

Abstract
Many large double standed DNA viruses employ high force-generating ATP driven molecular motors to package to high density their genomes into empty procapsids. Bacteriophage T4 DNA translocation is driven by a two-component motor consisting of the procapsid portal docked with a packaging terminase-ATPase. Fluorescence resonance energy transfer and fluorescence correlation spectroscopic (FRET-FCS) studies of a branched (Y-junction) DNA substrate with a procapsid-anchoring leader segment and a single dye molecule situated at the junction point reveal that the “Y-DNA” stalls in proximity to the procapsid portal fused to GFP. Comparable structure Y-DNA substrates containing energy transfer dye pairs in the Y-stem separated by 10 or 14 base pairs reveal that B-form DNA is locally compressed 22-24% by the linear force of the packaging motor. Torsional compression of duplex DNA is thus implicated in the mechanism of DNA translocation.
Keywords: Bacteriophage portal and terminase, DNA packaging, Fluorescence resonance energy transfer and fluorescence correlation spectroscopy (FRET-FCS)
Introduction
Nucleic acid translocation into an empty procapsid is a conserved assembly mechanism found among diverse DNA and RNA viruses (Kainov, Tuma, and Mancini, 2006). Many large double stranded (ds) DNA viruses employ high force-generating molecular motors to package their genomes into procapsids (Smith et al., 2001). In tailed dsDNA bacteriophages translocation is driven by a two-component motor consisting of a dodecameric portal situated at a unique procapsid vertex that is docked during packaging with a homopentameric terminase-ATPase (Figure1a). High resolution structures of both the phage T4 terminase large subunit gp17 and the portal protein gp20 are known or can be inferred (Driedonks et al., 1981; Lebedev et al., 2007; Simpson et al., 2000; Sun et al., 2008). The T4 portal is turbine-shaped with a hollow cylinder 14 nm long and 7 nm in diameter with a central channel of 3-4 nm through which DNA is pumped to high density (∼500 mg/ml) within the procapsid (100 nm long and 75 nm wide). Following packaging and addition of DNA end protection proteins (gp2), and neck and tail components to complete assembly, DNA is ejected through the portal central channel and tail to infect a susceptible bacterial host. The small gp16 T4 terminase subunits are associated with the gp17 terminase large subunits and are required for cutting and packaging the natural genomic substrate, a replicative DNA concatemer in vivo, or circular DNA in vitro (Black and Peng, 2006; Catalano; Kondabagil, Zhang, and Rao, 2006). However the small gp16 subunit is nonessential and in fact inhibitory for linear DNA packaging into purified procapsids in vitro. These features make phage T4 packaging a particularly favorable subject for study of the DNA translocation mechanism.
Figure 1.

A Texas Red single dye containing Y-DNA with a 2.7 kb leader sequence complexed with gp17 terminase-ATPase stalls in proximity to a GFP-fusion portal containing procapsid. a) A GFP-portal containing procapsid with anchored, stalled 2.7 kb TxY-DNA; b) diagram of the TxY-DNA structure and sequence, c) construction of the 2.7 kb leader Tx-Y-DNA, only later packaged.
Efficient packaging of DNA substrates into phage T4 procapsids by the terminase-ATPase large subunit in vitro allowed specific DNA structural requirements to be identified. Duplex DNAs of random sequence from 20 to 500 bp were packaged with nearly 100% efficiency. Dye and short, single-stranded end extensions were tolerated, whereas 20-base extensions, hairpin ends, 20-bp DNA–RNA hybrid, and 4-kb dsRNA substrates were not packaged at all. DNA substrates 60 bp in length with 10 internal mismatched basepairs were translocated with reduced efficiencies compared to fully duplexed DNA; while substrates with 20 mismatched basepairs, a related D-loop structure, or ones with an internal 20-base single-strand region were not. Strikingly, a single nick in 100 or 200 bp duplexes, irrespective of location, greatly reduced translocation efficiency, but singly nicked 500 bp or longer molecules were packaged as effectively as an unnicked control in a limit packaging reaction. Fluorescence correlation spectroscopy further revealed that a 100 bp nicked substrate did not remain stably bound to the terminase–procapsid. Two unbroken DNA strands are thus important for translocation, markedly so when no substantial packaged leader portion of the substrate DNA is available within the procapsid to anchor the substrate to the motor. Taken together, these observations suggested that DNA torsion is implicated in phage DNA packaging and led to a proposed torsional compression translocation mechanism (Oram, Sabanayagam, and Black, 2008). In this report we provide experimental evidence that the packaging motor exerts a linear force that locally compresses duplex DNA in a stalled translocation complex.
Results
Our previous work established that fluorescently labeled DNA and protein packaging components could be employed to measure packaging efficiency by fluorescence resonance energy transfer fluctuation correlation spectroscopy (FRET-FCS) as confirmed by the well-established nuclease protection assay (Sabanayagam et al., 2007). In this work fluorescent components are used to examine the structure of stalled DNA packaging intermediates. Our stalling assay employed a Y-DNA substrate with a single Texas Red dye (Tx) positioned at the branched junction (Figure1b). The procapsid contained either a wild-type portal or a portal with four to six GFP fusion proteins of the twelve portal monomers. Such particles had previously been shown to be active and, together with other active portal fusion containing procapsids, strongly argued against the hypothesis that the portal functioned as a rotary motor (Hendrix, 1978; Simpson et al., 2000), a conclusion in agreement with other recent work (Baumann, Mullaney, and Black, 2006; Hugel et al., 2007). Here we found that the 90 bp TxY-DNA was not packaged into the prohead, nor was FRET observed between the TxY-DNA and the GFP prohead, consistent with earlier observations that short looped DNAs (e.g. the Y-ends) or short DNAs containing nicks or non-duplex elements (e.g. the Y-stem and Y-junction) were not packaged or retained by the procapsid as determined by the nuclease (Figure 2 a) and FCS assays (Figure 3a,c) (Oram, Sabanayagam, and Black, 2008). However, when the TxY-DNA stem was joined to a 2.7 kb duplex leader to anchor the DNA substrate inside the procapsid (Figure 1c), a substantial portion of the 2.7 kb DNA was nuclease protected (Figure 2b). Additionally, with the leader DNA present, Tx-GFP FRET was observed as was packaging measured by FCS (Figure 3 b, d). Fluorescence correlation analysis of the long 2.7 kb TxY-DNA was compared with the short TxY-DNA in the packaging assay samples. A decrease in the linear diffusion constant showed that a substantial portion of the long - but not the short -substrate had been packaged (compare Figures 3c and d). The correlation curves shown in Figures 3c and d were fitted with single- and two-species homogeneous brightness diffusion models respectively (Oram, Sabanayagam, and Black, 2008). The diffusion coefficients and the respective contributions of each species including the error associated with the fit of the autocorrelation plots are presented in Table 1. The diffusion coefficient of the 2.7 kb TxY-DNA anchored into the GFP-portal was measured to be 1.7 μm2/sec, comparable to previous measurements (Sabanayagam et al., 2007). Figure 3b shows the FRET histogram when 2.7 kb TxY-DNA is packaged in the GFP-portal. From this measurement, the donor-to-acceptor distance (r) is estimated to be ∼ 5.8 nm, given a Förster distance of 4 nm for a GFP-Txred donor-acceptor pair (Duckworth et al., 2007). As previously concluded, evidently the 2.7 kb leader portion of the DNA substrate serves to introduce and anchor the DNA within the procapsid (Oram, Sabanayagam, and Black, 2008). The Y-junction portion of the 2.7 kb TxY-DNA is stalled in the vicinity of the portal-GFP fusion and Tx-GFP FRET was observed (Figure 3b), consistent with the known dimensions of the portal (Driedonks et al., 1981).
Figure 2.
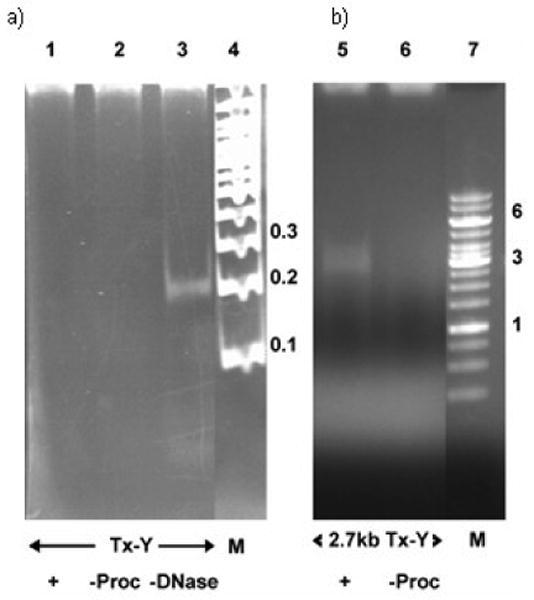
Nuclease assay shows the TxY-DNA without leader is not packaged a) lane 1 - all packaging components, lane 2 - procapsids omitted, lane 3 - DNAase omitted shows input DNA; lane - 4 Mol. Wt. markers in kb, b) Nuclease assay shows that 2.7 kb leader containing TxY-DNA is packaged, lane 5 - all packaging components, lane 6 - procapsids omitted, lane 7 - Mol. Wt. markers in kb.
Figure 3.
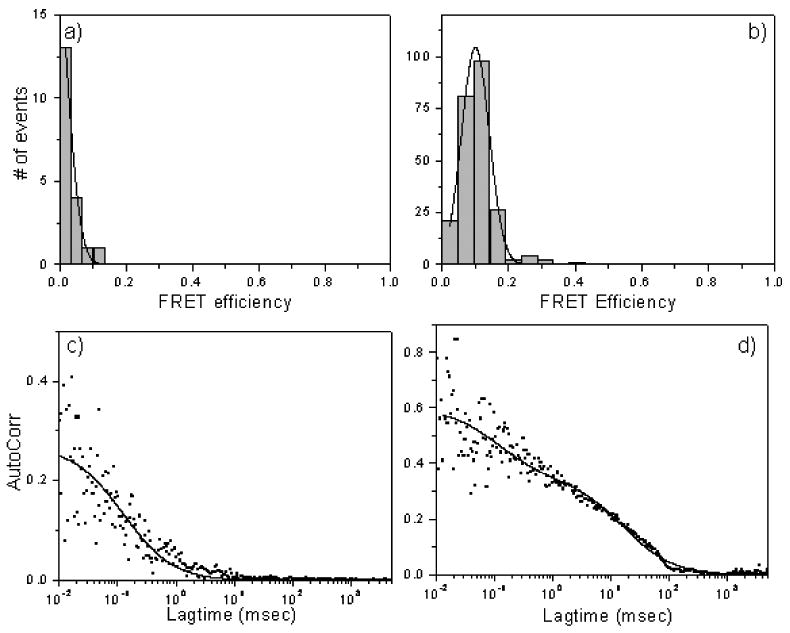
TxY-DNA stalls in proximity to GFP-containing portal by FRET. a) no FRET of TxY-DNA in absence of leader and absence of packaging; b) FRET showing GFP to Tx when 2.7 kb TxY-DNA is packaged. Autocorrelation plots of Tx-red channel are shown in panels c) and d).
Table 1.
Diffusion coefficients and respective fractional contributions for different constructs of Y-DNA under various packaging conditions.
| Sample | D1 (μm2/sec) | Contribution | D2 (μm2/sec) | Contribution |
|---|---|---|---|---|
| Tx-Y DNA | 240 ± 20 | |||
| Tx-Y DNA with leader (Packaged) | 1.7 ± 0.3 | 57 % | 250 ± 24 | 43 % |
| 2.7 kb AxCyY-DNA 10bp | 11 ± 1.1 | 33 % | 301 ± 14 | 67 % |
| 2.7 kb AxCyY-DNA 14bp | 11 ± 0.6 | 38 % | 309 ± 11 | 62 % |
| 2.7 kb AxCyY-DNA 10bp, Packaged | 4.5 ± 0.2 | 41 % | 215 ± 7 | 59 % |
| 2.7 kb AxCyY-DNA 14bp, Packaged | 4.6 ± 0.3 | 39 % | 202 ± 8 | 61 % |
| 2.7 kb AxCyY-DNA 10bp, Packaged, DNAse added | 185.5 ± 3 | |||
| 2.7 kb AxCyY-DNA 14bp, Packaged, DNAse added | 187 ± 4.3 |
In order to examine the molecular orientation of stalled DNA substrates, 92 bp Y-DNA packaging substrates were internally labeled with an energy transfer pair: a Cy3 (Cy; acceptor) was positioned in the duplex Y stem either 14 bp or 10 bp apart from an Alexa 488 (Ax; donor) located at the Y junction (Figure 4a). When AxCyY-DNAs were employed in packaging into wild-type procapsids, no packaging was observed by either the nuclease or FCS assays, similar to results with the TxY-DNA without the 2.7 kb leader. However, following ligation to a 2.7 kb stem extension to allow anchoring and stalling in the procapsid portal (Figure 4b), protection of a portion of the 2.7 kb AxCyY-DNAs was observed when procapsids and terminase were present, and in the absence of terminase DNA was completely degraded by nuclease (Figure 5). FRET-FCS measurements of 2.7 kb AxCyY-DNA substrates in water are shown in Figure 6a, b. Figure 7a and b shows the correlation curves for the 2.7 kb AxCyY-DNAs (10 bp or 14 bp). A single species diffusion model does not exactly fit the data which may indicate the heterogeneity of the environment in the reaction buffer. The autocorrelation plots of the packaged 2.7 kb AxCyY-DNAs (10 bp or 14 bp) fitted with a double-species model are presented in Figure 7c and d. The fitted parameters and errors associated with measured diffusion coefficients are tabulated in Table 1. Correlation spectroscopy of the packaged 2.7 kb AxCyY-DNAs revealed a portion with procapsid-like diffusion, consistent with the nuclease assay (Figure 7c, d). The diffusion coefficient of the anchored 2.7kb AxCyY-DNA (10 bp or 14 bp) into normal procapsids was measured to be ∼ 4.5 μm2/sec. However, although a portion of the added 2.7 kb leader DNA was protected from nuclease, nuclease treatment rapidly converted all of the fluorescence to high mobility, showing protection of a portion of the added 2.7 kb leader but none of its Y portion (Figure 7e, f). Thus this portion of the DNA is not in a stable packaging protected location, unlike fully packaged single or double dye end-labeled DNA substrates (Sabanayagam et al., 2007). The autocorrelation plots shown in Figure 7e and f were fitted using a single species diffusion model and the diffusion coefficients were measured to be ∼190 μm2/sec in both cases. Strikingly, when the FRET efficiencies of the packaged 2.7 kb AxCyY-DNAs were measured, it was now found in each case that there were two components: the 10bp spacing displayed ∼20% and ∼70% energy transfer efficiencies, whereas the 14 bp spacing showed ∼15% and ∼55% (Figure 6c, d). In the absence of terminase or procapsids in packaging buffer (data not shown) or in water (Figure 6a, b) only the single lower FRET efficiencies were observed. Therefore, we attribute the high efficiency FRET to the packaged DNAs and the low efficiency component to the freely diffusing 2.7 kb AxCyY-DNA substrates.
Figure 4.
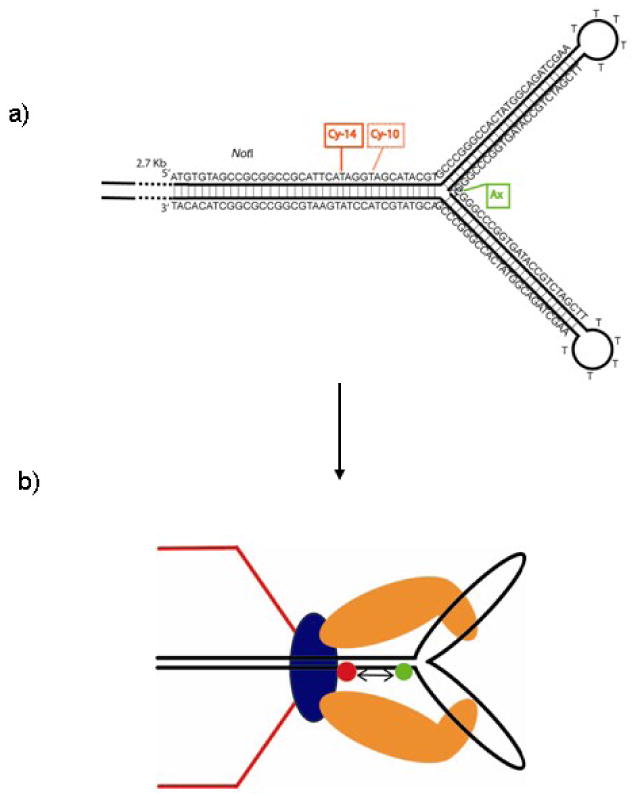
Diagram of a) structure and construction of the energy transfer dye pair 2.7 kb AxCyY-DNAs with 10 bp and 14 bp spacing; and b) normal procapsid portal with anchored, stalled 2.7 kb AxCyY-DNAs to measure packaging associated FRET change.
Figure 5.
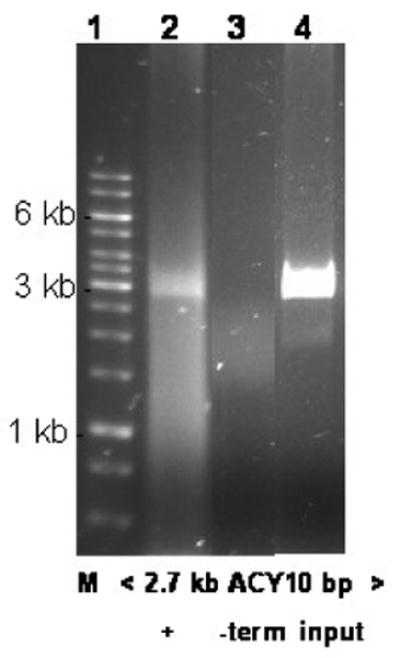
Nuclease assay following FRET showing protected 2.7 kb AxCyY-DNA (10 bp spacing). Lane 1 - Mol. Wt. markers in kbs; lane 2 - all packaging components; lane 3 - packaging omitting terminase; lane 4 - input DNA.
Figure 6.

FRET histograms of 2.7 kb AxCyY-DNAs with a) 10 bp and b) 14 bp interdye distances in water. FRET histograms in presence of packaging for c) 10 bp and d) 14 bp. Addition of nuclease to packaged 2.7 kb AxCyY-DNAs FRET histograms of e) 10 bp f) 14 bp DNAs.
Figure 7.
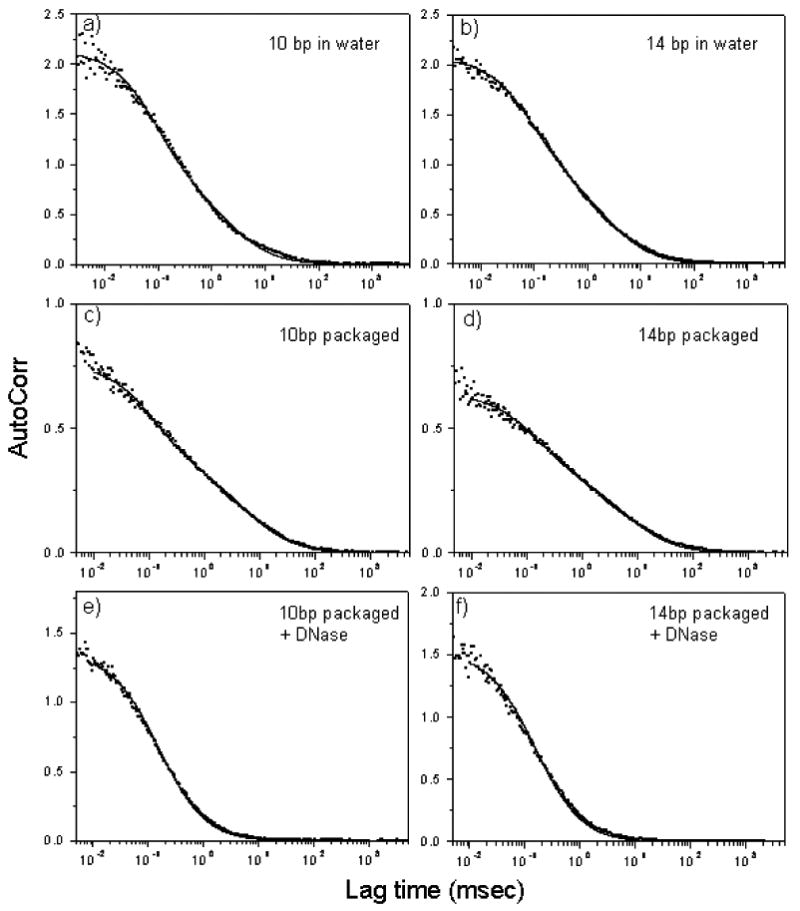
Autocorrelation plots to measure diffusion of the packaged and unpackaged 2.7 kb AxCyY-DNAs. Donor channels are shown in panels a), b), e) and f). Autocorrelation plots of acceptor channels are shown in panels c) and d).
For 10 bp AxCyY-DNA, in the absence of procapsids, we observed energy transfer of ∼ 20% (unpackaged form, Figure 6a) which resulted in a donor-to-acceptor distance (r) of 8.0 nm. For 10 bp AxCyY-DNA when packaged into the procapsid, we observed an energy transfer of ∼70% (Figure 6c). Consequently, for this case we estimated r = 6.1 nm. Thus, change in donor-to-acceptor distance after AxCyY-DNA (10 bp) is packaged into the procapsid is 2.1 nm which is equivalent to ∼ 24 % change in the interdye spacing. For 14 bp AxCyY-DNA, in the absence of procapsid, we observed energy transfer of ∼ 15% (unpackaged form, Figure 6b) which corresponds to a value of r = 8.4 nm. For the 14 bp AxCyY-DNA we observed energy transfer of ∼ 55% when packaged into the procapsid, corresponding to r = 6.6 nm (Figure 6d). Thus, the change in the donor-to-acceptor distance after AxCyY-DNA (14 bp) is packaged is 1.8 nm which is equivalent to ∼ 22% change in the interdye spacing. Hence, from the changes in the FRET efficiency and interdye distances, we calculate an average DNA compression of 22-24% that is associated with the packaging reaction.
It might be supposed that the FRET efficiency changes we attribute to compression of the rigid short B-form Y-stem segment are instead due to orientation changes in the dipole-dipole orientation of the dyes stalled in the portal channel relative to the free Y-DNA. First, we should note that the dyes employed for the FRET measurements are linked to the DNA by flexible linkers, making it unlikely that orientation changes could account for the FRET changes we observe. Additionally, we have measured the anisotropy of the donor molecule (Alexa 488) in the Y-DNA construct while packaged and unpackaged and find no change, since this value is R = 0.2 in either case (See Materials and Methods –Anisotropy Measurements). This suggests that the observed Förster distance changes in the donor-acceptor dyes bound to a rigid Y-DNA stem stalled in the procapsid are due to shortening of the interdye distance. This supports a structural change of the duplex DNA.
Additionally, structural changes in DNA apart from compression could possibly account for the measured distance changes. We note that our measurements are over a very short span of duplex DNA that is within the persistence length of B-DNA. Also, the two constructs of Y-DNA of identical stem sequence with 10 and 14 bp interdye spacing yields changes in the donor-to-acceptor distance of 24% and 22%, respectively. Given the constricted dimensions of the viral portal, it is unlikely that B-DNA with substantial bending or kinking could be accomodated. Still, if the observed distance change were due to bending or kinking of the short rigid stem segment, then we would expect to observe a greater difference in FRET efficiency between the two Y-DNA substrates because the two differently spaced dyes are rotated by ∼50% (4 bp) of a helical turn with respect to each other. Conversion of duplex DNA to two single strands, thereby collapsing the DNA segment and allowing even closer approach of the two dyes, appears even more unlikely.
Discussion
It might at first appear that a linear motor force should stretch DNA if it is stalled by a structure unable to translocate through the portal channel. Instead we observe a compression, suggesting that a linear motor force is pushing upon a constrained short (10-14 bp) DNA segment. As discussed above it is unlikely that the FRET differences we measure could result from packaging associated DNA bending, kinking, or melting rather than compression within such a short DNA segment -- indeed, short rigid dsDNAs often serve as FRET rulers (Deniz et al., 1999; Sabanayagam, Eid, and Meller, 2005). The narrow 3-4 nm diameter portal channel further constrains the DNA to maintain its linear conformation. Moreover, the magnitude of the changes we observe are incompatible with changes to the orientations of the DNA linked acceptor and donor dyes or to their local environments, consistent with direct anisotropy measurements (See Materials and Methods). While DNA stretching is thoroughly studied energetically, kinetically, and structurally (Bustamante et al., 2000), DNA compression is less well characterized experimentally because it is based on difficult to observe short range rigidity of B-DNA. Nevertheless, modeling suggests that up to ∼50% compression of B-DNA may occur without disrupting hydrogen bonding (Kosikov et al., 1999) – as in compressed A-form DNA - and moreover, measurements of reversible compression of B-DNA and the associated force required show that 22-24% compression can be readily achieved by considerably less than the measured T4 motor force of up to 60 pN (Fuller et al., 2007; Koenig et al., 2005).
Although we attribute our measured DNA compression to the obstruction of DNA translocation by the branched substrate, the observed DNA compression we believe reflects the normal functioning of the two-component motor that has successfully translocated the 2.7 kbp leader-anchor segment into the procapsid. We previously proposed that the portal grips the DNA and the terminase imparts a linear force that is stored as DNA torsion until DNA release by the portal (Oram, Sabanayagam, and Black, 2008), consistent with active participation of the portal in terminase-powered translocation (Cuervo et al., 2007; Lebedev et al., 2007). Such a mechanism would account for the strong inhibitory effect of DNA nicks in short substrates on translocation. Our current measurements offer direct evidence for this model by showing that the stalled motor exerts a linear force creating torsional compression of the B-DNA undergoing translocation. Possibly such a portal-DNA-grip-and-release mechanism could relate to the established role of the portal in sizing the amount of DNA to be packaged before termination in headful-packaging phages such as P22, SPP1, and T4 (Casjens et al., 1992; Tavares et al., 1992). Interestingly, recent measurements show that the Phi29 packaging motor moves DNA in 10 bp increments with four 2.5 bp steps, a difficult to explain non-integral number of base pairs, assuming that it is uncompressed B-DNA that is translocated through the portal channel (Moffitt et al., 2009); ∼25% compression might account for this non-integral step size.
Our findings and model are also compatible with the structures of the translocation complexes of the Phi6 and T4 phages and the mechanisms that have been proposed for these motor proteins. In both cases a linear motor force is thought to be applied by a circular pentameric or hexameric array of ATPase monomers that exert force either by large scale conformational swiveling of each monomer or by localized swiveling arm motions within the multimer in the phage T4 and Phi6 translocation complexes respectively. However in neither case is the translocating nucleic acid structure known, nor is it known how the nucleic acid is engaged by the motor. Although both proposed mechanisms are generally consistent with our observations and model, we suggest a more active role of the portal in a grip-and-release mechanism working together with the terminase motor protein. This is supported by the evidence for an active role of the SPP1 portal in translocation already discussed. We propose in addition that structural change in the DNA substrate we have measured in the stalled substrate is intimately involved in the translocation mechanism.
ATP-driven helicases and translocases are known to produce marked structural changes in DNA that accompany translocation (Lionnet et al., 2006). Profound changes to DNA structure are also produced by transcription motor proteins. For example, DNA structural changes by RNA polymerase called scrunching differ from those we measure here because they result from pulling rather than pushing on DNA, are measured over longer range (30 bp), and involve DNA melting rather than short range linear DNA compression (Kapanidis et al., 2006). Our experiments are novel in suggesting that local DNA compression may be associated with two-component motors that translocate DNA by drawing upon its spring-like properties. Such DNA “crunching” could be an important element of other DNA transactions and DNA motors where force is locally exerted. We aim to study the kinetics of DNA compression by the packaging motor during active translocation, along with the associated motor protein dynamics further by FRET-FCS and single-molecule fluorescence technologies.
Materials and methods
Fluorescent DNA construction
Y-DNAs were constructed from HPLC-purified dye-labeled oligonucleotides (Integrated DNA Technologies (IDT) Inc.) by annealing complementary single strands, followed by gel electrophoresis and purification from gel slices by extraction of the dye containing duplex DNAs (Qiagen Inc. Qiaex II kit isolation). The Texas Red dye (Tx) in TxY-DNA was joined to the 90 base oligonucleotide sequence shown by an XN linker to the 5′ terminal C (Figure 1b). The Cy3 (Cy) and Alexa488 (Ax) dyes were joined to the 92 base oligonucleotides shown in Figure 4a by three carbon linkers of Ax to the 5′ terminal C and amino six carbon linkers of Cy3 to internal T residues on the complementary oligonucleotide strand, consistent with FRET measurements of the annealed and purified double dye Y duplexes. The single dye TxY-DNA (90 bases) and double dye AxCyY-DNAs (92 bases) were cut with restriction endoncucleases EagI and NotI respectively, and then ligated to pBR322 DNA linearized with EagI, followed by PstI digestion and gel extraction and purification to yield the Y-DNAs with the 2.7 kb pBR322 derived leaders shown in Figure 1c and Figure 4a.
DNA Packaging Components
Unmodified wild-type empty large procapsids (elps) were concentrated from the T4 procapsid producing mutant infected bacteria. Procapsid purification employed differential centrifugation, glycercol gradient ultracentrifugation, and FPLC-DEAE chromatography as previously described (Black and Peng, 2006). Portal fusion protein gp20-GFP containing procapsids were produced by supplying gp20-GFP in trans from an expression vector to terminase and portal-deficient phage mutant infected bacteria as previously described (Baumann, Mullaney, and Black, 2006). The same purification as of wild-type procapsids was followed and the FPLC-DEAE chromatographed empty small procapsids (esps) - only esps are produced because of the modified gp20-GFP fusion protein containing portal - were converted to the more stable gp20-GFP elps by exhaustive low ionic strength dialysis at 4° C and then characterized for packaging activity by FCS and nuclease assay as previously described (Ray et al., 2009). The gp17 terminase large subunit was purified to near homogeneity from the chitin binding domain-intein fusion protein as previously described (Baumann and Black, 2003). DNA packaging was carried out as previously described (Black and Peng, 2006), except that polyethylene glycol (PEG 20,000) was added to 2% (wt/vol) final concentration to the packaging assay mixture for FCS measurements (Black and Peng, 2006).
DNA Packaging Assays
A nuclease protection assay measured the amount of packaged DNA by gel electrophoresis after incubation at 37° C for 30 minutes with pancreatic DNAase, followed by release of the packaged procapsid DNA by incubation at 65° for 30 minutes in the presence of EDTA, proteinase K, and 1% SDS as described previously (Black and Peng, 2006). The packaged EthBr stained DNAs were quantitated using UVP software to integrate gel DNA band density in comparison to known quantities of standard DNA ladders (Fermentas). For nuclease assay of packaging following FCS measurements, the sample was removed from the microscope observation stage and processed by the nuclease assay described above to determine the packaging that had been achieved during the FCS measurements (Figure 2 and Figure 5). The ∼20% recovery of Y-DNA substrates underestimates packaging compared to FCS diffusion measurements due to loss of packaging extract on the FCS viewing stage and possibly also because of some leakage of the anchoring 2.7 kbp segment from the procapsid following nuclease removal of the stalled exposed Y portion of the substrate. Packaging was assayed in addition by change in diffusability of the added dye-labeled DNA as compared to the added DNA without packaging (omitting either terminase or procapsids). Following FCS and FRET-FCS measurements at room temperature, FCS and FRET-FCS assays of packaging were carried out as described previously (Oram, Sabanayagam, and Black, 2008; Sabanayagam et al., 2007). FCS measurement of packaging after nuclease treatment employed <5 minutes of pancreatic DNAase addition (1 μl of 5 mg/ml/16μl packaging mixture) at room temperature assay followed by FRET-FCS (Figure 6e, f) and by FCS (Figure 7e, f).
FCS and FRET-FCS Measurements and Analyses
FCS measurements were performed using a Picoquant MicroTime 200 confocal microscope (Picoquant microtime system coupled to an Olympus IX71 microscope). The excitation laser (λex ∼ 470 nm) was reflected by a dichroic mirror (centered at 476 nm) to a high numerical aperture (NA) oil objective (100×, NA 1.3) and focused onto the solution sample. The fluorescence signals were collected through the same objective and dichroic mirror. An additional longpass filter was used to eliminate the scattered laser light. Fluorescence responses from the donor and the acceptor molecules were separated by a beam splitter and detected by two avalanche photodiode detectors (APD) using the method of time-correlated single photon counting and the Time-Tagged Time-Resolved (TTTR) mode of the TimeHarp 200 board. High quality bandpass (Chroma) filters were used for recording donor and acceptor fluorescence in two separate detection channels. FCS measurements were performed in a constant detection volume. PicoQuant Symphotime software was used to generate the autocorrelation curves and in analyzing the FRET data. The autocorrelation curves were fitted with the following diffusion model. The autocorrelation function for n fluorescent species traversing a 3D Gaussian volume with radius ω0 and half axial z0 is:
where τ is the lag time, N is the number of molecules in the volume and fi are the fractions of the corresponding diffusion coefficients Di (Lakowicz, 2006). The collected single photon data was binned by 1 msec bin in each channel (donor or acceptor) which resulted in intensity-time traces and count-rate histograms. Threshold values in each channel were used to identify the single molecule bursts from the corresponding background signal level. Significant signals from adjacent intervals were binned together to get a sum photon number for each burst. Thus, fluorescence bursts were recorded simultaneously in donor and acceptor channels and FRET efficiencies were calculated using E = nA/(nA + γnD) where nD and nA are the sums of donor counts and acceptor counts for each burst, taking into account the possible difference in the detection efficiency (γ) in two separate channels (Schuler, Lipman, and Eaton, 2002). The donor-to-acceptor distance (r) in terms of efficiency of energy transfer (E) and Förster Distance (R0) is given by r = R0[1/E − 1]1/6. We have used the value of R0 of 6.749 nm for the Alexa 488 (donor) and Cy3 (acceptor) pair for estimating the donor-to-acceptor distance (Elangovan et al., 2003). We assume that orientation factor changes in the donor-acceptor fluorophores bound to a rigid Y-DNA stem can account for at most a minor portion of the observed Förster Distance changes ((Lakowicz, 2006; Sabanayagam et al., 2007)
Anisotropy Measurements
The anisotropy (R) measurements were performed in a microcuvette with a Picoquant Fluorimeter by the L-format or single channel method. We have used laser excitation with a narrow (10 nm, Chroma) bandpass excitation filter. The emission from the sample was collected with 90° geometry with 500-540 nm bandpass (Chroma) emission filter. We use two subscripts to indicate the orientation of the excitation and emission polarizers respectively. IHV represents horizontally polarized excitation and vertically polarized emission. We determined the G-factor which is the ratio of the sensitivities of the detection system for vertically and horizontally polarized light.
The anisotropy is given by
When AxCyY-DNAs were employed in packaging into wild-type procapsids without 2.7 kb leader, the value of R was measured to be R = 0.2 for Alexa 488 in this construct. As documented in the manuscript following ligation to a 2.7 kb stem extension to allow anchoring and stalling in the procapsid portal, protection of a portion of the 2.7 kb AxCyY-DNAs was observed when procapsids and terminase were present. We have obtained an equal anisotropy of R = 0.2 of Alexa 488 when 2.7 kb AxCyY is stalled in the procapsid portal. These anisotropy measurements indicate that the orientation of the dye in the Y-DNA construct did not alter while stalled in the procapsid.
Acknowledgments
We thank J-F Allemand for helpful discussion and Judith Black for figures. This work was supported by NIH grants AI011676 and NHGRI HG002655.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Krishanu Ray, Email: krishanu@cfs.umbi.umd.edu.
Chandran R. Sabanayagam, Email: csabanyagam@desu.edu.
Joseph R. Lakowicz, Email: joe@cfs.umbi.umd.edu.
References
- Baumann RG, Black LW. Isolation and characterization of T4 bacteriophage gp17 terminase, a large subunit multimer with enhanced ATPase activity. J Biol Chem. 2003;278(7):4618–27. doi: 10.1074/jbc.M208574200. [DOI] [PubMed] [Google Scholar]
- Baumann RG, Mullaney J, Black LW. Portal fusion protein constraints on function in DNA packaging of bacteriophage T4. Mol Microbiol. 2006;61(1):16–32. doi: 10.1111/j.1365-2958.2006.05203.x. [DOI] [PubMed] [Google Scholar]
- Black LW, Peng G. Mechanistic coupling of bacteriophage T4 DNA packaging to components of the replication-dependent late transcription machinery. J Biol Chem. 2006;281(35):25635–43. doi: 10.1074/jbc.M602093200. [DOI] [PubMed] [Google Scholar]
- Bustamante C, Smith SB, Liphardt J, Smith D. Single-molecule studies of DNA mechanics. Curr Opin Struct Biol. 2000;10(3):279–85. doi: 10.1016/s0959-440x(00)00085-3. [DOI] [PubMed] [Google Scholar]
- Casjens S, Wyckoff E, Hayden M, Sampson L, Eppler K, Randall S, Moreno ET, Serwer P. Bacteriophage P22 portal protein is part of the gauge that regulates packing density of intravirion DNA. J Mol Biol. 1992;224(4):1055–74. doi: 10.1016/0022-2836(92)90469-z. [DOI] [PubMed] [Google Scholar]
- Catalano CE. Viral Genome Packaging Machines: Genetics, Structure, and Mechanism. Eurekah.com and Kluwer Academic/Plenum Publishers; [Google Scholar]
- Cuervo A, Vaney MC, Antson AA, Tavares P, Oliveira L. Structural rearrangements between portal protein subunits are essential for viral DNA translocation. J Biol Chem. 2007;282(26):18907–13. doi: 10.1074/jbc.M701808200. [DOI] [PubMed] [Google Scholar]
- Deniz AA, Dahan M, Grunwell JR, Ha T, Faulhaber AE, Chemla DS, Weiss S, Schultz PG. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of Forster distance dependence and subpopulations. Proc Natl Acad Sci U S A. 1999;96(7):3670–5. doi: 10.1073/pnas.96.7.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driedonks RA, Engel A, tenHeggeler B, van D. Gene 20 product of bacteriophage T4 its purification and structure. J Mol Biol. 1981;152(4):641–62. doi: 10.1016/0022-2836(81)90121-2. [DOI] [PubMed] [Google Scholar]
- Duckworth BP, Zhang Z, Hosokawa A, Distefano MD. Selective labeling of proteins by using protein farnesyltransferase. Chembiochem. 2007;8(1):98–105. doi: 10.1002/cbic.200600340. [DOI] [PubMed] [Google Scholar]
- Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M, Periasamy A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods. 2003;29(1):58–73. doi: 10.1016/s1046-2023(02)00283-9. [DOI] [PubMed] [Google Scholar]
- Fuller DN, Raymer DM, Kottadiel VI, Rao VB, Smith DE. Single phage T4 DNA packaging motors exhibit large force generation, high velocity, and dynamic variability. Proc Natl Acad Sci U S A. 2007;104(43):16868–73. doi: 10.1073/pnas.0704008104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW. Symmetry mismatch and DNA packaging in large bacteriophages. Proc Natl Acad Sci U S A. 1978;75(10):4779–83. doi: 10.1073/pnas.75.10.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugel T, Michaelis J, Hetherington CL, Jardine PJ, Grimes S, Walter JM, Falk W, Anderson DL, Bustamante C. Experimental test of connector rotation during DNA packaging into bacteriophage phi29 capsids. PLoS Biol. 2007;5(3):e59. doi: 10.1371/journal.pbio.0050059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kainov DE, Tuma R, Mancini EJ. Hexameric molecular motors: P4 packaging ATPase unravels the mechanism. Cell Mol Life Sci. 2006;63(10):1095–105. doi: 10.1007/s00018-005-5450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, Ebright RH. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science. 2006;314(5802):1144–7. doi: 10.1126/science.1131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig A, Hebraud P, Gosse C, Dreyfus R, Baudry J, Bertrand E, Bibette J. Magnetic force probe for nanoscale biomolecules. Phys Rev Lett. 2005;95(12):128301. doi: 10.1103/PhysRevLett.95.128301. [DOI] [PubMed] [Google Scholar]
- Kondabagil KR, Zhang Z, Rao VB. The DNA translocating ATPase of bacteriophage T4 packaging motor. J Mol Biol. 2006;363(4):786–99. doi: 10.1016/j.jmb.2006.08.054. [DOI] [PubMed] [Google Scholar]
- Kosikov KM, Gorin AA, Zhurkin VB, Olson WK. DNA stretching and compression: large-scale simulations of double helical structures. J Mol Biol. 1999;289(5):1301–26. doi: 10.1006/jmbi.1999.2798. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR. Principles of Fluorescence Spectroscopy. Third. Springer; 2006. [Google Scholar]
- Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, Dodson EJ, Tavares P, Antson AA. Structural framework for DNA translocation via the viral portal protein. Embo J. 2007;26(7):1984–94. doi: 10.1038/sj.emboj.7601643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T, Dawid A, Bigot S, Barre FX, Saleh OA, Heslot F, Allemand JF, Bensimon D, Croquette V. DNA mechanics as a tool to probe helicase and translocase activity. Nucleic Acids Res. 2006;34(15):4232–44. doi: 10.1093/nar/gkl451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Chemla YR, Aathavan K, Grimes S, Jardine PJ, Anderson DL, Bustamante C. Intersubunit coordination in a homomeric ring ATPase. Nature. 2009;457(7228):446–50. doi: 10.1038/nature07637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram M, Sabanayagam C, Black LW. Modulation of the packaging reaction of bacteriophage t4 terminase by DNA structure. J Mol Biol. 2008;381(1):61–72. doi: 10.1016/j.jmb.2008.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray K, Oram M, Ma J, Black LW. Portal control of viral prohead expansion and DNA packaging. Virology. 2009 doi: 10.1016/j.virol.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabanayagam CR, Eid JS, Meller A. Using fluorescence resonance energy transfer to measure distances along individual DNA molecules: corrections due to nonideal transfer. J Chem Phys. 2005;122(6):061103. doi: 10.1063/1.1854120. [DOI] [PubMed] [Google Scholar]
- Sabanayagam CR, Oram M, Lakowicz JR, Black LW. Viral DNA packaging studied by fluorescence correlation spectroscopy. Biophys J. 2007;93(4):L17–9. doi: 10.1529/biophysj.107.111526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler B, Lipman EA, Eaton WA. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 2002;419(6908):743–7. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- Simpson AA, Tao Y, Leiman PG, Badasso MO, He Y, Jardine PJ, Olson NH, Morais MC, Grimes S, Anderson DL, Baker TS, Rossmann MG. Structure of the bacteriophage phi29 DNA packaging motor. Nature. 2000;408(6813):745–50. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature. 2001;413(6857):748–52. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]
- Sun S, Kondabagil K, Draper B, Alam TI, Bowman VD, Zhang Z, Hegde S, Fokine A, Rossmann MG, Rao VB. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell. 2008;135(7):1251–62. doi: 10.1016/j.cell.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Tavares P, Santos MA, Lurz R, Morelli G, de Lencastre H, Trautner TA. Identification of a gene in Bacillus subtilis bacteriophage SPP1 determining the amount of packaged DNA. J Mol Biol. 1992;225(1):81–92. doi: 10.1016/0022-2836(92)91027-m. [DOI] [PubMed] [Google Scholar]