi | X | |||
| Translation | ||||
| EF2K | X | |||
| eIF4B | X | |||
| rps6 | X | X | X | |
| Cell Cycle Regulation | ||||
| Bub1 | X | |||
| Myt1 | X | |||
| Emi2 | X | |||
| Migration | ||||
| L1 | X | |||
| Filamin A | X | |||
| P27 kip | X | |||
| Survival | ||||
| DAPK | X | |||
| nNos | X | |||
| ATF-4 | X | |||
| Nutrient Signaling | ||||
| Raptor | X | |||
| LKB1 | X | |||
| CCTB | X | X | ||
| Tuberin | X | |||
| Metabolism | ||||
| GSK3 | X | |||
| AS160 | X | |||
| Ion transport | ||||
| NHE1 | X | X | ||
| Mitochondrial Integrity | ||||
| Nur-77 | X | |||
| Bad | X | X | X | |
To date, there has been little consideration of the RSKs as downstream mediators of steroid hormone non-genomic effects or of their ability to facilitate steroid receptor-mediated gene expression. Therefore, in this review we will explore the mechanisms associated with RSK in tumorigenesis and their relationship to steroid hormones and their receptors. We will present and discuss the evidence that RSK contributes to hormone-linked tumorigenesis in specific tissues. We will also discuss RSK-mediated tumorigenesis in tissues wherein our understanding of steroid hormone contributions is currently emerging, but is not yet clear. Perhaps by analyzing mechanisms of RSK and steroid hormone signaling in these tumors, we will open up new avenues of investigation and importantly, identify new therapeutic targets.
RSK in Breast Cancer
RSK has been shown by numerous groups to be a key regulator of breast cancer proliferation. Our lab was the first to report the importance of RSK in breast cancer proliferation together with the discovery of the first RSK-specific inhibitor, SL0101 [14]. We found that RSK2 is over-expressed in 50% of human breast cancer tissues, compared to normal tissue [14]. Since then many other groups have reported similar findings in breast and other tumor models. Law et al. recently showed that inhibition of the insulin-like growth factor 1 receptor (IGF-1R)/insulin receptor (IR) reduced proliferation and RSK activity in tamoxifen-resistent MCF-7 cells [13]. In human breast tumors high levels of the phosphorylated RSK substrate ribosomal protein S6 (rpS6), were directly correlated with IGF-1R/IR levels, and were associated with poor patient survival [13]. Thus, high levels of activated RSK promote tumorigenesis. Furthermore, ~56% of human breast tumors showed phosphorylated rpS6 indicating that RSK may be highly active. The ability of RSK to phosphorylate rpS6 has been controversial but recent studies have shown that rpS6 phosphorylation at Ser-235/236 is MAPK-dependent and has been attributed to RSK [25]. RpS6 is one of many RSK substrates associated with proliferation and tumorigenesis (Table 2).
The ability of RSK to regulate survival, anchorage-independent growth, and transformation in breast cancer was recently confirmed by Xian et al [15]. Their findings indicate that Fibroblast Growth Factor Receptor-1 (FGFR1)-mediated transformation of MCF-10A cells is dependent on RSK. FGFR1 is upregulated in many invasive lobular carcinomas (ILC), which, while ER+, may not respond very well to the ER antagonist, tamoxifen [26]. Inhibitors of FGFR1 can suppress growth of the ILC cell line, MDA-MB-134, suggesting that FGFR1 signaling is essential for proliferation of these tumors. Knockdown of RSK1 and inhibition of RSK, with the specific small molecule inhibitor chloromethylketone (CMK), reduced proliferation, suppressed colony formation in soft agar, and decreased survival of FGFR1-expressing cells. Together these data strongly support the hypothesis that the RSKs are important in breast cancer etiology.
RSK4 is a putative tumor suppressor in breast cancer [19, 27]. Thakur et al., found that overexpressing RSK4 in MDA-MB-231 cells suppressed colony formation in soft agar, tumor formation, and metastasis. Curiously, RSK4 levels were found to be elevated in MMTV-c-Myc transgenic mice [27, 28]. Expression of myc, a cell cycle regulator [29, 30], is upregulated very quickly following estrogen treatment and is essential for estrogen-mediated proliferation in breast cancer cells [31, 32]. Mammary tumors that form in c-Myc transgenic mice are neither invasive nor metastatic and it is hypothesized that c-myc overexpression upregulates RSK4, which then suppresses aggressive expansion [27]. Consistent with this hypothesis, c-myc was shown to stimulate RSK4 promoter activity in a luciferase reporter assay [27]. Our knowledge of RSK4 remains limited. RSK4 may have tumor suppressor functions in some cancer types, but the paucity of data on this kinase suggests that further studies are necessary before specific conclusions can be drawn.
The growing body of literature implicating RSK in breast cancer supports the hypothesis that RSK is an important therapeutic target. We have found that treatment with the RSK-specific inhibitor, SL0101 (20 μṂ; 48hṛ, reduced proliferation in the immortalized human breast cancer cell line, MCF-7, but did not effect proliferation of the non-tumorigenic breast cell line, MCF-10A (Figure 3A, [14]). Consistent with these findings, silencing RSK2 also reduced proliferation in MCF-7 cells. The mechanism by which RSK2 regulates proliferation in breast cancer cells is not well understood. However, significant evidence is emerging that indicates RSK regulates several key breast cancer-associated proteins. For example, we have found that RSK2 stimulates the transcriptional activity of estrogen receptor α (ERα) [33–36] which is known to be important in the etiology of many breast cancers. Estrogens can stimulate RSK activity, and RSK2 enhances ERα-mediated transcription by phosphorylation and by physical association [33]. The interaction of ERα and RSK can be disrupted by tamoxifen. This process may be dependent on the ERK1/2 pathway. Additionally, we have found that RSK2 regulates expression of the oncogene, cyclin D1, which is a co-activator of ERα and overexpressed in approximately 50% of human breast tumors [37, 38]. The importance of cyclin D1 as an oncogene is highlighted by the finding that overexpression of the protein is sufficient to induce formation of mammary tumors in transgenic animals [39]. Although the ERK1/2 pathway is known to regulate cyclin D1 levels, we identified that cyclin D1 is a key RSK2 target in breast cancer cells [38]. Consistent with findings in human tissue, we found that MCF-7 cells overexpress cyclin D1 as compared to MCF-10A cells by approximately 5-fold based on normalization to the housekeeping protein, Ran (Figure 3B). SL0101 (50 μṂ; 4hṛ reduced cyclin D1 levels in MCF-7 cells by 70% at the protein level and 40% at the mRNA level (Figure 3C, [38]). Importantly, SL0101 did not affect cyclin D1 expression in MCF-10A cells (Figure 3C) suggesting that RSK regulation of cyclin D1 is confined to transformed cells. SL0101 inhibits the kinase activity of RSK1 and RSK2 in in vitro kinase assays, but RSK2 is primarily responsible for the regulation of cyclin D1 levels [38]. We also found forced nuclear localization of RSK2 drives cyclin D1 expression in the absence of activation of any other signal transduction pathway [38]. These results suggest that nuclear RSK2 is able to act as an oncogene in breast cancer.
Figure 3. RSK regulates proliferation and cyclin D1 levels in breast cancer cell lines.
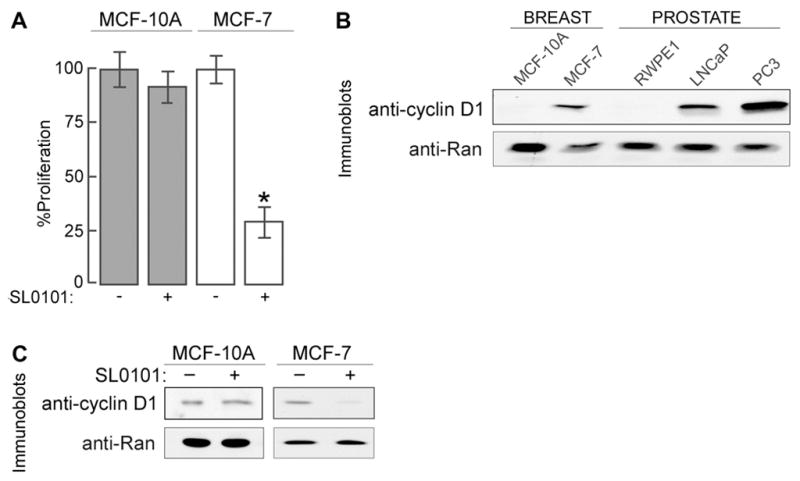
A) Cells were treated with vehicle (−) or 20 μM SL0101, and cell viability was measured after 48 hr of treatment. Values are % of the growth observed in vehicle-treated cells. Columns, mean (n=2, in triplicate); bars, SD. *, p=0.005, Student’s t test B) Lysates of the normal human cell lines, MCF-10A and RWPE1, and of the human cancer cell lines, MCF-7, LNCaP and PC-3 were prepared from cells grown in the appropriate media as recommended by ATCC. C) Cells were treated with vehicle (−) or 50 μM SL0101 for 4 hr before lysis. To permit detection of cyclin D1 the total protein loaded differed between cell lines. Equal loading of the lysate within a cell line is shown by the anti-Ran immunoblot.
We have also identified a mechanism by which RSK regulates mRNA localization and translation via stress granules in breast cancer cells [38]. Normal mammary and breast cancer cells form cytoplasmic RNA complexes called stress granules under either oxidative stress or serum-deprivation stress. In general, stress granules form under conditions in which translation initiation has been reduced or inhibited [40]. These granules recruit selected mRNAs and associated proteins from polyribosomes, for storage, or for triage through processing bodies [41]. Stress granules are thought to aid cell survival by acting as sites of translational repression and to facilitate post-stress recovery by acting as reservoirs of poly(A)+ RNA. In breast cells subjected to stress, endogenous RSK2 localizes to stress granules and controls recruitment of other key stress granule proteins in the complex [38]. In response to stress, RSK interacts directly with the essential stress granule protein, TIA-1, driving stress granule formation. This regulation is physiologically important, because loss of RSK2, via specific knockdown or inhibition, prevents stress granule formation and decreases cell survival in response to stress. In nutritionally-stressed breast cancer cells, addition of mitogen triggers the dissolution of stress granules. Once released from sequestration in the granules, RSK2 accumulates in the nucleus, where it induces cyclin D1 expression in transformed cell lines, driving entry into the cell cycle. RSK2 has not previously been implicated as a regulatory component in the stress response and RSK2-mediated regulation of translation and cellular stress are understudied. However, there is now a significant amount of evidence suggesting that the involvement of RSK2 in translation may be crucial for understanding its role in tumor cell survival. In addition, several recent studies suggest that stress granule formation protects tumor cells from chemotherapy and radiation-induced stress [42, 43]. Interestingly, cyclin D1 mRNA has been found in stress granules [44] suggesting that this mechanism may effectively protect mRNAs necessary for proliferation in breast cancer. RSK2 is an essential regulator of stress granules in breast cancer cells, and therefore RSK2 inhibition may increase tumor cell death in response to standard treatments.
Another mechanism by which RSK may regulate breast cancer cell proliferation is through phosphorylation of the transcription factor YB-1 at Ser-102 [45]. YB-1 regulates expression of numerous proteins associated with tumorigenesis via direct interaction with the promoter regions of target genes [46, 47]. Not only is YB-1 overexpressed in breast and other cancers, but overexpression of YB-1 is sufficient to drive mammary tumor formation in mice [45, 48–51]. Conversely, knockdown of YB-1 suppresses tumor cell proliferation [49]. Phosphorylation of YB-1 at Ser-102 is essential for nuclear translocation of the protein as well as the interaction of YB-1 with target genes [52]. Therefore, RSK-dependent phosphorylation at Ser-102 may be essential for YB-1 function. Interestingly, YB-1 nuclear localization and ERα regulation appear to be connected [53]. However, the mechanism and physiological outcome of this relationship are not yet clear. The observation that RSK participates in ERα and YB-1 signaling suggests that the potential connection between YB-1 and ERα may be mediated by RSK.
RSK2 and Prostate Cancer
We found that RSK2 is over-expressed in ~50% of prostate cancer tissues compared with normal tissue and benign prostate hyperplasia [12]. RSK inhibition via SL0101 (20 μṂ; 48hṛ reduced proliferation in LNCaP and PC3 prostate cancer cell lines but not in the untransformed prostate cell line, RWPE1 (Figure 4A). SL0101 (50 μṂ; 4hṛ also reduced cyclin D1 levels in prostate cancer cell lines compared to untransformed cells (Figure 4B). These results are consistent with those observed in breast cell lines. An isoform of cyclin D1 has been found to be overexpresed in prostate cancer and has been shown to stimulate proliferation in prostate cancer cells [54]. We have also found cyclin D1 to be overexpressed in LNCaP and PC3 prostate cancer cells as compared to RWPE1 cells (Figure 3B). Thus cyclin D1 appears to be implicated in the transformation of prostate and breast cells. RSK2 indirectly regulates androgen receptor (AR)-mediated transcription in LNCaP cells [12]. The AR is known to be important in the etiology of prostate cancer [55, 56]. Therefore, RSK regulates both AR and ERα in cancer suggesting a relationship between RSK and general steroid-receptor signaling.
Figure 4. RSK regulates proliferation and cyclin D1 levels in prostate cancer cell lines.

A) RWPE1, LNCaP, and PC3 cells were treated as in Figure 3A. Columns, mean (n = 2 in quadruplicate); bars, SD. *, p = 0.005, Student’s t test. B) RWPE1, LNCaP, and PC3 cells were treated as in Figure 3C.
RSK3 and Ovarian Cancer
Ovarian cancers are among the most lethal malignancies in women [57]. Interestingly, 40%–60% of ovarian tumors express ERα but less than 20% of tumors respond to anti-estrogen treatments in the clinic [58–60]. These observations suggest that growth factor-activated pathways like the ERK1/2-RSK pathway, rather than genomic hormone signaling, may be important in some ovarian tumors [61, 62]. Non-genomic hormone-mediated signaling is thought to play a role in growth factor pathways via the orphan G-protein coupled receptor 30 (GPR30) [63]. In ovarian cancer cells estrogens and G1, the GPR30 agonist, were shown to activate the EGFR pathway, resulting in upregulation of c-Fos expression. RSK is a key regulator of c-Fos levels and function [64, 65], which suggests a link between estrogen signaling and RSK-mediated c-Fos activity in ovarian cancer.
In contrast to standard view of RSKs as tumor promoters, Bignone et al. found that RSK3 suppresses growth in multiple ovarian cancer cell lines [18]. Overexpression of RSK3 reduced proliferation and colony formation in soft agar compared to a control. RSK3 levels were found to be high in normal ovarian tissue and decreased in cancer cell lines and in ≥ 50% of the sporadic human tumors of various stages and grades. These findings suggest that RSK3 may function as a tumor suppressor in some ovarian cancer. However, given our limited knowledge of RSK3, more studies analyzing RSK3 function are necessary.
RSK2 and Multiple Myeloma
ERα and ERβ are both expressed in multiple myeloma cell lines [66]. The exact role of these receptors in multiple myeloma is not known, but interestingly, these tumor cells undergo apoptosis following exposure to anti-estrogens like tamoxifen [66–68], suggesting a role for estrogen signaling in multiple myeloma. Furthermore, a recent analysis of gene expression in plasma cells from 74 multiple myeloma patients showed a significant increase in CCND1 levels [69]. CCND1, which encodes the oncogene cyclin D1, is regulated, in part, by estrogens [70] suggesting that estrogen signaling may promote myeloma via cyclin D1 expression. As discussed above, RSK2 is a key regulator of cyclin D1 expression in some cancer cell lines. In addition to elevated CCND1 levels, increased expression of the fibroblast growth factor receptor 3 (FGFR3) was found in plasma cells of multiple myeloma patients [69]. Mutations in FGFR3, a receptor-tyrosine kinase, occur in approximately 15% of multiple myeloma cases [71]. Constitutively active FGFR3 was shown to phosphorylate RSK2 at Tyr-529 in Ba/F3 cells, a murine pro-B cell line, enhancing RSK interaction with inactive ERK1/2 [72]. This association is thought to increase the subsequent activation of RSK2 by ERK1/2 [73]. Inhibition of RSK with the inhibitor fluoromethyl ketone (fmk) induced apoptosis in FGFR3- expressing primary and immortalized human myeloma cells. In subsequent studies this group has shown that FGFR3 interacts with RSK2 at Trp-332, enhancing both Tyr-529 and Tyr-707 phosphorylation and promoting RSK activation [74, 75] (Figure 2). In FGFR3 transformed bone marrow transplantation studies, survival of animals with RSK2-depleted marrow was prolonged compared with control animals. Together, these findings suggest that RSK2 plays a significant role in hematopoietic transformation. The contribution of estrogen signaling in multiple myeloma is not clear. However, the role of estrogens in bone cells is becoming increasingly well understood [76]. Estrogen signaling has a protective effect on osteoblasts, mediated in part via regulation of essential cytokines, thus supporting healthy bone formation [77]. Additionally, RSK2-mediated phosphorylation of activating transcription factor 4 (ATF-4) promotes osteoblast proliferation [78]. Perhaps, some of the RSK-dependent transformation effects in hematopoetic cells are mediated by hormone signaling.
Activation of RSK via Tyr-529 may be mediated by estrogen signaling in some cells. In 293T and COS7 cells, which do not express FGFR3, RSK2 is phosphorylated at Tyr-529 by Src family kinases, Src and Fyn, as determined by in vitro kinase assay [79, 80]. Src family kinases have been implicated in the development of multiple human cancers including those associated with RSK, which include breast, prostate, lung, melanoma, ovarian, and gastric cancers [81]. In response to estrogen, Src is activated via its interaction with ERα and the scaffold protein MNAR [2]. Thus, activation of Src by estrogen may enhance RSK2 activity in some tumors. Activation of RSK by Src has yet to be investigated in any tumor type.
RSK in Non Small Cell Lung Cancer (NSCLC)
Expression of ERα and ERβ have been shown, in multiple studies, to be elevated in human lung tumors [82–85]. These observations suggest that estrogens might play a role in lung cancer development and may explain why female non-smokers have a higher risk of developing lung adenocarcinomas than male non-smokers [82]. Estrogen as a proliferative driving force in lung cancer is supported by observations that estrogen promotes tumor progression in mouse models of lung adenocarcinoma [86].
RSK has been implicated in lung cancer cell survival, via an anti-apoptotic mechanism. Several groups have shown that RSK activation or overexpression inhibited cell death via inactivation of the Bcl-2 homology 3-only proapoptotic protein, Bad [16, 87, 88]. RSK directly phosphorylated Bad at Ser-75 (Ser-75; human, Ser-112; mouse), which resulted in the sequestration of Bad by 14-3-3 and inhibition of its association with the apoptosis-inducing Bcl-xl [87, 89]. Estrogen signaling may play a role in RSK-mediated phospho-Bad induced survival. Fernando et al. showed that estradiol-activated RSK1, immunoprecipitated from MCF-7 cells, phosphorylated Bad by in vitro kinase assay [90]. Supporting this finding, estrogen treatment has also been shown to induce Bad phosphorylation in skeletal muscle cells [91]. Additionally, in NSCLC cells Amphiregulin and Insulin like-growth factor type 1 (IGF1) stimulated RSK-mediated Bad phosphorylation [22]. These cells often secrete Amphiregulin and IGF1 [92, 93], which cooperate to prevent serum starvation-induced apoptosis [94]. Silencing RSK1, or overexpression of a catalytically inactive RSK2, in the NSCLC cell line H322 inhibited the ability of AR/IGF1 to prevent apoptosis due to serum-starvation. Thus, RSK-mediated Bad phosphorylation is a well-established anti-apoptotic mechanism that may be regulated by estrogen signaling in NSCLC.
There are other signaling events, in addition to Bad phosphorylation, by which RSK regulates apoptosis in response to estrogens. Mitochondrial regulation of apoptosis is ultimately facilitated by activation of the pro-apoptotic proteins Bcl2 homologous antagonist killer protein (Bak) and Bcl2-associated X protein (Bax). Bak expression and therefore, mitochondrial integrity and survival, may be regulated by estrogen signaling [95]. Upon activation, Bak and Bax permeabilize the mitochondrial membrane permitting the release of cytochrome c into the cytosol and apoptosis [96]. Bak and Bax are regulated through interactions with binding partners that sequester them and prevent them from compromising the mitochondria. Dehan et al. showed that RSK phosphorylates and regulates the stability of the pro-apoptotic protein Bcl-2 interacting mediator of death- extra long (BimEL) [23]. BimEL is one of three Bim splice variants that are thought to regulate Bax-mediated apoptosis [23, 96]. RSK-dependent phosphorylation promotes BimEL interaction with the F-box potein bTrCP, facilitating BimEL degradation and inhibition of apoptosis. Knockdown of RSK1/2 was found to stabilize BimEL levels and induce apoptosis in the lung cell lines, HCC87 and H1650 cells. Consistent with these findings, downregulation of BimEL by siRNA suppressed apoptosis in RSK1/2 knockdown cells, confirming that RSK1/2 can regulate apoptosis via BimEL in lung cancer cells [23]. Estrogen activation of RSK in lung cancer cells could therefore inhibit apoptosis by protecting mitochondrial activity.
RSK and Melanoma
Several studies have shown that estrogen receptors are expressed in melanoma tumors and cell lines [97–99] and a small study of 14 patients has shown that ERα and ERβ mRNA and ERβ protein are expressed in neoplastic skin cells [100]. However, the role of steroid-hormone activated receptors in melanocytes and melanoma is controversial. Tamoxifen treatment reduces growth of some melanoma cell lines [101], and overall survival was increased for melanoma patients receiving tamoxifen in initial clinical trials [102, 103]. Subsequent clinical testing did not confirm these findings. Thus, the role of steroid hormone signaling in melanoma is not clear. RSK1 has been implicated in melanoma proliferation suggesting that estrogen signaling in melanoma may be connected to RSK activation. In melanoma cells, Eisenmann et al., showed that RSK1 was hyperactivated leading to the phosphorylation of Bad at Ser-75 and increased cell survival [24]. Thus RSK may regulate apoptosis via Bad phosphorylation in skin cancer.
RSK has also been implicated in other mechanisms of melanoma formation and metastasis. RSK1 phosphorylation and inactivation of the tumor suppressor Tuberin at Ser-1798 led to activation of mTOR and enhanced proliferation [104]. In contrast to these findings, RSK1 has been proposed to have tumor suppressor activity in some melanoma cells via phosphorylation of the Ser/Thr kinase tumor suppressor Liver Kinase B1 (LKB1) at Ser-431. RSK may be able to both suppress and promote tumorigenesis in melanoma; its function is likely dependent on additional signaling inputs that are not yet known.
RSK has also been implicated in regulation of migration in melanoma cells. Filamin A, a RSK substrate, is a membrane localized cytoskeletal protein essential for migration in some melanoma cell lines[105–107]. Additionally, RSK1-mediated phosphorylation of p27 at Thr-198 stimulates migration of melanoma cells [108]. RSK-mediated migration may promote tumor cell invasion in melanoma and other cancer cells via regulation of pro-invasion genes; such as matrix metallo-proteinases and protease receptor complex proteins [109]. These findings suggest that RSK may play an important role in metastases of melanoma and other cancer cells, but it is not clear if RSK-mediated metastases occur in response to estrogen signaling.
RSK2 and Osteosarcoma
ERα, ERβ, and Progesterone Receptor (PR) expression and signaling have been linked to osteosarcoma proliferation [110, 111]. The majority of human osteosarcomas possess elevated levels of the transcription factor c-Fos. ERα can stimulate c-Fos expression via an estrogen dependent mechanism [112, 113]. There is a substantial body of evidence suggesting c-Fos is a protooncogene capable of initiating RSK2-dependent transformation of osteoblasts [114–117]. c-Fos transgenic mice crossed with RSK2 null mice (H2-c-fosLTR/Rsk2−/y mice) produce offspring whose tumors have increased levels of apoptosis and decreased proliferation compared to c-Fos transgenic animals expressing wild-type RSK2. [65]. These findings suggest that RSK2 is essential for survival of c-Fos-induced osteosarcomas. Both expression of total c-Fos protein and phosphorylation of c-fos at Ser-362 were abolished in the H2-c-fosLTR/Rsk2−/y mice. Phosphorylation of Ser-362 by RSK2 is essential for c-Fos transactivation because it stabilizes c-fos protein [64, 65, 118]. Thus, RSK2 may regulate osteosarcoma development via control of c-Fos activation and stability. Furthermore, the expression of ERα and ERβ in osteosarcomas suggests that hormone signaling enhances RSK-mediated tumor formation via induction of c-Fos expression.
Interestingly, mutations in the human Rsk2 gene result in truncated forms of RSK2 protein, causing diverse skeletal and cognitive defects collectively known as Coffin-Lowry Syndrome (CLS) (reviewed in [119, 120]). These findings suggest that RSK2 may be an important regulator of bone development whose hyperactivation could contribute to bone tumors. The transcriptional activity of ATF-4, a critical regulator of bone formation [121, 122], was significantly impaired in osteoblasts isolated from Rsk2-deficient mice [78]. Thus, RSK2 may control osteoblasts via ATF4. Importantly, the skeletal deformities observed in ATF4−/− and Rsk2−/− mice are nearly identical [78]. These data suggest that RSK2 phosphorylation of ATF-4 is crucial for normal skeletal development. Though this mechanism is untested in osteosarcomas, it is possible that overexpression or hyperactivation of RSK may drive tumor formation via ATF-4. ATF-4 is upregulated in primary human breast cancer tissue compared with paired normal samples [123]. ATF-4 levels were increased specifically near necrotic areas of the tumor, as shown by immunostaining, and could be induced in breast cancer cell lines under anoxic conditions. ATF-4 may also be regulated by estrogen [124]. A recent microarray analysis of mouse uterine tissue showed a 2-fold increase in ATF-4 mRNA following 12hr of estradiol treatment. Further study of estrogen signaling in RSK-mediated regulation of ATF-4 in breast cancer would be of interest.
RSK and Big MAPK1/Extracellular Regulated Kinase 5 (BMK1/ERK5) in Angiogenesis
Angiogenesis is essential for tumor progression as tumors must develop neovasculature to procure oxygen and nutrients for survival [125]. Recent studies indicate that angiogenesis might be under the control of steroid hormone receptors in cancer [126–128]. Numerous investigations have shown that the only adult human system to undergo angiogenesis during homeostasis is the female reproductive tract, suggesting a connection between estrogens and angiogenic potential [129, 130]. There are several putative mechanisms for estrogen-mediated angiogenesis. Estrogen treatment increases expression of vascular endothelial growth factor (VEGF), a master regulator of angiogenesis, in mouse mammary tumors [131, 132]. Estrogens can also mediate Nitric oxide production, which is essential for VEGF-dependent angiogenesis [129]. Interestingly, RSK regulation of angiogenesis is thought to occur downstream of ERK5 (also known as BMK1), a key regulator of tumor vascularization [133–135] and a member of the MAPK superfamily. Deletion of ERK5 in Lewis lung carcinoma or in B16F10 melanoma xenografts resulted in smaller and fewer blood vessels than the control xenografts [136]. Inhibition of ERK5 signaling prevented bFGF-mediated RSK activation in endothelial cells. Loss of ERK5 in endothelial cells decreased phosphorylation of the RSK substrate, rpS6 and reduced tumor growth and angiogenesis [136]. Similarly, re-expression of ERK5 in the area of tumor injection in ERK5 knockout mice restored rpS6 phosphorylation and angiogenesis. These findings suggest that RSK-mediated phosphorylation of rpS6 and related tumor angiogenesis are dependent on ERK5 signaling. The possibility that estrogen signals promote angiogenesis via activation of ERK5 and RSK has not yet been explored, but the hypothesis is consistent with current observations.
Conclusion
Steroid hormone-activated receptors are now well established in the etiology of many cancers; including classical hormone-dependent tumors like breast and prostate, and unexpected malignancies like melanoma. It is likely that the role steroid receptors play will vary between tumor types, acting in some cases through non-genomic effects and in other cases through genomic/transcriptional regulation of key proteins. Steroid hormone receptor signaling can activate the ERK1/2-RSK pathway via a non-genomic mechanism (Figure 5). Activation of RSK inhibits mitochondrial-mediated apoptosis and increases proliferation. Thus, non-genomic steroid hormone signaling to RSK may promote transformation in multiple tumor types. Importantly, active RSK can stimulate higher steroid receptor transcriptional activity as has been shown for the androgen and estrogen receptors Therefore, we postulate that in some RSK-mediated tumors hormone signaling may drive proliferation and survival by stimulating both genomic and non-genomic pathways. This dual signaling response may increase expression and/or activity of oncogenic target proteins like c-Fos and cyclin D1, resulting in tumor formation and progression.
Figure 5. Major mechanisms proposed to regulate hormone-dependent transformation in some tumors.
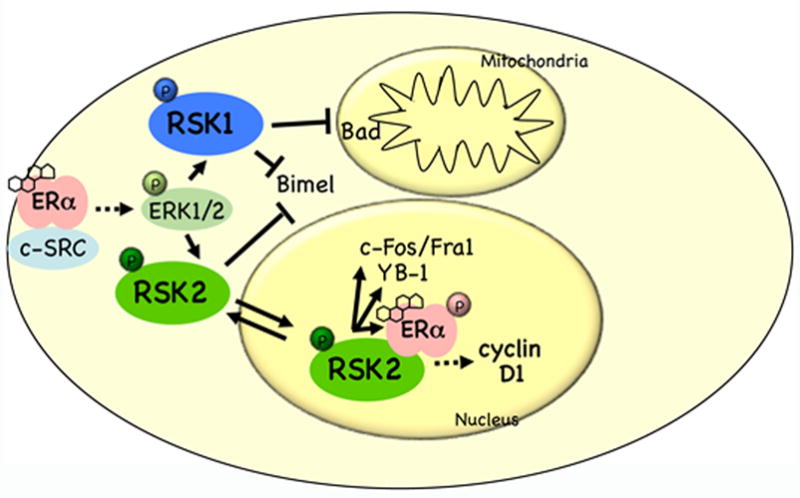
Estrogens stimulate ERα complex formation with cytoplasmic signaling proteins like MNAR, cas, and c-SRC. This complex activates the ERK1/2 signal transduction pathway, and phosphorylation of RSK. RSK1 signaling inhibits apoptosis via phosphorylation and inactivation of the pro-apoptotic protein Bad. RSK2 can translocate to the nucleus where it regulates nuclear targets that drive proliferation. In the nucleus RSK directly phosphorylates ERα stimulating its transcriptional activity.
The recent discovery of RSK-specific inhibitors has the potential to dramatically advance our knowledge of RSK-mediated mechanisms in cancer and to test the effects of RSK inhibition in pre-clinical studies. There are currently no isoform-specific RSK inhibitors, but development of these tools is the next logical step in the process of understanding RSK function and the clinical implications of RSK. Given the preponderance of data linking steroid hormone signaling to RSK-associated cancers, these inhibitors would be of particular value in the study and treatment of hormone-dependent tumors.
Acknowledgments
This work was supported by GM084386 (D.A.L), T32CA009109-30 (T.S.K.E.M), Patients and Friends of the UVa Cancer Center (D.A.L), Swing Fore the Cure (D.A.L), Virginia Kincaid Cancer Center Research Fund (D.A.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shupnik MA. Crosstalk between steroid receptors and the c-Src-receptor tyro ine kinase pathways: implications for cell proliferation. Oncogene. 2004;23:7979–7989. doi: 10.1038/sj.onc.1208076. [DOI] [PubMed] [Google Scholar]
- 2.Cheskis BJ, Greger J, Cooch N, McNally C, McLarney S, Lam HS, Rutledge S, Mekonnen B, Hauze D, Nagpal S, et al. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids. 2008;73:901–905. doi: 10.1016/j.steroids.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 3.Fox EM, Andrade J, Shupnik MA. Novel actions of estrogen to promote proliferation: integration of cytoplasmic and nuclear pathways. Steroids. 2009;74:622–627. doi: 10.1016/j.steroids.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 5.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 6.Grant S. Cotargeting survival signaling pathways in cancer. J Clin Invest. 2008;118:3003–3006. doi: 10.1172/JCI36898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–346. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 9.Adjei NRaA. Inhibitors of Raf kinase and MEK signaling. Update on cancer therapeutics. 2007;2:111–118. [Google Scholar]
- 10.Carriere A, Ray H, Blenis J, Roux PP. The RSK factors of activating the Ras/MAPK signaling cascade. Front Biosci. 2008;13:4258–4275. doi: 10.2741/3003. [DOI] [PubMed] [Google Scholar]
- 11.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark DE, Errington TM, Smith JA, Frierson HF, Jr, Weber MJ, Lannigan DA. The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res. 2005;65:3108–3116. doi: 10.1158/0008-5472.CAN-04-3151. [DOI] [PubMed] [Google Scholar]
- 13.Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, Park E, Gee JM, Finlay P, Jones HE, et al. Phosphorylated insulin-like growth factori/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008;68:10238–10246. doi: 10.1158/0008-5472.CAN-08-2755. [DOI] [PubMed] [Google Scholar]
- 14.Smith JA, Poteet-Smith CE, Xu Y, Errington TM, Hecht SM, Lannigan DA. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005;65:1027–1034. [PubMed] [Google Scholar]
- 15.Xian W, Pappas L, Pandya D, Selfors LM, Derksen PW, de Bruin M, Gray NS, Jonkers J, Rosen JM, Brugge JS. Fibroblast Growth Factor Receptor 1-Transformed Mammary Epithelial Cells Are Dependent on RSK Activity for Growth and Survival. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan Y, Ruan H, Demeter MR, Comb MJ. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J Biol Chem. 1999;274:34859–34867. doi: 10.1074/jbc.274.49.34859. [DOI] [PubMed] [Google Scholar]
- 17.Dummler BA, Hauge C, Silber J, Yntema HG, Kruse LS, Kofoed B, Hemmings BA, Alessi DR, Frodin M. Functional characterization of human RSK4, a new 90-kDa ribosomal S6 kinase, reveals constitutive activation in most cell types. J Biol Chem. 2005;280:13304–13314. doi: 10.1074/jbc.M408194200. [DOI] [PubMed] [Google Scholar]
- 18.Bignone PA, Lee KY, Liu Y, Emilion G, Finch J, Soosay AE, Charnock FM, Beck S, Dunham I, Mungall AJ, et al. RPS6KA2, a putative tumour suppressor gene at 6q27 in sporadic epithelial ovarian cancer. Oncogene. 2007;26:683–700. doi: 10.1038/sj.onc.1209827. [DOI] [PubMed] [Google Scholar]
- 19.Thakur A, Rahman KW, Wu J, Bollig A, Biliran H, Lin X, Nassar H, Grignon DJ, Sarkar FH, Liao JD. Aberrant expression of X-linked genes RbAp46, Rsk4, and Cldn2 in breast cancer. Mol Cancer Res. 2007;5:171–181. doi: 10.1158/1541-7786.MCR-06-0071. [DOI] [PubMed] [Google Scholar]
- 20.Myers AP, Corson LB, Rossant J, Baker JC. Characterization of mouse Rsk4 as an inhibitor of fibroblast growth factor-RAS-extracellular signal-regulated kinase signaling. Mol Cell Biol. 2004;24:4255–4266. doi: 10.1128/MCB.24.10.4255-4266.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho YY, Yao K, Kim HG, Kang BS, Zheng D, Bode AM, Dong Z. Ribosomal S6 kinase 2 is a key regulator in tumor promoter induced cell transformation. Cancer Res. 2007;67:8104–8112. doi: 10.1158/0008-5472.CAN-06-4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurbin A, Coll JL, Dubrez-Daloz L, Mari B, Auberger P, Brambilla C, Favrot MC. Cooperation of amphiregulin and insulin-like growth factor-1 inhibits Bax- and Bad-mediated apoptosis via a protein kinase C-dependent pathway in non-small cell lung cancer cells. J Biol Chem. 2005;280:19757–19767. doi: 10.1074/jbc.M413516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dehan E, Bassermann F, Guardavaccaro D, Vasiliver-Shamis G, Cohen M, Lowes KN, Dustin M, Huang DC, Taunton J, Pagano M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell. 2009;33:109–116. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisenmann KM, VanBrocklin MW, Staffend NA, Kitchen SM, Koo HM. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein bad. Cancer Res. 2003;63:8330–8337. [PubMed] [Google Scholar]
- 25.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jirstrom K, Ryden L, Anagnostaki L, Nordenskjold B, Stal O, Thorstenson S, Chebil G, Jonsson PE, Ferno M, Landberg G. Pathology parameters and adjuvant tamoxifen response in a randomised premenopausal breast cancer trial. J Clin Pathol. 2005;58:1135–1142. doi: 10.1136/jcp.2005.027185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thakur A, Sun Y, Bollig A, Wu J, Biliran H, Banerjee S, Sarkar FH, Liao DJ. Anti-invasive and antimetastatic activities of ribosomal protein S6 kinase 4 in breast cancer cells. Clin Cancer Res. 2008;14:4427–4436. doi: 10.1158/1078-0432.CCR-08-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thakur A, Xu H, Wang Y, Bollig A, Biliran H, Liao JD. The role of X-linked genes in breast cancer. Breast Cancer Res Treat. 2005;93:135–143. doi: 10.1007/s10549-005-4516-0. [DOI] [PubMed] [Google Scholar]
- 29.Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer . 2005;12(Suppl 1):S47–59. doi: 10.1677/erc.1.00993. [DOI] [PubMed] [Google Scholar]
- 30.Hanson KD, Shichiri M, Follansbee MR, Sedivy JM. Effects of c-myc expression on cell cycle progression. Mol Cell Biol. 1994;14:5748–5755. doi: 10.1128/mcb.14.9.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson PH, Pon RT, Shiu RP. Inhibition of c-myc expression by phosphorothioate antisense oligonucleotide identifies a critical role for c-myc in the growth of human breast cancer. Cancer Res. 1991;51:3996–4000. [PubMed] [Google Scholar]
- 32.Dubik D, Shiu RP. Mechanism of estrogen activation of c-myc oncogene expression. Oncogene. 1992;7:1587–1594. [PubMed] [Google Scholar]
- 33.Clark DE, Poteet-Smith CE, Smith JA, Lannigan DA. Rsk2 allosterically activates estrogen receptor alpha by docking to the hormone-binding domain. EMBO J. 2001;20:3484–3494. doi: 10.1093/emboj/20.13.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joel PB, Traish AM, Lannigan DA. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem. 1998;273:13317–13323. doi: 10.1074/jbc.273.21.13317. [DOI] [PubMed] [Google Scholar]
- 35.Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol. 1998;18:1978–1984. doi: 10.1128/mcb.18.4.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joel PB, Traish AM, Lannigan DA. Estradiol and phorbol ester cause phosphorylation of serine 118 in the human estrogen receptor. Mol Endocrinol. 1995;9:1041–1052. doi: 10.1210/mend.9.8.7476978. [DOI] [PubMed] [Google Scholar]
- 37.Sutherland RL, Musgrove EA. Cyclins and breast cancer. J Mammary Gland Biol Neoplasia. 2004;9:95–104. doi: 10.1023/B:JOMG.0000023591.45568.77. [DOI] [PubMed] [Google Scholar]
- 38.Eisinger-Mathason TS, Andrade J, Groehler AL, Clark DE, Muratore-Schroeder TL, Pasic L, Smith JA, Shabanowitz J, Hunt DF, Macara IG, et al. Codependent functions of RSK2 and the apoptosis-promoting factor TIA-1 in stress granule assembly and cell survival. Mol Cell. 2008;31:722–736. doi: 10.1016/j.molcel.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–671. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 40.Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10:1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 43.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–441. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 44.Lopez de Silanes I, Galban S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M. Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol Cell Biol. 2005;25:9520–9531. doi: 10.1128/MCB.25.21.9520-9531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stratford AL, Fry CJ, Desilets C, Davies AH, Cho YY, Li Y, Dong Z, Berquin IM, Roux PP, Dunn SE. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 2008;10:R99. doi: 10.1186/bcr2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finkbeiner MR, Astanehe A, To K, Fotovati A, Davies AH, Zhao Y, Jiang H, Stratford AL, Shadeo A, Boccaccio C, et al. Profiling YB-1 target genes uncovers a new mechanism for MET receptor regulation in normal and malignant human mammary cells. Oncogene. 2009;28:1421–1431. doi: 10.1038/onc.2008.485. [DOI] [PubMed] [Google Scholar]
- 47.Stratford AL, Habibi G, Astanehe A, Jiang H, Hu K, Park E, Shadeo A, Buys TP, Lam W, Pugh T, et al. Epidermal growth factor receptor (EGFR) is transcriptionally induced by the Y-box binding protein-1 (YB-1) and can be inhibited with Iressa in basal-like breast cancer, providing a potential target for therapy. Breast Cancer Res. 2007;9:R61. doi: 10.1186/bcr1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Habibi G, Leung S, Law JH, Gelmon K, Masoudi H, Turbin D, Pollak M, Nielsen TO, Huntsman D, Dunn SE. Redefining prognostic factors for breast cancer: YB-1 is a stronger predictor of relapse and disease-specific survival than estrogen receptor or HER-2 across all tumor subtypes. Breast Cancer Res. 2008;10:R86. doi: 10.1186/bcr2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee C, Dhillon J, Wang MY, Gao Y, Hu K, Park E, Astanehe A, Hung MC, Eirew P, Eaves CJ, et al. Targeting YB-1 in HER-2 overexpressing breast cancer cells induces apoptosis via the mTOR/STAT3 pathway and suppresses tumor growth in mice. Cancer Res. 2008;68:8661–8666. doi: 10.1158/0008-5472.CAN-08-1082. [DOI] [PubMed] [Google Scholar]
- 50.Faury D, Nantel A, Dunn SE, Guiot MC, Haque T, Hauser P, Garami M, Bognar L, Hanzely Z, Liberski PP, et al. Molecular profiling identifies prognostic subgroups of pediatric glioblastoma and shows increased YB-1 expression in tumors. J Clin Oncol. 2007;25:1196–1208. doi: 10.1200/JCO.2006.07.8626. [DOI] [PubMed] [Google Scholar]
- 51.Bergmann S, Royer-Pokora B, Fietze E, Jurchott K, Hildebrandt B, Trost D, Leenders F, Claude JC, Theuring F, Bargou R, et al. YB-1 provokes breast cancer through the induction of chromosomal instability that emerges from mitotic failure and centrosome amplification. Cancer Res. 2005;65:4078–4087. doi: 10.1158/0008-5472.CAN-04-4056. [DOI] [PubMed] [Google Scholar]
- 52.Sutherland BW, Kucab J, Wu J, Lee C, Cheang MC, Yorida E, Turbin D, Dedhar S, Nelson C, Pollak M, et al. Akt phosphorylates the Y-box binding protein 1 at Ser102 located in the cold shock domain and affects the anchorage-independent growth of breast cancer cells. Oncogene. 2005;24:4281–4292. doi: 10.1038/sj.onc.1208590. [DOI] [PubMed] [Google Scholar]
- 53.Fujii T, Kawahara A, Basaki Y, Hattori S, Nakashima K, Nakano K, Shirouzu K, Kohno K, Yanagawa T, Yamana H, et al. Expression of HER2 and estrogen receptor alpha depends upon nuclear localization of Y-box binding protein-1 in human breast cancers. Cancer Res. 2008;68:1504–1512. doi: 10.1158/0008-5472.CAN-07-2362. [DOI] [PubMed] [Google Scholar]
- 54.Burd CJ, Petre CE, Morey LM, Wang Y, Revelo MP, Haiman CA, Lu S, Fenoglio-Preiser CM, Li J, Knudsen ES, et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A. 2006;103:2190–2195. doi: 10.1073/pnas.0506281103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suzuki H, Ueda T, Ichikawa T, Ito H. Androgen receptor involvement in the progression of prostate cancer. Endocr Relat Cancer. 2003;10:209–216. doi: 10.1677/erc.0.0100209. [DOI] [PubMed] [Google Scholar]
- 56.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem. 2004;91:483–490. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 57.La Vecchia C. Epidemiology of ovarian cancer: a summary review. Eur J Cancer Prev. 2001;10:125–129. doi: 10.1097/00008469-200104000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Hatch KD, Beecham JB, Blessing JA, Creasman WT. Responsiveness of patients with advanced ovarian carcinoma to tamoxifen. A Gynecologic Oncology Group study of second-line therapy in 105 patients. Cancer. 1991;68:269–271. doi: 10.1002/1097-0142(19910715)68:2<269::aid-cncr2820680209>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 59.Scambia G, Benedetti-Panici P, Ferrandina G, Distefano M, Salerno G, Romanini ME, Fagotti A, Mancuso S. Epidermal growth factor, oestrogen and progesterone receptor expression in primary ovarian cancer: correlation with clinical outcome and response to chemotherapy. Br J Cancer. 1995;72:361–366. doi: 10.1038/bjc.1995.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rao BR, Slotman BJ. Endocrine factors in common epithelial ovarian cancer. Endocr Rev. 1991;12:14–26. doi: 10.1210/edrv-12-1-14. [DOI] [PubMed] [Google Scholar]
- 61.Pohl G, Ho CL, Kurman RJ, Bristow R, Wang TL, Shih Ie M. Inactivation of the mitogen-activated protein kinase pathway as a potential target-based therapy in ovarian serous tumors with KRAS or BRAF mutations. Cancer Res. 2005;65:1994–2000. doi: 10.1158/0008-5472.CAN-04-3625. [DOI] [PubMed] [Google Scholar]
- 62.Nakayama N, Nakayama K, Yeasmin S, Ishibashi M, Katagiri A, Iida K, Fukumoto M, Miyazaki K. KRAS or BRAF mutation status is a useful predictor of sensitivity to MEK inhibition in ovarian cancer. Br J Cancer. 2008;99:2020–2028. doi: 10.1038/sj.bjc.6604783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Ando S, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67:1859–1866. doi: 10.1158/0008-5472.CAN-06-2909. [DOI] [PubMed] [Google Scholar]
- 64.Chen RH, Juo PC, Curran T, Blenis J. Phosphorylation of c-Fos at the C-terminus enhances its transforming activity. Oncogene. 1996;12:1493–1502. [PubMed] [Google Scholar]
- 65.Chen RH, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci U S A. 1993;90:10952–10956. doi: 10.1073/pnas.90.23.10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Renoir JM, Bouclier C, Seguin A, Marsaud V, Sola B. Antioestrogen-mediated cell cycle arrest and apoptosis induction in breast cancer and multiple myeloma cells. J Mol Endocrinol. 2008;40:101–112. doi: 10.1677/JME-07-0143. [DOI] [PubMed] [Google Scholar]
- 67.Gauduchon J, Seguin A, Marsaud V, Clay D, Renoir JM, Sola B. Pure antiestrogen-induced G1-arrest in myeloma cells results from the reduced kinase activity of cyclin D3/CDK6 complexes whereas apoptosis is mediated by endoplasmic reticulum-dependent caspases. Int J Cancer. 2008;122:2130–2141. doi: 10.1002/ijc.23310. [DOI] [PubMed] [Google Scholar]
- 68.Otsuki T, Yamada O, Kurebayashi J, Moriya T, Sakaguchi H, Kunisue H, Yata K, Uno M, Yawata Y, Ueki A. Estrogen receptors in human myeloma cells. Cancer Res. 2000;60:1434–1441. [PubMed] [Google Scholar]
- 69.Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, Sanderson R, Yang Y, Wilson C, Zangari M, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99:1745–1757. doi: 10.1182/blood.v99.5.1745. [DOI] [PubMed] [Google Scholar]
- 70.Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R, Cancemi M, Caporali S, Caristi S, Scafoglio C, et al. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol. 2004;24:7260–7274. doi: 10.1128/MCB.24.16.7260-7274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, Bergsagel PL. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang S, Dong S, Gu TL, Guo A, Cohen MS, Lonial S, Khoury HJ, Fabbro D, Gilliland DG, Bergsagel PL, et al. FGFR3 Activates RSK2 to Mediate Hematopoietic Transformation through Tyrosine Phosphorylation of RSK2 and Activation of the MEK/ERK Pathway. Cancer Cell. 2007;12:201–214. doi: 10.1016/j.ccr.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roux PP, Richards SA, Blenis J. Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates extracellular signal-regulated kinase docking and RSK activity. Mol Cell Biol. 2003;23:4796–4804. doi: 10.1128/MCB.23.14.4796-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kang S, Elf S, Dong S, Hitosugi T, Lythgoe K, Guo A, Ruan H, Lonial S, Khoury HJ, Williams IR, et al. FGFR3 associates with and tyrosine-phosphorylates p90RSK2, leading to RSK2 activation that mediates hematopoietic transformation. Mol Cell Biol. 2009 doi: 10.1128/MCB.00998-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poteet-Smith CE, Smith JA, Lannigan DA, Freed TA, Sturgill TW. Generation of constitutively active p90 ribosomal S6 kinase in vivo. Implications for the mitogen-activated protein kinase-activated protein kinase family. J Biol Chem. 1999;274:22135–22138. doi: 10.1074/jbc.274.32.22135. [DOI] [PubMed] [Google Scholar]
- 76.Rickard DJ, Subramaniam M, Spelsberg TC. Molecular and cellular mechanisms of estrogen action on the skeleton. J Cell Biochem Suppl. 1999;32–33:123–132. doi: 10.1002/(sici)1097-4644(1999)75:32+<123::aid-jcb15>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 77.Zallone A. Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann N Y Acad Sci. 2006;1068:173–179. doi: 10.1196/annals.1346.019. [DOI] [PubMed] [Google Scholar]
- 78.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117:387–398. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- 79.Kang S, Dong S, Guo A, Ruan H, Lonial S, Khoury HJ, Gu TL, Chen J. Epidermal growth factor stimulates RSK2 activation through activation of the MEK/ERK pathway and src-dependent tyrosine phosphorylation of RSK2 at Tyr-529. J Biol Chem. 2008;283:4652–4657. doi: 10.1074/jbc.M709673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kang S, Dong S, Guo A, Ruan H, Lonial S, Khoury HJ, Gu TL, Chen J. EGF stimulates RSK2 activation through activation of the MEK/ERK pathway and Src-dependent tyrosine phosphorylation of RSK2 at Y529. J Biol Chem. 2007 doi: 10.1074/jbc.M709673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 82.Schwartz AG, Wenzlaff AS, Prysak GM, Murphy V, Cote ML, Brooks SC, Skafar DF, Lonardo F. Reproductive factors, hormone use, estrogen receptor expression and risk of non small-cell lung cancer in women. J Clin Oncol. 2007;25:5785–5792. doi: 10.1200/JCO.2007.13.3975. [DOI] [PubMed] [Google Scholar]
- 83.Kaiser U, Hofmann J, Schilli M, Wegmann B, Klotz U, Wedel S, Virmani AK, Wollmer E, Branscheid D, Gazdar AF, et al. Steroid-hormone receptors in cell lines and tumor biopsies of human lung cancer. Int J Cancer. 1996;67:357–364. doi: 10.1002/(SICI)1097-0215(19960729)67:3<357::AID-IJC9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 84.Chaudhuri PK, Thomas PA, Walker MJ, Briele HA, Das Gupta TK, Beattie CW. Steroid receptors in human lung cancer cytosols. Cancer Lett. 1982;16:327–332. doi: 10.1016/0304-3835(82)90014-3. [DOI] [PubMed] [Google Scholar]
- 85.Beattie CW, Hansen NW, Thomas PA. Steroid receptors in human lung cancer. Cancer Res. 1985;45:4206–4214. [PubMed] [Google Scholar]
- 86.Hammoud Z, Tan B, Badve S, Bigsby RM. Estrogen promotes tumor progression in a genetically defined mouse model of lung adenocarcinoma. Endocr Relat Cancer. 2008;15:475–483. doi: 10.1677/ERC-08-0002. [DOI] [PubMed] [Google Scholar]
- 87.Bertolotto C, Maulon L, Filippa N, Baier G, Auberger P. Protein kinase C theta and epsilon promote T-cell survival by a rsk-dependent phosphorylation and inactivation of BAD. J Biol Chem. 2000;275:37246–37250. doi: 10.1074/jbc.M007732200. [DOI] [PubMed] [Google Scholar]
- 88.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 89.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 90.Fernando RI, Wimalasena J. Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras-dependent nongenomic pathways requiring signaling through ERK and Akt. Mol Biol Cell. 2004;15:3266–3284. doi: 10.1091/mbc.E03-11-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boland R, Vasconsuelo A, Milanesi L, Ronda AC, de Boland AR. 17beta-estradiol signaling in skeletal muscle cells and its relationship to apoptosis. Steroids. 2008;73:859–863. doi: 10.1016/j.steroids.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 92.Fernandes AM, Hamburger AW, Gerwin BI. Production of epidermal growth factor related ligands in tumorigenic and benign human lung epithelial cells. Cancer Lett. 1999;142:55–63. doi: 10.1016/s0304-3835(99)00166-4. [DOI] [PubMed] [Google Scholar]
- 93.Werner H, LeRoith D. The role of the insulin-like growth factor system in human cancer. Adv Cancer Res. 1996;68:183–223. doi: 10.1016/s0065-230x(08)60354-1. [DOI] [PubMed] [Google Scholar]
- 94.Hurbin A, Dubrez L, Coll JL, Favrot MC. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. J Biol Chem. 2002;277:49127–49133. doi: 10.1074/jbc.M207584200. [DOI] [PubMed] [Google Scholar]
- 95.Leung LK, Do L, Wang TT. Regulation of death promoter Bak expression by cell density and 17 beta-estradiol in MCF-7 cells. Cancer Lett. 1998;124:47–52. doi: 10.1016/s0304-3835(97)00430-8. [DOI] [PubMed] [Google Scholar]
- 96.Fletcher JI, Huang DC. Controlling the cell death mediators Bax and Bak: puzzles and conundrums. Cell Cycle. 2008;7:39–44. doi: 10.4161/cc.7.1.5178. [DOI] [PubMed] [Google Scholar]
- 97.Cohen C, DeRose PB, Campbell WG, Schlosnagle DC, Sgoutas D. Estrogen receptor status in malignant melanoma. Am J Dermatopathol. 1990;12:562–564. doi: 10.1097/00000372-199012000-00005. [DOI] [PubMed] [Google Scholar]
- 98.Fisher RI, Neifeld JP, Lippman ME. Oestrogen receptors in human malignant melanoma. Lancet. 1976;2:337–339. doi: 10.1016/s0140-6736(76)92592-7. [DOI] [PubMed] [Google Scholar]
- 99.Walker MJ, Beattie CW, Patel MK, Ronan SM, Das Gupta TK. Estrogen receptor in malignant melanoma. J Clin Oncol. 1987;5:1256–1261. doi: 10.1200/JCO.1987.5.8.1256. [DOI] [PubMed] [Google Scholar]
- 100.de Giorgi V, Mavilia C, Massi D, Gozzini A, Aragona P, Tanini A, Sestini S, Paglierani M, Boddi V, Brandi ML, et al. Estrogen receptor expression in cutaneous melanoma: a real-time reverse transcriptase-polymerase chain reaction and immunohistochemical study. Arch Dermatol. 2009;145:30–36. doi: 10.1001/archdermatol.2008.537. [DOI] [PubMed] [Google Scholar]
- 101.Gill PG, De Young NJ, Thompson A, Keightley DD, Horsfall DJ. The effect of tamoxifen on the growth of human malignant melanoma in vitro. Eur J Cancer Clin Oncol. 1984;20:807–815. doi: 10.1016/0277-5379(84)90220-7. [DOI] [PubMed] [Google Scholar]
- 102.Cocconi G, Bella M, Calabresi F, Tonato M, Canaletti R, Boni C, Buzzi F, Ceci G, Corgna E, Costa P, et al. Treatment of metastatic malignant melanoma with dacarbazine plus tamoxifen. N Engl J Med. 1992;327:516–523. doi: 10.1056/NEJM199208203270803. [DOI] [PubMed] [Google Scholar]
- 103.Cocconi G. High-dose cisplatin with dacarbazine and tamoxifen in the treatment of metastatic melanoma. Cancer. 1992;70:902. doi: 10.1002/1097-0142(19920815)70:4<902::aid-cncr2820700430>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 104.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101:13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ohta Y, Hartwig JH. Phosphorylation of actin-binding protein 280 by growth factors is mediated by p90 ribosomal protein S6 kinase. J Biol Chem. 1996;271:11858–11864. doi: 10.1074/jbc.271.20.11858. [DOI] [PubMed] [Google Scholar]
- 106.Woo MS, Ohta Y, Rabinovitz I, Stossel TP, Blenis J. Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol Cell Biol. 2004;24:3025–3035. doi: 10.1128/MCB.24.7.3025-3035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Byers HR, Etoh T, Doherty JR, Sober AJ, Mihm MC., Jr Cell migration and actin organization in cultured human primary, recurrent cutaneous and metastatic melanoma. Time-lapse and image analysis. Am J Pathol. 1991;139:423–435. [PMC free article] [PubMed] [Google Scholar]
- 108.Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA, Slingerland JM. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A. 2009;106:9268–9273. doi: 10.1073/pnas.0805057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, Cohen MS, Johansen JV, Winther BR, Lund LR, et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol Cell. 2009;35:511–522. doi: 10.1016/j.molcel.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dohi O, Hatori M, Suzuki T, Ono K, Hosaka M, Akahira J, Miki Y, Nagasaki S, Itoi E, Sasano H. Sex steroid receptors expression and hormone-induced cell proliferation in human osteosarcoma. Cancer Sci. 2008;99:518–523. doi: 10.1111/j.1349-7006.2007.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Walker MJ, Chaudhuri PK, Beattie CW, Das Gupta TK. Steroid receptors in malignant skeletal tumors. Cancer. 1980;45:3004–3007. doi: 10.1002/1097-0142(19800615)45:12<3004::aid-cncr2820451221>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 112.Duan R, Porter W, Safe S. Estrogen-induced c-fos protooncogene expression in MCF-7 human breast cancer cells: role of estrogen receptor Sp1 complex formation. Endocrinology. 1998;139:1981–1990. doi: 10.1210/endo.139.4.5870. [DOI] [PubMed] [Google Scholar]
- 113.Weisz A, Rosales R. Identification of an estrogen response element upstream of the human c-fos gene that binds the estrogen receptor and the AP-1 transcription factor. Nucleic Acids Res. 1990;18:5097–5106. doi: 10.1093/nar/18.17.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu JX, Carpenter PM, Gresens C, Keh R, Niman H, Morris JW, Mercola D. The proto-oncogene c-fos is over-expressed in the majority of human osteosarcomas. Oncogene. 1990;5:989–1000. [PubMed] [Google Scholar]
- 115.Dony C, Gruss P. Proto-oncogene c-fos expression in growth regions of fetal bone and mesodermal web tissue. Nature. 1987;328:711–714. doi: 10.1038/328711a0. [DOI] [PubMed] [Google Scholar]
- 116.Hoyland J, Sharpe PT. Upregulation of c-fos protooncogene expression in pagetic osteoclasts. J Bone Miner Res. 1994;9:1191–1194. doi: 10.1002/jbmr.5650090808. [DOI] [PubMed] [Google Scholar]
- 117.Candeliere GA, Glorieux FH, Prud’homme J, St-Arnaud R. Increased expression of the c-fos proto-oncogene in bone from patients with fibrous dysplasia. N Engl J Med. 1995;332:1546–1551. doi: 10.1056/NEJM199506083322304. [DOI] [PubMed] [Google Scholar]
- 118.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 119.Young ID. The Coffin-Lowry syndrome. J Med Genet. 1988;25:344–348. doi: 10.1136/jmg.25.5.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hanauer A, Young ID. Coffin-Lowry syndrome: clinical and molecular features. J Med Genet. 2002;39:705–713. doi: 10.1136/jmg.39.10.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yu VW, Gauthier C, St-Arnaud R. Inhibition of ATF4 transcriptional activity by FIAT/gamma-taxilin modulates bone mass accrual. Ann N Y Acad Sci. 2006;1068:131–142. doi: 10.1196/annals.1346.027. [DOI] [PubMed] [Google Scholar]
- 122.Yu VW, Ambartsoumian G, Verlinden L, Moir JM, Prud’homme J, Gauthier C, Roughley PJ, St-Arnaud R. FIAT represses ATF4-mediated transcription to regulate bone mass in transgenic mice. J Cell Biol. 2005;169:591–601. doi: 10.1083/jcb.200412139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ameri K, Lewis CE, Raida M, Sowter H, Hai T, Harris AL. Anoxic induction of ATF-4 through HIF-1-independent pathways of protein stabilization in human cancer cells. Blood. 2004;103:1876–1882. doi: 10.1182/blood-2003-06-1859. [DOI] [PubMed] [Google Scholar]
- 124.Hong SH, Nah HY, Lee JY, Gye MC, Kim CH, Kim MK. Analysis of estrogen-regulated genes in mouse uterus using cDNA microarray and laser capture microdissection. J Endocrinol. 2004;181:157–167. doi: 10.1677/joe.0.1810157. [DOI] [PubMed] [Google Scholar]
- 125.Tonini T, Rossi F, Claudio PP. Molecular basis of angiogenesis and cancer. Oncogene. 2003;22:6549–6556. doi: 10.1038/sj.onc.1206816. [DOI] [PubMed] [Google Scholar]
- 126.Nakamura J, Savinov A, Lu Q, Brodie A. Estrogen regulates vascular endothelial growth/permeability factor expression in 7,12-dimethylbenz(a)anthracene-induced rat mammary tumors. Endocrinology. 1996;137:5589–5596. doi: 10.1210/endo.137.12.8940388. [DOI] [PubMed] [Google Scholar]
- 127.Rubanyi GM, Johns A, Kauser K. Effect of estrogen on endothelial function and angiogenesis. Vascul Pharmacol. 2002;38:89–98. doi: 10.1016/s0306-3623(02)00131-3. [DOI] [PubMed] [Google Scholar]
- 128.Johns A, Freay AD, Fraser W, Korach KS, Rubanyi GM. Disruption of estrogen receptor gene prevents 17 beta estradiol-induced angiogenesis in transgenic mice. Endocrinology. 1996;137:4511–4513. doi: 10.1210/endo.137.10.8828515. [DOI] [PubMed] [Google Scholar]
- 129.Losordo DW, Isner JM. Estrogen and angiogenesis: A review. Arterioscler Thromb Vasc Biol. 2001;21:6–12. doi: 10.1161/01.atv.21.1.6. [DOI] [PubMed] [Google Scholar]
- 130.Spyridopoulos I, Sullivan AB, Kearney M, Isner JM, Losordo DW. Estrogen-receptor-mediated inhibition of human endothelial cell apoptosis. Estradiol as a survival factor. Circulation. 1997;95:1505–1514. doi: 10.1161/01.cir.95.6.1505. [DOI] [PubMed] [Google Scholar]
- 131.Dabrosin C, Palmer K, Muller WJ, Gauldie J. Estradiol promotes growth and angiogenesis in polyoma middle T transgenic mouse mammary tumor explants. Breast Cancer Res Treat. 2003;78:1–6. doi: 10.1023/a:1022133219353. [DOI] [PubMed] [Google Scholar]
- 132.Hyder SM, Huang JC, Nawaz Z, Boettger-Tong H, Makela S, Chiappetta C, Stancel GM. Regulation of vascular endothelial growth factor expression by estrogens and progestins. Environ Health Perspect . 2000;108(Suppl 5):785–790. doi: 10.1289/ehp.00108s5785. [DOI] [PubMed] [Google Scholar]
- 133.Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol. 2002;22:270–285. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.English JM, Pearson G, Hockenberry T, Shivakumar L, White MA, Cobb MH. Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J Biol Chem. 1999;274:31588–31592. doi: 10.1074/jbc.274.44.31588. [DOI] [PubMed] [Google Scholar]
- 135.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hayashi M, Fearns C, Eliceiri B, Yang Y, Lee JD. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005;65:7699–7706. doi: 10.1158/0008-5472.CAN-04-4540. [DOI] [PubMed] [Google Scholar]