

Abstract
Frontal impact, closed head trauma is a frequent cause of traumatic brain injury (TBI) in motor vehicle and sports accidents. Diffuse axonal injury (DAI) is common in humans and experimental animals, and results from shearing forces that develop within the anisotropic brain. Because the specific anisotropic properties of the brain are axis-dependent, the anatomical site where force is applied as well as the resultant acceleration, be it linear, rotational, or some combination, are important determinants of the resulting pattern of brain injury. Available rodent models of closed head injury do not reproduce the frontal impact commonly encountered in humans. Here we describe a new rat model of closed head injury that is a modification of the impact-acceleration model of Marmarou. In our model (the Maryland model), the impact force is applied to the anterior part of the cranium and produces TBI by causing anterior-posterior plus sagittal rotational acceleration of the brain inside the intact cranium. Skull fractures, prolonged apnea, and mortality were absent. The animals exhibited petechial hemorrhages, DAI marked by a bead-like pattern of β-amyloid precursor protein (β-APP) in damaged axons, and widespread upregulation of β-APP in neurons, with regions affected including the orbitofrontal cortex (coup), corpus callosum, caudate, putamen, thalamus, cerebellum, and brainstem. Activated caspase-3 was prominent in hippocampal neurons and Purkinje cells at the grey-white matter junction of the cerebellum. Neurobehavioral dysfunction, manifesting as reduced spontaneous exploration, lasted more than 1 week. We conclude that the Maryland model produces diffuse injuries that may be relevant to human brain injury.
Key words: activated caspase-3, β-amyloid precursor protein, diffuse axonal injury, frontal impact, impact acceleration, traumatic brain injury
Introduction
In humans, frontal impact, closed head trauma is arguably the most frequently encountered cause of traumatic brain injury (TBI), especially with motor vehicle, sports, and other types of accidents, and accounts for a disproportionate number of diffuse brain injuries (Adams et al., 1982; Adams et al., 1989; Cecil et al., 1998). Diffuse brain injury is often marked by subarachnoid hemorrhage, scattered intraparenchymal petechial hemorrhages, and diffuse axonal injury (DAI). DAI is a common primary lesion both in humans and in experimental animals, and results from shearing forces that develop within the anisotropic brain, especially when it is subjected to rotational acceleration (Gennarelli et al., 1981; Gennarelli et al., 1982; Prange et al., 2000).
Numerous factors govern the specific nature and anatomical distribution of the individual elements that make up diffuse brain injury, including the type of force applied, its severity, the anatomical site where it is applied, and whether it causes linear or rotational acceleration (LaPlaca et al., 2007). Of particular importance are the specific mechanical properties of the brain itself. The mechanical properties of the brain are markedly inhomogeneous (anisotropic), owing to the different properties of grey matter, white matter, and cerebrospinal fluid. The general organization of the mammalian brain (a cortical mantle overlying white matter tracts, surrounding ventricles and a centrally located thalamus, and basal ganglia) gives rise to specific anisotropic conditions that are axis-dependent. As a result, a given injury force can be expected to yield a different pattern and distribution of injuries when that force is administered in one axis or plane versus another.
Several rodent models of diffuse brain injury have been described (Cernak, 2005; Morales et al., 2005). The widely-used impact-acceleration model (Marmarou et al., 1994) produces dorsal-ventral linear acceleration. Recently-described angular acceleration models produce pure coronal acceleration (Ellingson et al., 2005; Fijalkowski et al., 2006; Fijalkowski et al., 2007; He et al., 2004; Xiao-Sheng et al., 2000). Although highly important, these models do not specifically reproduce the acceleration forces frequently encountered by the human brain involved in motor vehicle, sports, and other types of accidents, wherein frontal impact complicated by lateral rotation of the head imparts a complex combination of anterior-posterior linear acceleration plus sagittal rotational acceleration.
Here we describe a new rat model of frontal impact closed head injury. Our model is a modification of the impact-acceleration model of Marmarou (hereinafter called the Marmarou model) (Foda and Marmarou, 1994; Marmarou et al., 1994). However, in our model (hereinafter called the Maryland model), the impact force is directed horizontally and is applied to the anterior part of the cranium. Our model produces brain injury by causing anterior-posterior plus sagittal rotational acceleration of the brain inside the intact cranium, without any deformation or fracture of the skull. Here we report that this model produces diffuse injuries, including intra-axial and extra-axial hemorrhages, DAI, and widespread upregulation of β-amyloid precursor protein (β-APP) in neurons and caspase-3 activation, as well as persistent neurobehavioral dysfunction.
Methods
Frontal impact injury device
The energy for impact was applied via a steel ball (∼500 g; Small Parts, Inc., Miami, FL) accelerated by gravity by rolling down from a vertical height of 2.1 m (Fig. 1A). The path of the rolling ball was directed by a pair of parallel rails (two pieces of 19-mm-diameter tubular steel electrical conduit, joined 3 cm apart at their centers by nuts and bolts), bent to resemble the shape of a hockey stick. The long ends of the rails, held at a 66° angle from horizontal by a pedestal, curved gently into horizontally directed short ends, thus redirecting the trajectory of the ball horizontally. At the end of its run, the rolling ball entered into a collecting chamber (Fig. 1B), where it struck one end of a coupling arm, with the other end of the coupling arm engaging the malar processes of the rat bilaterally. The coupling arm passed through a hole in the back wall of the collecting chamber, with the rat lying horizontally opposite the hole on the outside of the chamber, protected from direct contact with the ball.
FIG. 1.

Apparatus for the application of the Maryland model of frontal impact closed head traumatic brain injury (TBI). (A) Schematic diagram depicting the essential elements of the injury apparatus, illustrating the key feature of a ball rolling down rails and being redirected from a vertical to a horizontal trajectory before striking the coupling arm attached to the anterior part of the rat's face. (B and C) Photographs of the rails, the collecting chamber, and the coupling arm of the device. (D) Photograph of the rat's skull with the malar eminences engaged by the brass rods of the coupling arm. (E and F) Lateral and superior views of the rat prior to injury.
The coupling arm was made of three parts, a solid cylinder (a delrin acetal rod 25 mm in diameter and 70 mm long; Small Parts, Inc.), one end of which was impacted by the ball and the other end of which had two holes 12 mm apart and 25 mm deep drilled into it to receive a pair of hollow brass rods (type C330 ASTM B135, 3.2 mm in diameter and ∼60 mm long; Small Parts, Inc.) (Fig. 1C). The coupling arm was constructed so that one rod protruded from the solid cylinder 3 mm farther than the other rod. The two brass rods, each with a small V-shaped groove on its outer end, engaged the exposed malar processes on each side, with the longer rod engaging the right malar prominence (Fig. 1D). The rods were gently secured to the snout using a rubber band. When the coupling arm was struck, the 3-mm offset induced sagittal rotational acceleration of the head, in addition to axial acceleration. When assembled and affixed to the snout, the coupling arm protruded ∼3 cm from the tip of the snout, thereby allowing horizontally directed frontal impact without damaging the rat's facial structures (Fig. 1E and F).
Injury procedure
Surgical procedures were approved by the Institutional Animal Care and Use Committee of the University of Maryland. Fasted male Long-Evans rats (250–275 g; Harlan, Indianapolis, IN) were anesthetized (60 mg/kg ketamine and 7.5 mg/kg xylazine given IP) and allowed to breathe air spontaneously. Core temperature was maintained at 37°C using a heating pad regulated by a rectal thermal probe (Harvard Apparatus, Holliston, MA).
Using conventional surgical techniques, the malar process on each side was exposed via infraorbital incisions. The grooves of the brass rods of the coupling arm were positioned on the malar processes and the rods were secured to the snout using a rubber band, with care being taken not to occlude the nasal passages. The rat was laid prone outside of the collecting chamber with the coupling arm protruding through the hole in the back wall of the collecting chamber, opposite the rails that directed the rolling ball. The rat was gently restrained in position using adhesive tape placed across the dorsal thorax (Fig. 1E and F).
For most experiments, the ball was positioned on the rails at a vertical height of 2.1 m to produce severe frontal impact TBI (50 rats). In a separate series, a vertical height of 0.25 m was used to produce mild TBI (5 rats), and for sham injury, the rat was operated on as described above, but no ball was dropped (10 rats). After release, the ball rolled down the rails and impacted the coupling arm, imparting an axial and sagittal rotational force to the skull. After injury, the coupling arm was removed and the infraorbital incisions were closed.
Oxygen saturation and heart rate were monitored pre- and post-injury using a pulse oximeter (Mouse Ox™; STARR Life Sciences Corp., Oakmont, PA) placed on a hindlimb. A tail artery catheter was used to monitor blood pressure (CyQ; CyberSense Inc., Nicholasville, KY) and blood gases (iSTAT; Heska Inc., Loveland, CO), except when neurobehavioral function was assessed, since tail surgery was found to interfere with performance.
After injury, the rats recovered and were observed for 1–7 days, after which they were euthanized using a lethal dose of sodium pentobarbital given IP. The animals were perfused with heparinized saline to remove blood from the intravascular compartment, followed by perfusion-fixation with 4% paraformaldehyde. The brain was harvested and photographed to document surface hemorrhages. Tissues were cryoprotected using 30% w/v sucrose for histology and immunohistochemistry.
Histology and immunohistochemistry
For routine histology, 10-μm cryosections were stained using hematoxylin and eosin.
β-APP was examined by labeling 40-μm floating cryosections. The sections were incubated in 0.3% hydrogen peroxide to block endogenous peroxidase activity for 30 min. After three washes in PBS, the sections were blocked with 0.2% Triton X-100 (Sigma, St. Louis, MO) in PBS containing 2% goat serum (Invitrogen, La Jolla, CA) for 2 h at room temperature, and then incubated overnight with primary antibody against the C-terminus of β-APP (CT695 1:5000 in blocking solution; Invitrogen, La Jolla, CA). The next day, the sections were washed in PBS and incubated with biotinylated secondary antibody (BA-1000 1:500 goat anti-rabbit; Vector Laboratories, Burlingame, CA) for 2 h. After washing in PBS, the sections were incubated in avidin-biotin solution (Vector Laboratories) and the color was developed in nickel-DAB chromogen solution (0.02% DAB and 2.5% NiSO4 in 0.175 M sodium acetate) activated with 0.01% hydrogen peroxide. The sections were rinsed, mounted, dehydrated, and cover-slipped with DPX mounting medium (Electron Microscopy Services, Fort Washington, PA).
Activated caspase-3 was examined by labeling 10-μm cryosections. The sections were mounted on slides, blocked in 2% goat serum with 0.2% Triton X-100 in PBS for 1 h, then incubated overnight with primary antibody directed against activated (cleaved) caspase-3 (#9661 1:200; Cell Signaling, Danvers, MA) at 4°C. The sections were washed three times in PBS, then incubated in the dark with fluorescent-labeled secondary antibody (1:500 goat anti-rabbit AlexaFlour555; Invitrogen). After 1 h, the slides were washed and cover-slipped with Prolong Antifade reagent with DAPI (P36931; Molecular Probes, Invitrogen). Control experiments included the omission of the primary antibody. Low- and high-power photomicrographs were taken using a CoolSNAP camera (Photometrics, Tucson, AZ), and images were adjusted for brightness and contrast using IPLab Software (Scanalytics, Inc., Fairfax, VA).
Neurobehavioral function
Spontaneous behavioral activity was assessed using quantified vertical exploration (Frey et al., 2009). The rats were placed in a freshly-cleaned acrylic glass cylinder (20 cm diameter × 20 cm high) and spontaneous behavior was recorded using a digital video camcorder. Vertical exploration was quantified as the number of seconds spent with both front paws elevated above shoulder height during the first 3 min spent inside the chamber.
Vestibulomotor function was evaluated using beam balance and forelimb extension. The beam balance test consisted of measuring the duration (60 sec maximum) of time that an animal could balance on a narrow (1.5-cm) beam elevated 30 cm above the ground (Wagner et al., 2007). The forelimb extension test consisted of holding the rat by its tail 30 cm above the ground and observing forepaw extension/flexion and symmetry (Kolb et al., 1985). All neurobehavioral tests were performed prior to injury (pre-injury baseline) and post-injury on days 1, 2, 3, and 7.
Statistical analysis
Unless otherwise noted, values given are mean ± SE. A paired t-test or ANOVA was used, as appropriate, to assess statistical significance, with Bonferroni comparisons. A p value <0.05 was considered to be statistically significant.
Results
Energy imparted
We compared the energy imparted by our device with that imparted in the Marmarou model (Foda and Marmarou, 1994; Marmarou et al., 1994). The Marmorou model utilizes a 450-g weight in vertical free-fall from 2 meters, which yields an impact energy of 8.8 joules (J).
In our model, we assumed a frictionless surface for the stainless steel ball rolling down the rails. The energy imparted by the rolling ball to the coupling arm (Eimpact) is equal to the total energy of the system (free-fall; Etotal) minus the rotational energy (Erotational) of the ball:
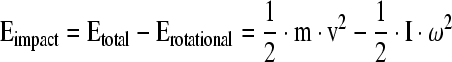 |
with:
v = velocity = (2·g·h)1/2, where g = 9.8 m/sec2
I = moment of inertia = 2/5·m·r2
ω = angular velocity = 2·π·f, where f = l /2·π·r/t
plus the following empirically-derived values:
m = mass of the ball = 0.535 kg
r = radius of the ball = 0.0255 m
h = vertical height of the fall = 2.1 m
l = length of the ramp = 2.57 m
t = time the ball travels down the ramp = 0.9 sec
thus:
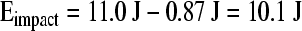 |
Physiological variables
Apnea was not apparent or was short-lived (<5 sec) in all rats. No deaths occurred. Injury did not significantly affect arterial blood gases, which were: pO2 = 78 ± 3 mm Hg, 52 ± 4 mm Hg, 7.3 ± 0.02 mm Hg before injury, and pCO2 =77 ± 7 mm Hg, 49 ± 6 mm Hg, and 7.3 ± 0.03 mm Hg after injury (7 rats). Mean arterial pressure, heart rate and oxygen saturation were not significantly changed during 1 h of monitoring post-injury (7 rats; Fig. 2). The rats awoke after anesthesia without any appreciable delay compared to sham controls.
FIG. 2.

Physiological variables during severe frontal impact traumatic brain injury (TBI) were stable. Pre- and post-injury measurements of mean arterial pressure (MAP) (A), heart rate (HR) (B), and oxygen saturation (pO2) as measured by pulse oximetry (C). The rats were injured at time 0 (data are for 7 rats per panel).
Neurobehavioral function
When placed in an acrylic glass cylinder, normal rats exhibit spontaneous vertical exploration (rearing), contacting the walls with their forepaws. However, spontaneous behavioral activity is typically blunted post-TBI (Wagner et al., 2007). To assess spontaneous activity, we used a test of quantified vertical exploration in which we recorded the time spent with both front paws elevated above shoulder height during a 3-min period of observation (rearing time).
In rats that sustained severe frontal impact TBI (2.1 m height), comparison of pre-injury values of rearing time with values recorded 24 h post-injury showed normal distributions, with a significant shift from 57 ± 17 to 25 ± 16 sec (p < 0.01 by paired t-test for 29 rats; Fig. 3A). In a separate group of 14 rats, serial measurements for 7 days after the same severe injury showed a statistically significant reduction in rearing time that did not return to pre-injury levels (Fig. 3B, ○). In 5 rats that sustained a mild injury (0.25 m height), a small reduction in rearing time was observed that did not reach statistical significance (Fig. 3B, ▪). Rats subjected to sham injury (9 rats) were essentially unaffected (Fig. 3B, □). Overall, this neurobehavioral test appeared to reflect a significant dose-response effect of frontal impact TBI (Fig. 3C).
FIG. 3.

Spontaneous vertical exploration is blunted by frontal impact traumatic brain injury (TBI). (A) Rearing time measured before (▪) and 24 h after (○) severe (2.1 m height) frontal impact TBI in the same rats. The means and standard deviations were 57 ± 17 and 25 ± 16 sec (p < 0.01 by paired t-test for 29 rats). Fit of these data to gaussian functions demonstrated normal distributions of this behavioral measure. (B) Spontaneous vertical exploration, measured before and over the course of 7 days post-injury, in three separate groups of rats that sustained severe injury (2.1 m height, ○), mild injury (0.25 m height, ▪), and sham injury (no hit, □). The data shown are mean ± standard error for 14, 5, and 9 rats, respectively, per injury group. Repeated measures analysis of variance (ANOVA) was performed for each group separately, with Bonferroni comparisons to pre-injury performance (**p < 0.01). (C) Spontaneous vertical exploration measured 24 h post-injury, normalized to pre-injury performance, in rats that sustained severe (2.1 m), mild (.25 m), and sham (0 m) frontal impact TBI (**p < 0.01 by one-way ANOVA, with Bonferroni comparisons to sham animals).
To determine whether the observed decrease in spontaneous vertical exploration was due to a vestibulomotor deficit or to post-injury stress or anxiety (Fromm et al., 2004; Vink et al., 2003), we assessed postural reflex and performance on the beam balance. The forelimb extension test is a postural reflex that provides a sensitive measure of central motor status, with abnormalities reflecting injuries to the pyramidal and extrapyramidal systems (Kolb and Whishaw, 1985). The beam balance is an integrated behavior that requires proper function of the sensorimotor cortex (Hamm, 2001). Both beam balance and the forelimb extension reflex, evaluated before injury and at 1, 2, 3, and 7 days after severe frontal impact TBI, were unaffected by injury (the same 14 rats with severe injury as those described above; data not shown), suggesting that the reduced vertical exploration that we observed was due to a reduction in spontaneous behavioral activity attributable to post-injury stress or anxiety (Fromm et al., 2004; Vink et al., 2003).
Pathology
Necropsy was performed in 25 rats 24–48 h after severe frontal impact TBI, following transcardiac perfusion to remove intravascular blood. No fractures of the malar eminences, other skull fractures, or injuries involving the cervical spine were identified. Macroscopic contusions of the orbitofrontal cortex (coup) were not observed, but macroscopic subarachnoid hemorrhage was identified in the posterior fossa behind the cerebellum (contrecoup) in 10/25 rats (Fig. 4A). Histological examination revealed frequent petechial hemorrhages, typically located in the white matter of the frontal and parietal lobes, in the rostral corpus callosum, in the deep nuclei, and in the brainstem (Fig. 4B–D), as well as occasional thrombosed veins (Fig. 4E).
FIG. 4.

Severe frontal impact traumatic brain injury (TBI) produces extra-axial and intra-axial hemorrhages. (A) Photograph of a brain 24 h after severe frontal impact TBI, showing subarachnoid hemorrhage involving the cerebellum and brainstem. (B–E) Sections stained with hematoxylin and eosin 24–48 h post-TBI, showing petechial hemorrhages in the white matter of the frontal cortex (B), the brainstem (C), and in the caudate (D). A thrombosed vein in the frontal lobe (E) is also shown; intravascular blood was removed by perfusion.
No macroscopic or microscopic hemorrhages were observed in the 5 rats with mild frontal impact TBI.
β-Amyloid precursor protein
β-Amyloid precursor protein (β-APP) is an important marker of brain injury, with two distinct patterns emerging after TBI. Diffuse axonal injury (DAI) is characterized by the accumulation of β-APP in injured axons in a pattern that resembles a string of beads (Blumbergs et al., 1995; Bramlett et al., 1997; Pierce et al., 1996; Stone et al., 2000). In addition, it is common to have diffuse upregulation of β-APP in neuronal perikarya (Itoh et al., 2009). Both of these abnormalities were found in the present study.
As previously observed, immunolabeling for β-APP tended to be more prominent on day 7 than earlier, on days 1–2, after severe frontal impact TBI (7 rats; Tables 1 and 2). Damaged axons exhibiting the classical beaded appearance were identified in various regions of the brain (Fig. 5). Damaged axons were found not only in the frontal cortex beneath the region the impact (coup), but also in the corpus callosum, hippocampus, caudate/putamen, cerebellum (contrecoup), and brainstem. Notably, damaged axons were not found in the thalamus of any specimen.
Table 1.
Axonal Labeling
| |
R74 |
R75 |
R81 |
R94 |
R96 |
R106 |
R107 |
|---|---|---|---|---|---|---|---|
| Rat no. | Early | Early | Early | Late | Late | Late | Late |
| Frontal cortex | ++ | + | ++ | + | ++ | +++ | ++ |
| Corpus callosum | + | 0 | + | + | + | +++ | 0 |
| Hippocampus | + | 0 | ++ | + | + | ++ | + |
| Caudate/putamen | + | + | 0 | + | ++ | ++++ | + |
| Thalamus | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cerebellum | + | 0 | + | + | ++ | ++ | + |
| Brainstem | + | 0 | ++ | + | ++ | +++ | ++ |
Values given are from visual evaluation based on a scale of 0 to ++++.
Early, 1–2 days after TBI; late, 7 days after TBI; TBI, traumatic brain injury.
Table 2.
Neuronal Labeling
| |
R74 |
R75 |
R81 |
R94 |
R96 |
R106 |
R107 |
|---|---|---|---|---|---|---|---|
| Rat no. | Early | Early | Early | Late | Late | Late | Late |
| Frontal cortex | ++ | ++ | ++ | ++ | ++ | +++ | +++ |
| Hippocampus | ++ | + | ++ | + | + | ++ | ++ |
| Caudate/putamen | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Thalamus | ++ | + | + | + | ++ | +++ | ++ |
| Cerebellum | ++ | + | ++ | ++ | +++ | ++++ | +++ |
| Brainstem | ++ | + | ++ | + | +++ | ++++ | +++ |
Values given are from visual evaluation based on a scale of 0 to ++++.
Early, 1–2 days after TBI; late, 7 days after TBI; TBI, traumatic brain injury.
FIG. 5.

β-Amyloid precursor protein (β-APP) identifies disrupted axons after severe frontal impact traumatic brain injury (TBI). (A–D) High-magnification images of the frontal cortex (A), corpus callosum (B), hippocampus (C), and brainstem (D), showing the beaded appearance of disrupted axons immunolabeled for β-APP. All images were taken 7 days after injury.
Prominent upregulation of β-APP was found in neuronal perikarya in various regions of the brain (Fig. 6). Upregulation was found in cells of the frontal cortex, as well as in the hippocampus, thalamus, cerebellum, and brainstem. Interestingly, the cells in the caudate/putamen showed no upregulation in any specimen, although damaged axons were identified in these regions (7 rats; Tables 1 and 2). This pattern was opposite that observed in the thalamus, where damaged axons were not found, but cells showed prominent upregulation of β-APP.
FIG. 6.

β-Amyloid precursor protein (β-APP) is upregulated in neurons after severe frontal impact traumatic brain injury (TBI). (A and B) Low-magnification images of sagittal sections immunolabeled for β-APP from a naïve rat and from a rat 7 days after injury. (C–F) High-magnification images of frontal cortex (C), thalamus (D), Purkinje layer of the cerebellum (E), and ventral brainstem (F), showing neuronal immunolabeling for β-APP. All images were taken 7 days after injury.
Activated caspase-3
Activated caspase-3 is another important marker of TBI. As previously observed, nuclear labeling for activated caspase-3 tended to correlate with cytoplasmic labeling for β-APP. After severe frontal impact TBI, caspase labeling was found in the cells of the cortex, both frontal and parietal, and in the hippocampus, thalamus, cerebellum, and brainstem (7 rats; Fig. 7). In the hippocampus, labeling for activated caspase-3 was especially prominent in the dentate gyrus and CA3 regions (Fig. 7A, B, and C). In the cerebellum, caspase labeling was especially prominent in Purkinje cells at the grey-white matter junction (Fig. 7A, D, and E).
FIG. 7.

Activated caspase-3 is upregulated in neurons after severe frontal impact traumatic brain injury (TBI). (A and B) Montage of images of a coronal section immunolabeled for activated caspase-3 from a rat 7 days after injury. Note the distinct labeling in the cerebral cortex, dentate gyrus of the hippocampus, and the Purkinje layer of the cerebellum. (B–E) Moderate- and high-magnification images of the hippocampus (B and C), and of the Purkinje layer (D and E) immunolabeled for activated caspase-3. All images were taken 7 days after injury.
Discussion
Diffuse brain injury continues to be a major cause of morbidity following motor vehicle, sports, and other types of accidents. Diffuse brain injury is characterized in part by stretch injury to axons resulting in DAI, which may be widely distributed. DAI is characterized by morphological and biochemical changes including axonal swelling, formation of axonal bulbs, and upregulation of β-APP (Adams et al., 1991; Meythaler et al., 2001; Onaya, 2002). In humans, the severity of diffuse brain injury determines whether patients recover, remain disabled, are vegetative, or die following head trauma.
At the time of injury, the brain experiences forces of linear, rotational, and angular acceleration resulting in injuries that depend on the site of impact of the traumatizing force, its direction, and its severity. The general organization of the mammalian brain gives rise to specific anisotropic conditions that are axis-dependent. The principal anatomical features that determine specific anisotropic conditions in rodent and human brains are similar, including the general organization and orientation of major white-matter tracts, which are determined by the location of the cortical mantle, the central position and orientation of the basal ganglia and thalamus, and related anatomical characteristics. The principal dissimilarity between rodent and human brains is the orientation of the brainstem relative to the anterior-posterior axis. Nevertheless, broad similarities between species dictate that, in any given plane, comparable anisotropic conditions will be encountered in rodent and human brains.
A rodent model that imparts frontal impact with lateral rotation, such as the Maryland model, is likely to reproduce salient features of TBI in humans that may not be reproduced in other models. Differences between different closed-head injury models will not be found in the types of cellular or molecular injury encountered, including petechial hemorrhages, DAI, β-APP upregulation, and caspase activation, among others, which are found in all relevant models. However, different models may produce these discrete injuries with different anatomical distributions. It may be that the opposite pattern of axonal versus neuronal β-APP in the caudate/putamen versus the thalamus represents such an axis-specific response. Also, it is likely that prominent involvement of the cerebellum, which is not reported in the Marmarou model, also represents an axis-specific response. To the extent that anatomical locations of injury in any given model mimic those encountered in humans, the more likely it is that the animal model will be useful for studying long-term neurofunctional sequelae in humans.
Comparison of rat models of closed head and diffuse brain injury
The Marmarou model
The Marmarou model is the best characterized, most widely regarded model of diffuse brain injury in the rat. A variant of this model has also been described (Cernak et al., 2004). The Marmarou model was designed to direct an impact force to the protected skull, to avoid direct focal cortical injuries. However, impact causes compressive deformation of the cranial vault in the dorsal-ventral axis, with “virtually all compression [being] the result of displacement of the vertex” (Marmarou et al., 1994). In the absence of protection, deformation of the skull results in fractures of the thin frontal and parietal bones. With the protection of a metal disc bonded to the dorsal aspect of the skull, the incidence of fractures is greatly reduced, but deformation estimated to be 0.3 mm or greater still occurs beneath the disc (Marmarou et al., 1994).
A significant portion of the brain injury seen in the Marmarou model is located immediately beneath the site of impact. Abnormal neurons are confined to the cortical areas under the site of impact, immediately beneath the metallic disc, with more normal-appearing neurons at the periphery (Foda and Marmarou, 1994). Although unarguably injured (Farkas et al., 2006), abnormal-appearing neurons beneath the disc may not die (Goda et al., 2002; Rafols et al., 2007). Similarly, pericapillary astrocytic swelling is observed, primarily in areas of the cerebral cortex located underneath the metallic disc in association with injured neurons, while capillaries, astrocytes, and neurons in the rest of the cerebral cortex are morphologically intact. These focal cortical injuries have been attributed to the coup. However, the presence of skull deformation in this model raises the possibility that injury beneath the site of impact results from direct percussion rather than from the coup, which strictly defined, is caused by the accelerated un-deformed skull striking the brain.
The Marmarou model is most significant for the associated diffuse brain injuries located remotely from regions beneath the disc (Foda and Marmarou, 1994). Perivascular edema is frequently observed in the thalamus and brainstem. Most importantly, DAI (retraction balls of severed axons) is prominent in long tracts of the brainstem and elsewhere. In the original description (Foda and Marmarou, 1994), neurofilament labeling revealed axonal abnormalities in the optic tracts, cerebral peduncles, superior cerebellar peduncles, the rubrospinal tract, corticospinal tracts in the pons and medulla oblongata, and in the pyramidal decussation in the medulla oblongata. Injured axons could also be observed, although to a lesser extent, in the corpus callosum, internal capsule, and the spinothalamic, gracilis, and cuneate tracts. Recent re-assessments of this model, utilizing either silver staining or immunolabeling for β-APP, reaffirmed that the most prominent pathologic event is DAI within long-association and subcortical projection fiber systems in the white matter of the cerebral hemispheres and brainstem (Kallakuri et al., 2003; Rafols et al., 2007). Upregulation of β-APP in neuronal perikarya and in reactive astrocytes near the region of injury is characteristic of diffuse injury in the Marmarou model (Rafols et al., 2007).
Another feature that distinguishes the Marmarou model is the high incidence of brainstem injury, manifesting as transient hypertension and prolonged apnea that give rise to high mortality, especially in animals without controlled ventilation.
The Gennarelli model
The Gennarelli model is an impact acceleration model, but is designed to produce pure coronal rotational acceleration of the brain within the intact cranium (Ellingson et al., 2005; Fijalkowski et al., 2006; Fijalkowski et al., 2007). A non-impact (inertial) angular acceleration model has also been described (He et al., 2004; Xiao-Sheng et al., 2000). These rat models employing pure coronal rotation were developed because of the finding that in a primate model, coronal rotation produced the greatest amount of diffuse injury (Gennarelli et al., 1982).
Models of pure coronal rotation produce evidence of DAI in the medulla oblongata, midbrain, upper cervical cord, and corpus callosum (He et al., 2004; Xiao-Sheng et al., 2000). Surprisingly, upregulation of β-APP was reportedly absent in the one study in which this was examined (Fijalkowski et al., 2007).
The Maryland model
The Maryland model is also an impact acceleration model that utilizes a similar magnitude of impact force as the (severe) Marmarou model. It is designed to produce linear plus rotational acceleration, with linear acceleration in the anterior-posterior direction and rotational acceleration in the sagittal plane. This form of injury is intended to reproduce frontal impacts common in motor vehicle, sports, and other types of accidents. In the Maryland model, the impact force is applied to the malar processes, which are firmly anchored to the skull base. Application of the impact force to this location accelerates the head without apparent deformation of the cranial vault. We observed no fractures in our model, but even if fractures involving the malar processes or the zygomas were to occur, this would not deform the cranial vault. Brain injury in the Maryland model results from the impact of the accelerated un-deformed cranial vault onto brain surfaces, combined with rotational shear strain that develops within the anisotropic brain.
The Maryland model is characterized by absence of cortical contusions, although petechial hemorrhages in white matter and deep nuclei were frequently encountered. Even the orbitofrontal cortex, the site of the coup, rarely showed any evidence of hemorrhage on the surface, although significant injury to this region occurred, as evidenced by subcortical hemorrhages and prominent upregulation of β-APP and nuclear activation of caspase-3. These observations contrast with findings in the Marmarou model, where hemorrhagic cortical contusions are prominent in the parietal cortex, beneath the site of impact.
Although axonal accumulation of β-APP has been the focus of most reports related to trauma, TBI also leads to overexpression of APP within neuronal cell bodies (Bramlett et al., 1997; Itoh et al., 2009; Van Den et al., 1999). Outside of the immediate site beneath the impact, the Marmarou model reportedly shows little neuronal upregulation of β-APP. By contrast, in the Maryland model, we found pronounced upregulation of β-APP in numerous cellular regions. It has been postulated that following trauma, the overexpression of β-APP may exceed the limit of normal processing capacity, possibly resulting in mis-metabolization of β-APP into potentially amyloidogenic fragments (Van Den et al., 2007). Also, caspase-3 activation was a prominent feature in our model and tended to occur in cellular regions where β-APP upregulation was prominent. Notably, caspase-3-mediated cleavage of β-APP may result in formation of amyloid β peptide, which has been postulated as a potential risk factor for later development of Alzheimer's disease (Stone et al., 2002).
The diffuse injuries produced in the Maryland model were associated with neurobehavioral abnormalities. Non-coerced vertical exploration was blunted for 1 week, but this was not due to injury to sensorimotor, pyramidal, or extrapyramidal systems, but appeared to be attributable to post-injury stress or anxiety (Fromm et al., 2004; Vink et al., 2003). Detailed neurocognitive testing was beyond the scope of the present investigation, but future study of neurocognitive performance will be of interest, given the widespread abnormalities involving neuronal β-APP and caspase-3 activation in the hippocampus, thalamus, cerebellum, and other locations.
Brainstem injury was not clinically severe in the Maryland model. There was no transient hypertension or prolonged apnea at the time of injury and mortality was nil, even though the animals were not artificially ventilated. Yet injury to the brainstem was always present, as indicated by petechial hemorrhages and prominent upregulation of β-APP in white matter tracts and neurons. Similarly, brainstem axonal injury without coma is observed in a rat angular acceleration model (Xiao-Sheng et al., 2000). Lesser brainstem injury in these models compared to the Marmarou model may be due to the difference in the way impact force is applied to the brainstem: in the Marmorou model the brainstem is accelerated down into the skull base, whereas in the Maryland model the brainstem is accelerated along its principal axis.
Injury to the cerebellum was particularly prominent in our model, presumably owing to a contrecoup mechanism. Cerebellar injury is also observed with a fluid percussion model of open head injury (Ai et al., 2007; Park et al., 2007), but to our knowledge, has not been reported with the Marmarou model. In the Maryland model, posterior fossa injury involving the cerebellum included frequent subarachnoid hemorrhage, and prominent upregulation of β-APP and activation of caspase-3 in Purkinje cells of the grey-white matter junction, a quintessential anisotropic boundary. Cerebellar injury is not a frequent focus of pre-clinical work on TBI, but nonetheless is a common occurrence in humans that markedly affects diverse functions, including speech, praxis, and ambulation.
Conclusion
The Maryland model is the first description of a rat model of closed head injury that mimics the frontal impact, sagittal rotation commonly encountered in human traumatic brain injury. In this model, we documented petechial hemorrhages, DAI, extensive upregulation of β-APP, and widespread activation of caspase-3 in the cortex, deep nuclei, cerebellum, and brainstem. Additional work will be required to further characterize this model, but the initial description presented here suggests that this model may offer unique pathophysiological features not encountered in previous models of closed head injury. Given the widespread diffuse injuries observed, we anticipate that future studies will uncover important types of neurological dysfunction not addressed here.
Acknowledgments
This work was supported by grants to J.M.S. from the Department of Veterans Affairs (Baltimore, MD), the National Heart, Lung, and Blood Institute (no. HL082517), the National Institute of Neurological Disorders and Stroke (no. NS061808 and NS060801), and the Christopher and Dana Reeve Foundation, and to V.G. from the National Institute of Neurological Disorders and Stroke (no. NS061934).
Author Disclosure Statement
No conflicting financial interests exist.
References
- Adams J.H. Doyle D. Ford I. Gennarelli T.A. Graham D.I. McLellan D.R. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Adams J.H. Graham D.I. Gennarelli T.A. Maxwell W.L. Diffuse axonal injury in non-missile head injury. J. Neurol. Neurosurg. Psychiatry. 1991;54:481–483. doi: 10.1136/jnnp.54.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J.H. Graham D.I. Murray L.S. Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann. Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- Ai J. Liu E. Park E. Baker A.J. Structural and functional alterations of cerebellum following fluid percussion injury in rats. Exp. Brain Res. 2007;177:95–112. doi: 10.1007/s00221-006-0654-9. [DOI] [PubMed] [Google Scholar]
- Blumbergs P.C. Scott G. Manavis J. Wainwright H. Simpson D.A. McLean A.J. Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury. J. Neurotrauma. 1995;12:565–572. doi: 10.1089/neu.1995.12.565. [DOI] [PubMed] [Google Scholar]
- Bramlett H.M. Kraydieh S. Green E.J. Dietrich W.D. Temporal and regional patterns of axonal damage following traumatic brain injury: a beta-amyloid precursor protein immunocytochemical study in rats. J. Neuropathol. Exp. Neurol. 1997;56:1132–1141. doi: 10.1097/00005072-199710000-00007. [DOI] [PubMed] [Google Scholar]
- Cecil K.M. Hills E.C. Sandel M.E. Smith D.H. McIntosh T.K. Mannon L.J. Sinson G.P. Bagley L.J. Grossman R.I. Lenkinski R.E. Proton magnetic resonance spectroscopy for detection of axonal injury in the splenium of the corpus callosum of brain-injured patients. J. Neurosurg. 1998;88:795–801. doi: 10.3171/jns.1998.88.5.0795. [DOI] [PubMed] [Google Scholar]
- Cernak I. Animal models of head trauma. NeuroRx. 2005;2:410–422. doi: 10.1602/neurorx.2.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernak I. Vink R. Zapple D.N. Cruz M.I. Ahmed F. Chang T. Fricke S.T. Faden A.I. The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol. Dis. 2004;17:29–43. doi: 10.1016/j.nbd.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Ellingson B.M. Fijalkowski R.J. Pintar F.A. Yoganandan N. Gennarelli T.A. New mechanism for inducing closed head injury in the rat. Biomed. Sci. Instrum. 2005;41:86–91. [PubMed] [Google Scholar]
- Farkas O. Lifshitz J. Povlishock J.T. Mechanoporation induced by diffuse traumatic brain injury: an irreversible or reversible response to injury? J. Neurosci. 2006;26:3130–3140. doi: 10.1523/JNEUROSCI.5119-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowski R.J. Ellingson B.M. Stemper B.D. Yoganandan N. Gennarelli T.A. Pintar F.A. Interface parameters of impact-induced mild traumatic brain injury. Biomed. Sci. Instrum. 2006;42:108–113. [PubMed] [Google Scholar]
- Fijalkowski R.J. Stemper B.D. Pintar F.A. Yoganandan N. Crowe M.J. Gennarelli T.A. New rat model for diffuse brain injury using coronal plane angular acceleration. J. Neurotrauma. 2007;24:1387–1398. doi: 10.1089/neu.2007.0268. [DOI] [PubMed] [Google Scholar]
- Foda M.A. Marmarou A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J. Neurosurg. 1994;80:301–313. doi: 10.3171/jns.1994.80.2.0301. [DOI] [PubMed] [Google Scholar]
- Frey L.C. Hellier J. Unkart C. Lepkin A. Howard A. Hasebroock K. Serkova N. Liang L. Patel M. Soltesz I. Staley K. A novel apparatus for lateral fluid percussion injury in the rat. J. Neurosci. Methods. 2009;177:267–272. doi: 10.1016/j.jneumeth.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm L. Heath D.L. Vink R. Nimmo A.J. Magnesium attenuates post-traumatic depression/anxiety following diffuse traumatic brain injury in rats. J. Am. Coll. Nutr. 2004;23:529S–533S. doi: 10.1080/07315724.2004.10719396. [DOI] [PubMed] [Google Scholar]
- Gennarelli T.A. Adams J.H. Graham D.I. Acceleration induced head injury in the monkey. I. The model, its mechanical and physiological correlates. Acta Neuropathol. Suppl. 1981;7:23–25. doi: 10.1007/978-3-642-81553-9_7. [DOI] [PubMed] [Google Scholar]
- Gennarelli T.A. Thibault L. Adams J.H. Graham D.I. Thompson C.J. Marcincin R.P. Diffuse axonal injury and traumatic coma in the primate. Ann. Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- Goda M. Isono M. Fujiki M. Kobayashi H. Both MK801 and NBQX reduce the neuronal damage after impact-acceleration brain injury. J. Neurotrauma. 2002;19:1445–1456. doi: 10.1089/089771502320914679. [DOI] [PubMed] [Google Scholar]
- Hamm R.J. Neurobehavioral assessment of outcome following traumatic brain injury in rats: an evaluation of selected measures. J. Neurotrauma. 2001;18:1207–1216. doi: 10.1089/089771501317095241. [DOI] [PubMed] [Google Scholar]
- He X.S. Xiang Z. Zhou F. Fu L.A. Shuang W. Calcium overloading in traumatic axonal injury by lateral head rotation: a morphological evidence in rat model. J. Clin. Neurosci. 2004;11:402–407. doi: 10.1016/j.jocn.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Itoh T. Satou T. Nishida S. Tsubaki M. Hashimoto S. Ito H. Expression of amyloid precursor protein after rat traumatic brain injury. Neurol. Res. 2009;31:103–109. doi: 10.1179/016164108X323771. [DOI] [PubMed] [Google Scholar]
- Kallakuri S. Cavanaugh J.M. Ozaktay A.C. Takebayashi T. The effect of varying impact energy on diffuse axonal injury in the rat brain: a preliminary study. Exp. Brain Res. 2003;148:419–424. doi: 10.1007/s00221-002-1307-2. [DOI] [PubMed] [Google Scholar]
- Kolb B. Whishaw I.Q. An observer's view of locomotor asymmetry in the rat. Neurobehav. Toxicol. Teratol. 1985;7:71–78. [PubMed] [Google Scholar]
- LaPlaca M.C. Simon C.M. Prado G.R. Cullen D.K. CNS injury biomechanics and experimental models. Prog. Brain Res. 2007;161:13–26. doi: 10.1016/S0079-6123(06)61002-9. [DOI] [PubMed] [Google Scholar]
- Marmarou A. Foda M.A. van den B.W. Campbell J. Kita H. Demetriadou K. A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. J. Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- Meythaler J.M. Peduzzi J.D. Eleftheriou E. Novack T.A. Current concepts: diffuse axonal injury-associated traumatic brain injury. Arch. Phys. Med. Rehabil. 2001;82:1461–1471. doi: 10.1053/apmr.2001.25137. [DOI] [PubMed] [Google Scholar]
- Morales D.M. Marklund N. Lebold D. Thompson H.J. Pitkanen A. Maxwell W.L. Longhi L. Laurer H. Maegele M. Neugebauer E. Graham D.I. Stocchetti N. McIntosh T.K. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience. 2005;136:971–989. doi: 10.1016/j.neuroscience.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Onaya M. Neuropathological investigation of cerebral white matter lesions caused by closed head injury. Neuropathology. 2002;22:243–251. doi: 10.1046/j.1440-1789.2002.00456.x. [DOI] [PubMed] [Google Scholar]
- Park E. Ai J. Baker A.J. Cerebellar injury: clinical relevance and potential in traumatic brain injury research. Prog. Brain Res. 2007;161:327–338. doi: 10.1016/S0079-6123(06)61023-6. [DOI] [PubMed] [Google Scholar]
- Pierce J.E. Trojanowski J.Q. Graham D.I. Smith D.H. McIntosh T.K. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and beta-amyloid peptide after experimental brain injury in the rat. J. Neurosci. 1996;16:1083–1090. doi: 10.1523/JNEUROSCI.16-03-01083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange M.T. Meaney D.F. Margulies S.S. Defining brain mechanical properties: effects of region, direction, and species. Stapp. Car. Crash. J. 2000;44:205–213. doi: 10.4271/2000-01-SC15. [DOI] [PubMed] [Google Scholar]
- Rafols J.A. Morgan R. Kallakuri S. Kreipke C.W. Extent of nerve cell injury in Marmarou's model compared to other brain trauma models. Neurol. Res. 2007;29:348–355. doi: 10.1179/016164107X204657. [DOI] [PubMed] [Google Scholar]
- Stone J.R. Okonkwo D.O. Singleton R.H. Mutlu L.K. Helm G.A. Povlishock J.T. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid beta peptide in traumatic axonal injury. J. Neurotrauma. 2002;19:601–614. doi: 10.1089/089771502753754073. [DOI] [PubMed] [Google Scholar]
- Stone J.R. Singleton R.H. Povlishock J.T. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res. 2000;871:288–302. doi: 10.1016/s0006-8993(00)02485-9. [DOI] [PubMed] [Google Scholar]
- Van Den H.C. Blumbergs P.C. Finnie J.W. Manavis J. Jones N.R. Reilly P.L. Pereira R.A. Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: an ovine head impact model. Exp. Neurol. 1999;159:441–450. doi: 10.1006/exnr.1999.7150. [DOI] [PubMed] [Google Scholar]
- Van Den H.C. Thornton E. Vink R. Traumatic brain injury and Alzheimer's disease: a review. Prog. Brain Res. 2007;161:303–316. doi: 10.1016/S0079-6123(06)61021-2. [DOI] [PubMed] [Google Scholar]
- Vink R. O'Connor C.A. Nimmo A.J. Heath D.L. Magnesium attenuates persistent functional deficits following diffuse traumatic brain injury in rats. Neurosci. Lett. 2003;336:41–44. doi: 10.1016/s0304-3940(02)01244-2. [DOI] [PubMed] [Google Scholar]
- Wagner A.K. Postal B.A. Darrah S.D. Chen X. Khan A.S. Deficits in novelty exploration after controlled cortical impact. J. Neurotrauma. 2007;24:1308–1320. doi: 10.1089/neu.2007.0274. [DOI] [PubMed] [Google Scholar]
- Xiao-Sheng H. Sheng-Yu Y. Xiang Z. Zhou F. Jian-ning Z. Diffuse axonal injury due to lateral head rotation in a rat model. J. Neurosurg. 2000;93:626–633. doi: 10.3171/jns.2000.93.4.0626. [DOI] [PubMed] [Google Scholar]