

INTRODUCTION
Estrogen is the major sex steroid affecting the growth, remodeling and homeostasis of the female skeleton. Estrogen loss in postmenopausal women leads to osteoporosis characterized by low bone mass, altered bone microarchitecture and increased risk of fracture, and is one of the most prevalent degenerative diseases affecting women in developed countries [1; 2]. Similarly, loss of estrogen due to ovariectomy (OVX) induces osteoporosis in mice, rats and primates [3; 4; 5; 6; 7; 8; 9]. In women, high bone turnover bone loss and a net increase in bone resorption are hallmarks of the osteoporosis resulting from postmenopausal estrogen deficiency. Yet the mechanisms by which estrogen loss leads to activation of the resorption process remain incompletely understood.
Recent studies indicate that osteocyte apoptosis may play a central role in signaling the activation and maintaining the progress of bone remodeling [10; 11; 12; 13; 14]. Best elucidated is the remodeling of bone microdamage, where localized apoptosis of osteocytes near microdamage sites plays a direct and controlling role in targeted bone remodeling induced by fatigue damage [11; 13]. Verborgt et al [11,13] showed that there is tight spatial and temporal coupling of osteocyte apoptosis to bone microdamage and subsequent bone remodeling, i.e., osteocytes close to microcracks undergo apoptosis, and this region of apoptosis coincides with the location of the subsequent osteoclastic response. Moreover, this apoptosis precedes the onset of bone resorption. Recently, Cardoso et al in our laboratory demonstrated that suppression of osteocyte apoptosis in bone completely prevented the onset of osteoclastic resorption due to fatigue, indicating that apoptotic cell death near microcracks sites is a key controlling step in the activation and/or targeting of osteoclastic resorption initiated by microdamage [14]. Spatial and/or temporal associations between osteocyte apoptosis and bone resorption have also been demonstrated at various skeletal sites following estrogen loss in humans and animals, as well as in response to other resorption-activating stimuli (e.g. disuse, glucocorticoids) [15; 16; 17; 18]. For example, Noble found a non-uniform distribution of apoptotic osteocytes among the femoral head and iliac crest, raising possibility of a functional relationship between programmed osteocyte death and bone turnover [10]. While Hedgecock demonstrated that regional variations in remodeling correlate with osteocyte apoptosis in rabbit tibia mid-shafts [12]. Moreover, Aguirre et al showed that osteocyte apoptosis induced by weightlessness precedes osteoclast recruitment and bone loss, with apoptotic osteocytes preferentially located at the site of subsequent bone resorption [18]. However, while osteocyte apoptosis has been associated with bone resorption in response to estrogen loss [19; 20], the close spatial, temporal and the potential for causal clinks between osteocyte apoptosis and subsequent bone resorption comparable to those observed in remodeling of bone microdamage have not been established. Therefore, in the current studies, we tested the hypotheses that 1) the osteocyte apoptosis and resorption that occur after estrogen loss are both spatially and temporally coupled, similar to microdamage-induced remodeling, and 2) that this osteocyte apoptosis plays a comparable role in the controlling the onset of the osteoclastic resorption induced by estrogen loss.
METHODS AND MATERIALS
Overview of the experimental design
Previous studies by Li et al from our laboratory demonstrated that following ovariectomy (OVX), C57B/6J mice (B6) undergo osteoclastic resorption not only in cancellous bone, but also at the endocortical surface of the femoral mid-diaphysis [21]. Moreover, this endocortical resorption process after OVX occurs in a temporally and spatially consistent pattern, with the resorption occurring principally in the posterior region. This consistency, coupled with the simple tubular structure of the femoral diaphysis offers distinct advantages for spatial analyses, particularly compared to the complex lattice architecture of cancellous bone, where OVX-induced bone loss studies have typically been focused. In the mid-diaphysis we were able to establish a well-defined anatomical coordinate system, which allows straightforward mapping measurements for spatial and temporal relationships between osteocyte apoptosis and bone remodeling responses after OVX. Accordingly, adult female mice were subjected to OVX to induce estrogen depletion, after which sites of osteocyte apoptosis and osteoclastic bone resorption were mapped from histological sections of the diaphyseal cortex at various times post-OVX. In a second experiment, we combined this approach with pharmacological inhibition of osteocyte apoptosis to test directly whether bone resorption following estrogen loss was dependent on osteocyte apoptosis.
Experiment 1: Spatial and temporal patterns of osteocyte apoptosis and bone resorption following ovariectomy Animal Model and Procedures
Forty-five adult (17-week old) female, C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were used in this study. Twenty animals underwent bilateral ovariectomy (OVX) under Avertin anesthesia. An additional 20 animals underwent sham surgery in which ovaries were exteriorized and replaced in the abdominal cavity. Animals were weighed at surgery and at sacrifice. At 3, 7,14 and 21 days following surgery, groups of OVX and SHAM animals (n=5 each) were anesthetized as above and blood samples were collected by retro-orbital bleeding, after which the animals were euthanized by cervical dislocation. These time points were used to examine tissues before the resorption activation (3 days), in the early osteoclastic resorptive phase (7 days), and at the periods of peak resorption in this model (14 & 21 days) [21].
Tissue collection and processing
Following sacrifice, uteri were harvested and weighed to confirm estrogen loss. Femora were dissected, cleaned of soft tissue and fixed in 10% neutral buffered formalin for 48 hr at 4°C. Right femora were decalcified in formic acid for 3 days with daily changes then embedded in paraffin. Mid-diaphyseal 5-micron cross-sections were cut, adhered to charged slides and used for immunohistochemistry (IHC). Left femora remained undecalcified for cortical bone histomorphometry or microstructural analysis by confocal microcrcopy. These bones were bulk-stained in Villanueva’s bone stain, embedded in PMMA following procedures we have described elsewhere [21; 22], and mid-diaphyseal cross sections were cut (150 micron thickness) using a Leica 1600 Sawing Microtome (Leica Instruments, Nussloch, Germany) and polished to 30-micron thickness.
Osteocyte apoptosis
Osteocyte apoptosis was assessed principally by immunohistochemical staining for cleaved (activated) caspase-3, an effector caspase required for regulated cell death [23]; morphological assessment of pyknotic osteocytes in Toluidine Blue stained sections was used to obtain secondary confirmation of cell death as we have reported previously [11, 14]. Immunohistochemistry: Sections were deparaffinized, rehydrated and treated with 3% hydrogen peroxide to block endogenous peroxidase activity. Antigen retrieval was performed using a methanol-sodium hydroxide based solution for 30 minutes at room temperature (DeCal Retrieval Solution, Biogenex, San Ramon, CA), after which non-specific tissue binding was blocked by incubating tissue sections in 10% rabbit serum in PBS for 30 minutes at room temperature. Sections were incubated overnight at 4 °C in a humidified chamber with rabbit anti-mouse cleaved caspase-3 primary antibody (#9661, Cell Signaling Technologies, Danvers MA) at 1:50 dilution in Antibody Diluent (DakoCytomation, Carpinteria, CA). Detection was performed using a goat anti-rabbit secondary antibody labeled with a Horseradish Peroxidase conjugate and developed with a DAB substrate chromogen system (DakoCytomation, Carpinteria, CA) with 5% fast-green was used as a counter-stain. Optimal dilution for the primary antibody was determined using internal positive control tissues (growth plate) that were treated in identical fashion to experimental bone samples. For negative staining controls, immunostaining was performed with normal rabbit serum instead of primary antibody.
Measurement of osteocyte apoptosis
Apoptotic (caspase-positive) and non-apoptotic (caspase-negative) osteocytes (or pyknotic and intact osteocytes) were counted under brightfield microscopy at 400X magnification using a 10mm × 10mm eyepiece grid reticule. Apoptotic osteocytes were counted through the entire cortical width for each of eight sampling regions around the bone cross section defined by the principal anatomical axes and at 45 degree increments from them. Each sampling region was further divided into 10 sub-regions of equal area extending from the periosteal to the endocortical surface (Figure 1). Numbers of caspase-positive and negative osteocytes were determined, and osteocyte apoptosis was expressed as a percentage of total cells in each region of interest.
Figure 1.
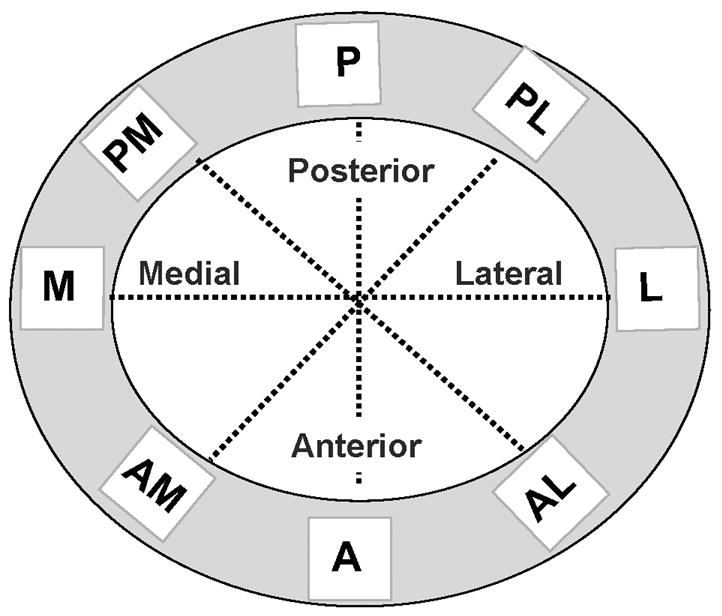
Schematic diagram summarizing the sampling method used to measure osteocyte apoptosis in the cortex of femoral mid-diaphysis (cortex depicted in gray). Data were collected along the anterior (A), posterior (P), medial (M) and lateral (L) anatomical axes and at 45 degree intervals to these axes. Measurements were made along the entire cortical width for each axis, from the periosteal to endocortical surface.
Histomorphometric assessment of bone resorption
Bone resorption was assessed based on eroded surfaces (Er.Pm/B.Pm, %) on both periosteal and endocortical surfaces using an OsteoMeasure system (Osteometrics, Atlanta, GA) connected to a Zeiss Axioskop microscope.
Confocal microscopy of osteocytes and lacunar-canalicular organization
Undecalcified sections from 5 additional baseline control mice were examined using a confocal laser scanning microscope with 514 nm wavelength excitation (TCS-SP, Leica Microsystems, Wetzlar, Germany) to assess for morphological differences between osteocytes in different cortical bone regions. A 63X magnification oil immersion objective lens was used to view fluorescent Villanueva staining. Measurements of the lacunar-canalicular system were performed on stacks of 10 sequential confocal images taken at 0.5-μm intervals in order to recreate a comparable thickness to histology slides. All images were imported into quantitative image software (IMAQ Vision Builder; National Instruments, Austin TX) with optimized threshold settings. At least 75 lacunae per region of interest per bone were measured to determine the number of lacunae and canaliculi, lacunar perimeter and area.
Experiment 2: Inhibition of osteocyte apoptosis: Effects on bone resorption after ovariectomy In vivo apoptosis inhibition
Caspase inhibitors prevent apoptosis both in vitro and in vivo. and have been used successfully in animal models to attenuate cell death and minimize tissue damage following ischemic injury to brain, gut and heart [24; 25; 26; 27]. Moreover, Cardoso et al from our laboratory recently demonstrated that in vivo pan-caspase inhibition with QVD prevented bone microdamage-induced osteocyte apoptosis, and thereby prevented the activation of bone remodeling [14]. The irreversible pan-caspase inhibitor used in this study, QVD (QVD-OPh: quinolyl-valyl-O-methylaspartyl-[-2, 6-difluorophenoxy]-methyl ketone; MP Biomedicals, Livermore, CA) is more potent than the previous generation of apoptosis inhibitors, and eliminates the potential for kidney damage associated with long-term in vivo use of earlier caspase inhibitors containing a fluoromethyl ketone (-fmk) leaving group [27; 28].
Experimental groups (n=6/group) were as follows: OVX mice that received the pan-caspase inhibitor (OVX+ QVD), SHAM+QVD, SHAM+Veh and OVX+Veh. QVD or Vehicle treatments were initiated 1 day prior to surgery and continued until sacrifice 14 days later. Animals received QVD (20 mg/kg/day) or a weight-equivalent volume of vehicle (DMSO) by intraperitoneal injection. The daily dose of QVD was divided in two, and each half dose was administered every 12 hours. This dosing regimen was continued for the entire duration of the experiment, as in our earlier studies of osteocyte apoptosis and microdamage remodeling [29].
Analyses
The methods of analysis were the same as described for Experiment 1 except that tissues were also stained with an additional apoptosis marker, phosphorylated histone H2AX (rabbit-anti mouse H2AX antibody AB3369, Chemicon, Temecula, CA) that reflects cell death but is not directly dependent on the caspases. H2AX becomes phosphorylated when double-strand breaks in DNA are exposed in apoptosis [30].
Statistical Analyses
Differences in the caspase-positive osteocyte numbers among experimental groups were tested using one-way ANOVA and repeated separately for each time point examined. Differences in osteocyte apoptosis between OVX and sham at each anatomical region were tested using the Mann-Whitney test. Results were expressed as mean ± SD for each group. Analyses were performed using the GraphPad Prism 3.0 software program [GraphPad Software, San Diego, CA].
RESULTS
General Changes after OVX
Ovariectomy resulted in an decreases in uterine weight of approximately 26, 81, 85 and 90 percent at 3, 7, 14 and 21 days post-OVX (p< 0.01) confirming the effectiveness of the surgical procedure. By 3 weeks post-OVX, mice also showed 23 percent increase in body weight and a 2-fold increase in gonadal fat relative to age-and weight-matched sham controls (p < 0.005). Uterine and body weight changes were the comparable after treatment with QVD or vehicle in Experiment 2.
Experiment 1: Osteocyte apoptosis after OVX
Ovariectomy caused an approximately 2-fold increase in overall osteocyte apoptosis within the femoral cortex (p<0.01). However, this osteocyte apoptosis was not uniformly distributed throughout the diaphyseal cortex. Rather, osteocyte apoptosis occurred overwhelmingly in the posterior cortical regions, where caspase positive osteocytes numbers were 4–7 fold higher than in the comparable regions of sham controls. Representative data (7 days post-OVX) for the distribution of caspase-positive osteocytes are summarized in Figure 2; similar distributions were present at all time periods (see online supplement for summary data at all time periods and anatomical locations). All trends for osteocyte apoptosis were confirmed in studies where cell death was assessed from counts of pyknotic osteocytes (data not shown). Furthermore, within the posterior cortical regions osteocyte apoptosis was concentrated in the inner one-half of the cortex.
Figure 2.
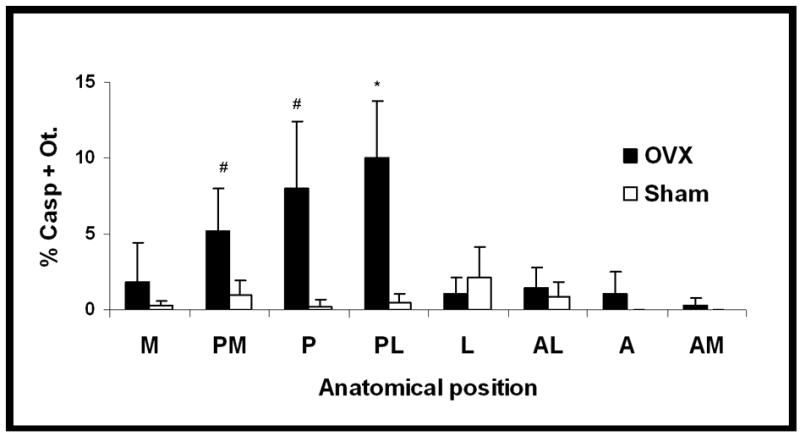
Osteocyte apoptosis (% Casp + Ot.) around the femoral mid-shaft after ovariectomy. Osteocyte apoptosis was dramatically and significantly increased in the posterior cortical regions (PM, P, PL) after OVX (#p < 0.005; *p < 0.0005), but not elsewhere in the cortex. Data are shown for 7 days post-OVX, with similar results observed for all time periods (see text for details).
The increased numbers of apoptotic osteocytes in the posterior femoral cortex were seen by day 3 after OVX, and decreased slowly thereafter. Osteocyte apoptosis remained increased at 14 days post-OVX (p< 0.01; Figure 3), but was not significantly different from control levels at 21 days after OVX.
Figure 3.
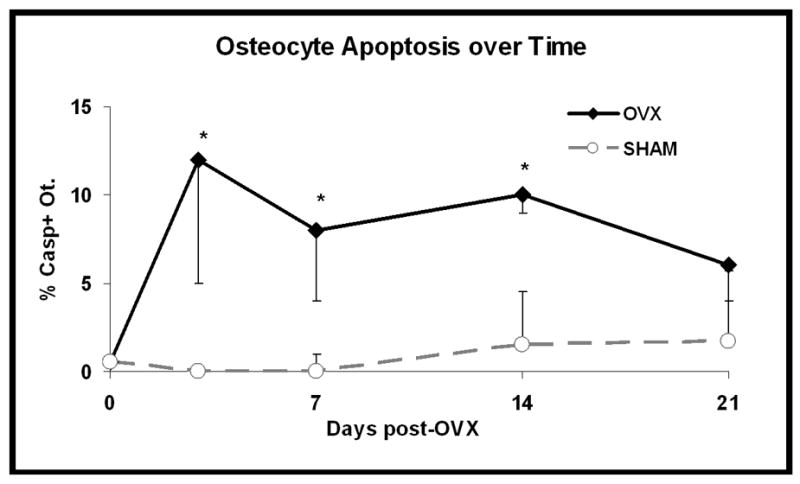
Changes in osteocyte apoptosis over time-course of the experiment for the combined posterior cortical region (PM + P + PL). Osteocyte apoptosis was significantly increased by 3 days after ovariectomy and remained elevated at 7 and 14 post-OVX (*p < 0.005); osteocyte apoptosis was not significantly different from sham control at 21 days after OVX (p>0.2).
Resorption after OVX
In contrast to the early rise in osteocyte apoptosis, significant increases in endocortical resorption over controls were not observed until 14 days post-OVX; resorption remained similarly elevated at 21 days post-OVX. (p <0.01; Figure 4). Most importantly, the endocortical resorption following OVX occurred solely at surfaces adjacent to the regions of elevated osteocyte apoptosis.
Figure 4.

Endocortical resorption (%Er.Pm/B.Pm) in the femoral diaphysis of OVX and Sham mice. Resorption was increased significantly compared to Shams at 14 and 21 days post-OVX (*p < 0.002).
Confocal Microscopy of Osteocyte Morphology
Confocal microscopy images revealed baseline morphological difference in the osteocytes of the posterior cortical regions, (the apoptosis-prone regions after OVX) compared to osteocytes elsewhere in the bone. Osteocyte lacunae in the posterior cortical regions were both irregularly shaped and fewer in number than in the anterior cortical region (Figure 7 and Table 1). Most notably, canalicular connectivity among osteocytes was significantly lower in the posterior (apoptosis-prone) region, with canalicular density per osteocyte lacuna in the posterior cortex reduced almost 20 percent compared (p<0.0001) compared to the anterior cortex.
Figure 7.
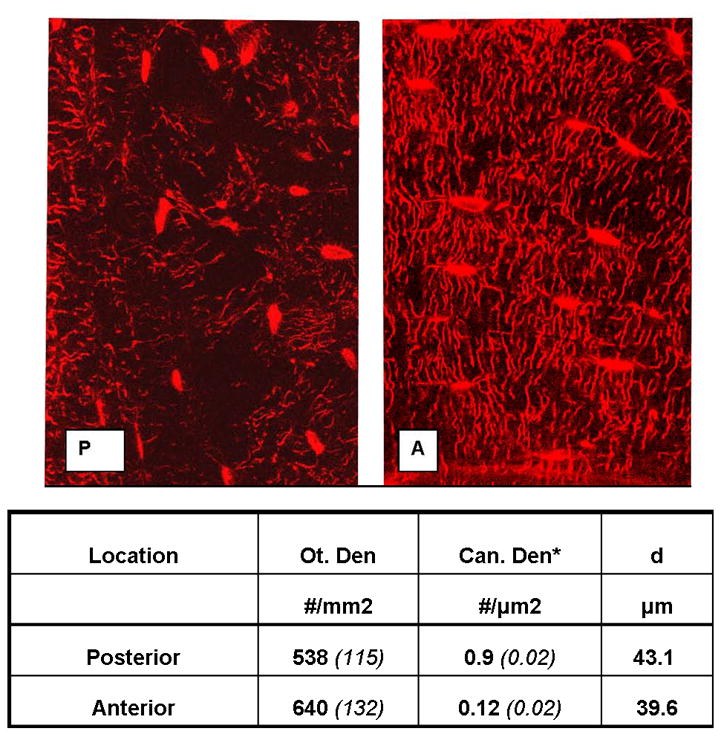
Confocal photomicrographs showing osteocytes, their lacunae and canaliculi from the posterior (P) and anterior (A) cortical regions of mouse mid-femoral diaphyses (regions of high osteocyte apoptosis and low osteocyte apoptosis post-OVX, respectively). Osteocytes in the posterior cortex are more irregularly spaced, fewer in number and less interconnected than those in the anterior cortex.
Table 1.
Summary morphometric data for differences between osteocytes in the anterior and posterior regions of the mouse femoral diaphysis (Ot. Den=osteocyte density, Can. Den=canalicular density, d=canalicular length, *p< 0.001).
| Location | Ot. den #/mm2 | Can. den* #/μm2 | d μm |
|---|---|---|---|
| Posterior | 538 (115) | 0.9 (0.02) | 43.1 |
| Anterior | 640 (132) | 0.12 (0.02) | 39.6 |
Experiment 2: Apoptosis Inhibition, Osteocytes and Resorption
Treatment with QVD days after OVX suppressed osteocyte apoptosis such that the level in QVD-treated bones was equivalent to those in Sham groups (Figure 5). Comparable results were observed for caspase-3 and for H2AX. Results for the H2AX staining were similar to the caspase data (%H2AX+ staining Ot: OVX+Veh = 7.85±2.14%; OVX+QVD = 2.58±3.36%; SHAM+Veh = 1.92±1.69%; SHAM+QVD = 2.91±2.4%). In this study changes in osteocyte apoptosis are reported only for the posterior cortex as Experiment 1 revealed that this is was location of osteocyte apoptosis increase following OVX (see online supplement for osteocyte apoptosis data at all anatomical sites and from all treatment groups).
Figure 5.
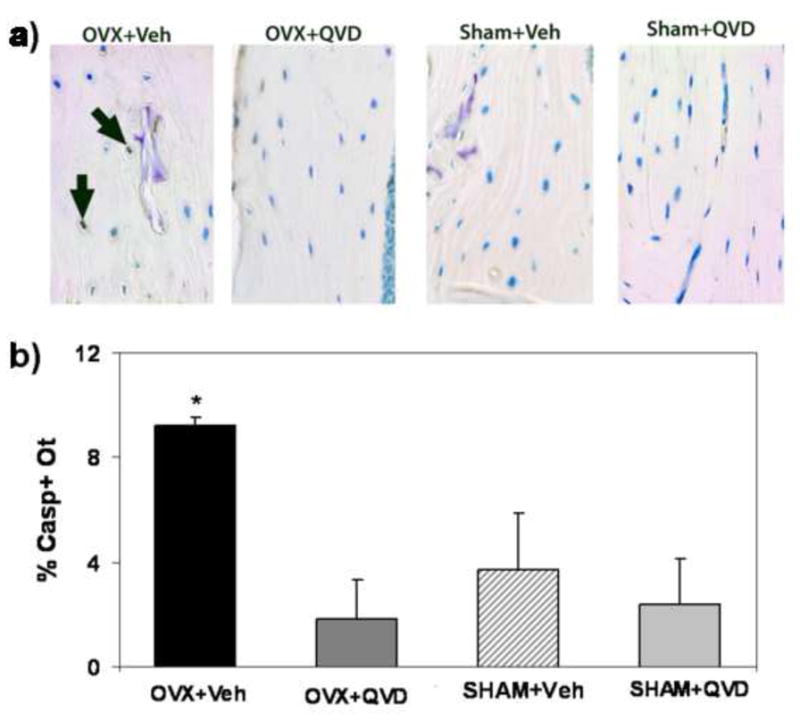
a. Photomicrographs showing immunohistochemical staining for activated caspase 3 in sections from OVX mice (OVX+Vehicle) and OVX mice treated with the pan-caspase inhibitor QVD, as well as bones from sham control animals. The OVX+Veh image shows strong staining for caspase-3 (arrows), which is absent from the OVX+ QVD and Veh-treated SHAM-treated bones (photomicrograph field width = 225 μm). b: Summary data for % Casp + Ot. among treatment groups showing that QVD treatment prevented the OVX-induced increase in osteocyte apoptosis (*p < 0.005).
As in Experiment 1, significantly OVX increased endocortical resorption at surfaces adjacent to the regions of osteocyte apoptosis. Treatment with QVD completely prevented this OVX-induced increase in osteoclastic activity, with erosion levels equivalent to the sham control groups (p= 0.43; Figure 6).
Figure 6.
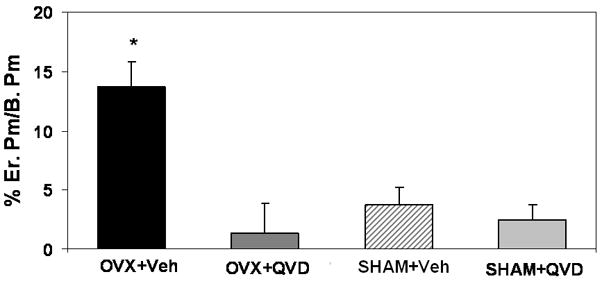
Endocortical resorption in mouse femoral diaphyses after ovariectomy with and without the pan-caspase inhibitor QVD Treatment with the apoptosis inhibitor prevented the OVX-induced increase endocortical resorption.
DISCUSSION
The current study demonstrates that following estrogen loss osteocyte apoptosis occurs in cortical bone within a discrete and highly consistent area of the cortex that coincides with the region of subsequent increased bone resorption [21]. Previous studies have demonstrated that increased osteocyte apoptosis occurs after estrogen withdrawal, but did not examine the microstructural location of the apoptotic cells or their relationship to subsequent bone resorption [19; 20]. In addition, the increased osteocyte apoptosis seen precedes resorption. This tight spatial and temporal coupling between osteocyte apoptosis and resorption after estrogen loss appears globally similar to the relationship between microdamage-induced osteocyte apoptosis and osteoclastic resorption [11; 13; 14; 31].
Parfitt [32] argued that bone remodeling could be either a stochastic or targeted process. Targeted remodeling occurs when a microscopic region of bone is damaged or has otherwise outlived its functional lifespan. This region is then resorbed and replaced by a basic multicellular unit (BMU) specifically formed at that site. Targeted remodeling to repair microdamage in bone was originally proposed by Frost, and confirmed subsequently in numerous experimental studies in animal models where damage is created by fatigue loading [11; 33; 34; 35; 36; 37]. Recent studies point to osteocyte injury and apoptosis at those bone microdamage sites as the biological “target” for osteoclastic resorption. Bentolila and co-workers found that intracortical resorption foci following fatigue loading occur in regions of pyknotic osteocyte morphology [31]. Verborgt and coworkers demonstrated that osteocytes at microdamage sites undergo apoptosis, and this apoptosis precedes and then co-localizes with the location subsequent to bone resorption [11; 13], a finding later confirmed by Noble and coworkers [38]. Most recently, Cardoso demonstrated conclusively that osteocyte apoptosis plays a direct and controlling role in the activation and targeting of a microdamage remodeling response [14]. They showed that pharmacological suppression of osteocyte apoptosis after bone fatigue in vivo using a pan-caspase inhibitor (as in experiment 2 of this investigation) prevented both the osteocyte apoptosis and the activation of new bone remodeling activity
In contrast to targeted remodeling, Parfitt posited that stochastic remodeling occurs without an identifiable tissue focus or target for resorption, as exemplified by the resorption increases seen after estrogen loss, glucocorticoid treatment and disuse [15; 18; 19; 39]. However, our current studies reveal that in cortical bone the increased bone resorption following ovariectomy occurs in a preferential (non-random) manner and that moreover, the sites of subsequent resorption mirror the distribution of increased osteocyte apoptosis. This temporal and spatial coupling between osteocyte apoptosis and bone resorption is highly similar to that seen in targeted repair of bone microdamage. These data argue that bone remodeling in response to estrogen deficiency may also be a targeted remodeling process, albeit one in which the target for osteoclasts is not a matrix damage site but is nevertheless still dying osteocytes.
The ability of the pan-caspase inhibitor Q-VD-OPh to prevent the increase of bone resorption following OVX provided evidence for a functional link between osteocyte apoptosis and the initiation of bone remodeling due to estrogen withdrawal. Because caspase action is essential for most apoptotic cell death, these enzymes are logical targets for pharmacological modulation of apoptosis [27; 28; 40; 41; 42; 43]. However, caspase inhibitors lack cell type specificity and so raise obvious questions about whether impaired resorption was due to actions on cells other than osteocytes, in particular by directly suppressing the differentiation or activity of osteoclasts. A specific caspase-3 inhibitor was shown to impair osteoblast recruitment from the bone marrow stromal cells, suggesting that some osteoblast apoptosis is needed to maintain recruitment from the precursor pool [44]. In contrast, a recently published study has demonstrated that pan-capsase inhibitors do not directly affect osteoclast recruitment or function [14]. Furthermore, the use of pan-caspase inhibition either in vivo or in vitro did not alter osteoclast forming potential of the bone marrow or the response of osteoclast precursors to m-csf or RANKL. In the current studies, use of QVD in sham animals did not significantly affect the low baseline level of endocortical resorption in B6 mice (Figure 6), confirming that the inhibitor did not directly affect osteoclast function. Only in estrogen-deprived animals did the caspase inhibitor suppress increases in osteocyte apoptosis and in osteoclastic activity. Thus, our findings point to osteocytes, and not other cell types, as the cells whose death appears to triggers the activation and targeting of bone resorption after estrogen loss. This concept received further strong support by recent work by Tatsumi and co-workers [45], who demonstrated that osteocyte death induced by selective gene-targeted ablation caused rapid bone loss.
Estrogen deficiency-induced bone loss has been widely studied in cancellous bone [5; 6; 46; 47; 48]. However, bone resorption after estrogen loss in mouse bone also occurs to a lesser degree at the diaphyseal endocortical surface [21; 48]. Moreover, compared to the complex, three-dimensional lattice architecture of the cancellous bone, the diaphyseal cortex is a tubular structure with a geometrically straightforward analysis that allowed us to identify the unique spatial and temporal relationships between estrogen loss and osteocyte apoptosis and the coupling between apoptosis and the bone remodeling response that ensues. While additional studies are needed to confirm the same cellular mechanisms in the more clinically critical cancellous bone compartment, evidence to date argues for similar fundamental control mechanisms for remodeling in all bone envelopes [31; 32; 49; 50; 51]. Thus, we posit that osteocyte apoptosis is a generalized response governing the activation and progression of bone resorption after estrogen loss.
While this study points to a functional link between estrogen loss, osteocyte apoptosis and the onset of bone resorption, how osteocyte signaling directs osteoclast formation and/or activity remains unclear. Osteoclasts belong to the monocyte-macrophage lineage. Osteoclasts, like other members of the professional phagocytic cell lineage, likely respond aggressively to remove apoptotic cells and debris in their tissue environment. Indeed recent studies by Kogianni [52] show that apoptotic debris from osteocyte-like MLO-Y4 cells stimulates resorption of newborn rat calvaria. However, phagocytosis of apoptotic cells and debris is mediated through direct contact signals that reside on the membranes of dying cells which identify them as targets for phagocytic elimination. In bone, movement of such cellular debris from damage sites to bone surfaces, where osteoclasts first form, would be limited by the low permeability of the lacuna-canalicular system to movement of large solutes [53; 54]. Diffusible signaling molecules, either from apoptotic osteocytes or more likely from the surviving osteocytes near sites of apoptosis, would seem more plausible as initial local modulators that promote osteoclastogenesis [13]. Since even in the regions showing highest apoptosis after estrogen loss only about 15–20 percent of the osteocytes die, both apoptotic and intact, non-apoptotic osteocytes are present in the regions that will undergo resorption. It seems reasonable to hypothesize that the surviving osteocytes in this region could detect their dying neighbors (perhaps through losses of connectivity or short-range movement of apoptotic debris), and that these intact osteocytes signal to the overlying bone lining cells to initiate osteoclast recruitment. Indeed, Verborgt [13] demonstrated that the non-apoptotic osteocytes surrounding sites of microinjury exhibit an active anti-apoptotic response showing that these cells are not mere bystanders and suggesting that they, as well as apoptotic osteocytes, may play a role in regulating the localized tissue response to a remodeling stimulus. Alternatively, as other investigators have hypothesized, the loss of viable osteocytes could lead to reduced local inhibition of bone resorption [45; 55; 56]. In any case, osteocytes and the onset of apoptosis are proving to be key elements in the signaling mechanisms that osteoclasts use to direct their resorptive activity to specific areas of bone.
Among the most striking findings of this study was that osteocyte apoptosis after estrogen loss occurred in a discrete and anatomically consistent region of the diaphyseal cortex (i.e., the posterior cortex). Why osteocytes in this region of bone were preferentially affected by estrogen withdrawal remains obscure at this time. However, the biology of this location suggests some intriguing possibilities. Images of sequential bone fluorochrome labeled mouse femurs obtained during growth and development studies demonstrate that the posterior cortical region, where osteocyte apoptosis occurred after OVX, is the earliest-formed tissue in this area of the diaphysis and hence contains the oldest osteocytes [57]. This region is also composed principally of compacted cancellous bone, while the anterior cortex (exhibiting low apoptosis after OVX) is principally lamellar. Enlow [58] noted that compacted cancellous bone regions were the first within the cortex to undergo intracortical resorption, and also noted that this resorption was associated with dying osteocytes. Confocal microscopic images of these bones showed that osteocytes in the compacted cancellous bone of the posterior cortex are structurally distinct from those in the lamellar bone (low apoptosis) bone regions. Specifically, these osteocytes have significantly fewer connections to their neighbors than osteocytes in the anterior cortex. Fewer connections argue for impaired fluid and solute transport to and from these cells [53; 59]. Older osteocytes that are less well connected to their neighbors than those in other bone areas suggest a reasonable possibility that these apoptosis-prone osteocytes will have accumulated more stress (e.g. due to oxidative damage) than their younger, better-connected counterparts. Estrogen is well established as an antioxidant capable of regulating the glutathione and thioredoxin pathways [38; 60; 61; 62; 63; 64; 65]. Loss of estrogen has been shown to accelerate aging effects on bone by decreasing the ability of cells to mount a defense against reactive oxygen species [38; 60; 61]. It seems reasonable to speculate that osteocytes in oldest tissue are more primed for apoptosis because of their age and cumulative life stress, and that removal of estrogen [61; 63] constitutes a greater relative insult to older osteocytes that may already be living “closer to the edge.”
In summary, we found that estrogen withdrawal results in osteocyte apoptosis in cortical bone that i) occurs in an anatomically consistent areas, within the cortex, ii) precedes and coincides with areas that subsequently undergo osteoclastic resorption, and iii) is required in order to initiate this resorptive response. These responses to estrogen loss parallel the highly orchestrated, osteocyte apoptosis-based control seen for targeted remodeling of bone microdamage. This concordance of events suggest that dying osteocytes, irrespective of the cause, become targets for osteoclast recruitment and resorption, and that apoptosis is a necessary and controlling factor in the bone remodeling process. Thus, osteocyte apoptosis may represent a common pathway for activating and targeting bone resorption in response to diverse stimuli.
Supplementary Material
Supplement 1: Osteocyte apoptosis (% Casp + Ot.) around the femoral mid-shaft after ovariectomy for all time periods examined.
Supplement 2: Effect of QVD on osteocyte apoptosis after ovariectomy at the femoral mid-shaft: Data for all anatomical locations and all treatment groups.
Acknowledgments
This work was supported by grants AR41210 and AR44927 from National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the National Space Biomedical Research Institute through NASA NCC 9–58. We are grateful to Damien Laudier for sharing his expertise of bone specimen preparation, sectioning and immunostaining. We also thank Dr. Shoshana Yakar and Dr. Brad Herman for their helpful discussions and insightful comments.
Footnotes
Conflict of Interest Statement: None of the authors has a conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Johnell O, Kanis JA, Oden A, Sernbo I, Redlund-Johnell I, Petterson C, De Laet C, Jonsson B. Mortality after osteoporotic fractures. Osteoporos Int. 2004;15(1):38–42. doi: 10.1007/s00198-003-1490-4. [DOI] [PubMed] [Google Scholar]
- 2.Pacifici R. Mechanisms of Estrogen Action in Bone. Principles of Bone Biology. 2002;1:693–705. [Google Scholar]
- 3.Fox J, Newman MK, Turner CH, Guldberg RE, Varela A, Smith SY. Effects of treatment with parathyroid hormone 1-84 on quantity and biomechanical properties of thoracic vertebral trabecular bone in ovariectomized rhesus monkeys. Calcif Tissue Int. 2008;82(3):212–20. doi: 10.1007/s00223-008-9108-7. [DOI] [PubMed] [Google Scholar]
- 4.Zaman G, Jessop HL, Muzylak M, De Souza RL, Pitsillides AA, Price JS, Lanyon LL. Osteocytes use estrogen receptor alpha to respond to strain but their ERalpha content is regulated by estrogen. J Bone Miner Res. 2006;21(8):1297–306. doi: 10.1359/jbmr.060504. [DOI] [PubMed] [Google Scholar]
- 5.Waarsing JH, Day JS, Verhaar JA, Ederveen AG, Weinans H. Bone loss dynamics result in trabecular alignment in aging and ovariectomized rats. J Orthop Res. 2006;24(5):926–35. doi: 10.1002/jor.20063. [DOI] [PubMed] [Google Scholar]
- 6.McNamara LM, Ederveen AG, Lyons CG, Price C, Schaffler MB, Weinans H, Prendergast PJ. Strength of cancellous bone trabecular tissue from normal, ovariectomized and drug-treated rats over the course of ageing. Bone. 2006;39(2):392–400. doi: 10.1016/j.bone.2006.02.070. [DOI] [PubMed] [Google Scholar]
- 7.Kalu DN. The ovariectomized rat model of postmenopausal bone loss. Bone Miner. 1991;15(3):175–91. doi: 10.1016/0169-6009(91)90124-i. [DOI] [PubMed] [Google Scholar]
- 8.Smith SY, Jolette J, Turner CH. Skeletal health: primate model of postmenopausal osteoporosis. Am J Primatol. 2009;71(9):752–65. doi: 10.1002/ajp.20715. [DOI] [PubMed] [Google Scholar]
- 9.Oshima S, Onodera S, Amizuka N, Li M, Irie K, Watanabe S, Koyama Y, Nishihira J, Yasuda K, Minami A. Macrophage migration inhibitory factor-deficient mice are resistant to ovariectomy-induced bone loss. FEBS Lett. 2006;580(5):1251–6. doi: 10.1016/j.febslet.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 10.Noble BS, Stevens H, Loveridge N, Reeve J. Identification of apoptotic changes in osteocytes in normal and pathological human bone. Bone. 1997;20(3):273–82. doi: 10.1016/s8756-3282(96)00365-1. [DOI] [PubMed] [Google Scholar]
- 11.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000;15(1):60–7. doi: 10.1359/jbmr.2000.15.1.60. [DOI] [PubMed] [Google Scholar]
- 12.Hedgecock NL, Hadi T, Chen AA, Curtiss SB, Martin RB, Hazelwood SJ. Quantitative regional associations between remodeling, modeling, and osteocyte apoptosis and density in rabbit tibial midshafts. Bone. 2007;40(3):627–37. doi: 10.1016/j.bone.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Verborgt O, Tatton NA, Majeska RJ, Schaffler MB. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J Bone Miner Res. 2002;17(5):907–14. doi: 10.1359/jbmr.2002.17.5.907. [DOI] [PubMed] [Google Scholar]
- 14.Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24(4):597–605. doi: 10.1359/JBMR.081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu G, Hentunen TA, Nars M, Harkonen PL, Vaananen HK. Estrogen protects primary osteocytes against glucocorticoid-induced apoptosis. Apoptosis. 2005;10(3):583–95. doi: 10.1007/s10495-005-1893-0. [DOI] [PubMed] [Google Scholar]
- 16.Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab. 2000;85(8):2907–12. doi: 10.1210/jcem.85.8.6714. [DOI] [PubMed] [Google Scholar]
- 17.Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104(10):1363–74. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21(4):605–15. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 19.Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS. The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res. 1998;13(8):1243–50. doi: 10.1359/jbmr.1998.13.8.1243. [DOI] [PubMed] [Google Scholar]
- 20.Tomkinson A, Reeve J, Shaw RW, Noble BS. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab. 1997;82(9):3128–35. doi: 10.1210/jcem.82.9.4200. [DOI] [PubMed] [Google Scholar]
- 21.Li CY, Schaffler MB, Wolde-Semait HT, Hernandez CJ, Jepsen KJ. Genetic background influences cortical bone response to ovariectomy. J Bone Miner Res. 2005;20(12):2150–8. doi: 10.1359/JBMR.050819. [DOI] [PubMed] [Google Scholar]
- 22.Laudier D, Schaffler MB, Flatow EL, Wang VM. Novel procedure for high-fidelity tendon histology. J Orthop Res. 2007;25(3):390–5. doi: 10.1002/jor.20304. [DOI] [PubMed] [Google Scholar]
- 23.Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–45. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 24.Iwata A, Harlan JM, Vedder NB, Winn RK. The caspase inhibitor z-VAD is more effective than CD18 adhesion blockade in reducing muscle ischemia-reperfusion injury: implication for clinical trials. Blood. 2002;100(6):2077–80. doi: 10.1182/blood-2002-03-0752. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Colbourne F, Sun P, Zhao Z, Buchan AM, Iadecola C. Caspase inhibitors reduce neuronal injury after focal but not global cerebral ischemia in rats. Stroke. 2000;31(1):176–82. doi: 10.1161/01.str.31.1.176. [DOI] [PubMed] [Google Scholar]
- 26.Patil K, Sharma SC. Broad spectrum caspase inhibitor rescues retinal ganglion cells after ischemia. Neuroreport. 2004;15(6):981–4. doi: 10.1097/00001756-200404290-00010. [DOI] [PubMed] [Google Scholar]
- 27.Yang L, Sugama S, Mischak RP, Kiaei M, Bizat N, Brouillet E, Joh TH, Beal MF. A novel systemically active caspase inhibitor attenuates the toxicities of MPTP, malonate, and 3NP in vivo. Neurobiol Dis. 2004;17(2):250–9. doi: 10.1016/j.nbd.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8(4):345–52. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- 29.Cardoso L, Laudier D, Majeska R, Schaffler M. Inhibition of osteocyte apoptosis prevents activation of bone remodeling after fatigue in vivo. 52nd Annual Meeting of the Orthopaedic Research Society.2005. [Google Scholar]
- 30.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275(13):9390–5. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 31.Bentolila V, Boyce T, Fyhrie D, Drumb R, Skerry T, Schaffler MB. Intracortical remodeling in adult rat long bones after fatigue loading. Bone. 1998;23:275–81. doi: 10.1016/s8756-3282(98)00104-5. [DOI] [PubMed] [Google Scholar]
- 32.Parfitt AM. Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem. 1994;55(3):273–86. doi: 10.1002/jcb.240550303. [DOI] [PubMed] [Google Scholar]
- 33.Burr DB. Targeted and nontargeted remodeling. Bone. 2002;30(1):2–4. doi: 10.1016/s8756-3282(01)00619-6. [DOI] [PubMed] [Google Scholar]
- 34.Frost H. Presence of Microscopic Cracks In Vivo In Bone. Henry Ford Hospital Medical Bulletin. 1960;8:25–35. [Google Scholar]
- 35.Barrett JG, Sample SJ, McCarthy J, Kalscheur VL, Muir P, Prokuski L. Effect of short-term treatment with alendronate on ulnar bone adaptation to cyclic fatigue loading in rats. J Orthop Res. 2007;25(8):1070–7. doi: 10.1002/jor.20395. [DOI] [PubMed] [Google Scholar]
- 36.Colopy SA, Benz-Dean J, Barrett JG, Sample SJ, Lu Y, Danova NA, Kalscheur VL, Vanderby R, Jr, Markel MD, Muir P. Response of the osteocyte syncytium adjacent to and distant from linear microcracks during adaptation to cyclic fatigue loading. Bone. 2004;35(4):881–91. doi: 10.1016/j.bone.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 37.Muir P, Sample SJ, Barrett JG, McCarthy J, Vanderby R, Jr, Markel MD, Prokuski LJ, Kalscheur VL. Effect of fatigue loading and associated matrix microdamage on bone blood flow and interstitial fluid flow. Bone. 2007;40(4):948–56. doi: 10.1016/j.bone.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Mann V, Huber C, Kogianni G, Collins F, Noble B. The antioxidant effect of estrogen and Selective Estrogen Receptor Modulators in the inhibition of osteocyte apoptosis in vitro. Bone. 2007;40(3):674–84. doi: 10.1016/j.bone.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 39.Parfitt AM. Hormonal influences on bone remodeling and bone loss: applicatin to the management of primary hyperparathyroidism. Ann Intern Med. 1996;125(5):413–5. doi: 10.7326/0003-4819-125-5-199609010-00009. [DOI] [PubMed] [Google Scholar]
- 40.Wiren KM, Toombs AR, Semirale AA, Zhang X. Osteoblast and osteocyte apoptosis associated with androgen action in bone: requirement of increased Bax/Bcl-2 ratio. Bone. 2006;38(5):637–51. doi: 10.1016/j.bone.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 41.Saintier D, Khanine V, Uzan B, Ea HK, de Vernejoul MC, Cohen-Solal ME. Estradiol inhibits adhesion and promotes apoptosis in murine osteoclasts in vitro. J Steroid Biochem Mol Biol. 2006;99(4–5):165–73. doi: 10.1016/j.jsbmb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- 43.Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, Cohen GM. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem J. 1996;315(Pt 1):21–4. doi: 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miura M, Chen XD, Allen MR, Bi Y, Gronthos S, Seo BM, Lakhani S, Flavell RA, Feng XH, Robey PG, Young M, Shi S. A crucial role of caspase-3 in osteogenic differentiation of bone marrow stromal stem cells. J Clin Invest. 2004;114(12):1704–13. doi: 10.1172/JCI20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, Ito M, Takeshita S, Ikeda K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5(6):464–75. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Nazarian A, Cory E, Muller R, Snyder BD. Shortcomings of DXA to assess changes in bone tissue density and microstructure induced by metabolic bone diseases in rat models. Osteoporos Int. 2008 doi: 10.1007/s00198-008-0632-0. [DOI] [PubMed] [Google Scholar]
- 47.Fuchs RK, Phipps RJ, Burr DB. Recovery of Trabecular and Cortical Bone Turnover Following Discontinuation of Risedronate and Alendronate Therapy in Ovariectomized Rats. J Bone Miner Res. 2008 doi: 10.1359/JBMR.080501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bouxsein ML, Myers KS, Shultz KL, Donahue LR, Rosen CJ, Beamer WG. Ovariectomy-induced bone loss varies among inbred strains of mice. J Bone Miner Res. 2005;20(7):1085–92. doi: 10.1359/JBMR.050307. [DOI] [PubMed] [Google Scholar]
- 49.Li CY, Jepsen KJ, Majeska RJ, Zhang J, Ni R, Gelb BD, Schaffler MB. Mice lacking cathepsin K maintain bone remodeling but develop bone fragility despite high bone mass. J Bone Miner Res. 2006;21(6):865–75. doi: 10.1359/jbmr.060313. [DOI] [PubMed] [Google Scholar]
- 50.Hernandez CJ, Majeska RJ, Schaffler MB. Osteocyte density in woven bone. Bone. 2004;35(5):1095–9. doi: 10.1016/j.bone.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. Faseb J. 2005;19(13):1842–4. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 52.Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23(6):915–27. doi: 10.1359/jbmr.080207. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, Ciani C, Doty SB, Fritton SP. Delineating bone’s interstitial fluid pathway in vivo. Bone. 2004;34(3):499–509. doi: 10.1016/j.bone.2003.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L, Wang Y, Han Y, Henderson SC, Majeska RJ, Weinbaum S, Schaffler MB. In situ measurement of solute transport in the bone lacunar-canalicular system. Proc Natl Acad Sci U S A. 2005;102(33):11911–6. doi: 10.1073/pnas.0505193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42(4):606–15. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin RB. Toward a unifying theory of bone remodeling. Bone. 2000;26(1):1–6. doi: 10.1016/s8756-3282(99)00241-0. [DOI] [PubMed] [Google Scholar]
- 57.Price C, Herman BC, Lufkin T, Goldman HM, Jepsen KJ. Genetic variation in bone growth patterns defines adult mouse bone fragility. J Bone Miner Res. 2005;20(11):1983–91. doi: 10.1359/JBMR.050707. [DOI] [PubMed] [Google Scholar]
- 58.Enlow DH. A study of the post-natal growth and remodeling of bone. Am J Anat. 1962;110:79–101. doi: 10.1002/aja.1001100202. [DOI] [PubMed] [Google Scholar]
- 59.Ciani C, Doty SB, Fritton SP. Mapping bone interstitial fluid movement: displacement of ferritin tracer during histological processing. Bone. 2005;37(3):379–87. doi: 10.1016/j.bone.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, Kousteni S, O’Brien CA, Bellido T, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282(37):27285–97. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, Urry ZL, Chambers TJ. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112(6):915–23. doi: 10.1172/JCI18859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu H, Wang H, Shenvi S, Hagen TM, Liu RM. Glutathione metabolism during aging and in Alzheimer disease. Ann N Y Acad Sci. 2004;1019:346–9. doi: 10.1196/annals.1297.059. [DOI] [PubMed] [Google Scholar]
- 63.Grassi F, Tell G, Robbie-Ryan M, Gao Y, Terauchi M, Yang X, Romanello M, Jones DP, Weitzmann MN, Pacifici R. Oxidative stress causes bone loss in estrogen-deficient mice through enhanced bone marrow dendritic cell activation. Proc Natl Acad Sci U S A. 2007;104(38):15087–92. doi: 10.1073/pnas.0703610104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tam NN, Ghatak S, Ho SM. Sex hormone-induced alterations in the activities of antioxidant enzymes and lipid peroxidation status in the prostate of Noble rats. Prostate. 2003;55(1):1–8. doi: 10.1002/pros.10169. [DOI] [PubMed] [Google Scholar]
- 65.Lee MY, Jung SC, Lee JH, Han HJ. Estradiol-17beta protects against hypoxia-induced hepatocyte injury through ER-mediated upregulation of Bcl-2 as well as ER-independent antioxidant effects. Cell Res. 2008;18(4):491–9. doi: 10.1038/cr.2008.42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 1: Osteocyte apoptosis (% Casp + Ot.) around the femoral mid-shaft after ovariectomy for all time periods examined.
Supplement 2: Effect of QVD on osteocyte apoptosis after ovariectomy at the femoral mid-shaft: Data for all anatomical locations and all treatment groups.