

Abstract
A simple and reliable continuous assay for measurement of α-mannosidase activity is described and demonstrated for analysis with two recombinant human enzymes using the new substrate resorufin α-D-mannopyranoside (Res-Man). The product of enzyme reaction, resorufin, exhibits fluorescence emission at 585 nm with excitation at 571 nm and has a pKa of 5.8, allowing continuous measurement of fluorescence turnover at or near physiological pH values for human lysosomal and Drosophila Golgi α-mannosidases. The assay performed using recombinant Drosophila Golgi α-mannosidase (dGMII) has been shown to give the kinetic parameters Km of 200 μM and Vmax of 11 nmol/min per nmol dGMII. Methods for performing the assay using several concentrations of the known α-mannosidase inhibitor swainsonine are also presented, demonstrating a potential for use of the assay as a simple method for high-throughput screening of inhibitors potentially useful in cancer treatment.
Keywords: continuous fluorescent enzyme assay, kinetic assay, GMII, Golgi mannosidase II, dGMII, Drosophila melanogaster mannosidase II, fluorogenic substrate, Res-Man, resorufin α-D-mannopyranoside
Introduction
The N-glycosylation pathway is a target for chemical intervention in a number of pathological conditions including cancer. Cells that have undergone oncogenic transformation often display abnormal cell surface oligosaccharides [1-4]. These changes in glycosylation are important determinants of tumor progression. Inhibition of the mannose trimming enzyme Golgi α–mannosidase II (GMII; mannosyl-oligosaccharide 1,3-1,6-α–mannosidase II; E.C. 3.2.1.114) is a potential route for blocking the changes in cell surface oligosaccharides [5,6]. GMII is the final glycoside hydrolase in the N-glycosylation pathway and is responsible for the formation of the “core” tri-mannose structure. GMII catalyses the hydrolysis of both an α-1,6 and an α-1,3 linked mannose from GlcNAc-Man5-GlcNAc2-Asn to form GlcNAc-Man3-GlcNAc2-Asn [7,8]. Subsequently a series of glycosyl transferases add a variety of carbohydrates (including N-acetyl glucosamine, galactose and sialic acid) to this tri-mannose core to form the completed complex carbohydrate structure.
The α1,3 α1,6-mannosidases including GMII and lysosomal α-mannosidase (LM, E.C.3.2.1.24) are members of the Family 38 glycoside hydrolases. These enzymes cleave between 2 mannose residues with a net retention of configuration and operate via a mechanism involving formation of a covalent glycosyl-enzyme intermediate. Lysosomal α-mannosidases are involved in degradation of complex sugars derived from glycoproteins within the low pH environment of the lysosome. Impairment of LM activity, through genetic mutation or environmental toxicity, leads to the buildup of partially processed oligosaccharides in large vacuolar structures within cells and has severe neurological consequences [9,10]. Initial clinical tests indicated that the GMII inhibitor swainsonine (which inhibits in the nanomolar range) had promise as a cancer treatment agent [8]. However, side effects, possibly caused by the inhibition of LM by swainsonine, severely limit its usefulness, necessitating the search for more specific inhibitors.
Golgi α-mannosidase II (GMII) is thus a target for a number of synthetic efforts aimed at providing more selective and effective inhibitors. The catalytic domain of the Drosophila melanogaster enzyme (dGMII) has been extensively studied due to the ease of obtaining high resolution structural data [11]. Structural details of the substrate cleavage events have been elucidated [12-14]. GMII employs a two-stage mechanism involving two carboxylic acids positioned within the active site, which act in concert: one as a catalytic nucleophile and the other as a general acid/base catalyst. Protonation of the exocyclic glycosyl oxygen of a substrate molecule leads to bond-breaking and simultaneous attack of the catalytic nucleophile to form a glycosyl enzyme intermediate [12]. Hydrolysis of the covalent intermediate by a nucleophilic water molecule gives an α-mannose product. Substrate rearrangement in the active site repositions the second mannose for a second round of cleavage [13, 14].
The pH optimum of dGMII is low (pH 5.7) [15]. An existing fluorogenic substrate, 4-methylumbelliferyl α-D-mannopyranoside (MU-Man) [16] cannot be used for continuous fluorometric measurement because of the high pKa value of the released product, 4-methylumbelliferone, which has a pKa of 7.8 [17]. This substrate does not allow for continuous measurement of activity, as they are not substantially fluorescent at these low pH values if 360 nm excitation is used. α-Mannosidase has also been assayed for previously using a chromogenic substrate, p-nitrophenyl α-D-mannopyranoside (PNP-Man) [15,16]. However, chromogenic substrates suffer from low sensitivity, and the absorbance readings of released PNP also cannot be recorded continuously at low pH, as PNP has a pKa of 7.1 [18]. Addition of a high pH stop buffer, such as 0.5 M sodium bicarbonate [15] or glycine-NaOH buffer [16], is required prior to measurement when using either substrate. 5-bromo-4-chloro-3-indolyl α-D-mannopyranoside (X-Man) is a chromogenic substrate that releases a precipitating blue dye upon cleavage, and while activity can be observed at low pH [27], the solid nature of the product makes activity measurements difficult to quantitate. In order to overcome these limitations, a long wavelength fluorescent substrate, resorufin α-D-mannopyranoside (Res-Man), was prepared and utilized to obtain continuous fluorometric measurement of α-mannosidase activity. This substrate releases the red fluorescent fluorophore Resorufin (EX:571; EM:585) with pKa of 5.8 [19]. Use of resorufin as the released product for low pH enzymes has previously been found effective in measuring cellulase activity continuously [20]. We present several examples of continuous α-mannosidase activity measurement from recombinant samples of purified α-mannosidase; α-mannosidase II from dGMII [15], and human lysosomal α-mannosidase (HLM) at pH 6.0. The results indicate that the new Res-Man substrate can be used to provide continuous fluorometric measurement, kinetic analysis, and inhibition screening of α-mannosidase activity at physiological pH.
Materials and Methods
Chemicals and Instruments
Resorufin, sodium salt was obtained from Sigma Chemical Co. (St. Louis, MO). Hydrochloric acid, acetic anhydride, and dry pyridine were obtained from Mallinckrodt Chemicals (Phillipsburg, PA). Hydrobromic acid (33 wt % solution in glacial acetic acid), silver carbonate, Amberlite IRC-50 ion-exchange resin, sodium methoxide (25 wt % solution in methanol), and anhydrous dichloromethane were obtained from Aldrich Chemical Co. (Milwaukee, WI). Sym-collidine was from Acros Organics (Morris Plains, NJ). All-chemicals were used without further purification. 1H-NMR spectra were obtained using a Varian Inova 300-MHz nuclear magnetic resonance instrument. Microtiter plates used were from Becton–Dickinson Bio-Products Division (BD-Falcon plate No. 351172; (96 well, clear, flat bottom). Fluorescence readings were obtained using a Perkin–Elmer HTS 7000 Plus Bio-Assay Reader and HTSoft Analysis Software.
Synthesis
Resorufin-free acid
Resorufin, sodium salt (3.00 g, 12.75 mmol) was dissolved in ice–water (150 mL) and concentrated hydrochloric acid (6 mL) was added with stirring to pH 2. After stirring at 0°C (3 h), the red-brown precipitate was filtered and washed with water until the filtrate was neutral. The resulting resorufin-free acid was dried in air and in vacuo overnight to yield a dark red solid (2.64 g, 97%), homogeneous by TLC analysis (9:1 CH2Cl2:MeOH, Rf = 0.43).
Resorufin alpha-D-mannopyranoside, tetra-O-acetate
Under an anhydrous N2 (g) atmosphere, a mixture of acetobromomannoside (prepared by per-acetylation of D-mannose using excess acetic anhydride and pyridine [21], followed by treatment with HBr in glacial acetic acid) (2.37 g, 5.77 mmol), anhydrous CH2Cl2 (20 mL), anhydrous acetonitrile (20 mL), resorufin (370 mg, 1.74 mmol), dry silver carbonate (855 mg, 3.10 mmol), and sym-collidine (275 μL) were allowed to stir under anhydrous conditions at room temperature overnight, protected from light. After this time, the reaction mixture was filtered through a Celite™ pad and the grey solids were washed with additional CH2Cl2 (30 mL). The clear orange filtrate was evaporated to near dryness and redissolved in CH2Cl2 (50 mL) washed with water (2 × 50 mL), saturated aqueous sodium bicarbonate solution (1 × 50 mL), water (1 × 50 mL), 1 N HCl/H2O (1 × 50 mL), 0.2N sodium thiosulfate solution (1 × 50 mL) and water (1 × 50 mL). The resulting organic layer was dried over anhydrous sodium sulfate, filtered, evaporated, and dried in vacuo overnight to yield an orange powder (3.65 g). Purification by column chromatography (SiO2, 70–230 mesh, 25 · 75 mm, irrigant = 9:1 CH2Cl2:EtOAc) produced a bright orange solid that was recrystallized from ethylacetate to give amorphous reddish crystals (0.89 g, 95%). (Rf = 0.16; irrigant = 9:1 CH2Cl2:EtOAc). 1H-NMR (CDCl3, 300 MHz): d 7.72 (d, 1H), 7.43 (d, 1H), 6.95 (dd, 1H), 6.83 (dd, 2H), 6.33 (d, 1H), 6.09 (d, 1H), 5.35 (m, 1H), 5.25 (d, 1H), 4.30 (dd, 1H), 4.10 (m, 2H), 3.5 (m, 1H), 2.18 (s, 3H), 2.10 (s, 3H), 2.06 (s, 3H), 2.01 (s, 3H).
Resorufin-α-D-mannoside
Resorufin mannoside, tetraacetate (417 mg, 0.50 mmol) was suspended in anhydrous methanol (15 mL) and sodium methoxide, 25 % wt/v solution in methanol (300 μL) was added (to pH 10). This reaction was stirred overnight at room temperature, and the resulting bright orange product was filtered, washed with minimum anhydrous methanol, and dried in vacuo overnight to produce an orange solid that was recrystallized from acetonitrile. Yield = 268 mg, 99% TLC (Rf = 0.83, irrigant = 7:3 EtOAc:MeOH). 1H-NMR (DMSO-d6, 300 MHz): d = 7.80 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 8.5 Hz, 1H), 7.17 (d, J = 0.8 Hz, 1H), 7.10 (dd, J = 7.8, 0.8 Hz, 1H), 6.79 (dd, J = 7.8, 0.84 Hz, 1H), 6.39 (d, J = 1.3 Hz, 1H), 5.61 (d, J = 6.5 Hz, 1H), 5.21 (dd, J = 13.7, 5.9 Hz, 2H), 5.02 (dd, J = 9.1, 5.9 Hz, 2H), 4.83 (d, J = 0.1 Hz, 1H), 4.65 (m, J = 7.8 Hz, 2H), 4.28 (d, J = 0.7 Hz, 1H), 3.63 (m, 4H), 3.45 (m, 3H), 3.10 (m, 6H).
Protein purification
The purification and characterization of full-length human lysosomal mannosidase (HLM) will be described in more detail in a subsequent publication (M. Venkatesan, D.A. Kuntz, D.R. Rose, manuscript submitted). Briefly, HLM containing a C-terminal His6 tag was expressed in a secreted form in Drosophila S2 cells using the DES expression system (Invitrogen, Carlsbad CA). 2.5 mM CoCl2 was added to the medium containing secreted HLM. This was batch bound to Chelating Sepharose Fast Flow (GE Biosciences, Montreal PQ). Bound protein was eluted with decreasing pH buffers added stepwise. The pooled dialyzed fractions were then resolved by cation exchange on SP HiTrap (GE Biosciences). HLM was eluted over a gradient of 0.2-0.6M NaCl. The enzyme was concentrated to 1 mg/mL before being snap-frozen in liquid nitrogen for storage.
dGMII purification was essentially as described previously [11] with an added anion-exchange step to further purify the enzyme. Briefly, the soluble catalytic domain of Drosophila GMII (residues 76-1108) containing an N-terminal His6 tag was expressed in a secreted form in S2 cells. The medium was batch bound to Blue F3GA agarose (Sigma, St. Louis MO). dGMII was eluted with 0.35 M NaCl and directly batch bound to Ni-NTA agarose (Qiagen, Montreal PQ) from which it was eluted with 30 mM imidazole. After dialysis the protein was further purified by salt elution from a MonoQ anion exchange column (GE Biosciences), followed by subsequent dialysis, concentration, and freezing in liquid N2.
Enzyme Assays
Continuous assay of α-mannosidase enzymes
For all assays, the enyzmes dGMII and HLM were diluted to 67 μg/mL in 100 mM sodium acetate buffer, pH 6.0 (reaction buffer). Each enzyme preparation was added in triplicate (10 μL) to wells in a clear, flat-bottomed 96-well polystyrene microtiter plate (BD Falcon No. 351172). 40 μL of reaction buffer was added to each sample. A blank sample containing no enzyme (50 μL reaction buffer) was also prepared in triplicate wells. A 0.5 mM substrate reagent solution was prepared by diluting a 5 mM DMSO stock 1:10 in reaction buffer. The substrate reagent solution (50 μL) was added to each well to a final concentration of 0.25 mM. Final concentrations were 6.7 μg/mL for both enzymes (55-60 nM). Fluorescence was measured using a Perkin-Elmer HTS 7000 Plus BioAssay Reader in top read mode (excitation filter = 550 nm, emission filter = 595 nm). Fluorescence was recorded at room temperature for 40 cycles with a cycle time of 1 min. All readings were performed in triplicate and averaged.
Continuous kinetic assay
Four Res-Man substrate reagent solutions (1.0, 0.60, 0.30, 0.20 mM) were prepared by diluting DMSO stocks 1:5 in reaction buffer. The substrate reagent solutions were added in triplicate (50 μL) to wells in a clear, flat-bottomed 96-well polystyrene microtiter plate (BD Falcon No. 351172). 40 μL of reaction buffer was added to each well. 10 uL of diluted dGMII was added to the above wells. Final substrate concentrations were 0.50, 0.30, 0.20, and 0.10 mM. Final enzyme concentration was 56 nM. Fluorescence was measured as above. Fluorescence was recorded at 30-s intervals for 210 s, and the mean fluorescence value of a blank (50 μL substrate reagent added to 50 μL reaction buffer, triplicate wells) was subtracted from the value of each sample well to normalize data at each time point. A standard curve was generated by plotting fluorescence of four concentrations of resorufin standards (50, 20, 10, 5.0 μM) prepared by diluting DMSO stocks in sodium acetate buffer and adding to microtiter plate wells in triplicate (100 μL). All readings from triplicate wells were averaged. The curve generated from the standards was used to convert raw fluorescence data into μmol/mL/min resorufin produced. Initial velocities were determined from the linear portion of this curve using GraFit™ and Microsoft Excel graphing software. Kinetic parameters Km and Vmax were calculated using GraFit™ software employing a nonlinear regression analysis.
Continuous assay using known mannosidase inhibitor
Ten microlitres of diluted dGMII was added to 27 wells in a clear, flat-bottomed 96-well polystyrene microtiter plate (BD Falcon No. 351172). Swainsonine (H2O stock) (Sigma 068K8721) was added to wells at 8 different concentrations in triplicate, and well volume adjusted to 50 μL with reaction buffer. A set of triplicate wells received no swainsonine, and well volume was adjusted to 50μL with reaction buffer. Final concentrations of swainsonine were 10.0 μM, 1.00 μM, 500 nM, 250 nM, 100 nM, 50.0 nM, 25.0 nM, 10.0 nM, and 0.00 nM. A 0.5 mM substrate reagent solution was prepared by diluting a 5 mM DMSO stock 1:10 in reaction buffer. Fifty micolitres of the substrate reagent solution was added to each well to give a final concentration of 0.25 mM. Final concentration of enzyme in all wells was 56 nM. Fluorescence was measured using a Perkin–Elmer HTS 7000 Plus BioAssay Reader in top read mode (excitation filter = 550 nm, emission filter = 595 nm). Fluorescence was recorded at room temperature for 30 cycles with a cycle time of 1 min. All readings were performed in triplicate and averaged. The mean fluorescence value of a blank (50 μL substrate reagent added to 50 μL reaction buffer, triplicate wells) was subtracted from the value of each sample well to normalize data at each time point. IC50 was determined as the concentration of inhibitor that results in an initial velocity 50% that of the sample containing no inhibitor. IC50 was used along with previously calculated Km to determine Ki.
Results and Discussion
Several fluorescent and chromogenic mannopyranosides are commercially available for use in detection of α-mannosidase activity, including 4-methylumbelliferyl-α -D-mannopyranoside (MU-Man), p-nitrophenyl α-D-mannopyranoside (PNP-Man), and 5-Bromo-4-chloro-3-indolyl α-D-mannopyranoside (X-Man). However, kinetic assays using these substrates are inconvenient. PNP-Man and X-Man hydrolysis release a chromogenic product, which is less sensitive than using fluorescence spectroscopy and can be difficult to quantitate if enzyme activity is low. In addition, quantitation of PNP requires addition of a high pH buffer (i.e. a “stop” buffer) before absorbance can be recorded, elevating the pH of the reaction mixture above the pKa of PNP, 7.1 [18]. In order to obtain kinetic data, aliquots must be removed and measured at defined intervals. The product of X-Man hydrolysis is a dark blue precipitate that is difficult to quantitate. Assays using MU-Man must be obtained either photometrically (resulting in reduced sensitivity), or, to perform more sensitive fluorescence readings, aliquots must be removed at several time points, at which a stop buffer is added [16], elevating the pH of the reaction mixture above 7.8, the pKa of the product, 4-methylumbelliferone (4-MU) [17]. 4-MU gives minimal fluorescence at pH 5.7, the reported optimum for dGMII [15]. Other methods of continuous quantitation using these substrates involve analysis of product turnover using cumbersome thin layer chromatography or HPLC analysis of aliquots removed at various time points. These methods are time-consuming and limit the ability to perform sensitive kinetic analyses of the enzymatic reaction at early time points of the reaction.
Previous success has been achieved in measuring activity of cellulases, which also have optimal activities at acidic pH, by using a substrate that releases the fluorophore resorufin, resorufin β-D-cellobioside [20]. Resorufin is a long-wavelength, red-fluorescent fluorophore (EX:571, EM:585) with a pKa of 5.8 [16]. As previously demonstrated [20], its low pKa relative to 4-methylumbelliferone allows resorufin to retain appreciable fluorescence at low pH values. Very good results were achieved previously using resorufin β-D-cellobioside (Res-CB) for continuous fluorometric determination of cellulase activity at pH 6.0 [20]. Thus, it was reasoned that a resorufin-based substrate for α-mannosidase, resorufin α-D-mannopyranoside could be effective in obtaining continuous fluorometric measurements and kinetic enzyme data for α -mannosidases at or near their physiological pH values using long-wavelength excitation and emission wavelengths, that help avoid (albeit to a limited to extent) background fluorescence from HTS library compounds and endogenous cell lysate components.
Synthesis of Resorufin α-D-Mannoside (Res-Man)
Our synthetic method was similar to those used for other resorufin glycosides (Figure 1) [22]. The starting material was the free phenol form of resorufin, prepared by acid treatment and crystallization of commercially available resorufin sodium salt (Sigma Chem. Co.). The glycosylation reaction utilized the peracetylated 1-deoxy-1-bromo derivative of D-mannose that could be prepared by sequential acetylation and treatment of the anomeric mixture of α- and β-Mannose pentaacetates with HBr in glacial acetic acid at 0°C. Using these methods, high yields of the reactive bromo-sugar product were produced. The anomeric configuration of the acetobromo-D-mannose produced was found to be a mixture of alpha and beta anomers, as evidenced by the 1H-n.m.r. analysis.
Figure 1.

Res-Man Synthetic Scheme.
Glycosylation was carried out utilizing a modified Koenigs-Knorr methodology [23] employing silver carbonate as catalyst and sym-collidine as proton scavenger to give the protected intermediate resorufin α-D-mannoside, tetraacetate. Zemplen deprotection [24] with catalytic sodium methoxide in methanol afforded the desired Res-Man substrate as a single product that crystallized from the reaction mixture in virtually quantitative yield.
The Res-Man substrate thus produced was found to be stable and could be stored for long periods of time at −18°C (> 6 mo.), dessicated in a powdered form, or in anhydrous DMSO solution (5 mM), without noticeable decomposition (TLC analysis).
Enzymatic Assays of recombinant α-mannosidases
Our results demonstrate the ability of this new substrate to provide continuous fluorometric measurement of α-mannosidase activity from different sources. At pH 6.0, fluorescence increase over time was observed in assays recombinant Golgi mannosidase II from Drosophila melanogaster (Fig. 2), which has an optimum pH of 5.7 [15] and from recombinant human lysosomal mannosidase, which has an optimum pH of 4.5 (Figure 2).
Figure 2.
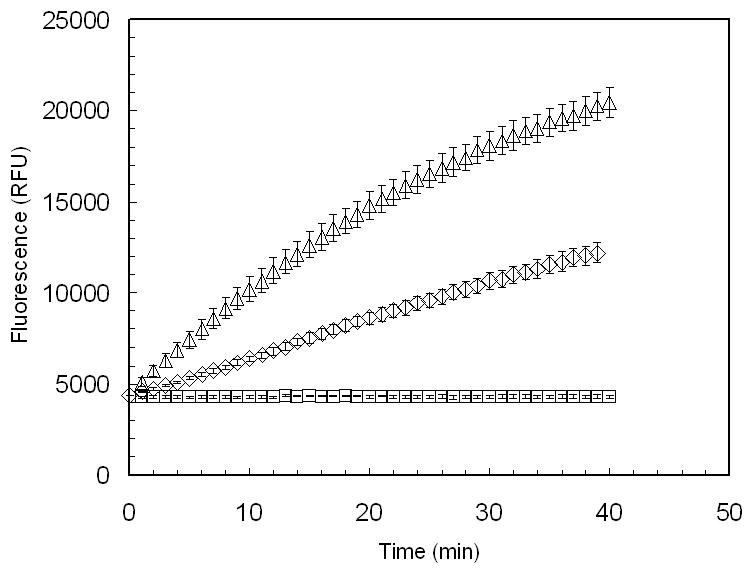
Continuous fluorescent analysis of recombinant golgi α-mannosidase II (dGMII) from Drosophila melanogaster (△) and recombinant human lysosomal mannosidase (HLM) (◊) using the substrate resorufin-α-D-mannopyranoside (Res-Man). Res-Man (500μM, diluted from 5mM DMSO stock using 100 mM sodium acetate buffer, pH 6.0) (50μL) was added to the enzyme samples (50 μL) to a final concentration of 250 μM. Enzyme samples were prepared in 100 mM sodium acetate buffer and diluted to a final concentration of (6.67 μg/mL) in the reaction mixture. Fluorescence of a blank (no-enzyme) sample (□) was also analyzed over the same time period. Fluorescence emission was measured at 590 nm with excitation at 550 nm for 40 min at 1-min intervals. Data points are means, and error bars represent standard errors (n = 3).
Inhibition Assay
Inhibition of α-mannosidase has been indicated as important in controlling tumor progression [1-4, 25]. A sensitive continuous assay of α-mannosidase activity could prove invaluable in high-throughput screening of inhibitors. Activity of dGMII in the presence of swainsonine, a natural inhibitor of type II α-mannosidases [8, 15, 25], at several different concentrations was assayed using a continuous assay format. The results suggest that the Res-Man substrate would be suitable for inhibitor screening, as decreasing activity was observed with increasing swainsonine concentration (Figure 3), and complete inhibition of activity at concentrations greater than 1μM (Figure 3). Using the Res-Man substrate, IC50 was determined to be 100 nM, and Ki calculated to be 44.7 nM. IC50 of swainsonine with dGMII has been previously reported to be in the range of 12-20 nM [15] using PNP-Man as a substrate.
Figure 3.
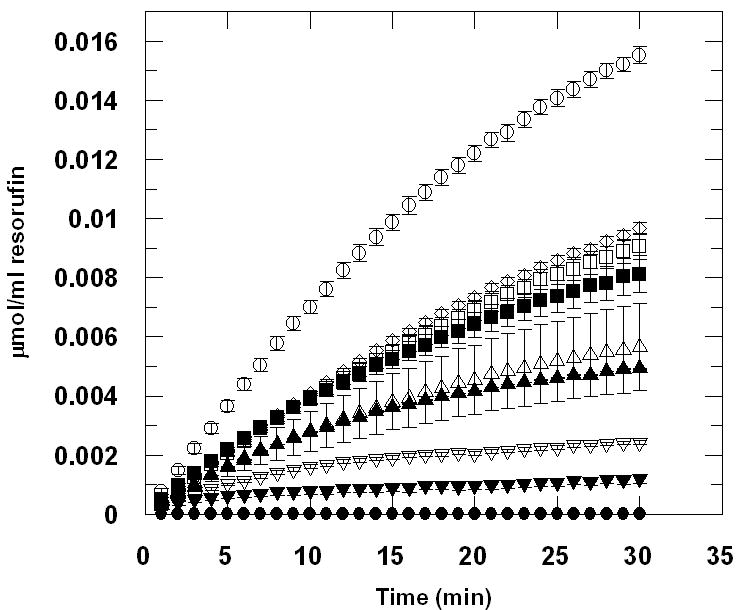
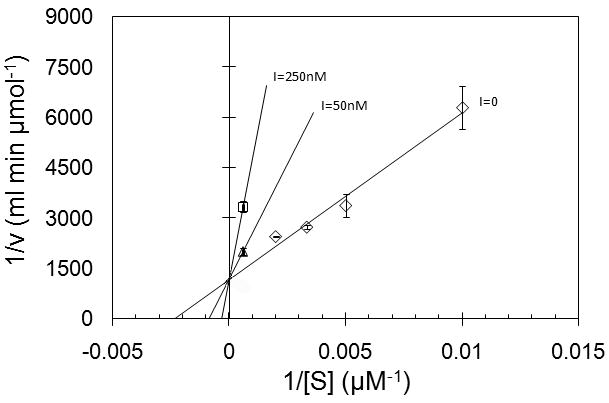
(a.) Inhibition of recombinant golgi α-mannosidase II (dGMII) from Drosophila melanogaster by swainsonine (SIGMA 068K8721), using the substrate resorufin- α -D-mannopyranoside (Res-Man). IC50 was determined to be 100nM and Ki was determined to be 44.7nM. DGMII was diluted to 6.67 μg/mL in 100mM Sodium Acetate, pH 6.0. Swainsonine was added at concentrations of 10μM (●), 1μM (▼), 500nM (▽), 250nM (▲), 100nM (△), 50nM (■), 25nM (□), and 10nM (◊). An enzyme sample containing no inhibitor (○) was also prepared. Res-Man (500μM, diluted from 5mM DMSO stock using sodium acetate buffer) (50μL) was added to the enzyme samples (50μL) to a final concentration of 250 μM. Fluorescence of all assays was recorded using EX: 550; EM: 595. Data were converted to μmol of resorufin product produced using a standard curve created by measuring fluorescence of several known concentrations of resorufin in sodium acetate buffer (pH 6.0). Data points are means, and error bars represent standard errors (n = 3). (b.) Lineweaver-Burk plots generated from kinetic assay using the substrate resorufin-α-D-mannopyranoside (Res-Man). The kinetic parameters Km and Vmax were determined via computer-assisted nonlinear regression analysis using GraFit (Erithacus Software). Values for Km and Vmax for the uninhibited assay were determined by GraFit to be 202.05 μM and 0.00059442 μmol/ml/min, respectively. Inset shows competitive inhibition by swainsonine at 50 nM (△) and 250 nM (□) concentrations.
In previous studies, the substrate 4-methylumbelliferyl α-D-mannopyranoside (MU-Man) has been used for determination of α-mannosidase activity, using fluorescence endpoint measurements at various time points with the addition of a high pH “stop buffer” to terminate the reaction and increase the fluorescence of the released fluorophore [16]. This method has been used to determine the activity of α-mannosidase at low pH values (6.0) [16], albeit not in a continuous format [16]. In addition, the products from these reactions (4-MU, mannose) have also been measured by cumbersome thin layer chromatography or HPLC analysis techniques.
Analysis of the affinity of dGMII for MU-Man (Km of 5mM, (H. Strachan and K. Moremen, personal communication)) and PNP-Man (Km50-60 mM, unpublished) indicates that the previously used artificial substrates are poor mimics of the natural substrate (Km of 85 μM (H. Strachan and K. Moremen, personal communication)[see footnote ref.28].
A continuous kinetic assay using dGMII, performed using a pH 6.0 buffer, resulted in a Km of 200 (±73) μM and a Vmax of 11 (±1.5) nmol/min per nmol dGMII. using the Res-Man substrate (Figure 4). Thus this new substrate has an affinity at least 250-fold better for dGMII than the commonly used PNP-mannose substrate as well as being about 20-fold better than the 4-MU-Man substrate and is similar in affinity to naturally occurring substrates [28]. These data suggest that the continuous assay performed at pH 6.0 with the new Res-Man substrate can be used to obtain reliable kinetic data at or near physiological pH values. Fluorescence emission of the released resorufin product at pH 6.0 is somewhat reduced (approximately 30%) relative to its fluorescence at a higher pH [20]. However, previous assays with Res-CB [20] suggest that the kinetic parameters obtained are essentially unaffected relative to an assay performed using a stop buffer. Furthermore, since resorufin is a relatively pH-insensitive fluorophore, fluorescence emission and excitation wavelengths remain fairly uniform over a large range of pH values from 3-9 [26] which allow the same filter sets and instrumental setup methods to be used for assays run with different samples having a variety of pH values. It is noted that fluorescence measurements of free resorufin standards should be measured over the range of concentrations and at the same pH as those used for test samples, in order to obtain quantitative turnover rates at specific pH values.
Figure 4.
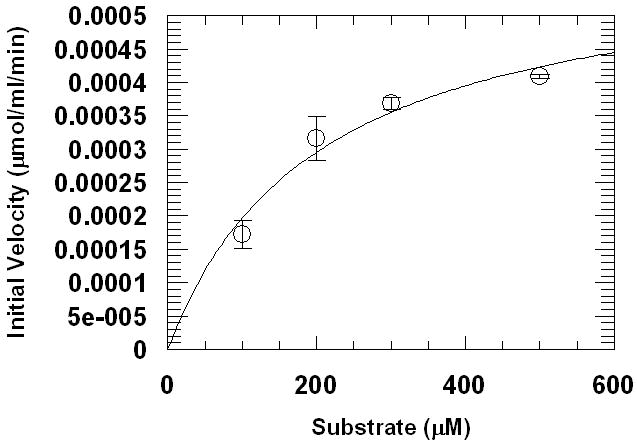
Computer-assisted nonlinear regression kinetic analysis of resorufin-α-D-mannopyranoside (Res-Man). Assay method was performed by adding recombinant golgi α-mannosidase II (dGMII) from Drosophila melanogaster (6.67 μg/mL final conc.) to four concentrations (0.50, 0.30, 0.20, 0.10 mM) of Res-Man in 100 mM sodium acetate buffer, pH 6.0. Fluorescence was recorded (EX: 550, EM: 595) at 30-s time intervals for 210 s. Data were converted to μmol of resorufin product produced using a standard curve created using known concentrations of resorufin in sodium acetate buffer (pH 6.0). Data points are means, and error bars represent standard errors (n = 3).
Uses of the new Res-Man substrate for continuous assay of α-mannosidase enzymes derived from additional species, towards various α-mannosidase isozymes, as well as analysis of activity and inhibition in live cell and live tissue formats are underway. Continued work with the Res-Man substrate holds the promise for analysis of α-mannosidase activities and identification of potential inhibitors or modulators of α-mannosidase activity that may prove useful for therapeutic intervention in a variety of important disease systems.
Acknowledgments
The authors wish to thank Heather Strachan and Professor Kelley Moremen at the Complex Carbohydrate Research Center and Department of Biochemistry and Molecular Biology, University of Georgia for data regarding the enzyme activities toward various mannosidase substrates. In addition, we would like to thank Drs. Anthony P. Guzikowski and Michael Strain for the help with NMR analyses. This work was funded in part by grant 1R43MH079542 from the National Institutes of Health – National Institute of Mental Health.
Abbreviations
- GMII
Golgi α1,3 α1,6-mannosidase II (E.C.3.2.1.114)
- dGMII
catalytic domain of Drosophila melanogaster GMII
- Res-Man
resorufin α-D-mannopyranoside
- pKa
acid dissociation constant
- Vmax
maximum velocity
- EX
excitation
- EM
emission
- Km
Michaelis-Menten constant
- MU-Man
4-methylumbelliferyl α-D-mannopyranoside
- PNP-Man
p-nitrophenyl α-D-mannopyranoside
- X-Man
5-bromo-4-chloro-3-indolyl- α-D-mannopyranoside
- PNP
p-nitrophenol
- CH2Cl2
dichloromethane
- MeOH
methanol
- Rf
retention factor
- TLC
thin-layer chromatography
- HCl
hydrochloric acid
- CDCl3
deuterochlorform
- Ac
acetate
- HOAc
acetic acid
- SiO2
silica
- EtOAc
ethylacetate
- NMR
nuclear magnetic resonance
- DMSO
dimethyl sulfoxide
- NAD
nicotinamide adenine dinucleotide
- 4-MU
4-methylumbelliferone
- Tris
trishydroxymethylaminomethane
- HPLC
high-pressure liquid chromatography
- HTS
High-Throughput Screening
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernacki RJ, Niedbala MJ, Korytnyk W. Glycosidases in cancer and invasion. Cancer Meta Rev. 1985;4:81–101. doi: 10.1007/BF00047738. [DOI] [PubMed] [Google Scholar]
- 2.Dennis JW, Granovsky M, Warren CE. Glycoprotein glycosylation and cancer progression. Biochim Biophys Acta. 1999;1473:21–34. doi: 10.1016/s0304-4165(99)00167-1. [DOI] [PubMed] [Google Scholar]
- 3.Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Nat Acad Sci USA. 2002;99:10231–10233. doi: 10.1073/pnas.172380699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dube DH, Bertozzi CR. Glycans in cancer and inflammation. Potential for therapeutics and diagnostics. Nat Rev Drug Disc. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 5.Goss PE, Baker ME, Carver JP, Dennis JW. Inhibitors of carbohydrate processing: A new class of anticancer agents. Clin Cancer Res. 1995;1:935–944. [PubMed] [Google Scholar]
- 6.Goss PE, Reid CL, Bailey D, Dennis JW. Phase IB clinical trial of the oligosaccharide processing inhibitor swainsonine in patients with advanced malignancies. Clin Cancer Res. 1997;3:1077–1086. [PubMed] [Google Scholar]
- 7.Harpaz N, Schachter H. Control of glycoprotein synthesis. Processing of asparagine-linked oligosaccharides by one or more rat liver Golgi α-D-mannosidases dependent on the prior action of UDP-N-acetylglucosamine: α-D-mannoside β 2-N-acetylglucosaminyltransferase I. J Biol Chem. 1980;255:4894–4902. [PubMed] [Google Scholar]
- 8.Moremen KW, Touster O, Robbins PW. Novel purification of the catalytic domain of Golgi α-mannosidase II. Characterization and comparison with the intact enzyme. J Biol Chem. 1991;66:16876–16885. [PubMed] [Google Scholar]
- 9.Broquist HP. The indolizidine alkaloids, slaframine and swainsonine: Contaminants in animal forages. Ann Rev Nutr. 1985;5:391–409. doi: 10.1146/annurev.nu.05.070185.002135. [DOI] [PubMed] [Google Scholar]
- 10.Tulsiani DR, Broquist HP, James LF, Touster O. Production of hybrid glycoproteins and accumulation of oligosaccharides in the brain of sheep and pigs administered swainsonine or locoweed. Arch Biochem Biophys. 1988;264:607. doi: 10.1016/0003-9861(88)90327-x. [DOI] [PubMed] [Google Scholar]
- 11.van den Elsen JM, Kuntz DA, Rose DR. Structure of Golgi α-mannosidase II: a target for inhibition of growth and metastasis of cancer cells. EMBO J. 2001;20:3008–3017. doi: 10.1093/emboj/20.12.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Numao S, Kuntz DA, Withers SG, Rose DR. Insights into the mechanism of Drosophila melanogaster Golgi α-mannosidase through the structural analysis of covalent reaction intermediates. J Biol Chem. 2003;278:48074–48083. doi: 10.1074/jbc.M309249200. [DOI] [PubMed] [Google Scholar]
- 13.Zhong W, Kuntz DA, Ember B, Singh H, Moremen KW, Rose DR, Boons GJ. Probing the substrate specificity of Golgi α-mannosidase II using synthetic oligosaccharides and a catalytic nucleophile mutant. J Am Chem Soc. 2008;13:8975–8983. doi: 10.1021/ja711248y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah N, Kuntz DA, Rose DR. Golgi α-mannosidase II cleaves two sugars sequentially in the same catalytic site. Proc Natl Acad Sci USA. 2008;105:9570–9575. doi: 10.1073/pnas.0802206105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabouille C, Kuntz DA, Lockyer A, Watson R, Signorelli T, Rose DR, van den Heuvel M, Roberts DB. The Drosophila GMII gene encodes a Golgi α-mannosidase II. J Cell Sci. 1999;112:3319–3330. doi: 10.1242/jcs.112.19.3319. [DOI] [PubMed] [Google Scholar]
- 16.Vázquez-Reyna AB, Bálcazar-Orozco R, Flores-Carreón A. Biosynthesis of glycoproteins in Candida albicans: Biochemical characterization of a soluble α-mannosidase. FEMS Microbio Lett. 1993;106:321–326. doi: 10.1111/j.1574-6968.1993.tb05983.x. [DOI] [PubMed] [Google Scholar]
- 17.Chernoglazov VM, Jafarova AN, Klyosov AA. Continuous photometric determination of endo-1,4,-β-D-cellobioside as a substrate. Anal Biochem. 1984;138:481–487. doi: 10.1016/0003-2697(89)90222-4. [DOI] [PubMed] [Google Scholar]
- 18.Sa Van H. Extracting organic compounds from aqueous solutions. European Patent EP0561760. 1993
- 19.Jefferson RA. Plant Promoter α-Glucuronidase Gene Construct. US patent 5,268,463. 1993
- 20.Coleman DJ, Studler MJ, Naleway JJ. A long-wavelength fluorescent substrate for continuous fluorometric determination of cellulase activity: resorufin β-D-cellobioside. Anal Biochem. 2007;371:146–153. doi: 10.1016/j.ab.2007.08.027. [DOI] [PubMed] [Google Scholar]
- 21.Goggin KD, Hammen PD, Kuntson KL, Lambert JF, Walinsky SW, Watson HA. Commercial synthesis of β-D-cellobiosyl bromide heptaacetate. J Chem Tech Biotech. 2004;60(3):253–256. doi: 10.1002/jctb.280600305. [DOI] [PubMed] [Google Scholar]
- 22.Haugland RP, Naleway JJ, Zhang YZ. Lipophilic fluorescent glycosidase substrates. US Patent 5,208,148. 1993 [Google Scholar]
- 23.Wimmer Z, Pechová L, Šaman D. Koenigs-Knorr synthesis of cycloalkyl glycosides. Molecules. 2004;9:902–912. doi: 10.3390/91100902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zemplen G, Biosen Abbau der reduzierenden., VII Konstitutions-Ermittlung der Maltose. Ber Dtsch Chem Ges. 1927;60B:1555. [Google Scholar]; Zemplen G, Pacsu E. Über die Verseifung acetylierter Zucker und verwandter Substanzen. Ber Dtsch Chem Ges. 1929;62:1613–1614. [Google Scholar]
- 25.Fiaux H, Kuntz DA, Hoffman D, Janzer RC, Gerber-Lemaire S, Rose DR, Juillerat-Jeanneret L. Functionalized pyrrolidine inhibitors of human type II α–mannosidases as anti-cancer agents: Optimizing the fit to the active site. Bioorg Med Chem. 2008;16:7337–7346. doi: 10.1016/j.bmc.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 26.Ryder AG, Power S, Glynn TJ. Fluorescence Lifetime Based pH Sensing Using Resorufin. Proc SPIE. 2003;4876:827–835. [Google Scholar]
- 27.Gallego del Sol F, Gomez J, Hoos S, Nagano CS, Cavada BS, England P, Calvete JJ. Energetics of 5-bromo-4-chloro-3-indolyl α -D-mannose binding to the Parkia platycephala seed lectin and its use for MAD phasing. Acta Cryst, Section F. 2005;61:326–331. doi: 10.1107/S1744309105004835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heather Strachan and Kelley Moremen at the Complex Carbohydrate Research Center and Department of Biochemistry and Molecular Biology, University of Georgia reported (personal communication) a value for the Km of 4-MU-Man used as a substrate when measuring inhibitory compounds from Geert-Jan Boon's lab using the same dGMII enzyme preparation of Km = 4.9 +/-2.6 mM with MU-Man (pH 5.5) at a total conc. range of 0.1875 - 3mM; and for Human Lysosomal Mannosidase (HLM) (pH 4.0) a Km = 0.8 =/-0.4 mM with MU-Man at a total conc. range of 0.1875 - 3mM. Attempts to measure the Km for PNP-mannose using dGMII indicated it was potentially in the range of 50-60 mM. However this is well above the solubility limit of PNP-mannose in aqueous solutions so an exact determination of the Km could not be determined. Measured Km's for several preferred natural substrates were found to be as follows: Km = 89.6 μM with GlcNAcMan5GlcNac2 for dGMII and Km= 85.8 μM for Man5GlcNac2 for HLM.