

Abstract
Rtt106p is a Saccharomyces cerevisiae histone chaperone with roles in heterochromatin silencing and nucleosome assembly. The molecular mechanism by which Rtt106p engages in chromatin dynamics remains unclear. Here, we report the 2.5 Å crystal structure of the core domain of Rtt106p, which adopts an unusual “double pleckstrin homology” domain architecture that represents a novel structural mode for histone chaperones. A histone H3-H4-binding region and a novel double-stranded DNA-binding region have been identified. Mutagenesis studies reveal that the histone and DNA binding activities of Rtt106p are involved in Sir protein-mediated heterochromatin formation. Our results uncover the structural basis of the diverse functions of Rtt106p and provide new insights into its cellular roles.
Keywords: Biophysics, Chromatin, DNA/Protein Interaction, Histones, Protein/Binding/DNA, Protein/DNA-Interactions, Protein/Domains, Protein/Structure
Introduction
Rtt106p (regulator of Ty1 transposition 106) is a histone chaperone of Saccharomyces cerevisiae that specifically binds histones H3 and H4 and participates in many chromatin-related processes (1–3). It was originally identified in a genetic screen for mutants that enhanced the retrotransposition of Ty1 (4). Recent studies have linked Rtt106p to at least three chromatin-related functions. Rtt106p participates in the replication-coupled nucleosome assembly pathway (3), which is critical for genome integrity in yeast. In addition, Rtt106p genetically interacts with elongation factors and is important for normal transcription-dependent histone H3 deposition at actively transcribed regions (5). Finally, Rtt106p interacts functionally and physically with Cac1p, the largest subunit of CAF-1 (chromatin assembly factor-1), which also includes Cac2p and Cac3p (6, 7), to affect the heterochromatin silencing (1, 2).
In budding yeast, telomeres, the silent mating-type loci (HMR and HML), and the rDNA locus are well characterized silenced chromatin domains (8, 9). The silent information regulator (Sir) proteins, Sir1p–Sir4p, are the major structural components of these silenced regions (8, 9). In double cac1Δrtt106Δ mutant cells, the spreading of Sir proteins along the nucleosomes is significantly reduced, and heterochromatin formation is defective (2). Hence, Rtt106p is proposed to be a histone chaperone connecting S phase to epigenetic inheritance. Nonetheless, despite the diverse functions of Rtt106p, the molecular mechanisms underlying Rtt106p-mediated heterochromatin silencing, and replication-coupled and transcription-coupled nucleosome assembly still remain elusive.
Rtt106p is a 455-residue, 52-kDa protein that consists of three regions: a long C-terminal acidic region similar to that also found in other histone chaperones, such as Nap1p (nucleosome assembly protein 1) (10, 11), Vps75p (vacuolar protein sorting 75) (12), and Asf1p (antisilencing protein 1) (13); a region (amino acids 195–301) homologous to the second pleckstrin homology (PH) domain of yeast Pob3p (Pol1-binding protein) middle domain (14); and an N-terminal region (amino acids 1–194) with no obvious sequence homology to other proteins. To understand how Rtt106p achieves its versatile functions, a structural investigation is necessary. Here we report the 2.5 Å crystal structure of the core domain of Rtt106p (Rtt106p-M, residues 65–320). The Rtt106p-M structure adopts an unusual “double PH” domain architecture that resembles the structure of the Pob3p middle domain (Pob3p-M). A conserved loop region in the second PH domain was discovered to be responsible for interaction between Rtt106p and the histone H3-H4 tetramer. Unexpectedly, another region clustered with conserved positively charged residues was found on the surface of Rtt106p-M and responsible for its interaction with double-stranded DNA (dsDNA).4 Mutation of those positively charged residues to neutral amino acids abolished DNA-Rtt106p interaction. In vivo assays demonstrated that the histone and DNA binding activities of Rtt106p are crucial for telomere silencing. Together, our structural, biochemical, and functional studies provided new insights into the functions of Rtt106p.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Rtt106p
The DNA fragments of full-length Rtt106p and Rtt106p-M (residues 65–320) were amplified from yeast genomic DNA (Saccharomyces cerevisiae, S288c) by PCRs. Then the DNA fragments were ligated into the NdeI/XhoI-cleaved plasmid pGEX-4T (Amersham Biosciences) and a modified pET-28a(+) vector with substitution of the thrombin cleavage site to a TEV cleavage site, yielding plasmid pGEX-Rtt106p, pTEV-Rtt106p, pGEX-Rtt106p-M, and pTEV-Rtt106p-M, respectively. Rtt106p-M mutants were generated by conventional PCR method using the pTEV-Rtt106p-M plasmid as template. The mutant Rtt106p-loopm represented mutations of ITRLT (residues 264–268) to five alanines; the mutant Rtt106p-loop1 represented mutations of ITR (residues 264–266) to three alanines; the mutant Rtt106p-loop2 represented mutations of RLT (residues 266–268) to three alanines; the mutant Rtt106p-loop3 represented mutations of TYSS (residues 260–263) to AYAA; the mutant Rtt106p-site1 represented mutations from RK (residues 86–87) to AA; and the mutant Rtt106p-site2 represented mutations from KK (residues 245–246) to AA. All of the recombinant Rtt106p proteins were produced in Escherichia coli BL21 (DE3). Generally, the protein expression was induced at A600 = 0.8–1.5 with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C for 40 h. The expressed recombinant protein was purified using Sepharose 4B (Amersham Biosciences) or nickel-chelating column (Qiagen) following the protocols. Then the fusion tag was removed by thrombin or TEV protease in 12 °C. The resulting protein was purified with HiLoad 16/60 Superdex 200 and HiTrap Q FF (5 ml) (GE Healthcare). The purified protein was dialyzed into buffer A (20 mm Tris-HCl, pH 7.0, 80 mm NaCl, 5 mm β-mercaptoethanol) and concentrated to 40–80 mg/ml.
To prepare the SeMet-derivative protein, Rtt106p was expressed in E. coli strain B834 (Novagen) using M9 medium supplemented with SeMet and six amino acids, including leucine, isoleucine, valine, phenylalanine, lysine, and threonine. The SeMet-derivative protein was purified by a procedure similar to that described above.
CD Spectroscopy
The CD spectra of Rtt106p-M and Rtt106p-M mutants were recorded at 298 K on a Jasco-810 spectropolarimeter. The spectra were recorded at wavelength between 190 and 250 nm using a 0.1-cm path length cell and 100 μg/ml protein in 50 mm phosphate-buffered saline, pH 7.5. A buffer-only sample was used as reference. The molar ellipticities [θ] were plotted versus wavelength, and the reference curve was subtracted from each curve.
Crystallization and Data Collection
Crystals of both the native and the SeMet-derivative Rtt106p-M were grown using the hanging drop vapor diffusion method at 285 K. The crystals suitable for x-ray diffraction of the SeMet-derivative protein were grown in 1.5 m dl-malic, pH 7.0, with a protein concentration of 20 mg/ml in buffer A. The crystals of the native protein were grown in 18% (w/v) polyethylene glycol 4000, 100 mm NaAc, pH 4.6, and 1% (v/v) jeffamine ED-2001, with a protein concentration of 40 mg/ml in buffer A.
A multiple-wavelength anomalous dispersion (MAD) data set was collected from a single crystal of SeMet-derivative protein at 100 K with cryoprotectant (1.2 m dl-malic and 20% glycerol) on beamline X12C at the National Synchrotron Light Source at Brookhaven National Laboratory. The data were collected at three wavelengths (λpeak = 0.9790 Å, λinflection = 0.9792 Å, and λremote = 0.9600 Å). The native data were collected at 100 K with cryoprotectant (15% (w/v) polyethylene glycol 4000, 80 mm NaAc, pH 4.6, and 20% glycerol) on beamline 3W1A of the Beijing Synchrotron Radiation Facility at the Institute of High Energy Physics, Chinese Academy of Sciences. Both MAD and native data were processed using HKL2000 (15) and programs in the CCP4 package (16).
Structure Determination and Refinement
Three of the six expected selenium positions were determined by SOLVE (17) using Bijvoet differences of the MAD data. The initial phases were calculated by RESOLVE (18) with the resolution ranging from 25 to 3.3 Å, and an initial model (165 of the 261 amino acids) was autobuilt. The model was further built and refined at 3.1 Å resolution using Refmac5 (19) and COOT (20) by manual model correction. Further cycles of refinement and model building were carried out until the crystallography R-factor and free R-factor converged to 21.5 and 26.0%, respectively. The CNS package (21) was used in this stage. The structure of SeMet-derivative protein was used as an initial search model for determining the native structure of Rtt106p-M by a standard molecular replacement method with MOLREP (22) in the CCP4 package. The final crystallography R-factor and free R-factor of native structure are 21.6 and 27.4%, respectively. TLS refinement (23) was executed in Refmac5 at the latest stage. The stereochemistry of the structure was checked by PROCHECK (24). For SeMet Rtt106p-M, 85.0% of residues fall in the most favored Ramachandran category, with 13.5% in the additional allowed category and 1.6% in the generously allowed category. For the native Rtt106p-M, 90.6% of residues fall in the most favored Ramachandran category, with 8.9% in the additional allowed category and 0.5% in the generously allowed category. Details about data collection and processing are presented in Table 1. Figures were prepared using PyMOL (DeLano Scientific LLC).
TABLE 1.
Crystallographic statistics
| Data collection | SeMeta | Nativea | ||
|---|---|---|---|---|
| Space group | F432 | P212121 | ||
| Cell dimensions: a, b, c (Å) | 262.50, 262.50, 262.50 | 46.42, 54.01, 109.51 | ||
| SeMet |
Native | |||
|---|---|---|---|---|
| Peak | Inflection | Remote | ||
| Wavelength(Å) | 0.9790 | 0.9792 | 0.9600 | 1.0000 |
| Resolution (Å)b | 50-3.10 (3.21-3.10) | 50-3.10 (3.21-3.10) | 50-3.10 (3.21-3.10) | 20-2.50 (2.60-2.50) |
| Rmerge | 10.3 (38.9) | 9.2 (33.9) | 9.8 (38.3) | 7.8 (26.3) |
| I/σI | 28.9 (6.8) | 34.7 (8.6) | 32.2 (7.6) | 21.5 (2.3) |
| Completeness (%) | 100 (100) | 100 (100) | 100 (100) | 97.0 (79.1) |
| Redundancy | 9.9 (9.8) | 11.5 (11.2) | 11.5 (11.1) | 6.3 (3.6) |
| Refinement | SeMet | Native |
|---|---|---|
| Resolution (Å) | 20-3.10 | 20-2.50 |
| No. reflections | 13,713 | 9277 |
| Rwork/Rfree | 21.5/26.0 | 21.6/27.4 |
| No. of atoms | ||
| Protein | 1750 | 1743 |
| BME/Cl− | 4/1 | |
| Water | 28 | 53 |
| B-factors | ||
| Protein | 44.0 | 39.9 |
| β-Mercaptoethanol/Cl− | 80.4/49.4 | |
| Water | 38.7 | 45.3 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.014 | 0.009 |
| Bond angles (degrees) | 1.792 | 1.399 |
a Data shown are statistics from one single crystal of SeMet Rtt106p-M or native Rtt106p-M.
b Values in parentheses are for the highest resolution shell.
Rtt106p-M and Histone Binding Assays
The genes of yeast histones H3 and H4 were cloned into the vector pRSF-Duet. The genes of yeast histones H2A and H2B were cloned into the vectors p29 (derived from pET-29a(+)) and pET-22b(+), respectively. The histones H3-H4 or histones H2A-H2B were co-expressed in the E. coli strain BL21-CodonPlus (DE3)-RIL, and the resulting H3-H4 tetramer or H2A-H2B dimer was without any fusion tag. Protein expression was induced at A600 = 0.5–0.7 with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 25 °C for 8 h. Cells were harvested at 5000 × g for 10 min, and then the pellets were suspended in 15 ml/200 ml culture of buffer B (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 5 mm imidazole, 0.2% Triton X-100, 10% glycerol). The cell suspensions were lysed by sonication on an ice-water bath. Then the lysate was spun at 15,000 × g for 20 min at 4 °C, and the supernatant was collected.
In Ni-NTA pull-down experiments, generally, a similar amount of purified recombinant Rtt106p-M or Rtt106p-M mutants was immobilized on 100 μl of Ni-NTA resin. After washing twice (1 ml each time) with buffer B, the beads were then incubated with 1 ml of the H3-H4 or H2A-H2B supernatant at 4 °C for 15 min, and the incubation was repeated with another 2 ml of H3-H4 or H2A-H2B supernatant. Then the beads were washed thoroughly by buffer B supplemented with 20 and 50 mm imidazole, respectively. Finally, the bound proteins were eluted from beads with 100 μl of buffer C (20 mm Tris-HCl, pH 8.0, 200 mm NaCl, 500 mm imidazole) and mixed with 100 μl of 2× SDS-PAGE loading buffer and boiled for 10 min at 90 °C. All of the samples were resolved by Tricine-SDS-PAGE (15%, w/v) and stained with Coomassie Brilliant Blue.
Recombinant yeast histones H3 and H4 were generated in bacteria as described (25) and refolded to H3-H4 tetramer. The pull-down assays assessing the interactions of Rtt106p-M or its mutants with purified H3-H4 tetramer were done in a way similar to that described above. Anti-Histone H3 monoclonal antibody (Upstate) was used in the subsequent Western blot assay.
Analysis of Rtt106p-DNA Interactions by Electrophoretic Mobility Shift Assay (EMSA)
The interactions of purified recombinant protein Rtt106p-M or Rtt106p-M mutants with DNA were assessed by the EMSA technique using the dsDNA segments ds001 (5′-GACATACTGCCCTTACAGCAAAGCTACTTTGTC-3′), AT-rich (5′-ATAATTTATATTTATTATTTTATTATAATTTAT-3′), and GC-rich (5′-GCGGCCCGCGCCCGCCGCCCCGCCGCGGCCCGC-3′), synthesized by Sangon (Shanghai). Two strands of complementary 33-bp single-stranded DNA were annealed to form dsDNA: ds001, AT-rich, and GC-rich, respectively. 0.2 nm 32P-labeled dsDNA was mixed with the indicated amount of wild type or mutated Rtt106p-M in buffer D (20 mm HEPES, pH 7.0, 80 mm NaCl, 1 mm dithiothreitol, and 2 mm EDTA). The reaction mixture was incubated at 4 °C for 2 h and analyzed by the electrophoresis in 8% native PAGE at 100 V using 0.5× TBE buffer at 4 °C.
Fluorescence Polarization Assays
Fluorescence polarization assays (FPAs) were performed in buffer D at 298 K using a SpectraMax M5 microplate reader system. The wavelengths of fluorescence excitation and emission were 490 and 524 nm, respectively. Each well of a 384-well plate contained 100 nm fluorescent-labeled (5′-FAM) DNA probe and different amounts of Rtt106p-M or Rtt106p-M mutants (concentrations from 0 to about 250 μm) with a final volume of 80 μl. For each assay, DNA-free controls (Rtt106p-M or Rtt106p-M mutants only) were included. The fluorescence polarization P (in mP units) was calculated with the equation,
 |
The fluorescence polarization change ΔP (in mP units) was fit to the equation,
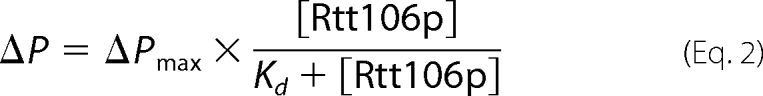 |
The sequences of 5′-FAM-labeled DNA probe are described in supplemental Table S1.
URA3 Silencing Assays
Yeast strains and plasmids are described in the supplemental material. Silencing at telomeric loci was scored as described previously (26). In brief, logarithmically growing cells whose genome contained a URA3 gene integrated near the right end of chromosome VII (URA3- TELVII-R) were serially diluted in 5-fold increments, spotted onto the YC plates with or without ∼0.1% 5-fluoroorotic acid (5-FOA), and incubated at 30 °C. Growth was documented at 20 h.
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation assays were performed as described previously (27) with some modification. Briefly, yeast cells were cross-linked with 1% formaldehyde and suspended in lysis buffer (50 mm HEPES, pH 7.5, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mm phenylmethylsulfonyl fluoride, protease inhibitor mixture). Cells were lysed using glass beads and were sonicated to shear the chromatin to fragment sizes of ∼200–500 base pairs. Cross-linked chromatin fragments were immunoprecipitated with antibodies that specifically recognized Myc epitope tags. Protein G/A-Sepharose beads (GE Healthcare) were then added into the samples, and the immunoprecipitated complexes were washed with lysis buffer, lysis buffer containing 500 mm NaCl, wash buffer (50 mm HEPES, pH 7.9, 300 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.5% Nonidet P-40, 0.1% sodium deoxycholate), and TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA). Next, the immunoprecipitated chromatin was eluted from beads with elution buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1% SDS). Formaldehyde cross-linking was reversed by incubating the eluates at 65 °C overnight. DNA from the eluates was treated with 100 μg/ml proteinase K and purified with the QIAquick PCR purification kit (Qiagen).
Immunoprecipitated fractions and whole-cell extracts containing DNA were analyzed by PCR. Quantitative PCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems). Primers used in the PCRs were analyzed for the appropriate range of linearity and efficiency in order to accurately evaluate DNA occupancy by the protein (percentage of immunoprecipitation/input). Relative enrichment values of Sir2p (silent information regulator 2) were normalized to the internal control ACT1, and these in turn were normalized to the corresponding input whole-cell extract.
RESULTS
Crystal Structure of the Rtt106p Core Domain
The 2.5 Å crystal structure of Rtt106p-M (residues 65–320) from yeast was determined by MAD phasing and molecular replacement (Fig. 1A and supplemental Fig. S1), with crystallographic statistics summarized in Table 1. Rtt106p-M, which is a monomer in one asymmetric unit, comprises 14 strands and five helices and displays a tandem PH domain architecture (residues 65–198 and 218–301 (Figs. 1A and 2)). The PH domains closely resemble the standard PH domain, which is a seven-stranded antiparallel β-barrel capped at the C-end with a helix. In the first PH domain (PH1), the β-barrel contains two β-sheets. Four β-strands (S1, S2, S3, and S4) form the first β-sheet, and four additional β-strands (S1, S5, S6, and S7) form the second β-sheet, with the S1 strand linking the two. A short α-helix (H1) links S4 and S5, and an additional α-helix (H2) is inserted between the S7 strand and the β-barrel-capping helix (H3). The architecture of the second PH domain (PH2) is similar to the first one but with no insertion element between the last S14 strand and the capping helix (H5). A region (residues 206–217) between the two domains and the last 18 residues of the protein are invisible in the density map, possibly because they are disordered in their native state (Fig. 1A). The two PH domains are not independent but are intimately associated with each other, primarily through hydrophobic residues (Met125, Leu129, Pro130, Pro132, Tyr139, Phe141, Pro155, Val157, Pro203, Phe204, His221, Thr232, Tyr234, Leu236, His239, Ile241, Phe244, Ile248, and Leu250) that are buried in the interface (Fig. 1D). These residues are mainly in the second β-sheet of PH1 (S5–S7) and the first β-sheet of PH2 (S8–S11), and they are highly conserved in type among Rtt106p homologues, suggesting an evolutionarily conserved role (Fig. 2). This result clearly shows the structural feature of Rtt106p: a double PH domain architecture at the N terminus and a primarily acidic C-terminal tail.
FIGURE 1.

Structure of Rtt106p-M. A, a stereo view of Rtt106p-M. Disordered residues between two PH domains and in the C terminus are modeled as dashed lines. B, the electronic potential surface of Rtt106p-M. The orientations of B (left) and A are the same. The ellipse highlights the positively charged ridge. C, the conserved residues comprising the positively charged ridge. D, conserved and type-conserved residues at the interface of the two PH domains. C and D have the same orientations as A.
FIGURE 2.

Sequence alignment of the double PH domains among Rtt106p and its homologues. The following sequences were aligned using ClustalW2 (available on the World Wide Web): S. cerevisiae Rtt106p (gi|6324123|NP_014193.1); Vanderwaltozyma polyspora Rtt106 (gi|156837701|XP_001642870.1); Candida glabrata Rtt106 (gi|50289433|XP_447148.1); Ashbya gossypii Rtt106 (gi|45185065|NP_982782.1), and Kluyveromyces lactis Rtt106 (gi|50307819|XP_ 453903.1). The secondary structures and residue numbers of Rtt106p-M are shown at the top. The residues comprising the positively charged ridge for dsDNA binding are designated with a black triangle below; the hydrophobic residues at the interface of the two PH domains are designated with an orange triangle; and residues comprising the crucial loop for H3-H4 binding are designated with a star. The three highly conserved negatively charged residues that form a negative patch near the conserved loop are denoted with a blue triangle.
The electrostatic potential surface of Rtt106p-M shows a positively charged ridge region stretching across one side of the two PH domains (Fig. 1, B and C). Nine positively charged residues (Arg86, Lys87, Lys88, Lys108, Lys134, Arg226, Lys229, Lys245, and Lys246) make up the positive charge of the surface. These amino acids are highly conserved, suggesting some biological roles for this ridge. In fact, together with the biochemical results (see details below), we conclude that Rtt106p binds dsDNA via this positively charged region.
Rtt106p-M Shares Structural Similarities with Pob3p-M
The overall sequence identity between Rtt106p and the Pob3p is low (15%). The second PH domain of Rtt106p-M had been proposed to be homologous to that of Pob3p-M, but the homology between the first PH domains of them had not been noticed. From the crystal structure of Rtt106p-M, we found that its whole structure was highly similar to Pob3p-M (Protein Data Bank code 2GCL). Both adopt a double PH architecture with a 2.7 Å root mean square deviation over their Cα atoms (Fig. 3A). In a structure-based, optimized sequence alignment, the sequence identity between the first PH domains of the two proteins is 14%, whereas that of the second PH domains is 26% (supplemental Fig. S2). The crystal structure shows that the second PH domain of Rtt106p (residues 218–301) and that of Pob3p-M are indeed much alike, each comprising a standard PH domain with a seven-stranded antiparallel β-barrel and a cap helix, with a 1.2 Å root mean square deviation over the Cα atoms of the two structures (Fig. 3C). The structure also showed that the first PH domain of Rtt106p (residues 65–198) is similar to its counterpart in Pob3p-M, and the root mean square deviation over the Cα atoms of the two structures is 2.8 Å (Fig. 3B). There are two major differences in the first PH domains of Rtt106p-M and Pob3p-M. The first is that the inserted element between the S7 strand and the capping helix (H3) of the Rtt106p-M PH1 domain is an α-helix (H2), whereas in the Pob3p-M structure there are two strands (S8 and S9) linked by a helix (H2) (Fig. 3D and supplemental Fig. S2). The second is that the long, nine-residue loop linking the S6 and S7 strands of the Rtt106p-M PH1 domain is replaced by two short strands in the Pob3p-M structure (Fig. 3D and supplemental Fig. S2). The angle formed by the tandem PH domains of Rtt106p-M is slightly smaller than that of Pob3p-M. These differences may explain why attempts to identify a molecular replacement solution using the Pob3p-M structure as a search model were unsuccessful.
FIGURE 3.

Structure comparison of Rtt106p-M and Pob3p-M. A, overall structure superposition of Rtt106p-M (red) and Pob3p (cyan). B, superposition of the first PH domains. C, superposition of the second PH domains. D, highlighted are the major structural differences between the first PH domains of Rtt106p-M and Pob3p-M. A–C have the same orientations as in Fig. 1.
The overall electrostatic potential surface of Rtt106p-M is different from that of Pob3p-M. However, on the surface of the Pob3p-M structure, a similar positively charged region could also be found at the same position as on the Rtt106p-M surface (supplemental Fig. S3A), implying that the two proteins may have similar functions through these regions.
The Loop between S12 and S13 in Rtt106p Is Crucial for Histone H3-H4 Binding
To assess the interaction between Rtt106p-M and histones, we tested the binding affinities of purified Rtt106p-M for the H3-H4 tetramer and H2A-H2B dimer in cell lysates. The results of pull-down assays clearly showed that Rtt106p-M specifically bound the H3-H4 tetramer but not the H2A-H2B dimer in vitro (Fig. 4A). Analysis of the Rtt106p crystal structure and sequence alignments of Rtt106p homologues from different yeast species showed a high degree of conservation in a solvent-exposed loop region between S12 and S13 (Figs. 1A and 2), which is probably involved in the interaction between Rtt106p and the H3-H4 tetramer. Therefore, we made four Rtt106p-M mutants, Rtt106p-loopm (five solvent-exposed residues 264ITRLT268 mutated to AAAAA), Rtt106p-loop1 (residues 264ITR266 mutated to AAA), Rtt106p-loop2 (residues 266RLT268 mutated to AAA), and Rtt106p-loop3 (residues 260TYSS263 in the S12 strand mutated to AYAA), and tested their abilities to bind H3-H4 tetramer (Fig. 4B). These mutations did not affect protein stability as assessed by CD spectra (Fig. 4C). The loop region mutants (Rtt106p-loopm, Rtt106p-loop1, and Rtt106p-loop2) had significantly diminished interaction with the H3-H4 tetramer, whereas the mutant Rtt106p-loop3, with mutations in the residues next to the loop, did not show any changes in interaction (Fig. 4D). These results suggest that the loop between S12 and S13 (residues 264ITRLT268) plays an important role in the interaction of Rtt106p-M with the H3-H4 tetramer.
FIGURE 4.

Physical interaction between Rtt106p-M and histones. A, Ni-NTA pull-down analysis of Rtt106p-M with histones H3-H4 and H2A-H2B in cell lysates. B, highlighted are the mutated residues in Rtt106p-M loop mutants. C, the CD spectra of Rtt106p-M and its mutants. D, Ni-NTA pull-down analysis of Rtt106p-M mutants with histones H3-H4 in cell lysates. The final samples were resolved by Tricine-SDS-PAGE (15%, w/v) and stained with Coomassie Brilliant Blue (CB).
To assess whether Rtt106p-M has different H3-H4 binding activity in the full-length context, we cloned and expressed full-length Rtt106p and found no significant differences between the H3-H4 binding abilities of Rtt106p-M and the full-length protein (supplemental Fig. S4). We could not obtain H3-H4 tetramer with H3 Lys56 highly acetylated in vitro, so we have not yet addressed how the acetylation of H3 Lys56 enhances the interaction between H3-H4 and Rtt106p (3).
The Conserved Positively Charged Surface of Rtt106p-M Is Responsible for dsDNA Binding
Rtt106p functions in chromatin-related processes, such as replication and transcription. We noticed that the positively charged ridge region of Rtt106p that covers one side of the two PH domains consisted of highly conserved arginine and lysine residues (Figs. 1, B and C, and 2). These residues are commonly found on the DNA-binding surfaces of histones (28) and other DNA-binding proteins (29, 30) and are involved in their interaction with DNA. We therefore tested if Rtt106p was able to bind DNA, in addition to its histone H3-H4 binding activity. We assessed the in vitro dsDNA binding ability of Rtt106p-M by EMSA and FPA. As the protein/DNA ratio increased, the amount of shifted DNA, or the value of fluorescence polarization (in mP units), increased correspondingly (Fig. 5, A and B). The dissociation constant (Kd) between Rtt106p-M and dsDNA was ∼22 μm, as determined by the FPA (supplemental Table S2). These results revealed that in addition to the previously identified H3-H4 binding activity, Rtt106p-M also interacts with dsDNA directly in vitro.
FIGURE 5.

Rtt106p-M interacts with dsDNA. A, results of an EMSA assessing the interaction of Rtt106p-M and the Rtt106p-site1 and Rtt106p-site2 mutants with a 32P-labeled 33-bp dsDNA, named ds-001, and used at 0.2 nm. Rtt106p-M concentrations are as follows: 0 μm (lane 1), 72 μm (lane 8). Lanes 2–8, 2-fold dilutions from right to left from 72 μm. Lane 9, 165 μm Rtt106p-site1; lane 10, 168 μm Rtt106p-site2. B, FPAs of Rtt106p-M and mutants with a 5′-FAM-labeled 33-bp dsDNA (with the same sequence as ds-001). The data were fitted according to Equation 2. C, EMSA of Rtt106p-M interaction with AT-rich and GC-rich dsDNA sequences. Lane 2, Rtt106p-M at 144 μm; lanes 4–6, 36, 72, and 144 μm, respectively; lanes 8–10, 36, 72, and 144 μm, respectively. D, FPA of Rtt106p-M interaction with AT-rich and GC-rich dsDNA. Rtt106p-M exhibited similar binding affinity to AT-rich and GC-rich dsDNA. E, FPA of Rtt106p-M interaction with dsDNA oligonucleotides of different lengths.
We investigated whether Rtt106p-M interacted with dsDNA via the positively charged region by preparing two mutants, Rtt106p-site1 (R86A/K87A) and Rtt106p-site2 (K245A/K246A), and examining their dsDNA binding abilities. Mutations in either Rtt106p-site1 or Rtt106p-site2 did not appear to affect overall protein structure as examined by CD analysis (Fig. 4C), but their dsDNA binding ability was almost completely abolished (Fig. 5, A and B). These results indicated that residues Arg86, Lys87, Lys245, and Lys246 were crucial for the ability of Rtt106p to bind dsDNA and that the positively charged ridge was responsible for dsDNA interaction.
We also examined if Rtt106p-M had a sequence preference. As shown in Fig. 5, C and D, no obvious preference for an AT- or GC-rich sequence was observed. The Kd values for Rtt106p-M binding to AT- or GC-rich dsDNA were ∼23 and ∼24 μm, respectively. We also tested the binding affinities of Rtt106p-M to dsDNA oligonucleotides of different lengths. Rtt106p-M bound dsDNA longer than 12 bp with similar Kd (12–20 bp, about 25–27 μm), and the Kd increased significantly to ∼42 μm when binding 10-bp dsDNA (Fig. 5E and supplemental Table S2). Single-stranded DNA oligonucleotide was also tested, and the Kd increased to ∼78 μm, indicating a much weaker affinity of Rtt106p-M for single-stranded DNA (supplemental Fig. S5). These results provided the first biochemical evidence that Rtt106p binds dsDNA with no sequence preference.
Rtt106p-M Binds Histone H3-H4 and dsDNA through Independent Regions
The regions found to be crucial for histone binding and DNA binding are spatially close, so we examined whether these two regions functionally affected each other. EMSA and FPA results showed that the mutant Rtt106p-loopm, which was deficient in histone binding, retained dsDNA binding activity similar to wild type Rtt106p-M (Figs. 5B and 6A), with a Kd estimated to be ∼24 μm by FPA. This indicated that the loop region that is crucial for histone binding did not participate in DNA binding. We then tested the H3-H4 binding ability of the Rtt106p-site1 and Rtt106p-site2 mutants that were defective in DNA binding. Pull-down assay results showed that the two mutants could still bind histone H3-H4 tetramer of cell lysates (Fig. 6B). When purified histone H3-H4 was used in a pull-down assay, Rtt106p-M and the DNA-binding deficient mutants displayed similar histone binding activities (Fig. 6C). These data indicated that the regions on Rtt106 for histone binding and DNA binding are functionally independent of each other.
FIGURE 6.

Rtt106p-M binds dsDNA and histone H3-H4 through unrelated regions. A, EMSA of Rtt106p-loopm interacting with dsDNA with similar affinity as Rtt106p-M. The ds-001 probe was used at 0.2 nm. Rtt106p-loopm concentrations were as follows: 0 μm (lane 1) and 144 μm (lane 10). Lanes 2–10, 2-fold dilutions from right to left from 144 μm. B, pull-down assays of Rtt106p-site1 and -site2 mutants interacting with H3-H4 tetramer in cell lysates. C, pull-down assays of Rtt106p-site1 and -site2 mutants interacting with purified H3-H4 tetramer with similar affinity as wild type Rtt106p-M. CB, Coomassie Brilliant Blue.
DNA and Histone Binding Activities of Rtt106p Are Involved in Telomeric Heterochromatin Formation in Vivo
Rtt106p and CAF-1 cooperatively mediate heterochromatin formation by contributing to the spreading of Sir proteins during the early stages of heterochromatin formation (2). Defects in formation of heterochromatin and aberrant spreading of Sir proteins were observed in cac1Δrtt106Δ double deletion strain but not in cac1Δ or rtt106Δ single mutant strain (2, 31). The precise role of Rtt106p in these processes has not yet been elucidated. To assess whether the histone binding and DNA binding activities of Rtt106p are involved in heterochromatin silencing, we constructed mutant strains cac1Δrtt106Δ, cac1Δrtt106-loop1, cac1Δrtt106-loop2, cac1Δrtt106-site1, and cac1Δrtt106-site2.
First, we examined the effect of RTT106 mutations on the transcriptional state of genes near the telomere. Experimentally, a URA3 gene was placed near the right telomere of chromosome VII. If telomere silencing (also called the telomere position effect) was intact (32), as in wild type cells, URA3 expression would be low, allowing cells to grow on plates containing 5-FOA. However, if the telomere position effect was disrupted, as in the sir2Δ control strain, URA3 expression would be greatly elevated, resulting in toxicity on a 5-FOA plate (Fig. 7A) (32). The single CAC1 deletion slightly reduced yeast growth on 5-FOA plates (Fig. 7A), suggesting that Cac1p modestly contributes to telomere silencing. Inactivation of Rtt106p did not lead to a growth defect compared with wild type, but simultaneous deletion of CAC1 and RTT106 phenocopied the loss of telomere position effect of SIR2 deletion (Fig. 7A). This suggested that Cac1p and Rtt106p are redundant in regulating heterochromation formation. Interestingly, in the cac1Δ background, RTT106 mutants that were deficient in histone binding or dsDNA binding displayed a significant decrease in telomeric silencing compared with wild type. These results suggested that both the histone binding and dsDNA binding abilities of Rtt106p are required for its function in the formation of telomeric heterochromatin.
FIGURE 7.

Rtt106p-histone and -DNA interactions are involved in telomeric heterochromatin formation. A, wild type, sir2Δ, cac1Δrtt106Δ, and rtt106 mutant cells with a URA3 gene integrated at the subtelomeric regions of chromosome VII (TELVII) were serially diluted (5-fold) and spotted onto YC medium with or without 5-FOA, followed by incubation at 30 °C. Photographs were taken after 20 h. B, a sketch map showing the primers designed according to the sequence near the telomere at chromosome IX. C, 13Myc-Sir2p binding to subtelomeric loci was assayed by chromatin immunoprecipitation using anti-Myc antibody in wild type, cac1Δrtt106Δ, and rtt106 mutant cells. Average relative Sir2p enrichments were shown for each primer set. The quantitative PCR data were normalized to an internal control (ACT1) and the input DNA. The results are the average of three independent chromatin immunoprecipitation assays with error bars shown for the S.E. for three independent experiments.
We next quantitatively evaluated the abundance of Myc-tagged Sir2p at subtelomeric regions in both wild type and mutant cells. Experimentally, we designed a series of primers according to the sequence near the telomere at chromosome IX (Fig. 7B) and performed chromatin immunoprecipitation analysis. The association of Sir2p with telomeric heterochromatin was significantly reduced in all double mutant cells compared with wild type or with cac1Δ, rtt106Δ single mutant cells (Fig. 7C). These data were in agreement with the silencing assay (Fig. 7A) and further demonstrated that both the histone binding and DNA binding activities of Rtt106p are essential for its synthetic effect with CAF-1 on Sir-mediated telomeric heterochromatin formation. The functional deficiency caused by RTT106 mutations was not due to reduced levels of mutant proteins expressed in yeast cells because we tested the cell extracts by Western blot (supplemental Fig. S6).
DISCUSSION
Rtt106p-M displays a tandem PH domain architecture, with the two PH domains intimately associated with each other. Separating the PH domains would expose the hydrophobic interacting face to solvent, causing instability of the domains. In fact, when we cloned and expressed the two PH domains separately, they aggregated into oligomers (data not shown). Therefore, Rtt106p-M should be viewed as one double PH domain, similar to the structure of Pob3p-M (14).
The overall structure of Rtt106p-M has no similarity to the known structures of histone chaperones in the Protein Data Bank, such as Nap1p, Vps75p, Asf1p, and SET (also named TAF-1) (33–39). However, a common feature can be found among Rtt106p-M and the other histone chaperone structures: a variant of the antiparallel β-sheet, which is proposed to be a histone recognition motif (38, 40). Asf1p uses this antiparallel β-sheet region to interact with histones H3 and H4 (38, 41), and SET is also suggested to use a conserved β-sheet for histone binding (34). In the Rtt106p-M structure, a highly conserved loop between S12 and S13 in the second antiparallel β-sheet (S12–S14) of PH2 was identified as crucial for binding the histone H3-H4 tetramer (Fig. 4, B and D, and supplemental Fig. S8). The solvent-exposed residues on this β-sheet are either highly conserved or have the same type in Rtt106p homologues, and they form a mainly neutral, although partially acidic, surface (supplemental Fig. S8). This surface closely juxtaposes the crucial loop and may also contribute to the interaction between Rtt106p-M and the H3-H4 tetramer. The S12 strand is at the edge of the antiparallel β-sheet formed by strands S12–S14, and the crucial loop is much like an extension of the S12 strand. Therefore, Rtt106p-M might be able to capture a β-strand of histones with the edge composed of the S12 strand and the extended loop in a manner similar to Asf1p. On the other side of the loop, three conserved residues (Glu133, Glu230, and Asp287) constitute an acidic patch, through which Rtt106p-M might neutralize the positive charge of histones when the loop binds histones (Fig. 2 and supplemental Fig. S8). Interacting with histones through an antiparallel β-sheet appears to be a common feature of a cluster of histone chaperones, and this view is further supported by our experimental results.
The sequence homology among the second PH domains of Rtt106p-M, Pob3p, and SSRP1 had already been identified (42). The crystal structure of Rtt106p-M revealed an unexpected similarity between the first PH domains of Rtt106p-M and Pob3p-M. Pob3p is a subunit of the S. cerevisiae FACT complex (facilitates chromatin transcription, an essential chromatin reorganizing factor and a heterodimer of Spt16p and Pob3p), and SSRP1 is the human homologue of Pob3p (43–45). Our analysis of the crystal structure of Rtt106p-M and careful sequence alignment of the three proteins led us to propose that Rtt106p is evolutionarily related to the other two proteins and that their middle regions adopt a similar double PH structure (supplemental Figs. S2 and S7). Beyond the middle double PH domain, Pob3p and SSRP1 each have an additional conventional PH domain at the N terminus (Protein Data Bank code 3F5R), and SSRP1 has a DNA binding HMG box in its C-terminal region (supplemental Fig. S7) (46). Pob3p and SSRP1 are structurally and functionally more closely related, and Rtt106p might be evolutionarily related to them but function in different pathways. It is interesting that the mammalian homologue(s) of Rtt106p has not yet been identified.
Rtt106p physically interacts with dsDNA through a positively charged ridge region stretching across one side of the two PH domains (Figs. 1B and 5). A similar positively charged region is found on the surface of Pob3p-M (supplemental Fig. S3A), so Pob3p may also bind DNA through this region. Our preliminary EMSA results appeared to support this idea (supplemental Fig. S3B). Instead of the DNA-binding region stretching across one side of the two PH domains in Rtt106p, the second PH domain is responsible for histone H3-H4 binding (Figs. 4 and 6) (3). Coincidentally, SSRP1 can also bind histone H3-H4 (47, 48). Because the second PH domains of Rtt106p, Pob3p, and SSRP1 are more similar in primary and tertiary structure, we speculate that both Pob3p and SSRP1 may also interact with histones through this domain.
Histone chaperones share the common characteristic of histone binding activity (49), whereas NAP-1 family histone chaperone SET (34) and Rtt106p seem to be the rare ones that have DNA binding activity. The histone-binding region and the DNA-binding region of Rtt106p are physically separated (Fig. 1 and supplemental Fig. S8), but they are equally important for Rtt106p function. This study indicates that both the histone and DNA binding activities of Rtt106p are involved in epigenetic silencing by cooperatively affecting the spreading of heterochromatin with CAF-1 (Fig. 7) (1, 2). In S. cerevisiae, the assembly of telomeric heterochromatin is a stepwise process. Initially, Sir4p is recruited through sequence-specific DNA-binding proteins, and then Sir2p is recruited through Sir4p and deacetylates the N-terminal tails of nearby histones H3 and H4. Subsequently, Sir3p and Sir4p are recruited to adjacent nucleosomes. As the Sir proteins are recruited repeatedly, they spread along the nucleosomes to establish heterochromatin (8, 9). Rtt106p and CAF-1 have no influence in the initial recruitment of Sir4p but affect the subsequent recruitment and spreading of Sir proteins (2). As shown by this study, defects in either Rtt106p histone or DNA binding caused abnormal telomeric heterochromatin formation. Rtt106p may participate in heterochromatin formation in two ways. First, because the nucleosome is the basal element of chromatin and Rtt106p plays a crucial role in the nucleosome assembly process, when its histone or DNA binding ability is abolished, it may cause defects in nucleosome assembly that lead to abnormalities in heterochromatin formation. Alternatively, because Rtt106p physically interacts with Sir4p (2), Rtt106p may be recruited to the initiation site of telomeric heterochromatin by Sir4p, where the histone and DNA binding functions of Rtt106p stabilize the Rtt106p-Sir4p-containing complex. Then Rtt106p, together with CAF-1, may facilitate the recruitment of Sir2p by Sir4p and the subsequent spreading of Sir proteins. Eliminating the histone or DNA binding abilities of Rtt106p would make the Rtt106p-Sir4p-containing complex unstable and influence recruitment of Sir2p (supplemental Fig. S9). Either model proposed above requires further experimental support. Nevertheless, our results have revealed the pivotal roles of the histone and DNA binding activities of Rtt106p in heterochromatin silencing and deepened our understanding of the role that Rtt106p plays in this process.
Rtt106p is a versatile protein, involved in replication- and transcription-coupled nucleosome assembly and heterochromatin silencing. Because these processes are chromatin-based and chromatin primarily consists of DNA and histones, the histone and DNA binding activities of Rtt106p are likely to be essential for its diverse functions. We have elucidated the histone- and DNA-binding regions of Rtt106p and found that they play important roles in telomeric heterochromatin formation. Our findings have uncovered the structural basis for the histone and DNA binding activities of Rtt106p and provide new insights into its cellular function.
Supplementary Material
Acknowledgments
We thank Dr. Ruiming Xu for critical editing of the manuscript. We thank Dr. Ruiming Xu, Dr. Weimin Gong, and Jia Wei for helping with data collection of the SeMet Rtt106p-M at beamline X12C at the National Synchrotron Light Source of the Brookhaven National Laboratory, NY. We also thank Dr. Yuhui Dong and Dr. Zengqiang Gao for assistance with data collection of the native Rtt106p-M at beamline 3W1A of the Beijing Synchrotron Radiation Facility at the Institute of High Energy Physics, Chinese Academy of Sciences. We thank Dr. Xuebiao Yao, Dr. Changlin Tian, Dr. Wei Zhao, Xiajing Tong, Zhaofeng Luo, Zhiqiang Zhu, Huihao Zhou, Qinglin Wu, Jiahai Zhang, Yu Qiu, Weiwei Wang, Fudong Li, Chao He, Xuecheng Zhang, and Huijuan Yu for helpful discussions about the experiments and manuscript.
This work was supported by Chinese National Fundamental Research Project Grants 2006CB806500 and 2006CB910200, Chinese National Natural Science Foundation Grants 30670426 and 30830031, National High-Tech R&D Program Grants 2006AA02A315 and 2006AA02A318, Chinese Academy of Sciences Grant KSCX2-YW-R-60, and Chinese Ministry of Education Grant 20070358025.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S3 and Figs. S1–S9.
The atomic coordinates and structure factors (codes 3GYP and 3GYO) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- dsDNA
- double-stranded DNA
- PH
- pleckstrin homology
- MAD
- multiple-wavelength anomalous diffraction
- FPA
- fluorescence polarization assay
- EMSA
- electrophoretic mobility shift assay
- TEV
- tobacco etch virus protease
- Ni-NTA
- nickel-nitrilotriacetic acid
- mP
- millipolarization
- SeMet
- selenomethionine
- 5-FOA
- 5-fluoroorotic acid
- Tricine
- N-tris(hydroxymethyl)methylglycine.
REFERENCES
- 1.Huang S., Zhou H., Katzmann D., Hochstrasser M., Atanasova E., Zhang Z. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13410–13415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang S., Zhou H., Tarara J., Zhang Z. (2007) EMBO J. 26, 2274–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Q., Zhou H., Wurtele H., Davies B., Horazdovsky B., Verreault A., Zhang Z. (2008) Cell 134, 244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scholes D. T., Banerjee M., Bowen B., Curcio M. J. (2001) Genetics 159, 1449–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imbeault D., Gamar L., Rufiange A., Paquet E., Nourani A. (2008) J. Biol. Chem. 283, 27350–27354 [DOI] [PubMed] [Google Scholar]
- 6.Enomoto S., McCune-Zierath P. D., Gerami-Nejad M., Sanders M. A., Berman J. (1997) Genes Dev. 11, 358–370 [DOI] [PubMed] [Google Scholar]
- 7.Kaufman P. D., Kobayashi R., Stillman B. (1997) Genes Dev. 11, 345–357 [DOI] [PubMed] [Google Scholar]
- 8.Grunstein M. (1998) Cell 93, 325–328 [DOI] [PubMed] [Google Scholar]
- 9.Rusche L. N., Kirchmaier A. L., Rine J. (2003) Annu. Rev. Biochem. 72, 481–516 [DOI] [PubMed] [Google Scholar]
- 10.Ishimi Y., Yasuda H., Hirosumi J., Hanaoka F., Yamada M. (1983) J. Biochem. 94, 735–744 [DOI] [PubMed] [Google Scholar]
- 11.Ishimi Y., Hirosumi J., Sato W., Sugasawa K., Yokota S., Hanaoka F., Yamada M. (1984) Eur. J. Biochem. 142, 431–439 [DOI] [PubMed] [Google Scholar]
- 12.Selth L., Svejstrup J. Q. (2007) J. Biol. Chem. 282, 12358–12362 [DOI] [PubMed] [Google Scholar]
- 13.Le S., Davis C., Konopka J. B., Sternglanz R. (1997) Yeast 13, 1029–1042 [DOI] [PubMed] [Google Scholar]
- 14.VanDemark A. P., Blanksma M., Ferris E., Heroux A., Hill C. P., Formosa T. (2006) Mol. Cell 22, 363–374 [DOI] [PubMed] [Google Scholar]
- 15.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 16.(1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 17.Terwilliger T. C., Berendzen J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terwilliger T. C. (2003) Acta Crystallogr. D Biol. Crystallogr. 59, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 20.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 21.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 22.Vagin A., Teplyakov A. (2000) Acta Crystallogr. D Biol. Crystallogr. 56, 1622–1624 [DOI] [PubMed] [Google Scholar]
- 23.Winn M. D., Murshudov G. N., Papiz M. Z. (2003) Methods Enzymol. 374, 300–321 [DOI] [PubMed] [Google Scholar]
- 24.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 25.Wittmeyer J., Saha A., Cairns B. (2004) Methods Enzymol. 377, 322–343 [DOI] [PubMed] [Google Scholar]
- 26.Xu F., Zhang Q., Zhang K., Xie W., Grunstein M. (2007) Mol. Cell 27, 890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Govind C. K., Zhang F., Qiu H., Hofmeyer K., Hinnebusch A. G. (2007) Mol. Cell 25, 31–42 [DOI] [PubMed] [Google Scholar]
- 28.Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 29.Chen C. Y., Ko T. P., Lin T. W., Chou C. C., Chen C. J., Wang A. H. (2005) Nucleic Acids Res. 33, 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masse J. E., Wong B., Yen Y. M., Allain F. H., Johnson R. C., Feigon J. (2002) J. Mol. Biol. 323, 263–284 [DOI] [PubMed] [Google Scholar]
- 31.Monson E. K., de Bruin D., Zakian V. A. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 13081–13086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottschling D. E., Aparicio O. M., Billington B. L., Zakian V. A. (1990) Cell 63, 751–762 [DOI] [PubMed] [Google Scholar]
- 33.Park Y. J., Luger K. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muto S., Senda M., Akai Y., Sato L., Suzuki T., Nagai R., Senda T., Horikoshi M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 4285–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berndsen C. E., Tsubota T., Lindner S. E., Lee S., Holton J. M., Kaufman P. D., Keck J. L., Denu J. M. (2008) Nat. Struct. Mol. Biol. 15, 948–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park Y. J., Sudhoff K. B., Andrews A. J., Stargell L. A., Luger K. (2008) Nat. Struct. Mol. Biol. 15, 957–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y., Meeth K., Jiang E., Luo C., Marmorstein R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 12206–12211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.English C. M., Adkins M. W., Carson J. J., Churchill M. E., Tyler J. K. (2006) Cell 127, 495–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daganzo S. M., Erzberger J. P., Lam W. M., Skordalakes E., Zhang R., Franco A. A., Brill S. J., Adams P. D., Berger J. M., Kaufman P. D. (2003) Curr. Biol. 13, 2148–2158 [DOI] [PubMed] [Google Scholar]
- 40.Park Y. J., Luger K. (2008) Curr. Opin. Struct. Biol. 18, 282–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Natsume R., Eitoku M., Akai Y., Sano N., Horikoshi M., Senda T. (2007) Nature 446, 338–341 [DOI] [PubMed] [Google Scholar]
- 42.Ponting C. P. (2002) Nucleic Acids Res. 30, 3643–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orphanides G., LeRoy G., Chang C. H., Luse D. S., Reinberg D. (1998) Cell 92, 105–116 [DOI] [PubMed] [Google Scholar]
- 44.Orphanides G., Wu W. H., Lane W. S., Hampsey M., Reinberg D. (1999) Nature 400, 284–288 [DOI] [PubMed] [Google Scholar]
- 45.Wittmeyer J., Formosa T. (1997) Mol. Cell Biol. 17, 4178–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yarnell A. T., Oh S., Reinberg D., Lippard S. J. (2001) J. Biol. Chem. 276, 25736–25741 [DOI] [PubMed] [Google Scholar]
- 47.Belotserkovskaya R., Oh S., Bondarenko V. A., Orphanides G., Studitsky V. M., Reinberg D. (2003) Science 301, 1090–1093 [DOI] [PubMed] [Google Scholar]
- 48.Belotserkovskaya R., Saunders A., Lis J. T., Reinberg D. (2004) Biochim. Biophys. Acta 1677, 87–99 [DOI] [PubMed] [Google Scholar]
- 49.Eitoku M., Sato L., Senda T., Horikoshi M. (2008) Cell Mol. Life Sci. 65, 414–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.