

Abstract
Cortical contusion injury can result in the partial loss of ipsilateral CA3 neurons within 48 h, leading to a proportional reduction in the number of afferent fibers to CA1 stratum radiatum. While the loss of afferent input to CA1 exhibits a remarkable, albeit incomplete, recovery over the next few weeks, little is known about the functional status of presynaptic afferents during the depletion and recovery phases following injury. Here, we prepared hippocampal slices from adult Sprague Dawley rats at 2, 7, and 14 days after lateral cortical contusion injury and measured fiber volley (FV) amplitudes extracellularly in CA1 stratum radiatum. Field excitatory post-synaptic potentials (EPSPs) were also measured and plotted as a function of FV amplitude to assess relative synaptic strength of residual and/or regenerated synaptic contacts. At 2 days post-injury, FV amplitude and synaptic strength were markedly reduced in the ipsilateral, relative to the contralateral, hippocampus. FV amplitude in ipsilateral CA1 showed a complete recovery by 7 days, indicative of a post-injury sprouting response. Synaptic strength in ipsilateral CA1 also showed a dramatic recovery over this time; however, EPSP-to-FV curves remained slightly suppressed at both the 7 and 14 day time points. Despite these deficits, ipsilateral slices retained the capacity to express long-term potentiation, indicating that at least some mechanisms for synaptic plasticity remain intact, or are compensated for. These results are in agreement with anatomical evidence showing a profound deafferentation, followed by a remarkable re-enervation, of ipsilateral CA1 in the first few weeks after traumatic brain injury. Although plasticity mechanisms appear to remain intact, synaptic strength deficits in CA1 could limit information throughput in the hippocampus, leading to persistent memory dysfunction.
Key words: cortical contusion injury, deafferentation, excitatory postsynaptic potential, fiber volley, Schaffer collaterals
Introduction
It is estimated that 1.5 million individuals suffer traumatic brain injury (TBI) each year in the United States (Langlois et al., 2005), which is associated with costly health problems and high mortality and morbidity in previously healthy populations. TBI involves primary and secondary injury cascades resulting in delayed neuron dysfunction, synapse loss, and cell death (Sullivan et al., 1998; Biegon et al., 2004; Scheff et al., 2005). These TBI-related cascades have been implicated in cytoskeletal damage, mitochondrial dysfunction, and altered signal transduction (Hall et al., 2004; Bayir et al., 2007; Singh et al., 2007).
In addition to neocortical damage that is obvious following different experimental models of TBI, subcortical structures, such as the hippocampus, are also affected (Baldwin et al., 1997; Grady et al., 2003; Anderson et al., 2005). Hippocampal involvement may be responsible for injury-related cognitive deficits. Experimental models using a moderate-to-severe TBI have shown that the hippocampal CA3 subregion is highly vulnerable to damage, with as many as 60% of the neurons lost within the first 48 h (Baldwin et al., 1997). CA3 pyramidal neurons provide the major input to the CA1 region of the hippocampus and are critical for the rapid acquisition of novel spatial and contextual information (Nakazawa et al., 2002, 2003). To maintain information flow through the hippocampus, surviving CA3 collateral/commissural fibers exhibit extensive sprouting followed by the re-establishment of new synaptic contacts in CA1 (Scheff et al., 2005). Although cognitive function following TBI appears to decline and recover in parallel with the loss and rebirth of new CA3 synaptic contacts, much remains unknown concerning the functional state of residual and regenerated CA3-CA1 synaptic contacts.
Several rodent models of TBI exhibit reduced excitatory postsynaptic potentials (EPSPs) and impaired long-term potentiation (LTP) in ipsilateral CA1 (Miyazaki et al., 1992; Reeves et al., 1995; D'Ambrosio et al., 1998; Sick et al., 1998; Albensi et al., 2000; Sanders et al., 2000; Schwarzbach et al., 2006) during the first few weeks post-injury. These deficits have been linked to changes in several postsynaptic mechanisms, including glutamate receptor expression, dendritic spine morphology, and Ca2+/calmodulin kinase II levels, among other factors (Scheff et al., 2005; Atkins et al., 2006; Schwarzbach et al., 2006). However, few studies have directly investigated whether alterations in CA1 synaptic function arise directly from damage to CA3 presynaptic elements. Deafferentation of CA1 would be expected to cause a reduction in basal field EPSP amplitudes and limit the temporal and spatial summation necessary for the induction of LTP (McNaughton et al., 1978). Conversely, loss of presynaptic inputs can also trigger compensatory changes in postsynaptic neurons, resulting in the strengthening of residual synaptic contacts (Foster et al., 1991). Many other postsynaptic compensatory reactions to deafferentation have been reported as well.
To address the direct impact of CA3 afferent loss and recovery on CA1 synaptic function and plasticity, we prepared hippocampal slices from adult male rats at 2, 7, and 14 days after lateral cortical contusion injury and measured electrically evoked CA3-CA1 fiber volleys (FV) in CA1 stratum radiatum. The FV amplitude, or the presynaptic population spike, is proportional to the number of activated afferent fibers and is easily distinguished from field EPSPs in this hippocampal subregion. EPSP amplitudes were concomitantly measured and plotted against FV amplitudes to assess relative synaptic strength at surviving and/or re-established synaptic contacts. Possible changes in the induction of LTP were also investigated.
Methods
Adult male Sprague Dawley rats (250–275 g) (Harlan Laboratories, Indianapolis, IN) were used in this study. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky. Animals were housed two per cage on a 12 h light/dark cycle and provided access to pellet diet and water ad libitum. A total of 21 rats were subjected to a unilateral cortical impact utilizing an electronic controlled pneumatic impact device (ECPI, TBI0310, PSI, Fairfax Station, VA), as previously described (Sullivan et al., 2002). Briefly, each animal was anesthetized with 2% isofluorane and placed in a Kopf stereotaxic frame (Tujunga, CA) with the incisor bar set at −5. Body temperature of each rat was monitored and maintained at 36°C with a heating pad. Following a midline incision and retraction of the skin, a 6-mm-diameter craniotomy was made approximately midway between bregma and lamda with a Michele hand trephine (Miltex, NY). The skull disk was removed without disturbing the dura. The exposed brain was injured using the ECPI. The impact rod had a 5-mm-diameter beveled tip that was used to compress the cortex to a depth of 1.5 mm at 3.5 m/s. Following injury, Surgicel (Johnson & Johnson, New Brunswick, NJ) was placed over the injury site, and the skull disk was replaced and sealed with a thin layer of dental acrylic.
Hippocampal slice preparation
At 2, 7, and 14 days after TBI, rats were euthanized with CO2 and brains were removed and stored briefly in ice-cold, oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM) 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 0.5 CaCl2, 26 NaHCO3, and 10 dextrose (pH ∼7.4). Hippocampal slices (450-mm sections) from the ipsilateral (injured) and contralateral (uninjured) hemispheres were cut parallel to the alvear fibers using a McIlwain Tissue Chopper (Stoelting Co., Wood Dale, IL) and transferred to netting in separate Plexiglas holding chambers. Slices were bathed in oxygenated ACSF (containing 2 mM CaCl) at an interface with warm (32°C), humidified air and permitted to equilibrate for at least 1.5 h before transfer to an RC-22 recording chamber (Warner Instruments, Hamden, CT) for electrophysiological analysis.
Field EPSP recordings
Slices were submerged in oxygenated ACSF (32°C) and perfused at a rate of 1–2 mL/min for 15–20 min before the start of each recording session. The generation and measurement of field EPSPs were highly similar to the procedures used in our previous work (Norris et al., 1996, 1998). Briefly, Schaffer collaterals were activated with a bipolar platinum/iridium electrode located in stratum radiatum of area CA1 at or near the CA3 border. Stimulus intensity was controlled by a constant current stimulus isolation unit (World Precision Instruments, Sarasota, FL), and stimulus timing was controlled by Clampex 9.2 software (Molecular Devices, Sunnyvale, CA). Field EPSPs were recorded using a glass micropipette (1–6 M), filled with ACSF and containing a Ag/AgCl wire, positioned in stratum radiatum of CA1 approximately 1 mm away from the point of stimulation. Field potentials were amplified 100 ×, Bessel-filtered at 1 kHz, and digitized at 10 kHz using a Multiclamp 700B amplifier and a Digidata 1320 digitizer (Molecular Devices).
Fiber excitability and synaptic strength measures
For each slice, twin stimulus pulses (S1 and S2), separated by 50 ms, were delivered at seven different intensity levels (range 30–500 mA) at a rate of 0.1 Hz. Five field potentials at each stimulus level were averaged, and measurements of FV amplitude (in mV) and EPSP slope (mV/ms) were performed offline using Clampfit software (Molecular Devices, Sunnyvale, CA). The relative number of activated CA3 afferents in each slice was assessed by plotting average FV amplitudes against stimulation intensity. Averaged EPSP slope measures were then plotted against their corresponding FV amplitudes to estimate the strength of existing CA3-CA1 synaptic contacts. To estimate population spike threshold, the EPSP slope amplitude at which a population spike first appeared in the ascending phase of the field potential was calculated and averaged across five successive trials at the spike threshold stimulation level. For each animal, electrophysiological parameters were averaged across all slices within each hemisphere (usually one to four slices) and the n used for all statistical comparisons reflects the number of animals per post-injury time point.
Fiber excitability and synaptic strength curves for each hemisphere were fit with a sigmoidal equation of the form:
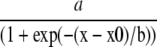 |
where a equals the maximal amplitude of the distribution, b equals the distribution slope, x equals the stimulation intensity (or FV amplitude), and x0 equals the stimulation intensity (or FV amplitude) required for half-maximal response amplitude. Fitted parameters were then compared across hemispheres at each post-injury time point using Z tests. Z values greater than |2| were considered significant. Paired-pulse facilitation of the EPSP slope (S2/S1 × 100) and population spike threshold was also compared across hemispheres using paired t tests, with significance at p < 0.05.
LTP measurements
For some slices, stimulation intensity was readjusted after the I/O curve to elicit an EPSP of ∼1 mV, and stimulus pulses were delivered at 0.033 Hz until a stable 20 min baseline was established. Very few slices from the ipsilateral hippocampus in the 2 day post-TBI group exhibited a 1 mV EPSP. In these cases, the stimulation intensity eliciting the maximal EPSP amplitude was used for LTP experiments. Two 100 Hz trains (1 s each, 10 s intertrain interval) were then delivered at the baseline stimulation intensity to induce LTP, followed by an additional 45 min baseline. Within each hemisphere, EPSP measures from the last 10 min of the post-LTP baseline were averaged across slices and compared to the pre-LTP baseline average and differences across hemispheres were determined by two-way repeated measures ANOVA, with significance at p < 0.05.
Because contralateral hippocampus can also undergo changes following TBI (Tran et al., 2006), which may contaminate interhemispheric comparisons, we carried out additional studies on sham animals (n = 3) that underwent craniotomy, but did not receive a cortical contusion injury. Electrophysiological parameters in sham animals were evaluated 7 days post-surgery.
Results
CA3 fiber excitability and CA1 synaptic strength at 2, 7, and 14 days post-TBI
Hippocampal slices were prepared from the ipsilateral (injured) and contralateral (non-injured) hemispheres of adult male rats at 2, 7, and 14 days post-TBI and CA3-CA1 synaptic connectivity was examined with electrophysiological techniques. Electrical pulses were delivered to Schaffer collaterals across a range of intensities (30–500 mA) and field potentials were recorded in CA1 stratum radiatum. Figure 1 shows representative CA1 field potentials collected from injured and uninjured hemispheres at each of the three post-injury time points.
FIG. 1.

Representative CA1 field potentials in hippocampal slices prepared at 2, 7, and 14 days post-TBI. (A) Representative field potential recorded in CA1 stratum radiatum showing the regions in which FV and EPSP slope measures were taken. (B–D) Representative field potentials from ipsilateral (Ipsi.) and contralateral (Contra.) hippocampal slices from 2, 7, and 14 day TBI rats in response to 100, 200, 300, and 400 mA orthodromic stimulus pulses. Each waveform represents the average of five responses at each stimulus intensity. Dashed lines indicate EPSP amplitude levels from corresponding ipsilateral hemispheres. Note that stimulus artifacts have been clipped. Calibration bars, 0.5 mV × 5 ms.
To assess the functional status of presynaptic afferents following injury, the FV amplitude (Fig. 1A) was measured at each stimulus intensity level and the resulting I/O curve for each hemisphere was fit with a sigmoidal equation (Table 1 and Fig. 2A–C). At 2 days post-TBI, the I/O curve for the injured hemisphere exhibited a downward shift, characterized by a significantly reduced maximal FV amplitude (Z = 6.00). By 7 and 14 days post-TBI, FV amplitudes in the injured hemisphere recovered to uninjured levels (Fig. 2B and C) and no significant differences in any curve parameters were observed (Table 1). Ipsilateral and contralateral slices taken from sham control animals at 7 days post-surgery exhibited highly similar FV properties relative to the 7 day TBI group (Table 1), suggesting that the recovery of FV excitability in the ipsilateral hemisphere following TBI is not attributable to a post-injury suppression of fiber excitability in the contralateral hemisphere. These findings are generally consistent with previous anatomical studies, which observed a striking loss, followed by a rapid recovery, of CA3-CA1 contacts within the first 2 weeks following TBI (Scheff et al., 2005).
Table 1.
FV Input/output Curve Parameters
| |
|
Max. FV (mV) |
FV curve slope |
½ Max. stimulation amplitude (Ma) |
|||
|---|---|---|---|---|---|---|---|
| Time | No. | Ipsi. | Contra. | Ipsi. | Contra. | Ipsi. | Contra. |
| 2 days | 5 | 0.64 ± 0.06a | 1.17 ± 0.06 | 93.1 ± 19.4 | 76.3 ± 11.0 | 236.5 ± 28.2 | 188.3 ± 14.0 |
| 7 days | 7 | 0.88 ± 0.05 | 0.88 ± 0.05 | 67.1 ± 11.8 | 65.1 ± 12.0 | 162.1 ± 14.5 | 157.0 ± 14.8 |
| 14 days | 9 | 0.93 ± 0.06 | 0.96 ± 0.05 | 83.5 ± 13.7 | 69.2 ± 10.4 | 207.5 ± 18.3 | 167.3 ± 12.9 |
| Shamb | 5c | 0.83 ± 0.06 | 0.91 ± 0.04 | 75.9 ± 13.9 | 52.3 ± 7.5 | 175.2 ± 17.3 | 150.7 ± 9.3 |
Values shown as mean ± SEM. Ipsi., ipsilateral; contra., contralateral; no., number of rats.
Significant difference from contralateral hemisphere.
Sham animals were evaluated 7 days post-surgery.
Number of slices per hemisphere in sham group (three rats overall).
FIG. 2.

Fiber excitability and synaptic strength curves in rat hippocampal slices prepared at 2, 7, and 14 days post-TBI. (A–C) I/O curves for mean ± SEM FV amplitudes in CA1 stratum radiatum of hippocampal slices from ipsilateral (Ipsi.) and contralateral (Contra.) hemispheres. (D–F) Mean ± SEM EPSP slope measures plotted against FV measures obtained in panels A–C. All curves were fitted with a three parameter sigmoidal equation.
To assess the relative strength of surviving synaptic contacts in the post-injury period, EPSP slopes (Fig. 1A) were measured and plotted against their corresponding FV amplitudes and then fitted with a sigmoidal equation (Table 2 and Fig. 2D–F). The results revealed a profound depression in CA1 synaptic strength in the injured hemisphere at 2 days post-TBI (max amplitude, Z = 10.64). Over the next 12 days, CA1 synaptic strength showed a remarkable increase (Fig. 2E and F), similar to the recovery observed for CA3 fiber excitability (Fig. 2B and C). However, unlike the FV, the recovery in synaptic strength was incomplete. At 7 days post-TBI (Fig. 2E), the synaptic strength curve in the injured hemisphere was still markedly shifted downward relative to the contralateral hemisphere (Z = 6.09). In addition, maximal EPSP amplitude was reduced (Z = 6.3) and one-half maximal FV amplitude was increased (Z = 2.4) in ipsilateral slices of the 7 day TBI group compared to ipsilateral slices from the 7 day sham group (Table 2). At 14 days post-TBI (Fig. 2F), synaptic strength was largely recovered. Nonetheless, a slight, but significant, downward shift was still exhibited by the injured hemisphere (Z = 3.42).
Table 2.
Synaptic Strength Curve Parameters
| |
|
Max. EPSP slope (mV/mS) |
Synaptic strength curve slope |
½ Max. FV amplitude (mV) |
|||
|---|---|---|---|---|---|---|---|
| Time | No. | Ipsi. | Contra. | Ipsi. | Contra. | Ipsi. | Contra. |
| 2 days | 5 | 0.31 ± 0.02a | 1.02 ± 0.06 | 0.14 ± 0.02 | 0.22 ± 0.04 | 0.33 ± 0.03 | 0.43 ± 0.05 |
| 7 days | 7 | 0.90 ± 0.06a,b | 1.33 ± 0.04 | 0.19 ± 0.03 | 0.15 ± 0.01 | 0.43 ± 0.04b | 0.38 ± 0.02 |
| 14 days | 9 | 0.93 ± 0.04a | 1.15 ± 0.05 | 0.18 ± 0.02 | 0.22 ± 0.02 | 0.42 ± 0.02 | 0.40 ± 0.03 |
| Shamc | 5d | 1.35 ± 0.04 | 1.31 ± 0.06 | 0.12 ± 0.01 | 0.16 ± 0.03 | 0.33 ± 0.02 | 0.36 ± 0.03 |
Values shown as mean ± SEM. Ipsi., ipsilateral; Contra., contralateral; no., number of rats.
Significant difference from contralateral hemisphere.
Significant difference from sham ipsilateral hemisphere.
Sham animals were evaluated 7 days post-surgery.
Number of slices per hemisphere in sham group (3 rats overall).
Paired pulse facilitation and population spike threshold at 2, 7, and 14 days post-TBI
To assess changes in presynaptic release properties following TBI, twin stimulus pulses (50 ms interpulse interval) were delivered during the I/O curve and paired pulse facilitation (PPF) was quantified as the increase in the EPSP slope from pulse 1 to pulse 2 (Fig. 3A). While PPF tended to be greatest in ipsilateral slices from 2 day post-TBI rats (Fig. 3B), no significant differences were observed for PPF in the ipsilateral hemisphere across the post-TBI time points. Furthermore, at no post-injury time point did PPF differ significantly across hemispheres, suggesting that the loss and recovery of CA3 synaptic contacts following injury is not associated with a remarkable change in excitation-neurotransmitter release coupling.
FIG. 3.

Paired pulse facilitation and population spike threshold in rat hippocampal slices prepared at 2, 7, and 14 days post-TBI. (A) Representative field potentials recorded in CA1 stratum radiatum during a paired pulse stimulation protocol. The twin pulses are separated by 50 mS. In CA1, the descending slope and amplitude of the EPSP generated by the second stimulus pulse are stereotypically larger relative to the EPSP elicited by the first pulse, a process known as paired pulse facilitation (PPF). The arrow points to the emergence of a CA1 population spike in the ascending phase of the field potential and provides an indication of neuronal excitability. The EPSP slope associated with the emergence of the population spike was used to calculate population spike threshold. (B) Mean ± SEM PPF values as a function of hemisphere (Ipsi. vs. Contra.) and post-injury time point. (C) Mean ± SEM population spike threshold values as a function of hemisphere and post-injury time point. n.m., pop spike threshold was not measured in 2 day ipsilateral slices, as these slices rarely exhibited a population spike.
Some studies have indicated that neuronal excitability is increased in CA1 following TBI as evidenced by a reduction in the population spike threshold in response to synaptic input (Reeves et al., 1995). This EPSP-to-spike (E-S) potentiation may serve as a compensatory mechanism to help maintain information flow through the hippocampus. To determine if depressed synaptic strength in the present study is offset by increased E-S potentiation, the EPSP slope amplitude associated with the initial appearance of a population spike was quantified and compared across hemispheres (Fig. 3A and C). In general, E-S potentiation was not observed with TBI (Fig. 3C). In fact, population spikes were rarely observed in ipsilateral slices from 2 day post-TBI rats (i.e., only 3 out of 10 of these slices exhibited a pop spike). However, in the 7 day TBI group, population spike threshold was mildly reduced (p = 0.08) in ipsilateral, relative to contralateral slices, suggesting a possible compensatory increase in CA1 neuronal excitability at this post-injury time point.
LTP induction at 2, 7, and 14 days post-TBI
Following the I/O curve, the stimulus intensity was readjusted in each slice to yield an EPSP of approximately 1 mV and a stable response baseline of at least 20 min was recorded before the delivery of two high frequency (100 Hz) trains for LTP induction. Because synaptic strength in the injured hemisphere was profoundly reduced at 2 days post-TBI, stimulus intensity for these slices was adjusted to yield the maximal EPSP amplitude. Despite the marked reduction in synaptic strength, slices from the injured hemisphere in the 2 day group still showed significant LTP (p < 0.05; Fig. 4A). Nonetheless, the magnitude of LTP in these slices was slightly, albeit non-significantly reduced (p = 0.08) compared to contralateral slices. As shown in Figures 4B and C, LTP induction at 7 and 14 days post-TBI was essentially identical across the two hemispheres, and no differences in EPSP slope were observed at any time point following high frequency stimulation. These values were also similar to LTP obtained in the ipsilateral (159.5 ± 5.7%) and contralateral (147.5 ± 8.9%) hemispheres of 7 day sham rats (Fig. 4D).
FIG. 4.

LTP in rat hippocampal slices prepared at 2, 7, and 14 days post-TBI. (A–C) Time plots showing mean ± SEM normalized EPSP slopes recorded in CA1 stratum radiatum of hippocampal slices prepared from ipsilateral (Ipsi.) and contralateral (Contra.) hemispheres. Arrows indicate the delivery of two 1 s duration 100 Hz trains (arrows) for LTP induction. n = 4–7 rats per post-injury time point. (D) Mean ± SEM LTP during the last 10 min of the post-100 Hz stimulus baseline (normalized to the pre-100 Hz stimulus baseline) as a function of hemisphere and post-injury time point.
Discussion
This is the first study to perform a careful characterization of electrophysiological changes in the rat hippocampal CA1 field following a moderate controlled cortical impact (CCI) injury. With the exception of an earlier study (Albensi et al., 2000), other evaluations of electrophysiological changes in the hippocampus have used a fluid percussion (FP) model of TBI. The major finding of this study is that CA3 fiber excitability and CA1 synaptic strength exhibit a marked depression, followed by a substantial recovery, in the first 2 weeks following cortical injury. These changes closely parallel the loss and recovery of CA3-CA1 synaptic contacts after TBI, which were shown previously through the use of unbiased stereology (Scheff et al., 2005). Moreover, surviving CA1-CA3 synaptic contacts maintained the capacity to express LTP in the 2 weeks following injury, suggesting that functional synaptic deficits may be milder than previously reported.
Using unbiased stereology, we previously showed in the cortical contusion model, that CA3 neuron loss 48 h after TBI was associated with a proportional reduction in the number of ipsilateral synaptic contacts in CA1 stratum radiatum (Baldwin et al., 1997). Synapse number in CA1 then recovered substantially over the next 2 weeks, consistent with a compensatory sprouting response in surviving CA3 neurons and/or neurons from other brain regions. The present work extended these anatomical findings by determining the functional status of the residual and newly sprouted presynaptic fibers in the post-injury period. Under nearly identical experimental conditions, we observed an approximately 50% reduction in the ipsilateral CA1 FV amplitude at 48 h post-TBI. Because the FV amplitude is directly proportional to the number of presynaptic fibers activated, this result confirms that the rapid loss of CA3-CA1 synaptic contacts shown in our earlier work stems mainly from the depletion of CA3 axons. At 7 and 14 days after the injury, the ipsilateral FV amplitude recovered to contralateral control levels consistent with the arrival of newly sprouted presynaptic fibers in the CA1 region. Together these results indicate that the structural re-enervation of CA1 in the post-insult period is closely paralleled by a functional re-enervation as well.
By plotting field EPSP slope measures against their corresponding FV amplitudes, we determined the extent to which residual and/or newly sprouted fibers establish or maintain functional connections with CA1 pyramidal neurons. Similar to the FV, the relative strength of residual synaptic contacts in ipsilateral CA1 was profoundly reduced 48 h after injury (Fig. 2D). Synaptic strength also showed a parallel recovery at the 7 and 14 day time points (Fig.2E and F). However, unlike the FV, which showed complete recovery over this time period, synaptic strength in ipsilateral CA1 still remained significantly depressed relative to the contralateral side. PPF, a general index of excitation-neurotransmitter release coupling, was not significantly altered at any time point in the post-injury period (Fig. 3B). The mechanistic basis for this synaptic deficit remains unclear but could involve alterations in Ca2+ dependent signaling pathways involved in synaptic plasticity, changes in dendritic spine properties, and/or reduced synapse number (Scheff et al., 2005; Atkins et al., 2006; Schwarzbach et al., 2006), among other factors. As recently suggested, these changes could result directly from a reduction in NMDA and AMPA type glutamate receptor function following injury (Schwarzbach et al., 2006).
We previously found that the number of CA3-CA1 synaptic contacts in stratum radiatum was reduced by 30% even up to 60 days post-injury (Scheff et al., 2005). If the number of functional presynaptic afferents is fully recovered at 1 week after injury, as shown in the present study, but the number of synapses in CA1 stratum radiatum is reduced, then it is possible that newly sprouted fibers are mistargeted to other hippocampal subregions. In addition to CA1 pyramidal neurons, Schaffer collateral/commissural fibers also enervate a variety of interneuronal types in strata pyramidale, oriens, and moleculare/lacunosum (Paxinos, 2004). Interestingly, a recent study (Witgen et al., 2005) observed an increase in inhibitory synaptic transmission in CA1 at 1 week after injury, but whether these inhibitory changes resulted from the mistargeting of newly sprouted CA3 axons was not investigated. Finally, aberrant sprouting of entorhinal axons is also observed throughout multiple layers in the CA1 region following CA3 neuronal loss (Shetty, 2002) and could be activated en passant by the orthodromic stimulation protocol used in the present study. The possibility that TBI-mediated deficits in CA3-CA1 excitatory transmission result from the aberrant sprouting of entorhinal neurons, or any other neuronal types, will require further investigation.
In contrast to several previous studies, we did not observe an LTP deficit in area CA1 of the ipsilateral hippocampus at 7 or 14 days post-injury. This discrepancy could be attributable to differences in the type of injury inflicted (i.e., FP, CCI, weight drop) and/or the animal model under investigation (rats vs. mice). Differences in LTP induction across laboratories may also be linked to differences in field depolarization levels during LTP induction. It is well established that LTP depends critically on the summed depolarization provided by co-activated synapses (McNaughton et al., 1978). This depolarization drives Ca2+ influx through voltage-operated sources, which in turn, recruits numerous Ca2+ dependent mechanisms for modulating the function and/or surface expression of glutamate receptors, among other things (Malenka and Bear, 2004). In many studies in which LTP is compared across two or more treatment groups—including most all TBI/LTP studies to date—the stimulation intensity used for inducing LTP is adjusted to the half-maximal intensity derived from an I/O curve (i.e., the stimulus intensity associated with a half-maximal synaptic response). This procedure is certainly appropriate when basal synaptic strength is similar across two treatment groups, as the stimulation intensity and level of depolarization during LTP induction is also roughly equivalent across groups. The problem occurs when basal synaptic strength is substantially different between the treatment groups—for example, when one group receives TBI and the other does not. In this case, adjusting the stimulus intensity to the half-maximal level imposes a functional disadvantage onto the group with lower basal synaptic strength. In other words, the half-maximal stimulation protocol is biased to find an LTP deficit in animals that also show depressed I/O curves (e.g., injured animals). It is therefore difficult to determine whether Ca2+ dependent plasticity mechanisms in injured animals are functionally intact or simply insufficiently stimulated under these conditions.
To minimize group differences in depolarization, we adjusted the stimulus intensity in all LTP experiments to a level sufficient to elicit a 1 mV EPSP. The exception was for ipsilateral slices from 2 day post-TBI rats, which rarely showed an EPSP that is ≥1 mV. For these slices, stimulus intensity associated with the maximal EPSP amplitude was used for baseline and LTP stimulation. Our results demonstrate that residual and/or regenerated synapses in ipsilateral CA1 are capable of expressing LTP within 2 days after injury. In fact, at the 1 and 2 week post-injury time points, we observed no interhemispheric differences in EPSP slope at any time following high frequency stimulation. Although ipsilateral slices did show reduced LTP at 2 days following TBI, this reduction was relatively small and did not reach statistical significance (p = 0.08).
These findings suggest that a variety of postsynaptic mechanisms required for LTP induction are functionally intact after injury. Another possibility, though, is that plasticity mechanisms are negatively affected by TBI, but compensated by unidentified mechanisms or processes. For instance, multiple forms of LTP, distinguished by their unique dependence on NMDA receptors and L-type Ca2+ channels, have been well characterized in hippocampus. An increase in L-type Ca2+ channels in CA1 neurons with brain aging (Campbell et al., 1996; Thibault and Landfield, 1996; Norris et al., 2008) is associated with an increase in L-channel dependent LTP (Shankar et al., 1998). Moreover, augmented L-channel LTP in aged animals appears to help mask deficits in NMDA receptor-dependent LTP (Boric et al., 2008). Whether similar changes in L-type Ca2+ channel function and/or expression compensate for NMDA-LTP deficits in brain-injured animals will require further investigation. Nevertheless, the injury-associated reduction in synaptic strength observed here, and in other studies, certainly indicates that there is a “throughput” deficit in ipsilateral CA1 after injury that could prevent endogenous patterns of synaptic activity from eliciting the depolarization necessary for LTP. An enduring deficit of this kind could underlie the persistent hippocampal-dependent memory deficits shown consistently in animal models and in humans following head trauma.
In summary, our results are in general agreement with anatomical evidence showing a profound deafferentation, followed by a remarkable re-enervation, of ipsilateral CA1 in the first 2 weeks after TBI. Functional presynaptic fibers appear to be fully recovered within 1 week post-injury, while relative CA3-CA1 synaptic strength remains slightly impaired for up to 2 weeks. The mechanistic bases for these synaptic changes and their contribution to persisting memory dysfunction in injured animals will require further investigation.
Acknowledgments
This work was supported by NIH grant AG21981and an award from the Kentucky Spinal Cord and Head Injury Trust.
Author Disclosure Statement
No competing financial interests exist.
References
- Albensi B.C. Sullivan P.G. Thompson M.B. Scheff S.W. Mattson M.P. Cyclosporin ameliorates traumatic brain-injury-induced alterations of hippocampal synaptic plasticity. Exp. Neurol. 2000;162:385–389. doi: 10.1006/exnr.1999.7338. [DOI] [PubMed] [Google Scholar]
- Anderson K.J. Miller K.M. Fugaccia I. Scheff S.W. Regional distribution of fluoro-jade B staining in the hippocampus following traumatic brain injury. Exp. Neurol. 2005;193:125–130. doi: 10.1016/j.expneurol.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Atkins C.M. Chen S. Alonso O.F. Dietrich W.D. Hu B.R. Activation of calcium/calmodulin-dependent protein kinases after traumatic brain injury. J. Cereb. Blood Flow Metab. 2006;26:1507–1518. doi: 10.1038/sj.jcbfm.9600301. [DOI] [PubMed] [Google Scholar]
- Baldwin S.A. Gibson T. Callihan C.T. Sullivan P.G. Palmer E. Scheff S.W. Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. J. Neurotrauma. 1997;14:385–398. doi: 10.1089/neu.1997.14.385. [DOI] [PubMed] [Google Scholar]
- Bayir H. Kagan V.E. Clark R.S. Janesko-Feldman K. Rafikov R. Huang Z. Zhang X. Vagni V. Billiar T.R. Kochanek P.M. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J. Neurochem. 2007;101:168–181. doi: 10.1111/j.1471-4159.2006.04353.x. [DOI] [PubMed] [Google Scholar]
- Biegon A. Fry P.A. Paden C.M. Alexandrovich A. Tsenter J. Shohami E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: implications for treatment of neurological and cognitive deficits. Proc. Natl. Acad. Sci. USA. 2004;101:5117–5122. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boric K. Munoz P. Gallagher M. Kirkwood A. Potential adaptive function for altered long-term potentiation mechanisms in aging hippocampus. J. Neurosci. 2008;28:8034–8039. doi: 10.1523/JNEUROSCI.2036-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell L.W. Hao S.Y. Thibault O. Blalock E.M. Landfield P.W. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. J. Neurosci. 1996;16:6286–6295. doi: 10.1523/JNEUROSCI.16-19-06286.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio R. Maris D.O. Grady M.S. Winn H.R. Janigro D. Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Res. 1998;786:64–79. doi: 10.1016/s0006-8993(97)01412-1. [DOI] [PubMed] [Google Scholar]
- Foster T.C. Barnes C.A. Rao G. McNaughton B.L. Increase in perforant path quantal size in aged F-344 rats. Neurobiol. Aging. 1991;12:441–448. doi: 10.1016/0197-4580(91)90071-q. [DOI] [PubMed] [Google Scholar]
- Grady M.S. Charleston J.S. Maris D. Witgen B.M. Lifshitz J. Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological estimation. J. Neurotrauma. 2003;20:929–941. doi: 10.1089/089771503770195786. [DOI] [PubMed] [Google Scholar]
- Hall E.D. Detloff M.R. Johnson K. Kupina N.C. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J. Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Langlois J.A. Marr A. Mitchko J. Johnson R.L. Tracking the silent epidemic and educating the public: CDC's traumatic brain injury-associated activities under the TBI Act of 1996 and the Children's Health Act of 2000. J. Head Trauma Rehabil. 2005;20:196–204. doi: 10.1097/00001199-200505000-00003. [DOI] [PubMed] [Google Scholar]
- Malenka R.C. Bear M.F. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- McNaughton B.L. Douglas R.M. Goddard G.V. Synaptic enhancement in fascia dentata: cooperativity among coactive afferents. Brain Res. 1978;157:277–293. doi: 10.1016/0006-8993(78)90030-6. [DOI] [PubMed] [Google Scholar]
- Miyazaki S. Katayama Y. Lyeth B.G. Jenkins L.W. DeWitt D.S. Goldberg S.J. Newlon P.G. Hayes R.L. Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 1992;585:335–339. doi: 10.1016/0006-8993(92)91232-4. [DOI] [PubMed] [Google Scholar]
- Nakazawa K. Quirk M.C. Chitwood R.A. Watanabe M. Yeckel M.F. Sun L.D. Kato A. Carr C.A. Johnston D. Wilson M.A. Tonegawa S. Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science. 2002;297:211–218. doi: 10.1126/science.1071795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K. Sun L.D. Quirk M.C. Rondi–Reig L. Wilson M.A. Tonegawa S. Hippocampal CA3 NMDA receptors are crucial for memory acquisition of one-time experience. Neuron. 2003;38:305–315. doi: 10.1016/s0896-6273(03)00165-x. [DOI] [PubMed] [Google Scholar]
- Norris C.M. Blalock E.M. Chen K.C. Porter N.M. Thibault O. Kraner S.D. Landfield P.W. Hippocampal “zipper” slice studies reveal a necessary role for calcineurin in the increased activity of L-type Ca(2+) channels with aging. Neurobiol. 2008 doi: 10.1016/j.neurobiolaging.2008.03.026. Aging Advance online publication. Retrieved May 7, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris C.M. Halpain S. Foster T.C. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2 + channels. J. Neurosci. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris C.M. Korol D.L. Foster T.C. Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. J. Neurosci. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G. The Rat Nervous System. Elsevier Academic Press; San Diego: 2004. [Google Scholar]
- Reeves T.M. Lyeth B.G. Povlishock J.T. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp. Brain Res. 1995;106:248–256. doi: 10.1007/BF00241120. [DOI] [PubMed] [Google Scholar]
- Sanders M.J. Sick T.J. Perez-Pinzon M.A. Dietrich W.D. Green E.J. Chronic failure in the maintenance of long-term potentiation following fluid percussion injury in the rat. Brain Res. 2000;861:69–76. doi: 10.1016/s0006-8993(00)01986-7. [DOI] [PubMed] [Google Scholar]
- Scheff S.W. Price D.A. Hicks R.R. Baldwin S.A. Robinson S. Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J. Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- Schwarzbach E. Bonislawski D.P. Xiong G. Cohen A.S. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus. 2006;16:541–550. doi: 10.1002/hipo.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar S. Teyler T.J. Robbins N. Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1. J. Neurophysiol. 1998;79:334–341. doi: 10.1152/jn.1998.79.1.334. [DOI] [PubMed] [Google Scholar]
- Shetty A.K. Entorhinal axons exhibit sprouting in CA1 subfield of the adult hippocampus in a rat model of temporal lobe epilepsy. Hippocampus. 2002;12:534–542. doi: 10.1002/hipo.10031. [DOI] [PubMed] [Google Scholar]
- Sick T.J. Perez-Pinzon M.A. Feng Z.Z. Impaired expression of long-term potentiation in hippocampal slices 4 and 48 h following mild fluid-percussion brain injury in vivo. Brain Res. 1998;785:287–292. doi: 10.1016/s0006-8993(97)01418-2. [DOI] [PubMed] [Google Scholar]
- Singh I.N. Sullivan P.G. Hall E.D. Peroxynitrite-mediated oxidative damage to brain mitochondria: protective effects of peroxynitrite scavengers. J. Neurosci. Res. 2007;85:2216–2223. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- Sullivan P.G. Keller J.N. Bussen W.L. Scheff S.W. Cytochrome c release and caspase activation after traumatic brain injury. Brain Res. 2002;949:88–96. doi: 10.1016/s0006-8993(02)02968-2. [DOI] [PubMed] [Google Scholar]
- Sullivan P.G. Keller J.N. Mattson M.P. Scheff S.W. Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J. Neurotrauma. 1998;15:789–798. doi: 10.1089/neu.1998.15.789. [DOI] [PubMed] [Google Scholar]
- Thibault O. Landfield P.W. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–1020. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- Tran L.D. Lifshitz J. Witgen B.M. Schwarzbach E. Cohen A.S. Grady M.S. Response of the contralateral hippocampus to lateral fluid percussion brain injury. J. Neurotrauma. 2006;23:1330–1342. doi: 10.1089/neu.2006.23.1330. [DOI] [PubMed] [Google Scholar]
- Witgen B.M. Lifshitz J. Smith M.L. Schwarzbach E. Liang S.L. Grady M.S. Cohen A.S. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]