

Summary
EGF induces signal transduction between EGFR and FAK, and FAK is required for EGF-induced cell migration. It is unknown, however, what factor mediates the interaction between EGFR and FAK and leads to EGF-induced FAK phosphorylation. Here we identify SRC-3Δ4, a splicing isoform of the SRC-3 oncogene, as a signaling adaptor that links EGFR and FAK and promotes EGF-induced phosphorylations of FAK and c-Src. We identify three PAK1-mediated phosphorylations in SRC-3Δ4 that promote the localization of SRC-3Δ4 to the plasma membrane and mediate the interactions with EGFR and FAK. Importantly, over-expression of SRC-3Δ4 promotes MDA-MB231-induced breast tumor metastasis. Our findings identify phosphorylated SRC-3Δ4 as a missing adaptor between EGFR and its downstream signaling molecule FAK, to coordinately regulate EGF-induced cell migration. Our study also reveals the new concept that a nuclear receptor coactivator can act in the periphery of a cell to directly mediate activation of an enzyme.
Keywords: SRC-3Δ4, EGF, EGFR, FAK, PAK1, phosphorylation, cell migration, metastasis
Introduction
Cell migration is an essential event in cell growth and cancer metastasis (Friedl and Wolf, 2003). Epidermal growth factor (EGF), a ligand of the EGF receptor (EGFR) proto-oncogene (Di Fiore et al., 1987), promotes cancer cell migration and metastasis by activating multiple downstream protein kinases, such as c-Src (Goi et al., 2000), p21-activted kinase 1 (PAK1) (Bokoch 2003), and focal adhesion kinase (FAK) (Sieg et al., 2000). PAK1 is activated by recruitment to EGFR through the adaptor protein Nck1 in response to EGF signaling (Galisteo et al., 1996). FAK, a non-receptor tyrosine kinase, is activated and auto-phosphorylated at Y397 by binding to integrin complexes (Schlaepfer and Mitra, 2004) (Fig. S1A). c-Src kinase next is recruited to the FAK complex by binding at phosphorylated Y397 through its SH2 domain (Eide et al., 1995); c-Src then phosphorylates FAK at multiple tyrosine sites including Y925 (Calalb et al., 1995). Phosphorylation at Y925 modifies the interaction of FAK with its associating partners, including Grb2 and paxillin, and is critical for FAK promotion of cell migration (Schlaepfer et al., 1994; Westhoff et al., 2004), tumor angiogenesis (Mitra et al., 2006), and tumor metastasis (Kaneda et al., 2008). Thus, FAK acts as a key integrator of the growth–factor pathway and the integrin signaling pathway to regulate cell motility (Sieg et al., 2000). Although EGF stimulates signal transduction between EGFR and FAK, no evidence for a direct interaction of these two proteins has been observed and it is unclear as to what factor(s) mediates the complex formation between EGFR and FAK and EGF-induced FAK phosphorylation. c-Src once was proposed to be the potential mediator since it interacts with both EGFR (Maa et al., 1995) and FAK (Eide et al., 1995). However, the fact that Y397 is not required for the interaction between EGFR and FAK excludes this possibility, since Y397 is the c-Src binding site on FAK (Eide et al., 1995). Therefore, a key molecule mediating EGF signal transduction from EGFR to FAK to facilitate a potent role in cell migration and metastasis remains undefined.
The steroid receptor coactivator 3 (SRC-3/AIB1) plays critical roles in promoting cancer cell proliferation, invasion, and metastasis (Torres-Arzayus et al., 2004; Zhou et al., 2005; Qin et al., 2008). A SRC-3 splice isoform with a deletion of exon 4 (SRC-3Δ4) was identified and found to be overexpressed in breast cancer cells and tumors (Reiter et al., 2001). In comparison with full-length SRC-3 protein (FL-SRC-3, Fig. S1B), SRC-3Δ4 protein lacks the N-terminal bHLH domain that contains a nuclear localization signal (NLS) (Li et al., 2007). Although it has been shown to coactivate nuclear receptors in transient transfection assays, the physiological and pathological functions of SRC-3Δ4 are unknown (Reiter et al., 2001; Reiter et al., 2003). Interestingly, two recent studies suggested that SRC-3 coactivator can modulate FAK intracellular localization and enhance its activation (Yoshida et al., 2005; Yan et al., 2008), but the underlying mechanisms have not been elucidated.
In this study, we identified SRC-3Δ4 as the missing adaptor protein that bridges the interaction between EGFR and FAK upon EGF stimulation. PAK1 kinase promotes SRC-3Δ4 membrane localization and its direct interaction with FAK and EGFR by phosphorylating SRC-3Δ4 on specific serine/threonine residues. Furthermore, knockdown of SRC-3Δ4 expression significantly decreases EGF-induced c-Src activation, FAK phosphorylation at Y925, and cell migration. In contrast, overexpression of SRC-3Δ4 promotes MDA-MB231 cell migration and MDAMB-231-induced breast tumor metastasis to the lymph node and lung.
Results
SRC-3Δ4 localizes in the lamellipodia of MDA-MB231 cells geographically with FAK
SRC-3 has been shown to alter FAK intracellular localization and enhance its activation (Yoshida et al., 2005; Yan et al., 2008), but the underlying mechanisms remain to be defined. As a first step in unraveling the underlying mechanism of SRC-3's function in this process, we examined the cellular localization of endogenous SRC-3 protein by immunofluorescent labeling using an antibody that recognizes both FL-SRC-3 and SRC-3Δ4 proteins. Interestingly, in MDA-MB231 breast cancer cells, we found that in addition to nuclear and cytosolic staining, SRC-3 localizes in the lamellipodia, the dynamic membrane structures at the leading edge of motile cells (Fig. S2A). SRC-3 distribution pattern in the lamellipodia closely correlates geographically with that of FAK and these two proteins have partial ‘compartmental localization’. Given that SRC-3Δ4 lacks a NLS signal and FL-SRC-3 localizes primarily in the nucleus (Amazit et al., 2007), we postulated that this splicing isoform could be the one that localizes to the lammellipodia. To test this, we generated a siRNA that specifically knocks down SRC-3Δ4 mRNA and protein by targeting the sequence spanning exon 3 and exon 5 (Fig. S1B and Fig. S2B). SRC-3Δ4 siRNA treatment dramatically decreased the immunostaining signal of SRC-3 protein in the lamellipodia but had no obvious effect on the nuclear signal (Fig. S2C), supporting that SRC-3Δ4 protein is the form localizing in the lamellipodia. In contrast, cells treated with SRC-3siRNA that targets both FL-SRC-3 and SRC-3Δ4 mRNAs have a globally decreased immunostaining signal of SRC-3 in the cell (Fig. S2D). In addition, exogenously expressed SRC-3Δ4 protein also localizes in the lamellipodia geographically with FAK. (Fig. S2E and 2G).
SRC-3Δ4 directly interacts with FAK through the RID region
The similar distribution pattern and partial ‘compartmental localization’ of SRC-3Δ4 and FAK in the lamellipodia suggests that these two proteins could function with each other. We confirmed an interaction between SRC-3Δ4 and FAK by reciprocal co-immunoprecipitation (co-IP) experiments using either a SRC-3 antibody or FAK antibody. While IP of total SRC-3 from MDA-MB231 cell extracts co-precipitated both FAK and PAK1 (Fig. 1A), SRC-3Δ4, but not FL-SRC-3, was shown to interact with FAK (Fig. 1B). To determine which region of FAK was required for the interaction with SRC-3Δ4, we co-expressed SRC-3Δ4 in 293T cells with either full-length FAK, the N-terminus of FAK, or the C-terminus of FAK and performed co-IP. The FERM and FAT domains of FAK are the two major binding regions for FAK-associated partners (Schlaepfer and Mitra, 2004). We found that the N-terminal region containing the FERM domain, but not the C-terminal region containing the FAT domain, interacts with SRC-3Δ4 (Fig. 1C). Consistent with the result of co-IP of endogenous proteins in MDA-MB231 cells (Fig. 1B), the interaction of FAK with SRC-3Δ4 is much stronger than that with FL-SRC-3 under exogenous overexpression conditions in 293T cells (Fig. 1C). In comparison with FL-SRC-3 protein, SRC-3Δ4 lacks the N-terminal bHLH region that contains a nuclear localization signal (NLS) (Fig. S1B). Therefore, the weaker ability of FL-SRC-3 to interact with FAK could be due to the predominant nuclear localization of FL-SRC-3 and /or the presence of the N-terminal bHLH region. To clarify this, we examined the interaction of FAK with FL-SRC-3K17A/R18A mutant that is localized in the cytoplasm due to K17A/R18A mutations in the NLS (Li et al., 2007). As shown in Fig. S3, in comparison with SRC-3Δ4, FL-SRC-3K17A/R18A also has much weaker interaction with FAK. This result indicates that the N-terminal bHLH region has inhibitory effect on the interaction of SRC-3 with FAK. To identify which region in SRC-3Δ4 is important for its interaction with FAK, an in vitro pull down assay was performed by incubating purified FAK protein immobilized on beads with GST-SRC-3Δ4 fragment fusion proteins. As shown in Fig. 1D, the RID region of SRC-3Δ4 interacts with FAK. Consistent with this, deletion of the RID region abolished the interaction of SRC-3Δ4 with FAK and fusion of the RID region with GFP conferred the ability of GFP-RID fusion protein to interact with FAK (Fig. 1E). Taken together, these results demonstrate that SRC-3Δ4 directly interacts with the FAK FERM domain through the RID region.
Figure 1.
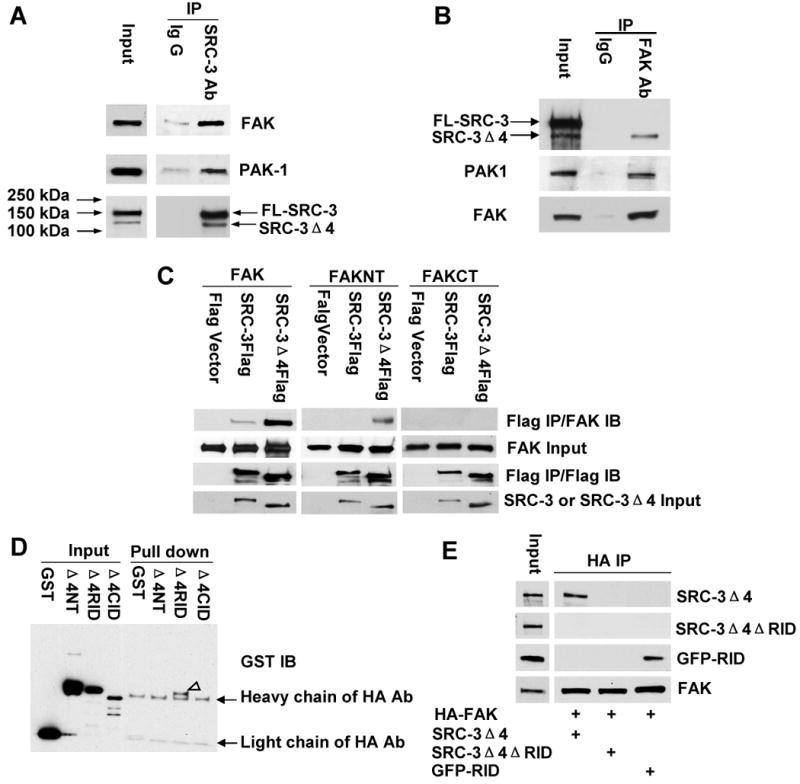
SRC-3Δ4 directly interacts with FAK through the RID region. The interaction among endogenous SRC-3, FAK, and PAK1 protein in MDA-MB231 cells was analyzed by co-immunoprecipitation (co-IP) using a SRC-3 Ab (A), a FAK Ab (B), or the corresponding control IgG, followed by Western blotting. (C). 293T cells were transfected with either a plasmid encoding full-length SRC-3 (SRC-3Flag), SRC-3Δ4Flag, or the Flag empty vector, together with either full-length FAK, the N-terminus of FAK (FAKNT, 1-402), or the C-terminus of FAK (FAKCT). IP was performed using anti-Flag conjugated agarose beads. (D). HA-FAK protein was expressed and purified using anti-HA beads. The N-terminal region of SRC-Δ4 (Δ4NT, aa 1-250), receptor interaction domain-containing region (Δ4RID, aa 450-700), and CBP interacting domain-containing region (Δ4CID, aa 701-921) were expressed as GST-fusion proteins in bacteria. Protein-protein pull down assay was performed by incubating each purified GST-SRC-Δ4 fragment fusion protein with HA-FAK immobilized on agarose beads. Western blot was probed with a GST antibody. Arrowhead indicates GST-Δ4RID protein pulled down by HA-FAK. (E). 293T cells were co-transfected with HA-FAK and one of the following constructs: SRC-3Δ4, SRC-3Δ4 with the RID region deleted (SRC-3Δ4ΔRID), or the RID region fused with GFP (GFP-RID). IP was done using a HA antibody. SRC-3Δ4 proteins were detected with either a Flag antibody (for SRC-3Δ4 and SRC-3Δ4ΔRID) or a GFP antibody (for GFP-RID).
SRC-3Δ4 promotes cell migration in a FAK-dependent manner
FAK is a key factor localizing in the lamellipodia and focal adhesions and promoting cell migration (Schlaepfer and Mitra, 2004). Localization of SRC-3Δ4 in the lamellipodia and the direct binding between SRC-3Δ4 and FAK imply that SRC-3Δ4 may play a role in cell migration by regulating FAK activity. Indeed, knockdown of SRC-3Δ4 significantly inhibits MDA-MB231 cell migration, which was demonstrated in both wound-healing (Fig. S4A) and transwell cell migration assays (Fig. 2A). In contrast, reintroduction of SRC-3Δ4 into SRC-3-null MEF cells significantly enhanced migration (Fig. 2B). Overexpression of SRC-3Δ4 significantly increased EGF-stimulated migration of HeLa cells, whereas knockdown of FAK kinase impaired the effect of SRC-3Δ4 (Fig. 2C). On the contrary, overexpressoion of FL-SRC-3 was incapable of significantly increasing EGF-stimulated migration of HeLa cells (Fig. S4B). As described above, the RID region of SRC-3Δ4 is required for interaction with FAK. Accordingly, deletion of the RID region abolished the ability of SRC-3Δ4 to enhance EGF-stimulated HeLa cell migration (Fig. 2D). In line with this, overexpression of GFP-RID significantly decreases EGF-induced MDA-MB231 cell migration (Fig. 2A). Taken together, our results demonstrate that SRC-3Δ4 promotes cell migration in a FAK-dependent manner.
Figure 2.
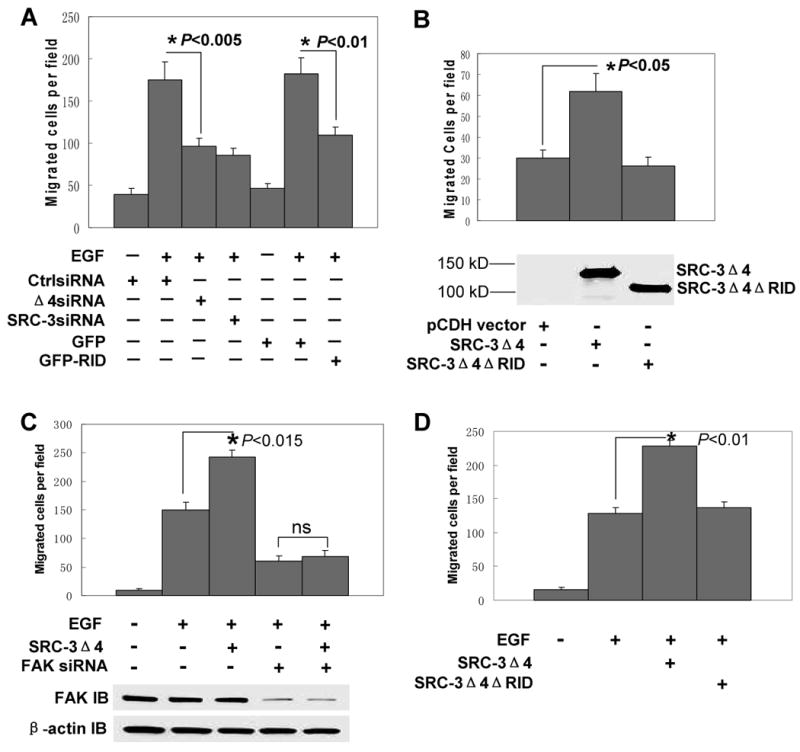
SRC-3Δ4 promotes cell migration in a FAK-dependent manner. (A). SRC-3Δ4 is important for EGF-induced MDA-MB231 cell migration. Transwell cell migration assay was performed using MDA-MB231 cells transfected with either CtrlsiRNA, SRC-3Δ4 siRNA, or SRC-3 siRNA targeting both FL-SRC-3 and SRC-3Δ4 mRNAs, or transfected with GFP-RID or GFP vector. 48 hrs post-transfection, cells were serum-starved overnight. EGF (50 ng/ml) was added to the media in bottom chamber during migration process. Values are means ± s.e of four separate experiments. “*” indicates significant difference (Student's t test). (B). Transwell cell migration assay in SRC-3-null MEF cells infected with lentiviruses expressing either pCDHSRC-3Δ4Flag (SRC-3Δ4), pCDHSRC-3Δ4ΔRIDFlag (SRC-3Δ4ΔRID), or the control lentiviral vector (pCDH vector). Values are means ± s.e. of three separate experiments. The expression of SRC-3Δ4 was analyzed by Western blotting using a Flag antibody. (C). Transwell cell migration assay in HeLa cells transfected with SRC-3Δ4 plasmid or together with FAK siRNA (25nM). 24 hrs post-transfection, cells were serum-starved overnight. EGF (50 ng/ml) was added to the media in bottom chamber during migration process. Values are means ± s.e of four separate experiments. “*” indicates significant difference. ns: no significance. Knockdown of FAK protein was determined by Western blotting using a FAK antibody. β-actin was used as a loading control. (D). HeLa cells were transfected with either SRC-3Δ4 or SRC-3Δ4ΔRID. Cell migration assay was performed as described in (C). Values are means ± s.e of four separate experiments.
SRC-3Δ4 mediates the interaction between EGFR and FAK and augments EGF-stimulated FAK and c-Src kinase phosphorylations
EGF activates c-Src and induces FAK phosphorylation on a functionally essential residue Y925; both events are critical for EGF-induced cell migration (Fincham and Frame, 1998; Meierjohann et al., 2006). Although EGF stimulates complex formation between EGFR and FAK, no direct interaction of these two proteins has been observed (Sieg et al., 2000). It was unclear what factor mediates the interaction between EGFR and FAK and FAK phosphorylation induced by EGF. Based on our observation that SRC-3Δ4 directly interacts with FAK and that SRC-3Δ4 promotes EGF-induced cell migration in a FAK-dependent manner, we asked whether SRC-3Δ4 functions as the mediator of EGF signal transduction to FAK. We first tested whether SRC-3Δ4 interacts with EGFR. As shown in Fig. 3A and 3B, SRC-3Δ4 interacted with EGFR and EGF stimulated SRC-3Δ4's interaction with EGFR, FAK, and PAK1. To determine whether SRC-3Δ4 directly interacts with EGFR and which region in SRC-3Δ4 is important for its interaction with EGFR, an in vitro pull down assay was performed by incubating purified EGFR protein immobilized on beads with GST-SRC-3Δ4 fragment fusion proteins. As shown in Fig.3C, the N-terminus of SRC-3Δ4 (Δ4NT) interacts with EGFR. Importantly, knockdown of SRC-3Δ4 in MDA-MB231 cells hindered EGF-induced interaction between EGFR and FAK (Fig. 3D), and greatly decreased EGF-stimulated FAK phosphorylation on Y925 (Fig. 3E). In addition, knockdown of SRC-3Δ4 decreased EGF-stimulated c-Src and downstream ERK1/2 phosphorylations, albeit to a lesser degree than that of FAK phosphorylation on Y925 (Fig. 3E). Unlike Y925, FAK phosphorylation on Y397 was not affected by either EGF stimulation or SRC-3Δ4 knockdown. In the absence of EGF stimulation, SRC-3Δ4 was incapable of promoting migration of serum-starved MDA-MB231 cells, and inhibition of EGFR by AG1478 eliminated the effect of SRC-3Δ4 promoting EGF-induced cell migration (Fig. S4C). To test whether the potential transcriptional co-activator function of SRC-3Δ4 is involved in cell migration, the effect of a SRC-3Δ4 transcriptionally inactive mutant with the deletion of the p300/CBP-interacting domain (CID, aa 833-899) on EGF-stimulated cell migration was determined. As expected, SRC-3Δ4ΔCID lost the transcriptional co-activity on ERα-regulated ERE-luciferase gene expression (Fig. S4D). However, it retained the capability of significantly increasing EGF-stimulated migration of HeLa cells (Fig. S4E). These results substantiate that SRC-3Δ4 plays a key role in mediating the EGF-induced interaction between EGFR and FAK, and in EGF signal transduction from EGFR to FAK to promote cell migration. The results also indicate that the primary mediation is exerted at the cell membrane rather than in the nucleus.
Figure 3.
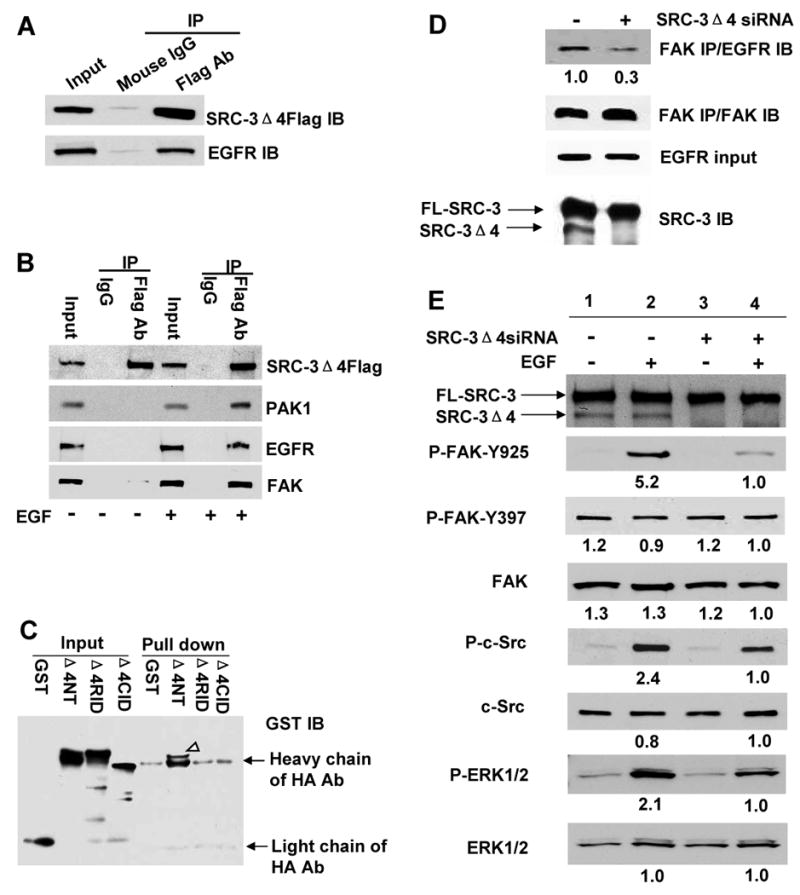
SRC-3Δ4 mediates EGF-induced interaction between EGFR and FAK and EGF-stimulated FAK and c-Src phosphorylations. (A). SRC-3Δ4 interacts with EGFR. SRC-3Δ4Flag was expressed in MDA-MB231 cells. The interaction between SRC-3Δ4 and endogenous EGFR was analyzed by co-IP using either a Flag antibody (Flag Ab) or control mouse IgG, followed by Western blotting. (B). EGF stimulates the interaction of SRC-3Δ4 with PAK1, EGFR, and FAK. HeLa cells were transfected with SRC-3Δ4Flag. Serum-starved cells were stimulated with either EGF (25 ng/ml) or vehicle for 15 min. IP was performed using either a Flag Ab or normal mouse IgG. (C). The N-terminus of SRC-3Δ4 (Δ4NT) interacts with EGFR. HA-EGFR protein was expressed and purified using anti-HA beads. Protein-protein pull down assay was performed by incubating each purified GST-SRC-3Δ4 fragment fusion protein with HA-EGFR immobilized on agarose beads, following the procedures as described in Fig. 1D. Western blot was probed with a GST antibody. Arrowhead indicates GST-Δ4NT protein pulled down by HA-EGFR. (D). Knockdown of SRC-3Δ4 in MDA-MB231 cells hinders EGF-induced interaction between EGFR and FAK. MDA-MB231 cells were transfected with SRC-3Δ4 siRNA or the silencer negative control. Two days post-transfection, cells were serum-starved for 24 hrs. Cells were then stimulated with EGF (25 ng/ml) for 15 min. IP was performed using a FAK Ab. Numbers below the Western blots in top panel represent the relative intensity of the protein bands quantitated by NIH Image J software. (E). Knockdown of SRC-3Δ4 in MDA-MB231 cells decreases EGF-stimulated c-Src phosphorylation, FAK phosphorylation on Y925, and downstream ERK1/2 phosphorylations. MDA-MB231 cells were treated with SRC-3Δ4 siRNA and stimulated with EGF as described in (D). EGF-induced phosphorylations of FAK, c-Src, and ERK1/2 were analyzed by Western blotting using each phospho-specific antibody described in the section of experimental procedures. Numbers below the Western blots represent the relative intensity of the protein bands. The band intensity in Lane 4 of each blot is set as “1.0”.
PAK1 promotes SRC-3Δ4 localization to the filopodia, its interaction with EGFR, and its function in cell migration
PAK1 is a kinase known to be activated by EGF and to play an important role in cell migration (Bokoch 2003). PAK1 is activated by binding to p21-GTPase including cdc42 and Rac-1. As described above, SRC-3Δ4 was found to interact with PAK1 (Fig. 1A) and this interaction was stimulated by EGF (Fig. 3B). We therefore asked whether PAK1 regulates SRC-3Δ4 plasma membrane localization and its function in cell migration. Exogenously-expressed SRC-3Δ4 primarily localized in the cytoplasm of HeLa cells (the left image, Fig. S5A), which is consistent with the lack of a NLS. Interestingly, activation of PAK1 by co-expression of cdc42 stimulated SRC-3Δ4 localization to the tip of the filopodia in HeLa cells (the middle image, Fig. S5A). In addition, constitutively-active PAK1 (CAPAK1) promoted the interaction between SRC-3Δ4 and EGFR, whereas expression of a PAK1 kinase-dead dominant-negative mutant (PAK1KD) (Tang et al., 1997) inhibited this interaction (Fig. S5B). Also, the PAK1KD inhibited the ability of SRC-3Δ4 to promote EGF-induced cell migration in HeLa cells (Fig. 4A), suggesting that PAK1 kinase activity is required for SRC-3Δ4's function in cell migration. As expected and similar to the effect of SRC-3Δ4 knockdown (Fig. 3E), expression of PAK1KD greatly decreased EGF-stimulated FAK phosphorylation at Y925 (Fig. S6).
Figure 4.
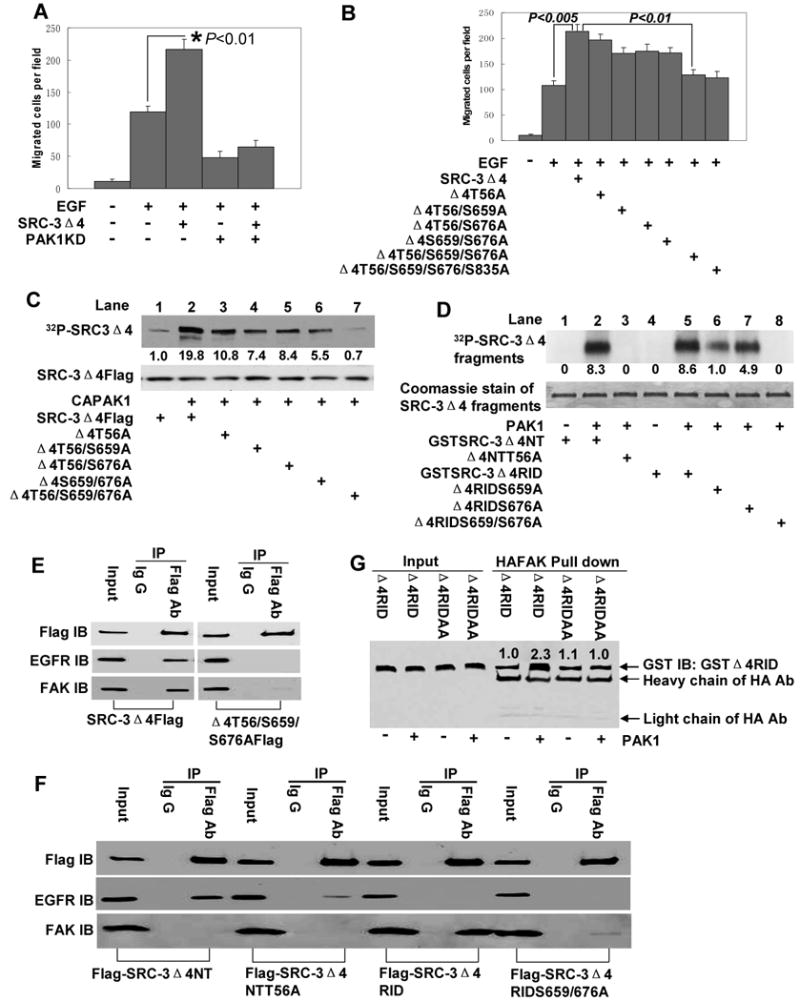
SRC-3Δ4 phosphorylation by PAK1 is critical for its function in cell migration by regulating the interaction of SRC-3Δ4 with EGFR and FAK (A). HeLa cells were transfected with either SRC-3Δ4 or PAK1KD, or both. Transwell cell migration assay was performed as described in Fig. 2C. Values are means ± s.e of four separate experiments. (B). Simultaneous mutations of T56, S659, and S676 to alanines significantly inhibits the function of SRC-3Δ4 in promoting EGF-induced HeLa cell migration. HeLa cells were transfected with SRC-3Δ4 or SRC-3Δ4 mutants with the potential PAK1 phosphorylation residue(s) replaced by alanine(s) as indicated in the figure. The effect of the mutation(s) on the function of SRC-3Δ4 in EGF-stimulated cell migration was determined by Transwell cell migration assay. Values are means ± s.e of four separate experiments. (C). Phosphorylation of SRC-3Δ4 by PAK1 in 293T cells. 293T cells were transfected with SRC-3Δ4Flag alone, or co-transfected with CAPAK1 and SRC-3Δ4Flag or its mutants with the potential PAK1 phosphorylation residue(s) replaced by alanine(s) as indicated in the figure. Cells were metabolically labeled with 32P-orthophosphate. SRC-3Δ4 proteins were immunoprecipitated using anti-Flag agarose beads. The phosphorylations of SRC-3Δ4 and its mutants were determined by autoradiography (upper panel) and the immunoprecipitated levels of SRC-3Δ4 and its mutants were determined by Western blotting using a Flag antibody (lower panel). Numbers below the upper panel represent the relative intensity of phosphorylation signals by setting the band intensity in Lane 1 as “1.0”. (D). In vitro phosphorylation of SRC-3Δ4 by PAK1 on T56, S659, S676. Purified GSTSRC-3Δ4 fragment proteins (Δ4NT, Δ4RID, and their mutants with PAK1 phosphorylation site(s) changed to alanine(s)) served as substrates of PAK1 kianse. Numbers below the upper panel represent the relative intensity of phosphorylation signals by setting the band intensity in Lane 6 as “1.0”. (E). Simultaneous mutations of T56, S659, and S676 to alanines almost abolish the interaction of SRC-3Δ4 with FAK and EGFR. HeLa cells were co-transfected with CAPAK1 and SRC-3Δ4Flag or its phosphorylation mutant as indicated. The interaction of SRC-3Δ4 with EGFR and FAK was determined by co-IP using a Flag Ab or the control IgG, followed by Western blotting. (F). SRC-3Δ4 interacts with EGFR through its N-terminus in which T56 plays essential role, and with FAK through its RID region in which S659 and S676 are critical. HeLa cells were co-transfected with CAPAK1 and Flag-SRC-3Δ4NT or Flag-SRC-3Δ4RID, or their corresponding phosphorylation mutants as indicated. Co-IP was performed using either Flag Ab or the control IgG. (G). Phosphorylation of SRC-3Δ4RID by PAK1 enhances the interaction of SRC-3Δ4RID with FAK in vitro. In vitro phosphosrylation was first performed by incubating active PAK1 kinase with purified GST-SRC-3Δ4RID or its mutant with S659 and S676 changed to alanines (Δ4RIDAA). Protein-protein pull down assay was then carried out by incubating the samples with HA-FAK protein immobilized on agarose beads. Western blot was probed with a GST antibody.
SRC-3Δ4 phosphorylation by PAK1 is critical for its function in cell migration by regulating the interaction of SRC-3Δ4 with EGFR and FAK
Based on the prediction by NetPhosK, a kinase-specific phosphorylation prediction program, there are four potential PAK1 phosphorylation sites in the SRC-3Δ4 protein: T56, S659, S676, and S835. Interestingly, although single-mutation of T56A or double-mutation of any two of these residues to alanines had no significant effect, simultaneous mutations of T56, S659, and S676 to alanines virtually eliminated the capacity of SRC-3Δ4 to promote EGF-induced HeLa cell migration (Fig. 4B). To determine whether these three residues are truly PAK1 phopshorylation sites, we performed PAK1 phosphorylation assays in both 293T cells and a cell free system. As shown in Fig. 4C, CAPAK1 dramatically induced SRC-3Δ4 phosphorylation (compare lane 2 with lane 1). Simultaneous mutation of T56A, S659A, and S676A completely abolished PAK1-induced SRC-3Δ4 phosphorylation (Fig. 4C, Lane 7), whereas a single-mutation of T56A or double-mutations of any two of these three residues to alanines only partially decreased PAK1-induced SRC-3Δ4 phosphorylation. To prove these residues are direct phosphorylation sites for PAK1, purified GST-fusion proteins of an N-terminal SRC-3Δ4 fragment containing T56 or the RID region containing S659 and S676 were used as substrates for an in vitro PAK1 kinase assay. As shown in Fig. 4D, T56A mutation completely abolished SRC-3Δ4NT phosphorlyation by PAK1 (compare Lane 3 with lane 2). Single mutation of either S659A or S676A partially decreased SRC-3Δ4RID phosphorylation, whereas double mutations of S659A and S676A completely abolished its phosphorlyaiton by PAK1. Taken together, these results substantiate that T56, S659, and S676 are direct PAK1 phosphorylation sites in SRC-3Δ4.
We then asked how these phosphorylation sites regulate the function of SRC-3Δ4 in cell migration. Since active PAK1 stimulates SRC-3Δ4 filopodia localization and the interaction of SRC-3Δ4 with EGFR (Fig. S5), we hypothesized that some combination of these phosphorylation sites are important for specific interactions of SRC-3Δ4 with both EGFR and FAK. Simultaneous mutations of T56A, S659A, and S676A abolished the interaction of SRC-3Δ4 with EGFR and FAK (Fig. 4E). As previously stated, the N-terminus and the RID region of SRC-3Δ4 interact with EGFR (Fig. 3C) and FAK (Fig. 1D and Fig.1E), respectively; S659 and S676 are located within the RID region, whereas T56 is located in the N-terminus of SRC-3Δ4. We postulated that SRC-3Δ4 interacts with EGFR through its N-terminus, in which T56 plays an important role, and with FAK through its RID region, in which S659 and S676 are critical. Indeed, this was the case. Fig.4F demonstrates that the N-terminus of SRC-3Δ4 (SRC-3Δ4NT) interacts with EGFR but not with FAK, and a T56A mutation reduced this interaction. Similarly, the RID region of SRC-3Δ4 (SRC-3Δ4RID) interacts with FAK but not with EGFR, and this interaction was greatly decreased by mutations of S659 and S676 to alanines. The importance of phosphorylations at S659 and S676 for the interaction of SRC-3Δ4RID with FAK was further supported by the fact that PAK1 kinase significantly enhanced the interaction of SRC-3Δ4RID with FAK in vitro, and this effect was lost when S659 and S676 were mutated to alanines in SRC-3Δ4RID (Δ4RIDAA, Fig. 4G). In addition, simultaneous mutations of T56A, S659A, and S676A eliminated the ability of SRC-3Δ4 to localize to the lamellipodia in MDA-MB231 cells (Fig. S2F). Taken together, these results clearly demonstrate that phosphorylations of SRC-3Δ4 by PAK1 are critical for its plasma membrane localization, its interaction with EGFR and FAK, and its function in promoting cell migration.
SRC-3Δ4 promotes mammary tumor metastasis to the lymph node and lung
Since cell migration is an essential event in cancer metastasis, we next determined the potential role of SRC-3Δ4 in metastasis. We generated an MDA-MB231 cell line stably over-expressing SRC-3Δ4 using lentiviral transduction (Fig. 5A). Overexpression of SRC-3Δ4 greatly enhanced EGF-stimulated FAK phosphorylaiton at Y925 and significantly increased MDA-MB231 cell migration and invasion (Fig. 5B and 5C), but did not affect cell growth (Fig. 5D, Top panel). We then investigated the function of SRC-3Δ4 in tumor metastasis using a mouse model capable of spontaneous metastasis. MDA-MB231 cells were orthotopically injected into the No.4 inguinal mammary fat pad of immuno-deficient nude mice. Overexpression of SRC-3Δ4 had no effect on MDA-MB231-induced mammary gland tumor growth (Fig. 5D, Lower panel). However, consistent with its important role in promoting cell migration and invasion, SRC-3Δ4 promoted mammary tumor metastasis to the lymph node (Fig. 5E) and lung (Fig. S7). As summarized in Table 1, SRC-3Δ4 did not influence primary tumor incidence, but increased thoracic lymph node metastasis. Thoracic lymph node metastasis occurred in 100% (9 out of 9) of the mice that acquired tumors in the SRC-3Δ4 group, as compared to 55% (5 out of 9) in the vector control group. The average areas of metastasis to lymph nodes in the SRC-3Δ4 group were significantly higher than that in the vector control group (Fig. 5E and 5F). Twelve weeks post-cell injection, 3 out of 9 mice in the SRC-3Δ4 group had detectable microscopic lung metastasis, whereas no lung metastasis was found in the vector control group (Fig. S7 and Table 1). The mammary origins of the tumors formed in the lymph nodes and lung were verified by immunohistological staining of breast-specific markers mammaglobin A (MGA, Fig. 5E) and CK19 (Fig. S7), respectively.
Figure 5.
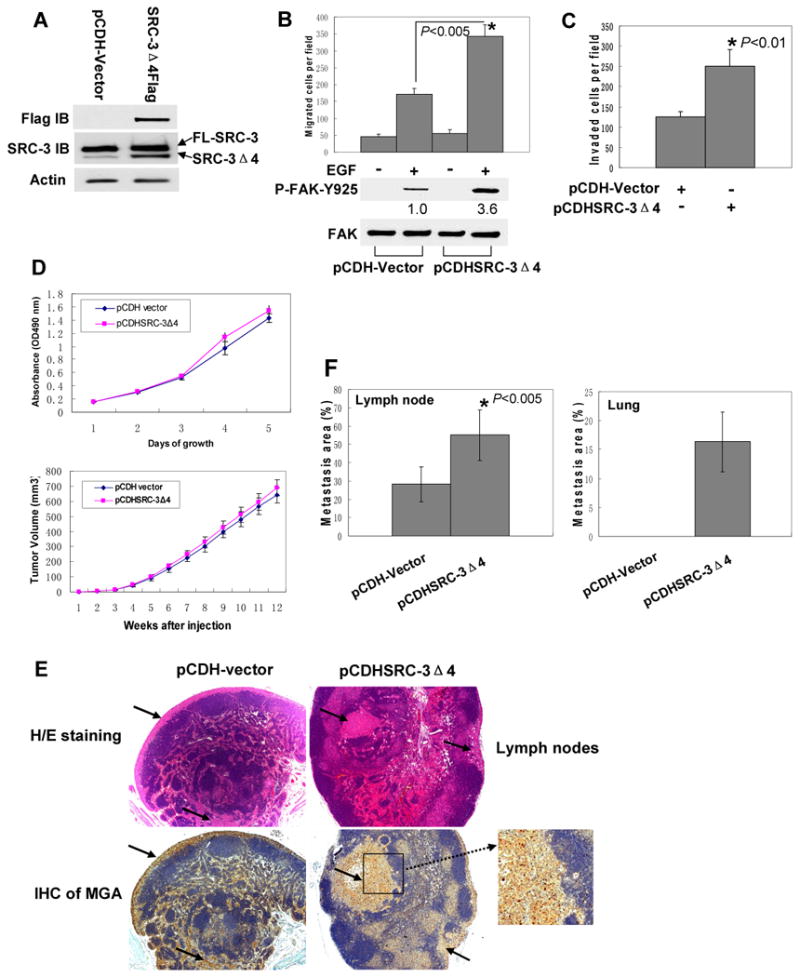
SRC-3Δ4 promotes mammary tumor metastasis to the lymph node and lung. (A). Western blots showing SRC-3Δ4 protein expression in MDA-MB231 cell lines with stable expression of either pCDH empty vector or pCDHSRC-3Δ4Flag. (B). SRC-3Δ4 promotes EGF-stimulated FAK phosphorylation on Y925 and increases MDA-MB231 cell migration. Serum starved MDA-MB231 cells with stable expression of either pCDH empty vector or pCDHSRC-3Δ4Flag were stimulated with EGF (25ng/ml) for 15 min. EGF-stimulated FAK phosphorylation on Y925 was determined by Western blotting using a phospho-specific FAK antibody. Numbers below the Western blots represent the relative intensity of the protein bands quantitated by NIH Image J software. For transwell cell migration assay, cells were first serum-starved overnight. EGF (50 ng/ml) was then added to the media in bottom chamber during migration process. Values are means ± s.e of three separate experiments. (C). Transwell matrigel cell invasion assay of MDA-MB231 cell lines stably expressing either pCDH vector or pCDHSRC-3Δ4. Values are means ± s.e of three separate experiments. (D). SRC-3Δ4 has no significant effect on MDA-MB231 cell growth and MDA-MB231-induced mammary tumor growth. Upper panel: MTT cell proliferation assay of MDA-MB231 cell lines stably expressing either pCDH vector or pCDHSRC-3Δ4. Values are means ± s.e of three separate experiments. Lower panel: primary tumor growth in the mammary gland injected with MDA-MB231 cells stably expressing either SRC-3Δ4 or pCDH vector. Values are means ± s.e of nine tumors. (E). Histological analysis of tumor metastasis in lymph node. Upper images: hematoxylin/eosin (H/E) staining of the brachial lymph nodes. Magnification: 25×. Lower images: immunohistochemical staining (IHC) of mammaglobin A (MGA). Magnification: 50×. Arrows indicate the areas with metastatic tumor cells, whereas the areas stained blue are the structures that are free of tumor metastasis. (F). The relative metastasis areas in the lymph node and lung were quantified using histomorphometry and the AxioVision software (Zeiss, Germany). Values are means ± s.e. Five mice in pCDH-vector group and nine mice in SRC-3Δ4 group had lymph node metastasis. Three mice in SRC-3Δ4 group, but none of pCDH-vector group, had lung metastasis.
Table 1.
Primary tumor incidence and metastasis to thoracic lymph nodes and lung. Numbers in columns 2, 3, and 4 indicate numbers of mice being analyzed. In column 5, two out of nine mice in control group and five out of nine mice in SRC-3Δ4 group were detected with circulating tumor cells in the blood.
| Cell lines (MDA-MB231) | Primary tumor incidence | Lymph node metastasis incidence | Microscopic lung metastasis incidence | Number of circulating tumor cells in blood (1 ml) |
|---|---|---|---|---|
| pCDH-Vector | 9/10 | 5/9 (55%) | 0/9 (0%) | 14 ±6 (n=2) |
| pCDHSRC-3Δ4 | 9/10 | 9/9 (100%) | 3/9 (33%) | 36 ±10 (n=5) |
Discussion
Adaptor or scaffold proteins facilitate signal transduction in kinase pathways by serving as platforms for upstream kinases to achieve access to their downstream targets that frequently are kinases as well (Morrison et al., 2003). For example, kinase suppressor of Ras (KSR) and β-arrestin 1 are important scaffolds in the ERK1/2 kinase pathway. FAK has been found to integrate integrin signaling and the growth-factor signal pathway to regulate cell growth and motility (Sieg et al., 2000). Although EGF stimulates complex formation between EGFR and FAK, no direct interaction of these two proteins has been observed previously (Sieg et al., 2000). It was unclear what factor mediates the interaction between EGFR and FAK and EGF-induced FAK phosphorylation. c-Src once was thought to be the potential mediator as it interacts with both EGFR (Maa et al., 1995) and FAK (Eide et al., 1995). However, the fact that Y397 is not required for the interaction between EGFR and FAK excludes this possibility, since Y397 is the c-Src binding site on FAK (Eide et al., 1995).
Here we identified SRC-3Δ4 as a signaling adaptor to mediate the interaction between EGFR and FAK and EGF signal transduction in promoting cell migration (Fig. 6). To our knowledge, this is the first report that a nuclear receptor coactivator functions as a direct adaptor in a cytoplasmic kinase pathway. Phosphorylations of SRC-3Δ4 by PAK1 facilitate its localization to the plasma membrane and its interaction with EGFR and FAK. PAK1 is known to be activated by EGF and to mediate EGF-like growth factor-induced cell migration by regulating actin cytoskeletal reorganization at the leading edge of cells (Galisteo et al., 1996; Adam et al., 1998). In addition, PAK1 is linked to FAK-mediated focal adhesion dynamics by the PAK1/PIX (PAK interacting exchange factor)/PKL (Paxillin kinase linker)/FAK connection (Manser et al., 1998; Brown et al., 2002). Our finding of the phosphorylation-dependent adaptor function of SRC-3Δ4 reveals an important mechanism by which PAK1 and FAK coordinately regulate EGF-induced cell migration.
Figure 6.

A molecular model for SRC-3Δ4's function as a unique EGF signaling adaptor promoting cell migration and cancer metastasis. PAK1 phosphorylates SRC-3Δ4 on T56 at the N-terminus and S659 and S676 within the RID region. Phosphorylations of SRC-3Δ4 promotes its localization to the plasma membrane region where it interacts with EGFR through the N-terminus, which is mediated by T56 phosphorylation, and with FAK through the RID region, which is mediated by phosphoryations of S659 and S676. SRC-3Δ4 mediates the interaction between EGFR and FAK, thereby promoting EGF-induced c-Src activation and FAK phosphorylation on Y925, which then drives cancer cell migration and metastasis.
Tumor angiogenesis plays critical roles in cancer progression and metastasis (Fidler, 2000). SRC-3 has been recently implicated in tumor angiogenesis. Disruption of the SRC-3 gene significantly inhibits tumor angiogenesis in both HER2/Neu induced mammary tumor and follicular thyroid cancer mouse models (Fereshteh et al., 2008; Ying et al., 2008). The underlying mechanisms by which SRC-3 promotes tumor angiogenesis, however, remain to be elucidated. FAK promotes cancer metastasis by multiple mechanisms (Zhao and Guan, 2009). One of them, as described above, is to integrate integrin signaling and the growth-factor signal pathway to promote cancer cell motility. Another important mechanism is to promote endothelial cell migration and tumor angiogenesis by regulating pro-angiogenic growth factors' signal pathways. FAK is activated by multiple pro-angiogenic growth factors such as VEGF and bFGF. It is unclear, however, how FAK is activated and tyrosine-phosphorylated upon VEGF and bFGF singaling. Growth factors including EGF, VEGF, and bFGF, stimulate signal transduction by the similar mechanism. These growth factors bind to and activate their membrane associated tyrosine kinase receptors, leading to the activation of downstream kinases including FAK. As such, our current findings raise an intriguing possibility that SRC-3Δ4 plays an important role in angiogenesis by mediating VEGF- and/or bFGF-induced FAK activation and phosphorylations by a similar mechanism to that in EGF signaling to FAK.
Accumulating evidence has shown that SRC-3 plays important roles in tumor metastasis. SRC-3 gene deletion (disruption of both FL-SRC-3 and SRC-3Δ4 mRNAs) suppresses both v-Ha-ras- and polyomavirus middle T (PyMT) transgene-induced breast tumor metastasis to the lung (Kuang et al., 2004, Qin et al., 2008), and prevents SV40 transgene-induced prostate tumors from developing into a metastatic stage (Chung et al., 2007). Cancer cell migration and invasion are two fundamental events during cancer metastasis. In cultured cells, SRC-3 was reported to promote cancer cell invasion by co-activating AP-1 and PEA-3-mediated matrix metalloproteinase (MMP) expression (Yan et al., 2008; Li et al., 2008; Qin et al., 2008). SRC-3 promotes FAK phosphorylation on Y397, probably by upregulating IRS2 gene expression (Yan et al., 2008). Nevertheless, all of these SRC-3 functions identified in cultured cell systems are associated with its transcriptional activity in the nucleus. Here we found that SRC-3Δ4 promotes cell migration and tumor metastasis by directly mediating EGF signal transduction from EGFR to c-Src and FAK. This is dependent upon SRC-3Δ4's localization to the plasma membrane that is stimulated by PAK1 phosphorylation. Interestingly, we found that specific knockdown of SRC-3Δ4 significantly inhibits MDA-MB231 cell migration, but there is no further significant decrease in cell migration when both FL-SRC-3 and SRC-3Δ4 were knocked down (Fig. 2A). In addition, overexpression of SRC-3Δ4, but not FL-SRC-3, significantly increases EGF-stimulated migration of HeLa cells (Fig. S4B). On the contrary, unlike FL-SRC-3, SRC-3Δ4 does not promote MMP13- and MMP2-luciferase activity (Fig. S8). Taken together, all of these findings demonstrate that FL-SRC-3 and SRC-3Δ4, two proteins derived from alterative splicing of a single gene transcript, cooperatively promote cancer cell migration and invasion and cancer metastasis by different mechanisms. FL-SRC-3 upregulates MMPs and IRS2 by acting as a transcriptional coactivator in the nucleus, whereas SRC-3Δ4, upon phopshorylations by PAK1, localizes to the plasma membrane to directly mediate EGF signal transduction to FAK and c-Src.
In summary, our study identifies an important function for SRC-3Δ4 in promoting cancer cell migration and tumor metastasis by acting as a direct signaling adaptor at the cell membrane in the EGF signal transduction pathway. Consequently, SRC-3Δ4 represents another potential therapeutic target for counteracting cancer metastasis.
Experimental Procedures
Immunofluorescence
Immunofluorescent staining was performed by following the procedures as described previously (Amazit et al., 2007). Images were captured with a Zeiss AxioVert S100 TV deconvolution microscope and a DeltaVison restoration microscopy system (Applied Precision, Inc.). The primary antibodies used were rabbit anti-SRC-3 (Calbiochem), mouse anti-FAK (BD Biosciences), anti-Flag M2 (Sigma), and PAK1 Ab (abcam).
Immunoprecipitation and Western blotting
The cell-based protein-protein interactions were analyzed by immunoprecipitation (IP) and Western blotting. The detailed procedures are described in Supplemental Experimental Procedures.
siRNA knockdown
SRC-3Δ4 siRNA is designed by targeting the junction sequence (5′-CAGGACAAGGGAAAAACTATT-3′) between exon 3 and exon 5 in SRC-3Δ4 mRNA and synthesized by Ambion. The silencer negative control #1 (Ambion) was used as the negative control for SRC-3Δ4 siRNA. SRC-3 siRNA on-target plus SMART pool, FAK siRNA on-target plus SMART pool, and non-targeting control siRNAs were purchased from Dharmacon. siRNAs were transfected with the TransIT-TKO Transfection Reagent (Mirus Bio Corporation) according to the manufacturer's instructions. Three days after siRNA transfection, cells were harvested for various assays.
Two-chamber transwell cell migration/invasion assay
Cell migration was analyzed using a modified two-chamber transwell system (BD Biosciences) following the manufacturer's instructions. Cells were detached by trypsin-EDTA and washed once with serum-free medium. Cells were then resuspended in serum-free medium. 0.5 ml of either complete culture media or serum-free media containing 50ng/ml of EGF was added to each bottom well. 1 × 105 cells were added in each transwell insert and allowed to migrate for 12 hrs (for MDA-MB231 cells) or 24 hrs (for HeLa cells) in a 37°C cell incubator. Cells in the upper surface of the transwell were removed using cotton swabs. Migrated cells attached on the undersurface were fixed with 4% paraformaldehyde for 10 min and stained with crystal violet solution (0.5% in water) for 10 min. Cells were counted under microscope at 100 × magnification.
Cell invasion assay was performed by following the same procedures in cell migration assay except that transwell inserts were pre-coated with growth factor-reduced matrigel (BD Biosciences).
Mammary tumor formation and metastasis
Animal work was done in accordance with a protocol approved by the Animal Care and Use Committee of Baylor College of Medicine. MDA-MB231 cells with stable overexpression of either SRC-3Δ4 or pCDH-Puro vector were trypsinized, washed twice with 1× PBS, and resuspended (107 cells/ml) in a 50:50 solution of serum-free DMEM and Matrigel (BD Biosciences). 100 μl of cell solution was injected orthotopically into the No. 4 inguinal mammary fat pad of anaesthetized athymic Ncr-Nu/Nu mice (NCI) at the age of 6-8 weeks. Mammary tumor outgrowth was monitored weekly by measuring tumor length (L) and width (W). Tumor volume is calculated as πLW2/6. The experiment was terminated 12 weeks after the tumor cell injection. At necropsy, 0.5 ml blood was collected by heart puncture for determining circulating tumor cells. The thoracic lymph nodes and lungs were harvested, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at a thickness of 4μM for histological examination.
Statistical Analysis
Results are expressed as mean ± s.e. Statistical significance was determined by a two-sided Student's t test. A P-value less than 0.05 is considered statistically significant.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. David Schlaepfer for providing FAK expression constructs, and Dr. Mien-Chie Hung for providing pcDNA3EGFR construct. We thank Dr. Jeff Rosen for help with the establishment of tumor metastasis animal model, Drs. Qin Feng, Ray-Chang Wu, Chao Li, and Jun Yan for providing reagents, Dr. Qin Jun for technical assistance, and Dr. Charles Foulds for critical reading of the manuscript. This work was supported by the grants NIH 5R01HD-8188, HD07857, 5P01DK59820, 2U19DK062434, Cancer Center P30CA125123, and the Welch Foundation to B.W.O, and NIH CA90970 to R.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, Kumar R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J Biol Chem. 1998;273:28238–28246. doi: 10.1074/jbc.273.43.28238. [DOI] [PubMed] [Google Scholar]
- Amazit L, Pasini L, Szafran AT, Berno V, Wu RC, Mielke M, Jones ED, Mancini MG, Hinojos CA, O'Malley BW, Mancini MA. Regulation of SRC-3 intercompartmental dynamics by estrogen receptor and phosphorylation. Mol Cell Biol. 2007;27:6913–6932. doi: 10.1128/MCB.01695-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Brown MC, West KA, Turner CE. Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol Biol Cell. 2002;13:1550–1565. doi: 10.1091/mbc.02-02-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AC, Zhou S, Liao L, Tien JC, Greenberg NM, Xu J. Genetic ablation of the amplified-in-breast cancer 1 inhibits spontaneous prostate cancer progression in mice. Cancer Res. 2007;67:5965–5975. doi: 10.1158/0008-5472.CAN-06-3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, Fleming TP, Hazan R, Ullrich A, King CR, Schlessinger J, Aaronson SA. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 1987;51:1063–1070. doi: 10.1016/0092-8674(87)90592-7. [DOI] [PubMed] [Google Scholar]
- Eide BL, Turck CW, Escobedo JA. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol Cell Biol. 1995;15:2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fereshteh MP, Tilli MT, Kim SE, Xu J, O'Malley BW, Wellstein A, Furth PA, Riegel AT. The nuclear receptor coactivator amplified in breast cancer-1 is required for Neu (ErbB2/HER2) activation, signaling, and mammary tumorigenesis in mice. Cancer Res. 2008;68:3697–3706. doi: 10.1158/0008-5472.CAN-07-6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler IJ. Angiogenesis and cancer metastasis. Cancer J. 2000;6 2:S134–141. [PubMed] [Google Scholar]
- Galisteo ML, Chernoff J, Su YC, Skolnik EY, Schlessinger J. The adaptor protein Nck links receptor tyrosine kinases with the serine-threonine kinase Pak1. J Biol Chem. 1996;271:20997–21000. doi: 10.1074/jbc.271.35.20997. [DOI] [PubMed] [Google Scholar]
- Fincham VJ, Frame MC. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. Embo J. 1998;17:81–92. doi: 10.1093/emboj/17.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- Galisteo ML, Chernoff J, Su YC, Skolnik EY, Schlessinger J. The adaptor protein Nck links receptor tyrosine kinases with the serine-threonine kinase Pak1. J Biol Chem. 1996;271:20997–21000. doi: 10.1074/jbc.271.35.20997. [DOI] [PubMed] [Google Scholar]
- Goi T, Shipitsin M, Lu Z, Foster DA, Klinz SG, Feig LA. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. Embo J. 2000;19:623–630. doi: 10.1093/emboj/19.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda T, Sonoda Y, Ando K, Suzuki T, Sasaki Y, Oshio T, Tago M, Kasahara T. Mutation of Y925F in focal adhesion kinase (FAK) suppresses melanoma cell proliferation and metastasis. Cancer Lett. 2008;270:354–361. doi: 10.1016/j.canlet.2008.05.042. [DOI] [PubMed] [Google Scholar]
- Kuang SQ, Liao L, Zhang H, Lee AV, O'Malley BW, Xu J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875–1885. doi: 10.1158/0008-5472.can-03-3745. [DOI] [PubMed] [Google Scholar]
- Li C, Wu RC, Amazit L, Tsai SY, Tsai MJ, O'Malley BW. Specific amino acid residues in the basic helix-loop-helix domain of SRC-3 are essential for its nuclear localization and proteasome-dependent turnover. Mol Cell Biol. 2007;27:1296–1308. doi: 10.1128/MCB.00336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Louie MC, Chen HW, Zou JX. Proto-oncogene ACTR/AIB1 promotes cancer cell invasion by up-regulating specific matrix metalloproteinase expression. Cancer Lett. 2008;261:64–73. doi: 10.1016/j.canlet.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci U S A. 1995;92:6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol Cell. 1998;1:183–192. doi: 10.1016/s1097-2765(00)80019-2. [DOI] [PubMed] [Google Scholar]
- Meierjohann S, Wende E, Kraiss A, Wellbrock C, Schartl M. The oncogenic epidermal growth factor receptor variant Xiphophorus melanoma receptor kinase induces motility in melanocytes by modulation of focal adhesions. Cancer Res. 2006;66:3145–3152. doi: 10.1158/0008-5472.CAN-05-2667. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–5984. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Qin L, Liao L, Redmond A, Young L, Yuan Y, Chen H, O'Malley BW, Xu J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol. 2008;28:5937–5950. doi: 10.1128/MCB.00579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter R, Oh AS, Wellstein A, Riegel AT. Impact of the nuclear receptor coactivator AIB1 isoform AIB1-Delta3 on estrogenic ligands with different intrinsic activity. Oncogene. 2004;23:403–409. doi: 10.1038/sj.onc.1207202. [DOI] [PubMed] [Google Scholar]
- Reiter R, Wellstein A, Riegel AT. An isoform of the coactivator AIB1 that increases hormone and growth factor sensitivity is overexpressed in breast cancer. J Biol Chem. 2001;276:39736–39741. doi: 10.1074/jbc.M104744200. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev. 2004;14:92–101. doi: 10.1016/j.gde.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Tang Y, Chen Z, Ambrose D, Liu J, Gibbs JB, Chernoff J, Field J. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol Cell Biol. 1997;17:4454–4464. doi: 10.1128/mcb.17.8.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Arzayus MI, Font de Mora J, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263–274. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]
- Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol. 2004;24:8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M, Yu-Lee LY, Tsai SY, Tsai MJ. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008;68:5460–5468. doi: 10.1158/0008-5472.CAN-08-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Willingham MC, Cheng SY. The steroid receptor coactivator-3 is a tumor promoter in a mouse model of thyroid cancer. Oncogene. 2008;27:823–830. doi: 10.1038/sj.onc.1210680. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Liu J, Samuel S, Cheng W, Rosen D, Naora H. Steroid receptor coactivator-3, a homolog of Taiman that controls cell migration in the Drosophila ovary, regulates migration of human ovarian cancer cells. Mol Cell Endocrinol. 2005;245:77–85. doi: 10.1016/j.mce.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, Ittmann M, Tsai SY, Tsai MJ. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976–7983. doi: 10.1158/0008-5472.CAN-04-4076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.