

Abstract
MicroRNAs (miRNAs) have attracted significant attention given their important roles in development, normal physiology, and disease states including cancer. Recent studies have identified specific miRNAs that regulate the cell cycle and documented that the loss or gain of miRNA-mediated cell-cycle control contributes to malignancy. miRNAs regulate classic cell-cycle control pathways by directly targeting proteins such as E2F transcription factors, cyclin-dependent kinases (Cdks), cyclins, and Cdk inhibitors (CKIs). Moreover, recent findings suggest that miRNAs themselves might be subject to cell-cycle dependent regulation. Together, these observations indicate that reciprocal control of RNA silencing and the metazoan cell-cycle impacts cellular behavior and disease.
Early evidence for small RNA-mediated cell-cycle control
MicroRNAs (miRNAs) are critical regulators of gene expression in multicellular eukaryotes. These ∼21 to 25 nucleotide non-coding RNAs are liberated from RNA polymerase II-transcribed precursors through a series of sequential endonuclease-mediated cleavage events and exert their regulatory effects by associating with a group of proteins termed the RNA-induced silencing complex (RISC). RISC is directed to target mRNAs via imperfect sequence complementarity between the miRNA and, in most cases, the 3′ untranslated region (UTR) of the target. In almost all studied examples, targeting of a transcript by RISC leads to diminished gene expression through inhibition of translation or accelerated turnover. Notably, this regulatory mechanism allows single miRNAs to target hundreds of transcripts and thus to coordinate complex programs of gene expression to influence cellular behavior [1].
Evidence for the importance of miRNAs in regulating cellular division and the cell cycle came with the discovery of the first miRNA, lin-4, in the nematode Caenorhabditis elegans. lin-4 loss-of-function causes improper reiteration of cell divisions normally associated only with the first larval developmental stage (L1) and a failure of cells to exit the cell cycle and terminally differentiate [2]. Let-7, the second identified miRNA, analogously regulates cell division events during the C. elegans L4 to adult transition [3]. The identification of let-7 led quickly to the discovery of highly conserved orthologs in other metazoans and the realization that small RNAs constitute a widespread system of gene regulation critical for appropriate cellular and developmental functions. miRNAs have since taken on added significance as numerous groups have demonstrated their prominent roles in various human diseases and especially in cancer [4]. In this article, we discuss how our understanding of the roles of miRNAs in human malignancies is illuminated by evidence implicating the cell cycle both as a target and as a regulator of the miRNA pathway.
miRNAs: regulators of cell cycle progression
Many individual components of the cell-cycle control machinery directly control or are targeted by individual miRNAs. Before delving into these pathways, two groups of miRNAs deserve special attention. A large body of evidence has implicated the let-7 family and the miR-15a/16-1 cluster both as important regulators of the cell cycle and as potential human tumor suppressors. The identified targets of these miRNAs illustrate well the mechanisms though which this class of regulatory RNAs exert their effects on cell cycle control.
The let-7 family
Early studies of let-7 in C. elegans revealed its critical role in cell cycle exit and terminal differentiation [3]. Mammalian let-7 miRNAs appear to have similar functions and accordingly, significant evidence exists supporting a tumor suppressor role for this family of miRNAs. The human genome encodes 12 let-7 homologs, produced from 8 distinct genomic loci. Four of these loci are located in regions known to be deleted in human cancers [5], and examination of human lung cancer samples revealed that low let-7 expression correlates with poor survival [6, 7]. Both in vitro and in vivo evidence for let-7-mediated tumor suppression has been established by multiple laboratories [8-13]. For example, expression of let-7 family members suppresses the in vitro and in vivo growth of mouse and human tumor cell lines. Furthermore, two laboratories recently demonstrated that virally-delivered let-7 reduced tumor number and size in a KRAS-driven model of lung adenocarcinoma [8, 9]. Interestingly, at least some let-7-expressing tumors escaped the tumor suppressor activity of this miRNA and grew at rates identical to controls, suggesting that cancer cells can become clonally resistant to the effects of let-7.
Our mechanistic understanding of let-7-mediated tumor suppression began with studies by Johnson et. al. which demonstrated that a C. elegans let-7 family member, miR-84, regulates let-60, the worm homolog of the human RAS oncoproteins [10]. Furthermore, let-7 directly downregulates human KRAS and NRAS expression through conserved 3′ UTR target sites. Additional insight was gained by the demonstration that let-7 regulates expression of the oncogene HMGA2. Moreover, chromosomal translocations in human cancers remove the HMGA2 3′ UTR and thus likely contribute to tumorigenesis by disrupting let-7-mediated regulation [11, 12]. Interestingly, Mayr and colleagues also noted that such translocations sometimes append the HMGA2 3′ UTR to tumor suppressor genes, suggesting that gains of let-7 regulatory function might cooperate with HMGA2 dysregulation to drive proliferation in malignancies [12]. let-7 can also negatively regulate the c-MYC proto-oncogene, providing an additional mechanism through which loss-of-function of this miRNA might contribute to tumorigenesis [14, 15]. To identify the mechanisms by which let-7 might globally regulate proliferation pathways, Johnson and colleagues overexpressed let-7 family members in liver cancer cells and noted an accumulation of cells in the G0 and G1 cell cycle stages [13]. Accordingly, microarray analysis and reporter assays identified numerous genes involved with promoting the G1 to S and G2 to M transitions including CDK6, CDC25A, and CCND2 (Cyclin D2) as direct let-7 targets [13]. Thus, compelling evidence implicates let-7 miRNAs as tumor suppressors through their activities as major regulators of pro-oncogenic pathways and cell-cycle progression.
The miR-15/16 cluster
The earliest evidence for a direct role for miRNA loss-of-function in human cancer pathogenesis came with the identification of miR-15a and miR-16-1 as long sought tumor suppressors at 13q14. Hemizygous and/or homozygous loss of 13q14 had previously been associated with numerous malignancies including chronic lymphocytic leukemia (CLL), mantle cell lymphoma, multiple myeloma, breast cancer, and high grade carcinomas of the prostate suggesting strongly that an undescribed tumor suppressor resided in this region. Using CLL patient lymphocytes to define a critical region of loss, Calin and colleagues identified the miR-15a/miR-16-1 cluster as a candidate. Indeed, miR-15 and miR-16 were down-regulated in the majority of assayed CLL samples [16]. Interestingly, a point mutation in the miR-16-1 primary transcript has been linked to diminished expression of the mature miRNA, suggesting another mechanism by which loss of function might occur [17]. These results were extended with the demonstration that BCL2, an anti-apoptotic regulator overexpressed in numerous hematopoietic malignancies including CLL, is a direct target of miR-15a and miR-16 [18]. Moreover, these miRNAs induce apoptosis and block tumorigenesis in a MEG-01 leukemia cell line xenograft model [19].
To better characterize the global regulatory effects of this miRNA cluster, Linsley et al. introduced miR-15a and miR-16 into colon carcinoma cell lines and evaluated the resulting expression changes by microarray. The downregulated transcripts were highly enriched for cell-cycle related genes, as indicated by gene ontology (GO) annotation [20]. Furthermore, enforced miR-16 expression induces G0/G1 accumulation by down regulating the expression of a number of cell-cycle related genes including CDK6, CARD10, and CDC27 [20]. Consistent with the notion that miRNAs coordinately regulate many transcripts to effect cellular changes, Calin et. al identified an overlapping but non-identical set of transcripts downregulated by miR-15/16 in MEG-01 leukemia cells that is enriched for GO terms relating to cell cycle regulation, checkpoints, and anti-apoptotic signaling [19]. Interestingly, transcripts containing AU-rich elements (AREs), mRNA destabilizing sequences found in the 3′ UTRs of many short-lived proto-oncogenes and cytokines, were also enriched in the set of downregulated genes [19]. This observation is consistent with the earlier demonstration that miR-16 is partially complementary to the core ARE sequence and is necessary for the accelerated decay of some ARE-containing transcripts [21]. These findings have uncovered a complex program of gene expression coordinated by miR-15/16 that significantly influences transit through the cell cycle.
Functional integration of miRNAs into cell-cycle control pathways
It is now apparent that miRNAs have been functionally integrated into many critical cell-cycle control pathways. As illustrated by the ensuing examples, the ability of miRNAs to fine tune these pathways significantly impacts the cell cycle in both physiologic settings as well as in disease states such as cancer where cell-cycle control is frequently subverted.
E2Fs, Myc, and Rb
Significant evidence has uncovered an important role for miRNA-mediated regulation of the E2F proteins, a family of transcription factors that function as key regulators of cell cycle progression and proliferation [22, 23]. Most E2Fs function similarly, by binding a consensus DNA sequence in conjunction with a DP family dimerization partner to modulate target gene transcription. Although all E2Fs are believed to bind the same target sequences, the family can be loosely divided into 2 groups based on function: the “activating” E2Fs (E2F1, E2F2, and E2F3) and the “repressive” E2Fs (E2F4 and E2F5). Three additional E2Fs have been identified, but their specific functions remain poorly characterized. Activating E2Fs mediate increased target gene transcription during the G1 to S transition whereas repressive E2Fs mediate target gene silencing during G0/G1. The pRB (Retinoblastoma protein), p107, and p130/Rbl2 (Retinoblastoma-like 2) “pocket proteins” confer cell-cycle dependent regulation to these E2F activities via cycles of their phosphorylation and dephosphorylation. During G0/G1, pocket proteins are hypophosphorylated and bind E2Fs to form complexes which lack transactivating function (activating E2Fs) and/or actively repress target gene transcription (repressive E2Fs). During the G1 to S transition, pocket proteins are hyperphosphorylated by cyclin–Cdk complexes and liberate E2F1, E2F2, and E2F3 to bind targets and activate the transcription of genes required for DNA synthesis and cell cycle progression. Concurrently, repressive E2Fs are shuttled to the cytoplasm owing to their nuclear export signals. Through this system, the E2Fs act as effectors of a cell-cycle dependent switch, termed the restriction point, after which cells are committed to DNA replication even in the absence of continuing proliferative signals.
Overexpression of activating E2Fs can promote either increased proliferation or, paradoxically, increased apoptosis [23]. Further, E2F1-deficient mice develop a wide variety of cancers, suggesting an in vivo tumor suppressor role for activating E2Fs. To reconcile these observations with the known functions of E2Fs in promoting proliferation, two models have been proposed. The first suggests that E2F1, E2F2 and E2F3 have unique functions and that E2F1, in particular, is most able to induce apoptotic responses [24]. The second model posits that thresholds of activating E2F activity exist via which low levels of E2F activation results in a non-proliferative state, moderate levels in proliferation, and high levels in a protective apoptotic response [25]. Although an area of continuing debate, it is clear that either mechanism depends on the sensitive modulation of E2F expression. It comes as little surprise, therefore, that miRNAs appear to be intimately involved in the regulation of E2Fs and pocket proteins.
An indication that miRNA-mediated regulation is important in controlling the activities of E2Fs came first from studies of the c-MYC proto-oncogene. c-Myc is a helix-loop-helix leucine zipper transcription factor with numerous functions and is known to be a potent transactivator of E2F1. O'Donnell and colleagues demonstrated that c-Myc activates the expression of the miR-17-92 cluster and that these miRNAs, in turn, inhibit E2F1 translation [26]. E2F1 is itself an activator of c-Myc transcription, so it appears from these data that the miR-17-92 cluster acts as a “brake” which prevents an otherwise runaway positive feedback cycle of c-Myc and E2F1 activation. Sylvestre and colleagues extended these findings by demonstrating that the miR-17-92 cluster also directly regulates E2F2 and E2F3 translation [27]. Intriguingly, work by this group and others established that the activating E2Fs, especially E2F3, can also induce expression of the miR-17-92 cluster [27, 28]. Analogously, Petrocca and colleagues demonstrated that E2F1 regulates the related miR-106b-25 cluster, revealing a parallel negative feedback circuit [29]. As E2Fs can activate their own transcription, this miRNA feedback system (Figure 1) provides a mechanism to protect cells from excess E2F activity which might otherwise culminate either in tumorigenesis or apoptotic cell death.
Figure 1. miR-17 family regulation of E2F activities.
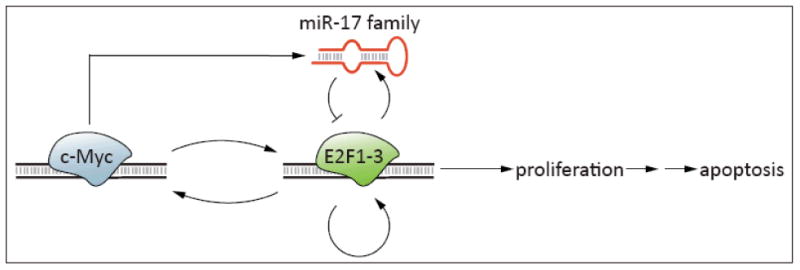
miR-17 family members are implicated in a complex regulatory circuit through which they modulate the effects of E2F transcription factors. The proto-oncoprotein c-Myc transcriptionally activates E2F1, E2F2 and E2F3 which, in turn, induce c-Myc expression. Likewise, E2Fs activate their own transcription. Unchecked, these reciprocal activations would result in a positive feedback cycle of E2F activity. Whereas moderate E2F activation results in cell proliferation, unconstrained activation can induce a protective apoptotic response. E2F signaling must therefore be tightly regulated and is held in check in part by the miR-17 family, whose members are induced both by c-Myc and by activating E2Fs.
The complexity of such regulatory mechanisms suggests that miR-17 family members might, depending on context, either promote or inhibit cellular proliferation. In the setting of low mitogenic stimulation, miR-17 might contribute to the maintenance of E2F signaling below the threshold required for proliferation. Consistent with anti-proliferative activity and a possible tumor suppressor role in some settings, allelic loss of the miR-17-92 cluster has been noted in several malignancies [30] and ectopic miR-17 expression reduces proliferation of some breast cancer cell lines [31]. By dampening abnormally high E2F activation, however, the miR-17-92 cluster might abrogate an apoptotic response and thereby function as an oncogene. Accordingly, amplification and overexpression of miR-17 family members has been noted in multiple hematopoietic and solid malignancies [32, 33]. Most available data suggest that, in vivo, the anti-apoptotic effects of miR-17 dominate and thereby endow the cluster with oncogene-like properties [34, 35]. Importantly, these effects likely involve the regulation of a complex network of targets extending well beyond the E2F family [36].
Recent evidence has also suggested that pocket proteins could be important targets of miRNA regulation. In adipocyte precursors, coordinated regulation of pocket proteins, in an event termed the “p130:p107 switch”, is critical for clonal proliferation associated with terminal differentiation. p130 is directly downregulated by miR-17 and as a consequence, upregulation of the miR-17-92 cluster promotes accelerated differentiation at this clonal expansion step [37]. As p130 also associates with repressive E2Fs to inhibit E2F activity in G0/G1, miR-17 might be capable of relieving this repression as well. Interestingly, two recent studies have extended these findings by describing miRNA regulation of another p130 function: repression of de novo DNA methyltransferases (DNMTs) in embryonic stem (ES) cells [38, 39]. In normal ES cells, miR-290 inhibits p130 expression, thereby allowing increased DNMT expression. DNMT activity, in turn, is critical for silencing of Oct4, Nanog, and other genes during differentiation, as well as for telomere homeostasis.
miRNA regulation of cyclins, Cdks and CKIs
Orderly progression through the cell cycle depends on cyclin-dependent kinase (Cdk)-mediated phosphorylation events. In early G1, D-type cyclins are synthesized and partner with Cdk4 and Cdk6. Late G1 is characterized by E-type cyclin expression and Cdk2 activity [40]. Activation of these kinases results in phosphorylation of critical regulators of cell cycle progression, such as pRb, which liberates activating E2Fs to drive S phase entry. The critical role for CDK activation in the decision to commit to cell division suggests that cells have finely tuned mechanisms by which to modulate these kinases. Indeed, in addition to the regulated synthesis and destruction of cyclins, Cdk activity can be adjusted by other mechanisms including phosphorylation and direct regulation by a class of proteins known as cyclin dependent kinase inhibitors (CKIs).
CKIs are grouped into 2 families based on homology and functional specificity. The Cip/Kip (Cdk interacting/Kinase inhibiting protein) family includes p21, p27, and p57. This family of regulators has been studied in significant detail and performs a wide variety of functions, reviewed elsewhere, in addition to cell cycle control [41]. Cip/Kip proteins function primarily by inhibiting Cdk2 activity, thereby inhibiting cell cycle progression. Regulation of Cip/Kips is complex; transcriptional, mRNA stability, translational, proteolytic, and subcellular localization mechanisms have all been implicated [40]. INK4 CKI family members (named for their ability to inhibit Cdk4) include p16, p15, p18, and p19 and function in early G1 by competing with D-type cyclins for binding to partner Cdks. In line with this biochemical role, p16 is a tumor suppressor in human malignancies and a key mediator of cellular senescence. Regulation of INK4 proteins is also complex and can occur at the transcriptional level, by differential splicing, and by modulation of transcript stability. Recently, miRNAs were identified as a new class of important regulators of both Cip/Kip and INK4 CKI proteins.
Hatfield and colleagues provided the earliest evidence for miRNA-mediated CKI regulation with the demonstration that Dicer null Drosophila melanogaster germline stem cells (GSCs) are blocked in the G1 to S transition [42]. As Dicer is required for miRNA biogenesis, this finding implied that one or more miRNAs are required for GSC progression into S phase. Intriguingly, Dicer-deficient GSCs exhibited increased expression of Dacapo, a fly ortholog of p21 and p27, suggesting that this protein is normally downregulated by miRNAs to promote cell-cycle progression. Accordingly, reduction of Dacapo expression rescues this cell-cycle arrest phenotype [42]. Using a glioblastoma (GBM) cell line, Gillies et. al. followed up on these findings and identified the human miR-221/miR-222 cluster as a direct regulator of p27 [43]. Parallel studies using distinct experimental approaches confirmed this regulatory relationship in a prostate cancer cell line, breast cancer cell line, thyroid carcinoma cell line, and primary GBM patient samples [44-46]. Enforced expression of miR-221/222 accelerated proliferation and conferred anchorage-independent growth, whereas suppression of this cluster induced G1 arrest in GBM and breast cancer cell lines. Moreover, the miR-221/miR-222 cluster is upregulated in a variety of human tumors, pointing to miR-221/222 regulation of p27 as a bona fide oncogenic pathway.
Recent data suggest that, like p27, p21 is also a target of miRNA regulation. p21 has long been known as a critical mediator of p53-induced cell cycle arrest during the G1 to S and G2 to M checkpoints. The finding, therefore, that miR-17 family members (which include miR-17, miR-20, miR-106, and miR-93) promote the G1 to S transition by targeting p21 linked a recognized oncogenic miRNA with a critical cell cycle checkpoint controller. Indeed, loss of function of the miR-17 family member miR-106b is sufficient to induce the accumulation of cells in G1 [47]. Conversely, transient transfection of this miRNA overrides DNA damage induced G1 arrest and enhances endoreduplication, a measure of failed G2/M arrest [47]. These findings were reinforced by the observation that miR-106b and miR-93 reduce Transforming Growth Factor β (TGFβ)-induced cell cycle arrest, which also requires p21 induction [29].
The INK4 family member p16 also appears to be subject to miRNA-mediated regulation, although the significance of this regulatory interaction remains unclear. Lal and colleagues demonstrated that miR-24-1, which is downregulated in senescing cells, directly represses p16 expression [48]. However, antisense inhibitors of miR-24 fail to induce senescence or cell cycle arrest and, likewise, miR-24 transfection did not accelerate proliferation. The interpretation of this result is challenging, as it likely reflects the reality that single miRNA–target interactions do not necessarily reflect global regulatory programs induced by miRNAs.
Accumulating evidence also implicates miRNAs in the control of cyclin expression. As mentioned above, let-7 regulates cyclin D2 whereas miR-34a, discussed in greater detail below, can regulate cyclin E2 expression. A third example is miR-122, an abundant hepatic miRNA which was initially the subject of much interest based on its requirement for efficient replication of hepatitis c virus [49]. Further work found miR-122 downregulated in many hepatic tumors and suggested that this miRNA might play a role in maintaining normal hepatocyte differentiation. Gramantieri and colleagues showed that miR-122a downregulates cyclin G1 expression in a hepatocellular carcinoma (HCC) derived cell line [50]. However, the interpretation of this finding is complicated by the currently poor understanding of the functions of G-type cyclins. Ccng1 deficient mice have reduced hepatic tumor formation after chemical insult [51], but the majority of available data implicate these cyclins in p53-dependent cell cycle arrest [52]. Thus, the in vivo significance of miR-122a-mediated cyclin G1 regulation remains unclear.
miRNAs further influence cell-cycle progression by controlling the expression of Cdks. Specifically, Cdk6 is a target of let-7, the miR-15/16 cluster, miR-34, and also miR-124a. Lujambio et. al. identified miR-124a in a screen to identify miRNAs whose expression in cancer cells is suppressed by CpG methylation; indeed, miR-124a is consistently epigenetically silenced in a variety of tumor types. Notably, miR-124a inhibition of Cdk6 translation detectably modulates downstream Rb phosphorylation, suggesting a functional role for this regulation [53].
miRNAs and the p53 network
p53 is a critical tumor suppressor which responds to an array of cellular stresses including DNA damage, hypoxia, and metabolic perturbations by inducing senescence, apoptosis, cell cycle arrest, or other cellular responses. Several groups recently demonstrated that the miR-34 family members miR-34a, miR-34b, and miR-34c are direct transcriptional targets of p53 [54-58]. In cell culture experiments, miR-34 induction contributes to important outcomes of p53 activation including G1 arrest, cellular senescence, and apoptosis. Moreover, the genomic locus encoding miR-34a, located at 1p36 in the human genome, is frequently deleted in diverse malignancies, suggesting that this miRNA might be a bona fide tumor suppressor. The identification of the full spectrum of targets downregulated by the miR-34 family awaits further study, but critical cell cycle effectors including Cdk4, Cdk6, cyclin E2, and E2Fs and anti-apoptotic proteins such as Bcl2 have been validated by one or more groups [54-56, 59]. Interestingly, miR-34 induction might contribute to the previously unclear p21-independent mechanism through which p53 induces G1 arrest [60]. In sum, numerous lines of evidence implicate miRNAs as major regulators of the cell cycle, and of the G1 to S transition in particular (Figure 2).
Figure 2. microRNAs that regulate the G1 to S transition.

The decision to commit to DNA replication is made at the G1 to S transition. This event depends on regulated phosphorylation of the Rb pocket protein (green), resulting in the liberation of E2Fs (green) to activate gene expression required for S phase entry. Rb phosphorylation, in turn, depends on the coordinate regulation of cyclins (blue), cyclin-dependent kinases (blue), and Cdk inhibitors (yellow). Numerous miRNAs, including those shown here, have been implicated in the direct or indirect regulation of this cellular decision. It is important to note that the depicted regulatory relationships illustrate the control of regulatory complexes by miRNAs and are not intended to depict specific miRNA-target pairs.
Regulation of miRNA function by cell cycle state
It is clear that miRNAs can regulate important components of the cell-cycle control machinery. However, exciting recent findings suggest that the converse might also be true: miRNA function might be modulated by cell-cycle state. Perhaps the simplest mechanism for such modulation would be the selective enhancement or inhibition of miRNA processing. Viswanathan and colleagues established that the mammalian RNA-binding protein Lin-28 and its homolog Lin-28B selectively inhibit the Drosha-dependent liberation of pre-let-7 miRNAs from their primary transcripts [61]. Intriguingly, evidence suggests that Lin-28B subcellular localization is regulated in a cell-cycle dependent manner. As revealed by examining a hepatocellular carcinoma cell line, Lin-28B is localized primarily to the cytoplasm during G1 but translocates to the nucleus during S phase [62]. As Drosha processing is restricted to the nucleus, regulation of Lin-28B subcellular localization provides a mechanism to link let-7 maturation to cell-cycle phase. Further investigation is necessary to examine whether let-7 biogenesis is indeed dynamically regulated during the cell-cycle and to elucidate the functional consequences of such restricted processing.
Both the stability and subcellular localization of miRNAs themselves, in select cases, are regulated in a cell-cycle dependent manner. Hwang and colleagues showed that miR-29a and miR-29b, encoded by a single primary transcript, are differentially expressed throughout the cell-cycle. miR-29b is expressed at low levels except during mitosis, whereas miR-29a is expressed constitutively. Interestingly, this regulation depends on rapid and selective miR-29b degradation in all cell-cycle phases except mitosis. Furthermore, a 6 nucleotide cis-regulatory element within miR-29b, but absent from miR-29a, directs nuclear enrichment of this miRNA during interphase [63]. These data provide the first documented example of regulated miRNA turnover and can be interpreted to suggest a yet undescribed nuclear role for miR-29b or, perhaps, an elegant mechanism which restricts its regulatory effects to M phase. A second, more speculative, mechanism by which intracellular trafficking of RNAs might confer cell cycle dependent miRNA activity is the regulated biogenesis of P-bodies (Box 1).
Perhaps the most surprising example of cell-cycle specific miRNA function comes from the recent discovery that miRNAs can upregulate translation. An initial study demonstrated that an AU-rich element (ARE) in the tumor necrosis factor alpha (TNFα) 3′ UTR can mediate translational upregulation through association with Fragile X mental retardation-related protein 1 (Fxr1) and Argonaute 2 (Ago2) proteins [64]. Moreover, this translational regulation occurred only in the setting of G0/G1 cell-cycle arrest induced by serum starvation or pharmacologic agents. As Ago2 is core RISC component and is guided to target mRNAs by miRNAs, these findings led to the hypothesis that miRNAs might also be involved in upregulating target translation during cell-cycle arrest. Vasudevan et. al. identified miR-369-3, specifically induced in serum-starved conditions, as complementary to the TNFα ARE. In reporter assays, sequence specific interaction of this miRNA with the TNFα ARE is required for cell cycle arrest-dependent translation activation [65]. Notably, this miRNA-dependent mechanism was extended to studies performed with let-7 and a synthetic miRNA. Although these observations have major implications for miRNA functions during the cell cycle, it remains to be demonstrated that a physiologic, endogenously-expressed miRNA target is upregulated under these conditions.
Concluding Remarks
In this article we have summarized our current understanding of the complex interplay between miRNAs and cell cycle control. Nevertheless, many important questions remain unanswered (Box 2). For example, though we have presented several clear examples of miRNAs regulating individual critical cell cycle effectors, many miRNAs function by modulating gene expression networks. Thus, it is likely that even the most important individual miRNA–target interactions reviewed here are single components of much larger regulatory programs. Indeed, it is in part this breadth of regulatory activity which makes miRNAs so attractive as potential therapeutic targets in human diseases such as cancer. Realizing this therapeutic potential will require a more complete understanding of the integration between miRNA-target networks and the cell cycle control circuitry.
Box 1. P bodies: a link between cell cycle state and miRNA function?
P bodies are 100 to 300nm cytoplasmic foci that were initially identified as sites of localization of factors required for mRNA decapping and 5′->3′ degradation. Further study demonstrated that P bodies also harbor proteins important in mRNA surveillance and translational repression, suggesting a more general role in post-transcriptional regulation of gene expression. In line with this function, dozens of RNA processing enzymes, RISC components, miRNAs, and miRNA targets have been localized to P bodies[66]. Downregulation of the P body component GW182 (trinucleotide repeat containing protein 6A) impairs miRNA-dependent target silencing, and substitutions within the Ago2 PAZ domain which disrupt P body localization prevent the translational inhibition of target mRNAs [67]. Based on these data and others a model has been proposed wherein miRNA-targeted mRNAs are trafficked to P bodies for accelerated turnover or sequestration from translation. However, Eulalio and colleagues demonstrated that P bodies are not required for miRNA-mediated silencing. Furthermore, P body formation itself appears to be dependent on mRNA silencing. These observations suggest that P bodies are likely to be consequences, rather than initiators, of miRNA-dependent mRNA silencing [68]. Future studies will better resolve these issues, but it is clear that P body localized factors play important roles in miRNA-mediated gene regulation.
Intriguingly, P body morphology and number vary based on cell-cycle state. As demonstrated by Yang and colleagues, P body number and size increase in G1, reach a maximum in G2, and eventually disassemble prior to mitosis. Likewise, serum starved fibroblasts contain few detectable P bodies, but serum stimulation potently increases P body size and number. This observation is especially fascinating in light of recent observations that serum starvation can induce the liberation and subsequent translation of mRNAs targeted to P bodies [69] or even initiate miRNA-dependent translational upregulation [65]. It is tempting to speculate, therefore, that programs of mRNA handling and miRNA function might be modulated by cell cycle state and that these changes are reflected in changes in P body morphology. Alternatively, P body assembly could be directly responsive to cell cycle state and thereby mediate the observed changes in miRNA functions as well as mRNA stability and translational efficiency.
Box 2. Outstanding Questions.
miRNAs can finely regulate hundreds of physiological targets to effect programs of cell action. In other cases, miRNA-mediated phenotypes appear to be due to the strong regulation of one or a few critical proteins. Given clear evidence for both these models, through which mechanism do specific miRNAs operate in regulating the cell cycle?
Since current technologies to detect global effects of miRNA regulation nearly always assess mRNA abundance rather than protein expression, it is likely that miRNA effects on translational efficiency are underappreciated. How accurate are our current models of miRNA regulatory effects given these limitations?
Dozens of miRNAs have been identified as cell cycle regulators, but far fewer have been convincingly integrated into classic cellular signaling pathways. What are the pathways and effector mechanisms, both transcriptional and post-transcriptional, that regulate miRNAs implicated in cell cycle control?
Are there specific developmental timepoints, tissues, or conditions in which miRNA-mediated regulation of the cell cycle is particularly important?
Given the great variability in signaling systems and miRNA and target mRNA expression patterns between tissues, how generalizable are results obtained in one experimental system to others?
Are there other, non-canonical, functions performed by miRNAs other than translational regulation of mRNAs?
Given the protean nature of miRNA-target networks, does modulation of miRNA function represent a viable therapeutic strategy for cancer and other human diseases characterized by loss of appropriate cell cycle control?
Acknowledgments
The Mendell laboratory receives support from the National Institutes of Health (R01 CA120185) and the Sol Goldman Pancreatic Cancer Research Center. R.R.C. is supported by the Medical Scientist Training program at Johns Hopkins and J.T.M. is a Rita Allen Foundation Scholar and a Leukemia and Lymphoma Society Scholar.
References
- 1.Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 2.Chalfie M, et al. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell. 1981;24:59–69. doi: 10.1016/0092-8674(81)90501-8. [DOI] [PubMed] [Google Scholar]
- 3.Reinhart BJ, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 4.Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Calin GA, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takamizawa J, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 7.Yanaihara N, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 8.Esquela-Kerscher A, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7 doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- 9.Kumar MS, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci U S A. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 11.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayr C, et al. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson CD, et al. The let-7 MicroRNA Represses Cell Proliferation Pathways in Human Cells. Cancer Res. 2007;67:7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 14.Kumar MS, et al. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 15.Sampson VB, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–9770. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 16.Calin GA, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calin GA, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 18.Cimmino A, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calin GA, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linsley PS, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol. 2007;27:2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jing Q, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 22.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 23.Iaquinta PJ, Lees JA. Life and death decisions by the E2F transcription factors. Curr Opin Cell Biol. 2007;19:649–657. doi: 10.1016/j.ceb.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeGregori J, et al. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziebold U, et al. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 2001;15:386–391. doi: 10.1101/gad.858801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Donnell KA, et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 27.Sylvestre Y, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007;282:2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 28.Woods K, et al. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007;282:2130–2134. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 29.Petrocca F, et al. E2F1-regulated microRNAs impair TGFβ-dependent cell cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–286. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hossain A, et al. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol. 2006;26:8191–8201. doi: 10.1128/MCB.00242-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ota A, et al. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 33.Volinia S, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsubara H, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007;26:6099–6105. doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- 35.He L, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Q, et al. miR-17-92 cluster accelerates adipocyte differentiation by negatively regulating tumor-suppressor Rb2/p130. Proc Natl Acad Sci U S A. 2008;105:2889–2894. doi: 10.1073/pnas.0800178105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinkkonen L, et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008;15:259–267. doi: 10.1038/nsmb.1391. [DOI] [PubMed] [Google Scholar]
- 39.Benetti R, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15:268–279. doi: 10.1038/nsmb.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vidal A, Koff A. Cell-cycle inhibitors: three families united by a common cause. Gene. 2000;247:1–15. doi: 10.1016/s0378-1119(00)00092-5. [DOI] [PubMed] [Google Scholar]
- 41.Besson A, et al. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 42.Hatfield SD, et al. Stem cell division is regulated by the microRNA pathway. Nature. 2005;435:974–978. doi: 10.1038/nature03816. [DOI] [PubMed] [Google Scholar]
- 43.Gillies JK, Lorimer IA. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle. 2007;6:2005–2009. doi: 10.4161/cc.6.16.4526. [DOI] [PubMed] [Google Scholar]
- 44.Galardi S, et al. miR-221 and miR-222 Expression Affects the Proliferation Potential of Human Prostate Carcinoma Cell Lines by Targeting p27Kip1. J Biol Chem. 2007;282:23716–23724. doi: 10.1074/jbc.M701805200. [DOI] [PubMed] [Google Scholar]
- 45.le Sage C, et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. Embo J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Visone R, et al. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer. 2007;14:791–798. doi: 10.1677/ERC-07-0129. [DOI] [PubMed] [Google Scholar]
- 47.Ivanovska I, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. 2008;28:2167–2174. doi: 10.1128/MCB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lal A, et al. p16(INK4a) translation suppressed by miR-24. PLoS ONE. 2008;3:e1864. doi: 10.1371/journal.pone.0001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jopling CL, et al. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 50.Gramantieri L, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67:6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 51.Jensen MR, et al. Reduced hepatic tumor incidence in cyclin G1-deficient mice. Hepatology. 2003;37:862–870. doi: 10.1053/jhep.2003.50137. [DOI] [PubMed] [Google Scholar]
- 52.Zhao L, et al. Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways. Mol Cancer Res. 2003;1:195–206. [PubMed] [Google Scholar]
- 53.Lujambio A, Esteller M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle. 2007;6:1455–1459. [PubMed] [Google Scholar]
- 54.Bommer GT, et al. p53-Mediated Activation of miRNA34 Candidate Tumor-Suppressor Genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 55.Chang TC, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He L, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raver-Shapira N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 58.Tarasov V, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 59.Tazawa H, et al. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A. 2007;104:15472–15477. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He L, et al. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viswanathan SR, et al. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo Y, et al. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene. 2006;384:51–61. doi: 10.1016/j.gene.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 63.Hwang HW, et al. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100. doi: 10.1126/science.1136235. [DOI] [PubMed] [Google Scholar]
- 64.Vasudevan S, Steitz JA. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 2007;128:1105–1118. doi: 10.1016/j.cell.2007.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vasudevan S, et al. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318:1931–1934. doi: 10.1126/science.1149460. [DOI] [PubMed] [Google Scholar]
- 66.Eulalio A, et al. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007;8:9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- 67.Liu J, et al. A role for the P-body component GW182 in microRNA function. Nat Cell Biol. 2005;7:1261–1266. doi: 10.1038/ncb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eulalio A, et al. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol. 2007;27:3970–3981. doi: 10.1128/MCB.00128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhattacharyya SN, et al. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]