

Abstract
Acute neurodegeneration is associated with high morbidity and mortality, and there are few effective treatments. Inflammation is central to the process of neuronal death, and within this field, the roles of the complement cascade have proven to be complex. The complement cascade is involved in triggering cell death and recruiting inflammatory cells to sites of inflammation, including the brain. However, complement might also have important neuroprotective roles that are only now coming to light. Recent evidence suggests that targeted activation of complement may be a potential avenue for treatment of stroke and other acute neurodegenerative diseases. Herein, we describe these novel neuroprotective roles of the complement cascade, focusing on signaling pathways that may be therapeutically targetable in acute neuronal injury.
Keywords: neuroprotection, neurodegeneration, acute neuronal injury, stroke, brain trauma, complement, inflammation, anaphylatoxins, membrane attack complex
Pathways of Neurodegeneration
Acute neurodegeneration resulting from such processes as ischemic and hemorrhagic stroke, brain trauma, neonatal ischemia-reperfusion injury, and spinal cord injury is associated with high morbidity and mortality with few effective treatments. Stroke alone is currently the third-leading cause of death in the United States, accounting for approximately 1 in 17 deaths annually (Box 1). In approaching these disorders, researchers are most limited by the restricted capacity of the central nervous system to regenerate. Effective neuroprotective treatments should thus be able to limit the extent of degeneration in patients immediately after injury, and such treatments are now being explored. Understanding the fundamental biology of neuronal injury and repair is central to the development of such novel neuroprotective therapies that could be of potential use in the treatment of these disorders.
Box 1. The Complement System.
Plasma components capable of killing bacteria were first described by Jules Bordet in 1896 at the Pasteur Institute and later named complement by Paul Ehrlich in his initial descriptions of the human immune system. Since that time, numerous biological roles of complement have been described and the high degree of the conservation of the complement pathways in both invertebrate and vertebrate organisms speak to both their ancient origin and their importance within the immune system.
Major Immunological Functions
direct lysis of bacteria and cells recognized as non-self
opsonization of bacteria and cells recognized as non-self, which promotes phagocytosis and immunological clearance by the spleen and liver
activation of macrophages, B cells, and T cells through cell surface complement receptors
Complement Pathways
The complement system is classified into three systems, the classical pathway, which was the first pathway described, the alternative pathway, and the mannose-binding lectin (MBL) pathway. Broadly, the classical pathway primarily acts to lyse cells and bacteria already recognized and opsonized by plasma antibodies as well as target these cells for immunological clearance. The alternative pathway acts independent of plasma antibodies and binds to cells and bacteria that do not express complement decay accelerating factor (DAF). Because all normal cells of the body express DAF, this pathway selectively targets non-self cells and bacteria. The MBL pathway recognizes lectins and ficolins, common bacterial surface proteins, and acts primarily to lyse bacteria and target them for immunological clearance. These pathways and their component proteins are illustrated in Figure 2.
Pathway Components
Classical Pathway: C3, Factor B
Alternative Pathway: C2, C4,
Mannose Binding Lectin Pathway: C1q, C1r, C1s, C2, C4
Membrane Attack Complex: C5, C6, C7, C8, C9
Evolution
Analogs of the human complement systems have been identified in all species of the kingdom animalia and the system is thought to have originated through divergence of a single complement C3 precursor. Current theories regarding the evolution of the system in both vertebrates and invertebrates have been recently reviewed (REVIEW).
Cell death in neurodegeneration occurs through two distinct mechanisms: apoptosis, an ordered and programmed process, and necrosis, an accidental and fairly unordered process 1. There is evidence that both of these processes occur simultaneously during a stroke. Necrosis is usually a disorderly process characterized by inflammation, release of cytoplasmic content to the extracellular space, and subsequent widespread tissue damage. Necrosis typically occurs in response to extreme or extensive cellular or tissue damage, such as that occurring after ischemia or trauma. By contrast, apoptosis is a highly conserved programmed cell death mechanism that morphologically consists of nuclear membrane fragmentation, chromatin compaction and fragmentation, and cell disintegration through membrane blebbing; the resultant apoptotic bodies are then quickly ingested and degraded by professional phagocytes and neighboring cells, preventing inflammation and tissue scarring 2-4.
Neuronal death in both acute (stroke, traumatic injury) and chronic (such as Alzheimer’s Disease, Parkinson’s Disease and Huntington’s Disease) neurodegenerative disorders involves a complex interplay between necrosis and apoptosis. These pathways appear to be predominantly mediated proximally by activated Caspase-1 (interleukin-1β converting enzyme; ICE) and are regulated by caspase-recruitment domain (CARD) proteins such as CARD-only protein (COP) and Rip2. The CARD domain is a recruitment domain through which procaspases aggregate and crossproteolyze to form active caspases. COP competitively binds Caspase-1 and prevents its aggregation and activation, while Rip-2 actively recruits and thus accelerates Caspase-1 aggregation and activation. Both of these proteins are highly expressed in the brain and are differentially expressed after acute and chronic neurodegeneration. Immediately after ischemic injury, COP is depleted while Rip-2 is upregulated, leading to increased aggregation and activation of Caspase 1. Caspase 1 activation leads to downstream activation of the effector caspase Caspase 3, which cleaves numerous downstream targets, leading ultimately to cell death (reviewed in 5). How upstream pathways, such as the inflammatory cascade and complement pathways, modulate this common caspase cascade remains a poorly understood aspect of neurobiology. There is evidence for the involvement of p53 pathways (reviewed in 6), BAX/Bcl-2 pathways (reviewed in 7), and superoxide dismutase pathways (reviewed in 8) but much remains to be eludicated. Understanding these upstream pathways will greatly enhance our potential for the development of new neuroprotective therapies.
While ischemia does lead directly to neuronal death, mostly through a process resembling classical necrosis, the damage of surrounding tissue (ischemic penumbra) is a potentially preventable event. The ischemic penumbra, the region surrounding the immediate infarct, is not only at risk from further or prolonged ischemia, which can potentially be resolved with the adept use of thrombolytic therapy, but is also at risk from secondary injury induced by inflammatory mediators released by the necrotic core. Indeed, recent studies have suggested that many of the neurons in the ischemic penumbra survive for hours or even days before death, suggesting that targeting these sub-acute processes could preserve significant neuronal function. Within the field, this gradual death has been referred to as ‘contagious apoptosis’, where release of diffusible factors such as interleukin-1β (IL-1β), tumor necrosis factor α (TNF-α) and reactive oxygen species (ROS) by an injured neuron of the central necrotic core leads to damage of surrounding neurons. This model has been used to describe the mechanism of widespread neuronal death after a wide range of injuries. This mode of secondary injury seems to be particularly important in acute injury, with subsequent calcium influx leading not only to direct neuronal apoptosis but also oligodendroglial apoptosis and subsequent demyelination. Thus, the theory of therapeutic design for acute neurodegeneration has recently focused on limiting this contagious apoptosis and thus limiting the degree of neuronal death by limiting the release of inflammatory factors from the core and limiting the extent of the resultant apoptotic response by the surrounding penumbra.
Inflammation is thus central to the process of neuronal death, but the precise roles of various inflammatory mediators, including cytokines, chemokines and immunological cells, is still being elucidated. Modulating the immune system could be an important therapeutic strategy in the treatment of acute neurodegenerative disorders. Within this field, the roles of the complement cascade, an important part of innate immunity, have proven to be particularly complex. Until recently, the complement cascade was believed to be a pathway primarily involved in promoting neuronal death. Recent evidence suggests, however, that complement pathways play important roles in neuronal survival and even in normal neuronal development. Herein, we review these novel roles of the complement cascade and focus on signaling pathways that might be therapeutically targetable in acute neuronal injury.
Complement – An Important Pathway in Innate and Adaptive Immunity
The complement cascade (diagrammed in Figure 2) is a pathway that is largely involved in the recruitment and chemotaxis of inflammatory cells and direct induction of cell death (reviewed in 9). This cascade is mediated by a number of soluble complement factors that are synthesized by the liver and circulate in the blood. These main complement factors are C1 through C9, while a number of additional factors also exist, including B and D. The complement cascade consists of three distinct sub-pathways that are distinguished by their trigger mechanisms, known as the alternative pathway, the classical pathway, and the mannose-binding-lectin pathway. These pathways converge with the final formation of the membrane attack complex (MAC).
Figure 2. Complement Cascades.
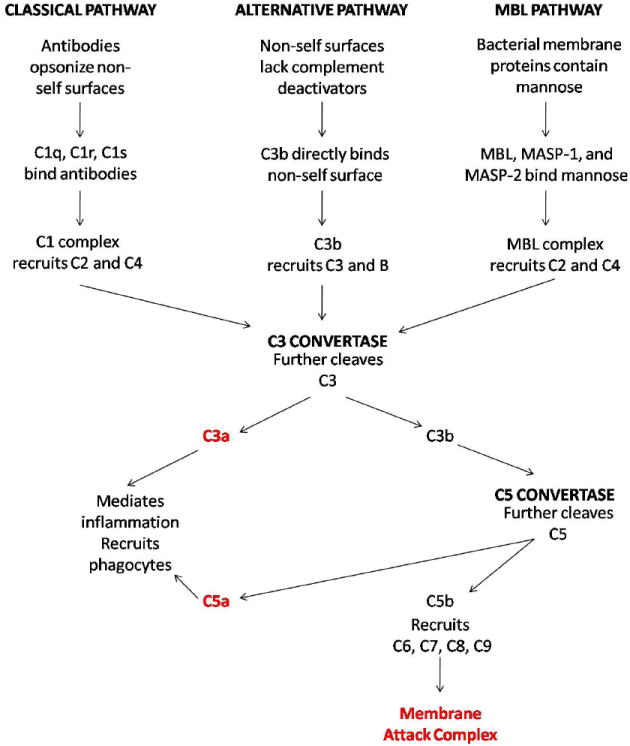
Three parallel complement cascades, the classical pathway, the alterative pathway, and the mannose-binding-lectin (MBL) pathway are activated by different immunological stimuli but ultimately converge on a common pathway that induces local inflammation, recruits phagocytes, and lyses cellular targets. A detailed description of these pathways is provided in Box 2.
The alternative pathway of complement activation involves the spontaneous cleavage of complement factor C3 into C3a and C3b. C3a acts a chemokine to recruit inflammatory mediators to the site of activation, while C3b acts as an opsonin, covalently binding to a target cell. Serum Factor B is then bound to C3b on the cell surface and is cleaved by Factor D to form a new complex, C3bBb, known as the C3 convertase. This complex then acts to rapidly catalyze the formation of more C3b to opsonize the surface of the cell, recruiting macrophages and other cells of innate immunity. Some of the newly converted C3b complexes with the C3 convertase to form C3bBbC3b, the alternative pathway C5 convertase. Normal cells possess a variety of complement-inactivating proteins that prevent this process, including decay accelerating factor (DAF) and CD59, thus allowing for selective activation of C3b on injured or diseased cells as well as foreign cells such as bacteria.
The classical pathway involves the binding of an antibody, either IgG or IgM, to a target cell, subsequently triggering the binding of C1q to the antibody. C1q then catalyzes the activation of C1s by C1r, leading to the formation of the C4 convertase. The C4 convertase activates C4 to C4a and C4b and C2 to C2a and C2b. C4b and C2a complex to form the classical pathway C3 convertase, C4b2a. Similar to C3bBb, C4b2a serves to catalyze the rapid formation of C3b to opsonize the cell surface and to form the classical pathway C5 convertase, which is formed by the C4b2a3b complex.
The mannose-binding-lectin pathway is similar to the classical pathway. It differs only insofar as a mannose-binding lectin and associated proteins, known as mannose-binding-lectin-associated serine proteases (MASPs), trigger the formation of C4a and C4b from C4. The rest of the pathway after the formation of C4a is identical to the classical pathway. The mannose-binding-lectin pathway primarily targets bacterial cells, which contain abundant mannose residues, and has little involvement within the context of mammalian cell targeting.
All three pathways converge with the formation of the C5 convertase. The C5 convertase serves to activate C5, forming C5a, a powerful chemokine, and C5b, which inserts into the cell membrane. C5b serves to recruit C6, C7, C8, and C9 to form the MAC, which forms pores within the membranes of target cells and induces cell lysis.
Physiological Roles of Complement in the Central Nervous System
Inflammation clearly plays a significant role in acute cerebral ischemic injury in several contexts, as discussed above. The complement cascade has proved to be of particular relevance in this regard (reviewed in 10). Complement activation might be an important part of immune-mediated neuronal toxicity, and complement inhibition has certainly been a therapeutic target for neurodegenerative disorders. This has primarily been driven by reports of complement activation following acute and chronic CNS injury. For example, activated complement elements, including C1q, C3b, C3d and C5b-C9, have been found to be localized in the immediate vicinity of neurons affected by traumatic head injury 11 Furthermore, C3 mRNA levels were elevated in the penumbra of the contusion, suggesting locally induced expression 11. Additionally, cerebral ischemic infarcts, as occur in ischemic strokes, are frequently found to include high concentrations of C9 and other components of the MAC membrane attack complex, with associated neutrophil infiltration. This is also associated with markedly elevated levels of complement factors in the cerebrospinal fluid (CSF) of patients following cerebrovascular accidents 12. C1q, the initiator of the classical pathway, has been shown to bind to the membranes of neurons in an antibody-independent manner, and neurons have particularly low levels of CD59 and DAF, molecules that normally confer cellular resistance to complement 13, 14. This could confer upon neurons an increased susceptibility to complement-based lysis as well as damage from recruited immune cells. There is also evidence that knocking out CD59 in mice confers increased susceptibility to neuronal death after trauma 15, further implicating complement pathways in this acute neurodegeneration. Thus, much evidence exists in the literature for the role of complement pathways in the promotion of neuronal death and generalized CNS inflammation. Much of this literature has been reviewed by Bonifati and Kishore 16.
Recent evidence suggests, however, that complement might actually have important neuroprotective roles that could be targetable therapeutically within the context of acute neurodegeneration and might also play an important physiological role in neurogenesis. Perhaps most striking is that many CNS cell types, including astroglia, oligodendroglia and neurons, have been shown to synthesize various components of the complement cascade de novo, a role previously thought to be served exclusively by the liver. Neurons have also recently been found to express various complement receptors, and the physiological roles of these receptors are just being uncovered. Complement activation might also promote oligodendrocyte regeneration and secretion of nerve growth factor. Also, complement deficiency leads to an increased susceptibility to mengingococcal meningitis and various infectious forms of encephalitis. Because of these recent findings that activation of the complement cascade can promote neuronal survival and tissue remodeling, inducing activation of the complement system in the brain might be an additional important therapeutic strategy in treating acute and chronic neurodegeneration.
That neurons express complement receptors and synthesize complement factors is a very recent and indeed startling finding. The first neuronal C5a receptor was discovered in 1998 and named nC5aR 17. Shortly thereafter, neuronal and glial expression of a C3a receptor was also found18. Subsequently, it has been shown that many neuronal subtypes, including dentate gyrus granule cells, hippocampal pyramidal hilar cells and cerebellar Purkinje cells constitutively express receptors for C3a and C5a and also express a number of other chemokine receptors, including CXCR2, CXCR3, CXCR4, CCR1, CCR3, CCR4, CCR5, CCR9 and CX3CR 18-20. Many neuronal cells also show inducible expression of these receptors that are responsive to numerous physiological and pathological stimuli, including acute injury and ischemia.
Complement Factor C5
Initial work with the receptor nC5aR suggested a pro-apoptotic role for C5a-mediated signaling. Very high concentrations of C5a induced intracellular calcium influx and induced DNA fragmentation and elevation of nuclear c-fos 17. However, the possible protective roles of C5a that have been recently elucidated are particularly noteworthy. While C5a is traditionally considered an anaphylatoxin with proinflammatory and chemotactic functions, several studies have implicated it in the development and survival of neurons. C5aR is expressed in cerebellar granule neurons, and C5a is a potent inhibitor of apoptosis in primary cultures of these neurons when used in lower concentrations than those used in demonstrating its pro-death functions 21. The physiological relevance of these higher and lower dose concentrations remains to be established although the signaling pathways they elucidate may be therapeutically targeted downstream of the C5aR. In vitro studies have shown that C5a is mitogenic for undifferentiated human neuroblastoma cells and might be neuroprotective for terminally differentiated cells against ischemia induced death. Murine CNS neurons express a C5a receptor that binds to C5a in vitro, and activation of this receptor leads to downstream induction of the heterotrimeric G protein Gαi and NF-κB signaling. For example, in a neuroblastoma cell model (SH-SY5Y cells), C5a induced mitogenic activity in undifferentiated SH-SY5Y cells in a dose-dependent fashion and had a distinctly protective effect in differentiated SH-SY5Y cells against Aβ-mediated neurotoxicity by enhancing cellular survival signals 22. LAN-5 human neuroblastoma have also been shown to constitutively express C5aR, and ligand binding induces a biphasic calcium response and downstream protein kinase C (PKC) and NF-κB activation through Gαi signaling that is neuroprotective 22. The neuronal C5aR could also regulate important signal transduction pathways in neurons that are as yet unidentified. For example, hereditary C5 deficiencies have been shown to exacerbate neurodegeneration following exposure to various injurious stimuli 23, 24. Recent evidence for C5a regulation of glutamate receptor functions in neurons might also be significant in its neuroprotective functionality. In an in vivo mouse model, C5a protected neurons from kainic-acid induced apoptosis. This protection was specifically associated with the inhibition of glutamate-mediated induction of caspase-3 25, 26. C5−/− knockout mice are also more susceptible to glutamate excitotoxicity and apoptosis than wild-type mice 27, and excitotoxicity has been implicated as an important means of neuronal death within the ischemic penumbra of strokes. C5a might also play important roles in neuronal development. Injecting C5a receptor agonists at the surface of the developing cerebellum in 11–12-day-old rats increased the thickness of the cortical layers, specifically inducing the proliferation of immature granule cells 28. This might have implications for inducing neuronal proliferation and regeneration after acute injury.
Complement Factor C3
Although less work has been done with C3a, several studies suggest that C3a might also have several protective roles. For example, C3a protects neurons against N-methyl-D-aspartate (NMDA) toxicity 29, and could thus be involved in modulating neuronal glutamate signaling. The NMDA receptor is known to modulate Ca2+ influx into neurons and thus mediate glutamate excitotoxicity. Thus, this might be evidence that C3a also protects against glutamate excitotoxicity. C3a has also been shown to promote secretion of nerve growth factor (NGF), an important neuronal survival and growth signal, by human microglial cells in vitro 30, 31 and by this mechanism could promote neuronal regeneration and repair. Other studies have implicated both C3a and C5a in the promotion of NGF production by astrocytes 32. Specifically, these anaphylatoxins upregulated the transcription of NGF within astrocytes. C3a might also promote basal and ischemia-induced neurogenesis. C3−/− knockout mice had a pronounced increase in infarct size and amount of brain tissue lost after ischemic injury compared with wild-type animals, and, in the same study, C3a was also shown to promote neuronal differentiation 33, 34. In another recent study, mice transgenically expressing C3a in the CNS (C3a/GFAP mice) were significantly resistant to mortality induced by endotoxic shock compared with wild-type and C3aR-knockout mice 35. Even in chronic neurodegeneration, C3a might provide a protective role. For example, in a mouse model of Alzheimer Disease, knocking out amyloid precursor protein (the protein implicated in the formation of the plaques characteristic of the disease) resulted in neuroprotection, with elevated levels of C3a in the cerebrospinal fluid 36. Like C5a, C3a might also be important in neuronal development as well. For example C3a can accelerate the normal migration of cerebellar granule cells 28. This could be relevant to the promotion of neuronal repopulation of an injured site after acute neurodegeneration. C3a is also essential in normal CNS synaptic elimination, and C3−/− knockout mice demonstrated defects in synaptogenesis 37.
The Membrane Attack Complex
At every level, complement pathways have proved to have very complex protective and injurious roles. In addition to the aforementioned neuroprotective roles of C3a and C5a, the MAC has been shown to promote oligodendrocyte and neuronal survival at sublytic concentrations. There appears to be a critical threshold of lysis for the MAC complex. Below this threshold, bound C5b-C9 are rapidly endocytosed by the cell and activate various intracellular signaling cascades that seem to be primarily protective. Postmitotic primary oligodendrocytes re-enter the cell cycle after being exposed to sublytic levels of MAC proteins in culture. Sublytic levels of MAC proteins also induce resistance to apoptosis induced by TNF-α by upregulating Bcl-2 and inhibiting Caspase-3 activation in oligodendroglia 38. Sublytic levels of MAC proteins might also inhibit BAD through the phosphoinositide 3-kinase (PI3K)–Akt pathway and inhibit Caspase-8 processing in oligodendroglia 39, 40. In addition to models of ischemia, sublytic levels of MAC proteins have also been associated with resistance to apoptosis in several models of human disease, including experimental autoimmune encephalomyelitis for multiple sclerosis and several models of colon cancer through various mechanisms, including the exposure of C-reactive protein binding sites on cell membranes 41. This might provide an important strategy for initiating regeneration of oligodendrocytes within an area of acute injury.
Other inflammatory cytokines and chemokines have also been shown to have some neuroprotective effects, although the field is small. IL-8, which binds CXCR1 and CXCR2, and growth-related oncogene-α2, which binds CXCR2, have also been shown to alter signal transduction in cerebellar Purkinje cells by inducing calcium influx in a neuroprotective manner. Reflecting possibly similar mechanisms, murine stromal cell-derived factor-1α, RANTES (CCL5), macrophage-derived chemokine and fractalkine, chemokines that bind to CXCR4, CCR-4 and CX3CR1 on neurons, regulate synaptic transmission of embryonic rat hippocampal neurons in the context of acute ischemia.
Thus complement pathways play a complex role in regulating neurogenesis, neuronal survival, and neurodegeneration. While some roles have been established for the major complement factors in all of these areas, much remains to be elucidated.
Therapeutic implications
Targeting complement pathways therapeutically could possibly be an exciting new approach to treating acute neurodegeneration. However, much work remains to be done in elucidating the precise mechanisms by which complement can be neuroprotective and which conditions promote its neuroprotective effects if such therapeutics are to have a hope of becoming a reality. While these protective roles of complement are novel and fascinating in their own right, one must be wary that they might be particularly difficult to modulate therapeutically. Small alterations in the concentrations of complement factors could tip the scale from neuroprotection to injury, and such a system would be difficult to control in the context of acute injury, where heterogeneity of response and degree of injury are common. Thus, targeting downstream signaling pathways might be of particular interest. Because both C3a and C5a operate through the activation of G-protein-coupled receptors that are coupled predominantly to the Gαi family, it will be worthwhile examining potential targets in this arena. This could be an additional interesting area of new research. Thus, in moving towards a therapeutic target, two goals must be accomplished: 1) the neuronal specificity of complement factors and their receptors must be examined and 2) the downstream signal transducers of complement pathways must be identified their neuron-specific functions elucidated.
There are a number of potential therapeutic strategies that are suggested by this early work on complement pathways in acute neurodegeneration. Some suggested therapeutic questions remain unanswered. For example, could injecting C3a into the CSF of trauma or stroke patients alleviate inflammation-induced cell death? Similar to various in vivo mouse models, C3a could have an extended neuroprotective effect in stroke or trauma that is complementary to tissue plasminogen activator (tPA), a currently used mainstream therapy. Alternatively, could C3a or C5a receptor agonists be used in the treatment of stroke or trauma? These potential therapies merit further cell biological and in vivo examination.
Another important question that these early studies raise is whether complement activation can actually trigger neuronal regeneration in productive ways. Therapeutic strategies to date have largely focused on neuroprotective approaches because productive neuroregeneration is a difficult problem. Regeneration of synaptic connections in the CNS in ways that restore neuronal circuit functionality remains an unsolved problem. Because complement is crucial for normal neuronal synaptogenesis, these pathways might also possess important potential for productive neuroregeneration as well.
The fact that both C3a and C5a inhibit glutamate excitotoxicity and related processes provides evidence that they could be important therapeutic targets in a number of both acute and chronic neurodegenerative processes in which glutamate excitotoxicity has been implicated as an important mechanism. This includes not only stroke and acute trauma, but also Alzheimer Disease, Parkinson Disease and others.
Additionally, we must answer the question of whether affecting normal complement pathways downstream of the complement receptor will promote neuroprotective effects similar to those induced by activated complement components. Investigation of the multiple pathways activated by complement factors in neurons and glia is still largely incomplete. Determination of the downstream effectors of the neuroprotective effects of complement factors will provide an immense new set of potential therapeutic targets.
The most difficult question perhaps is how to selectively target the neuroprotective or neuroregenerative effects of complement pathways without simultaneously inducing inflammation and cell-death. In the long term, this brings us back to the two major goals that must be accomplished: 1) the neuronal specificity of complement factors and their receptors must be examined and 2) the downstream signal transducers of complement pathways must be identified their neuron-specific functions elucidated. Towards accomplishing the first goal, we must focus on the G protein coupled receptors (GPCRs) that act as the targets of complement factors. GPCRs are highly cell type specific and thus present a selective therapeutic target. Elucidating the GPCRs that are activated by complement factors will be an important step towards selective therapeutic targeting of the neuroprotective pathways activated by complement. Towards accomplishing the second goal, we may be able to identify the specific neuroprotective and neuroregenerative pathways that complement factors activate and target these downstream directly. Again here, specifically because heterotrimeric G protein coupled receptors (GPCRs) are known to be highly cell-type specific, the identification of new neuron-specific GPCRs that activate these neuroprotective and neuroregenerative pathways will be an important therapeutic strategy.
Concluding remarks
Acute neurodegeneration is a leading cause of morbidity and mortality worldwide. Gaining an understanding of pathways involved in neurodegeneration in acute injury and stroke will provide important new targets for the development of novel therapeutics in a field with few existing options. Inflammatory pathways are central to the process of neuronal death in acute injury. Within this realm, complement pathways and their roles in neurodegeneration and neuroprotection are only now being uncovered. Until recently, complement was largely thought to promote neuronal death after acute injury. With the discovery of the first neuronal complement receptor in 1998, slowly over the past few years, the important roles of complement in neuroprotection and neuronal development have begun to be elucidated. It is now clear that all aspects of the complement cascade, from the anaphylatoxins C3a and C5a to the membrane attack complex C5-C9, play both neurodegenerative and neuroprotective roles. Understanding the molecular pathways of the neuroprotective roles of these complement factors and what the regulatory switches are that determine the balance between neurodegeneration and neuroprotection will be important avenues of investigation towards the development of novel therapies for acute neuronal injury (Box 2). Herein, we have reviewed the literature that exists to date in this nascent field with the hope that it will catalyze investigations into these novel neuroprotective roles of complement and lead to the development of novel therapies based on these future developments.
Box 2. Stroke.
Stroke is the third–leading cause of death in the United States and the second-leading cause of death worldwide
Tissue plasminogen Activator (tPA) is the only currently established clinical therapeutic available for the mitigation of stroke-induced neuronal death and indeed has only been proven to be effective within the first 3-4.5 hours after onset
There are no existing neuroprotective therapeutics for the treatment of stroke
Box 3. Outstanding questions.
Can complement activation be neuroprotective?
Can complement activation actually trigger neuronal regeneration in productive ways?
Can complement activation inhibit glutamate excitotoxicity, a pathologic process involved in a number of acute and chronic neurodegenerative diseases?
What are the downstream mediators of complement-mediated neuroprotection?
How can these pathways be targeted therapeutically?
Figure 1. Apoptosis Signaling Pathways.
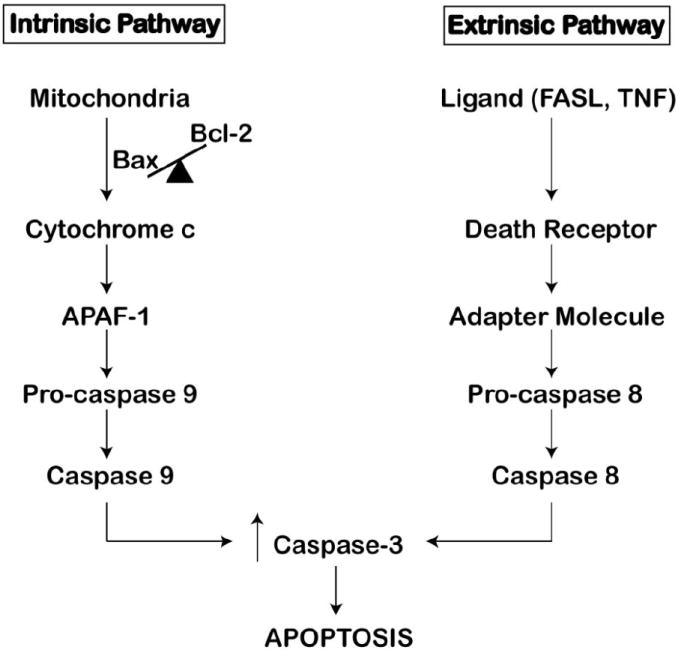
The Intrinsic pathway is triggered by a disruption in the BAX:Bcl-2 protein ratio at the mitochondrion. Increase in this ratio allows for the homodimerization of BAX, and subsequent Cytochrome c efflux into the cytoplasm activates APAF-1. APAF-1 triggers Caspase 9 activation and the subsequent caspase cascade. The extrinsic pathway is triggered by ligand binding to the death receptors. Receptor activation leads to downstream recruitment of adapter proteins, such as FADD, resulting in the activation of Caspase 8 and the subsequent caspase cascade. Both pathways converge on Caspase 3, leading to apoptosis.
Table 1.
Neuroprotective and Proliferative Actions of Complement Factors xx
| Complement Factor | Model System | Cell Type | Phenotype | Reference |
|---|---|---|---|---|
| C5a | Primary Culture | Cerebellar Granule Cells | Inhibited apoptosis | 21 |
| C5a | Cell Culture | Human Neuroblastoma Cells | Increased proliferation | 22 |
| C5a | Cell Culture | SH-SY5Y Neuroblastoma Cells | Increased proliferation and inhibited Aβ induced cell death | 22 |
| C5a | Cell Culture | Human LAN-5 Neuroblastoma Cells | Inhibited cell death | 22 |
| C5a | In vivo C5-/- mouse model | Hippocampal Pyramidal Cells | Lack of C5 increased susceptibility to kainic-acid induced apoptosis | 23, 24 |
| C5a | In vivo mouse model | Cortical Neurons | Inhibited kainic-acid induced apoptosis | 26,27 |
| C5a | In vivo C5-/- mouse model | Cortical Neurons | Lack of C5 increased susceptibility to glutamate excitotoxicity | 27 |
| C5a | In vivo mouse model | Cerebellar Cortical Neurons | Induced proliferation | 28 |
| C3a | Primary Culture | Cortical Neurons | Protects against NMDA toxicity | 26 |
| C3a | Primary Culture | Microglia | Promotes neuronal survival by induction of NGF expression in microglia | 30,31 |
| C3a | In vivo C3-/- mouse model | Cortical Neurons | Lack of C3 increased infarct size after ischemic injury | 33,34 |
| C3a | C3a/GFAP mouse model (overexpression) | CNS Neurons | Overexpression prevented endotoxic shock induced mortality | 35 |
| C3a | In vivo C3-/- mouse model | Cortical Neurons | Lack of C3 led to defects in synaptogenesis | 37 |
| Membrane Attack Complex | Cell Culture | Oligodendrocytes | Sublytic levels of MAC proteins protected against TNF-α induced apoptosis | 38, 39,40 |
| Membrane Attack Complex | In vivo experimental autoimmune encephalomyelitis mouse model | Oligodendrocytes | Sublytic levels of MAC proteins protected against TNF-α induced apoptosis | 41 |
Acknowledgments
This work was supported by the Paul and Daisy Soros Fellowship, the Huntington’s Disease Society of America Donald A. King Fellowship, and the American Medical Association Seed Grant (to V. Y.).
Abbreviations
- GFAP
glial fibrillary acidic protein
- MAC
membrane attack complex
- NGF
nerve growth factor
- NMDA
N-methyl-D-aspartate,
- TNF-α
tumor necrosis factor α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kanduc D, et al. Cell death: apoptosis versus necrosis (review) International journal of oncology. 2002;21:165–170. [PubMed] [Google Scholar]
- 2.Jacobson MD, et al. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 3.Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 4.Cohen JJ. Apoptosis. Immunology today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- 5.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. The New England journal of medicine. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 6.Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochemical and biophysical research communications. 2005;331:761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- 7.Plesnila N. Role of mitochondrial proteins for neuronal cell death after focal cerebral ischemia. Acta neurochirurgica. 2004;89:15–19. doi: 10.1007/978-3-7091-0603-7_3. [DOI] [PubMed] [Google Scholar]
- 8.Pong K. Oxidative stress in neurodegenerative diseases: therapeutic implications for superoxide dismutase mimetics. Expert opinion on biological therapy. 2003;3:127–139. doi: 10.1517/14712598.3.1.127. [DOI] [PubMed] [Google Scholar]
- 9.Rus H, et al. The role of the complement system in innate immunity. Immunologic research. 2005;33:103–112. doi: 10.1385/IR:33:2:103. [DOI] [PubMed] [Google Scholar]
- 10.Rus H, et al. The complement system in central nervous system diseases. Autoimmunity. 2006;39:395–402. doi: 10.1080/08916930600739605. [DOI] [PubMed] [Google Scholar]
- 11.Bellander BM, et al. Complement activation in the human brain after traumatic head injury. Journal of neurotrauma. 2001;18:1295–1311. doi: 10.1089/08977150152725605. [DOI] [PubMed] [Google Scholar]
- 12.Lindsberg PJ, et al. Complement activation in the central nervous system following blood-brain barrier damage in man. Annals of neurology. 1996;40:587–596. doi: 10.1002/ana.410400408. [DOI] [PubMed] [Google Scholar]
- 13.Singhrao SK, et al. Spontaneous classical pathway activation and deficiency of membrane regulators render human neurons susceptible to complement lysis. The American journal of pathology. 2000;157:905–918. doi: 10.1016/S0002-9440(10)64604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singhrao SK, et al. Differential expression of individual complement regulators in the brain and choroid plexus. Laboratory investigation; a journal of technical methods and pathology. 1999;79:1247–1259. [PubMed] [Google Scholar]
- 15.Stahel PF, et al. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. Journal of neuroinflammation. 2009;6:2. doi: 10.1186/1742-2094-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Molecular immunology. 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Farkas I, et al. A neuronal C5a receptor and an associated apoptotic signal transduction pathway. The Journal of physiology. 1998;507(Pt 3):679–687. doi: 10.1111/j.1469-7793.1998.679bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davoust N, et al. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201–211. doi: 10.1002/(sici)1098-1136(199905)26:3<201::aid-glia2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 19.Nataf S, et al. Kinetics of anaphylatoxin C5a receptor expression during experimental allergic encephalomyelitis. Journal of neuroimmunology. 1998;91:147–155. doi: 10.1016/s0165-5728(98)00169-6. [DOI] [PubMed] [Google Scholar]
- 20.Stahel PF, et al. TNF-alpha-mediated expression of the receptor for anaphylatoxin C5a on neurons in experimental Listeria meningoencephalitis. J Immunol. 1997;159:861–869. [PubMed] [Google Scholar]
- 21.Benard M, et al. Characterization of C3a and C5a receptors in rat cerebellar granule neurons during maturation. Neuroprotective effect of C5a against apoptotic cell death. J Biol Chem. 2004;279:43487–43496. doi: 10.1074/jbc.M404124200. [DOI] [PubMed] [Google Scholar]
- 22.O’Barr SA, et al. Neuronal expression of a functional receptor for the C5a complement activation fragment. J Immunol. 2001;166:4154–4162. doi: 10.4049/jimmunol.166.6.4154. [DOI] [PubMed] [Google Scholar]
- 23.Pasinetti GM, et al. Hereditary deficiencies in complement C5 are associated with intensified neurodegenerative responses that implicate new roles for the C-system in neuronal and astrocytic functions. Neurobiology of disease. 1996;3:197–204. doi: 10.1006/nbdi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 24.Tocco G, et al. Complement and glutamate neurotoxicity. Genotypic influences of C5 in a mouse model of hippocampal neurodegeneration. Molecular and chemical neuropathology / sponsored by the International Society for Neurochemistry and the World Federation of Neurology and research groups on neurochemistry and cerebrospinal fluid. 1997;31:289–300. doi: 10.1007/BF02815131. [DOI] [PubMed] [Google Scholar]
- 25.Mukherjee P, Pasinetti GM. The role of complement anaphylatoxin C5a in neurodegeneration: implications in Alzheimer’s disease. Journal of neuroimmunology. 2000;105:124–130. doi: 10.1016/s0165-5728(99)00261-1. [DOI] [PubMed] [Google Scholar]
- 26.Osaka H, et al. Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. Journal of cellular biochemistry. 1999;73:303–311. [PubMed] [Google Scholar]
- 27.Mukherjee P, et al. Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. Journal of neuroinflammation. 2008;5:5. doi: 10.1186/1742-2094-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benard M, et al. Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Molecular immunology. 2008;45:3767–3774. doi: 10.1016/j.molimm.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 29.van Beek J, et al. Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport. 2001;12:289–293. doi: 10.1097/00001756-200102120-00022. [DOI] [PubMed] [Google Scholar]
- 30.Heese K, et al. NF-kappaB modulates lipopolysaccharide-induced microglial nerve growth factor expression. Glia. 1998;22:401–407. doi: 10.1002/(sici)1098-1136(199804)22:4<401::aid-glia9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 31.Heese K, et al. Inflammatory signals induce neurotrophin expression in human microglial cells. Journal of neurochemistry. 1998;70:699–707. doi: 10.1046/j.1471-4159.1998.70020699.x. [DOI] [PubMed] [Google Scholar]
- 32.Jauneau AC, et al. Interleukin-1beta and anaphylatoxins exert a synergistic effect on NGF expression by astrocytes. Journal of neuroinflammation. 2006;3:8. doi: 10.1186/1742-2094-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahpeymai Y, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. The EMBO journal. 2006;25:1364–1374. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bogestal YR, et al. Signaling through C5aR is not involved in basal neurogenesis. Journal of neuroscience research. 2007;85:2892–2897. doi: 10.1002/jnr.21401. [DOI] [PubMed] [Google Scholar]
- 35.Boos L, et al. C3a expressed in the central nervous system protects against LPS-induced shock. Neuroscience letters. 2005;387:68–71. doi: 10.1016/j.neulet.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 36.Zhou J, et al. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. Journal of neurochemistry. 2008;106:2080–2092. doi: 10.1111/j.1471-4159.2008.05558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 38.Soane L, et al. Inhibition of oligodendrocyte apoptosis by sublytic C5b-9 is associated with enhanced synthesis of bcl-2 and mediated by inhibition of caspase-3 activation. J Immunol. 1999;163:6132–6138. [PubMed] [Google Scholar]
- 39.Soane L, et al. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J Immunol. 2001;167:2305–2311. doi: 10.4049/jimmunol.167.4.2305. [DOI] [PubMed] [Google Scholar]
- 40.Cudrici C, et al. C5b-9 terminal complex protects oligodendrocytes from apoptotic cell death by inhibiting caspase-8 processing and up-regulating FLIP. J Immunol. 2006;176:3173–3180. doi: 10.4049/jimmunol.176.5.3173. [DOI] [PubMed] [Google Scholar]
- 41.Li YP, et al. Sublytic complement attack exposes C-reactive protein binding sites on cell membranes. J Immunol. 1994;152:2995–3005. [PubMed] [Google Scholar]