

Abstract
Acetylcholine (ACh) regulates many key functions of the CNS by activating cell surface receptors referred to as muscarinic ACh receptors (M1–M5 mAChRs). Like other mAChR subtypes, the M4 mAChR is widely expressed in different regions of the forebrain. Interestingly, M4 mAChRs are coexpressed with D1 dopamine receptors in a specific subset of striatal projection neurons. To investigate the physiological relevance of this M4 mAChR subpopulation in modulating dopamine-dependent behaviors, we used Cre/loxP technology to generate mutant mice that lack M4 mAChRs only in D1 dopamine receptor-expressing cells. The newly generated mutant mice displayed several striking behavioral phenotypes, including enhanced hyperlocomotor activity and increased behavioral sensitization following treatment with psychostimulants. These behavioral changes were accompanied by a lack of muscarinic inhibition of D1 dopamine receptor-mediated cAMP stimulation in the striatum and an increase in dopamine efflux in the nucleus accumbens. These novel findings demonstrate that a distinct subpopulation of neuronal M4 mAChRs plays a critical role in modulating several important dopamine-dependent behaviors. Since enhanced central dopaminergic neurotransmission is a hallmark of several severe disorders of the CNS, including schizophrenia and drug addiction, our findings have substantial clinical relevance.
Introduction
Many of the important central actions of acetylcholine (ACh) are mediated by a family of cell surface receptors referred to as muscarinic ACh receptors (M1–M5 mAChRs) (Felder et al., 2000; Eglen, 2005; Wess et al., 2007; Langmead et al., 2008). Importantly, disturbances in the central muscarinic cholinergic system have been implicated in several pathophysiological conditions, including Alzheimer's and Parkinson's disease, depression, epilepsy, drug addiction, and schizophrenia (Felder et al., 2000; Eglen, 2005; Wess et al., 2007; Langmead et al., 2008).
To identify the specific mAChR subtypes that are involved in mediating the various central functions of ACh, we and others have generated and analyzed mutant mouse strains deficient in each of the five mAChR genes (Wess et al., 2007). Initial studies with whole-body M4 receptor knock-out (KO) mice suggested that M4 mAChRs are involved in modulating the activity of the central dopaminergic system (Gomeza et al., 1999; Felder et al., 2001; Zhang et al., 2002; Tzavara et al., 2004). Dopamine, by activating different dopamine receptor subtypes (D1–D5), plays a critical role in regulating many fundamental CNS processes, including locomotor control and drug reward mechanisms (Holmes et al., 2004; Dalley and Everitt, 2009; Le Foll et al., 2009). Moreover, disturbances in central dopaminergic neurotransmission are predicted to be involved in the pathogenesis of Parkinson's disease, drug addition, and schizophrenia (Di Chiara et al., 1994; Gainetdinov et al., 2001; Holmes et al., 2004; Dalley and Everitt, 2009; Le Foll et al., 2009).
The specific neuronal subpopulations involved in the interactions of the M4 mAChR with the central dopaminergic system have not been identified. Previous studies have shown that the M4 mAChR is widely distributed throughout the forebrain (Levey et al., 1991; Vilaró et al., 1993; Volpicelli and Levey, 2004). Interestingly, M4 receptors are coexpressed with D1 dopamine receptors in a specific subset of striatal medium spiny neurons that contain GABA as the major neurotransmitter and give rise to the so-called striatonigral pathway (Bernard et al., 1992; Di Chiara et al., 1994; Ince et al., 1997).
To investigate the physiological relevance of this subpopulation of neuronal M4 mAChRs, we used Cre/loxP technology to generate mutant mice that lack M4 mAChRs only in D1 dopamine receptor-expressing cells. We then analyzed these newly generated mutant mice in a series of biochemical and behavioral studies, with a particular focus on behavioral responses that involve the dopaminergic system.
Interestingly, the M4 mAChR mutant mice displayed several striking phenotypes, including enhanced hyperlocomotor activity and increased behavioral sensitization following treatment with psychostimulants. These behavioral changes were accompanied by specific biochemical and neurochemical changes. Moreover, the newly generated mutant mice displayed reduced cataleptic responses to first- or second-generation antipsychotic drugs. These novel observations indicate that M4 mAChRs expressed by D1 dopamine receptor-expressing neurons play a key role in modulating several key functions of the central dopaminergic system. Since an increase in dopaminergic neurotransmission has been implicated in the pathogenesis of schizophrenia, drug addiction, and several other CNS disorders, these findings should be of considerable general relevance.
Materials and Methods
Generation of D1-M4-KO mice.
To create mutant mice that lack M4 mAChRs only in D1 receptor-expressing neurons, we used Cre/loxP and Flp/frt technology. We first generated a targeting vector that was based on the pFlox vector (Herroeder et al., 2009). This vector is characterized by the presence of a tk/neo selection cassette that is flanked at either side by both frt and loxP sites (Fig. 1A). A mouse M4 mAChR receptor genomic clone was isolated from a 129Sv/J mouse genomic library as described previously (Gomeza et al., 1999). To generate the M4 receptor targeting vector, the 5′ arm (3.0 kb in length) was inserted into the SacII site 5′ of the first loxP site, and the 3′ arm (2.9 kb in length) was cloned into the XhoI site 3′ of the third loxP site. In the next step, a 2.2 kb fragment containing the entire M4 receptor coding sequence was inserted into the ClaI site between the second and third loxP sites of the targeting vector. The targeting construct was linearized by digestion with NotI and introduced into TC1 (129SvEv) embryonic stem (ES) cells via electroporation (Deng et al., 1996). Clones resistant to G418 were isolated as described previously (Yang et al., 1998). After properly targeted ES cells had been identified via PCR and Southern blotting analysis, the frt-flanked tk/neo cassette was deleted by electroporating properly targeted ES cells with the pCAGGS-FLPe plasmid (Gene Bridges) coding for FLPe DNA recombinase (Schaft et al., 2001). ES cell genotypes were determined by PCR analysis and Southern blotting analysis (see next paragraph). ES cells containing the floxed version of the M4 receptor gene and lacking the tk/neo cassette were microinjected into C57BL/6 blastocysts to generate male chimeric offspring, which in turn were mated with female C57BL/6 mice (Taconic) to generate heterozygous F1 mutant mice. Female mice harboring the floxed M4 receptor allele were crossed with the male D1-Cre transgenic mice (Lemberger et al., 2007). In D1-Cre transgenic mice, Cre expression is under the transcriptional control of the D1 dopamine receptor gene promoter (Lemberger et al., 2007). The resulting mutant mice were then intermated to generate mutant mice that lacked M4 mAChRs only in D1 receptor-expressing neurons (M4 fl/fl D1-Cre mice; also referred to as D1-M4-KO mice). This mating scheme also yielded floxed M4 mAChR mice (M4 fl/fl mice) lacking the D1-Cre transgene. These mice were used as control animals throughout all experiments. The mutant mice used for all experiments described in this study were backcrossed for at least four generations (N = 4) onto the C57BL/6 background (Taconic).
Figure 1.

Strategy used for the conditional deletion of the M4 mAChR gene in D1 dopamine receptor-expressing neurons. A, Partial restriction maps of the wild-type (WT) M4 mAChR genomic locus, targeting construct, recombinant targeted allele, and M4 mAChR-deleted locus. The approximate locations of the frt and loxP sites, the P1 and P2 probes used for Southern analysis (filled bars), and the primers (arrows) used for PCR genotyping studies are indicated. H, HindIII; N, NotI; B, BamHI; X, XhoI; S, SpeI; Sa, SacII. B, Southern blot analysis of genomic DNA from representative ES cell clones before FLPe-mediated deletion of the tk/neo cassette. SpeI- or BamHI-digested DNA was analyzed with the P2 and P1 probes, respectively. The 9.5 and 10.4 kb bands indicate the presence of the WT M4 receptor allele, whereas the 6.0 and 7.1 kb bands are diagnostic for the proper integration of the targeting construct. Note that ES cell clone 3 (marked with an asterisk) showed the proper targeting event. C, PCR genotyping analysis of ES cell DNA after FLPe-mediated deletion of the tk/neo cassette. The 71 and 210 kb bands indicate the presence of the WT and the floxed M4 receptor allele, respectively (use of primers 1 and 3, top). Analogously, the 233 and 267 bp bands are diagnostic for the presence of the WT and the floxed M4 receptor allele, respectively (use of primers 4 and 5, bottom). Note that ES cell clones 2, 6, and 8 were heterozygous for the floxed M4 receptor allele. The same strategy was used for the genotyping of mouse tail DNA. D, Real-time qRT-PCR analysis of M1, M2, and M4 mAChR mRNA expression in the striatum of control mice (M4 fl/fl) and D1–M4-KO (M4 fl/fl D1-Cre) mice. Data were normalized by the expression of cyclophilin A, which served as an internal control. Results are shown as relative gene expression levels compared to control mice (100%; means ± SEM; n = 4 per group). ***p ≤ 0.001 versus control. E, Confocal microscopy analysis of M4 mAChR and D1 dopamine receptor expression in the striatum. The three top panels demonstrate that M4 muscarinic and D1 dopamine receptor proteins were colocalized in the striatum of control mice. The three bottom panels indicate that striatal M4 mAChR staining was reduced to background levels in D1–M4-KO mice. F, Immunohistochemical localization of M4 mAChRs in selected brain regions. As expected, M4 mAChR staining was abolished throughout the brain in whole-body M4 receptor KO mice, indicative of the specificity of the M4 mAChR antibody used (top right). The corresponding WT mice gave the expected M4 mAChR staining pattern (top left). In brain sections from WT (control) mice (left), M4 mAChR staining was abundant in the striatum and in presumed axonal processes in the corpus callosum (high-power inset). In the corresponding sections from D1–M4-KO mice (bottom right), M4 mAChR staining was greatly reduced in the striatum, but was retained in other areas of the brain such as the corpus callosum (inset).
Southern blotting analysis.
Southern blotting studies were performed by standard techniques using 32P-labeled probes. The location of the P1 and P2 probes is indicated in Figure 1A. Both probes were generated via PCR using a mouse M4 mAChR genomic clone (Gomeza et al., 1999) as a template. The P1 probe (size: 605 bp) was generated by using the following PCR primes: forward, 5′-CAGCTCTTAGAGGAAGTGTTAGGCA-3′; reverse, 5′-GAGAAGCCTCTGCGATAGTCGTCAG-3′. The P2 probe (size: 526 bp) was obtained using the following PCR primes: forward, 5′-GCGTCTGTGCCTCTCACCTCTGA-3′; reverse, 5′-GTCTGGAACTTCTCAGACAGCTC-3′.
Mouse genotyping studies.
Mouse genotypes were determined by PCR analysis of mouse tail genomic DNA. All PCR primers used for ES cell and mouse genotyping studies are listed in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Real-time qRT-PCR analysis.
Relative gene expression levels were analyzed by real-time qRT-PCR (7900HT SDS; Applied Biosystems). Total RNA was isolated using the RNeasy Mini kit including a DNase I treatment step (Qiagen), and reverse transcription was performed using the SuperScribe III RT enzyme (Invitrogen). PCRs (25 μl total volume) included cDNA (∼20 ng of initial RNA), 100–120 nm of each primer, and 12.5 μl of 2× SYBR Green Master Mix (Applied Biosystems). The PCR conditions used were as follows: 50°C for 2 min followed by a denaturing step at 95°C for 10 min and by 40 cycles at 95°C for 15 s and 60°C for 1 min. Cyclophilin A was used as an internal control. The results were expressed as fold changes in expression of a particular RNA transcript relative to the internal standard (cyclophilin A) between D1-M4 KO and control mice (for primer sequences, see supplemental Table 2, available at www.jneurosci.org as supplemental material).
Immunohistochemistry.
Mouse brain tissue was fixed by perfusion with 4% paraformaldehyde. Frozen sections (50 μm thick) of mouse brain were processed for immunohistochemistry using an M4 receptor rabbit polyclonal antibody (1 μg/ml; Levey et al., 1991) and a D1 receptor rat monoclonal antibody (1:1000; Smiley et al., 1994). Following permeabilization and blocking using 0.1% Triton X-100 and 4% normal horse serum, brain sections were incubated with the primary antibodies overnight at 4°C. Sections were developed either by immunoperoxidase processing using diaminobenzidine (DAB), or by incubation with donkey anti-rabbit FITC (1:200; Jackson ImmunoResearch) and goat anti-rat Cy3 (1:200; Jackson ImmunoResearch). Fluorophore-labeled sections were rinsed and mounted using Vectashield mounting medium (Vector Laboratories). Nonspecific binding of the secondary antibodies was controlled for by omitting the primary antibodies. Confocal images were captured using a Zeiss LSM-510 laser scanning confocal microscope.
cAMP assay.
Crude mouse striatal membranes were prepared and cAMP assays were performed essentially as described previously (Olianas et al., 1997), except that cAMP was detected via ELISA. Reactions were performed at 30°C for 10 min in a 0.1 ml volume (20–30 μg of membrane protein per tube) in the presence of increasing concentrations (10−10 to 10−6 m) of the D1 receptor agonist, SKF82958 (Sigma), either in the absence or in the presence of carbachol (100 μm). Reactions were terminated by the addition of 0.4 ml of 0.1N HCl and centrifugation at 20,000 × g for 10 min. The supernatants were used for cAMP measurements using the cAMP Biotrak ELISA kit (GE Healthcare). Protein concentrations were determined by using the BCA protein assay kit (Pierce) with bovine plasma albumin as standard. Concentration–response curves were fitted by nonlinear regression using the using Prism software (version 4.0; GraphPad Software Inc.).
D1 dopamine receptor radioligand binding assay.
Mouse striata were homogenized using a Brinkman homogenizer in 6 ml of ice-cold lysis buffer containing 5 mm Tris-HCl, pH 7.4, and 5 mm MgCl2. Crude striatal membrane fractions were prepared by centrifugation at 27,000 × g for 20 min (4°C), resuspension of the pellet in 15 ml of binding buffer (50 mm Tris-HCl, pH 7.4), followed by recentrifugation and rehomogenization of the membrane pellet in binding buffer. Striatal membranes (∼200 μg of protein per tube) were incubated with increasing concentrations (0.06–2 nm) of the D1 receptor antagonist, [3H]-SCH23390 (85.0 Ci/mmol; PerkinElmer). Nonspecific binding was detected in the presence of butaclamol (4 μm). Binding mixtures (final volume: 1 ml) were incubated for 90 min at room temperature (22°C) and terminated via rapid filtration over Brandel GF/C filters. Binding data were analyzed by a nonlinear one binding site curve-fitting procedure using Prism software (version 4.0; GraphPad Software).
Mice used for behavioral studies.
For behavioral studies, mice were bred at the animal facilities of the Panum Institute, University of Copenhagen. Mouse genotypes were reconfirmed after the completion of experiments. The mice were housed in standard rodent cages (Macrolon type III), enriched with cardboard housing and nesting material. The animals were kept at room temperature (22–24°C) in a 12 h light/dark cycle (lights on at 6:00 A.M.) with ad libitum access to food and water. All experiments were performed in the middle of the light cycle between 8:00 A.M. and 5:00 P.M. The mice were allowed to acclimatize to the animal facility for at least 7 d before the start of experiments. Animals were taken to the experimental room at least 1 h before the experiment. Unless noted otherwise, all experiments were performed with 9- to 12-week-old male mice that were naive for all behavioral tests. All animal studies were conducted in accordance with the guidelines from the Animal Experimentation Inspectorate, Denmark, and the European Communities Council Directive 86/609/EEC, and were approved by the Animal Care and Use Committee of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland.
Drugs used for behavioral studies.
Cocaine hydrochloride and d-amphetamine were obtained from the Copenhagen University Hospital-Pharmacy. SKF82958 hydrobromide, quinpirole hydrochloride, and risperidone were purchased from Sigma-Aldrich and haloperidol (Serenase; 5 mg/ml) from Janssen-Cilag. All drugs were dissolved or diluted in saline, and solutions were adjusted to a pH range of 4.5–7.0 for subcutaneous or intraperitoneal injections (10 ml/kg).
SHIRPA behavioral test (primary screen).
Behavioral and physical characteristics of experimentally naive D1-M4-KO mice and control littermates were assessed using the SHIRPA primary screen procedure, which examines various reflexes and basic sensorimotor functions (Irwin, 1968; Rogers et al., 1997, 1999). Testing of each animal began by observing undisturbed behavior in a cylindrical clear Perspex viewing jar for 5 min. The mice were then transferred to a large Plexiglas cage to study motor behavior. This was followed by a sequence of manipulations using tail suspension where measurements of visual acuity, grip strength, body tone, and reflexes were recorded. The animals were handled firmly to record autonomic responses of skin color and heart rate and to measure body length. Limb tone, salivation, and provoked biting responses were also monitored. The primary screen was completed with the measurement of the righting reflex, contact-righting reflex, negative geotaxis, and a wire maneuver (to assess the animals' ability to grip with the hind legs when hung by the forelimbs). Throughout testing, incidences of abnormal behavior, fear, irritability, aggression, or vocalization were recorded. After testing of each mouse, the arena was cleaned and dried. Locomotor activity was measured for 1 h in locomotor activity boxes equipped with infrared light sources plus photocells.
Locomotor activity measurements.
Mice were injected subcutaneously with different drugs (SKF82958, quinpirole, amphetamine, or cocaine) and immediately transferred to activity test cages with a scant lining of fresh bedding. The test cages were situated in frames emitting 8 infrared lines 1.5 cm above the floor. The total number of beam breaks during a 1 h observation period was measured with the Photobeam Activity System-Home Cage (Ellegaard Systems).
Psychostimulant-induced behavioral sensitization.
To study behavioral sensitization, we paired repeated amphetamine (2 mg/kg, s.c.) or cocaine (10 mg/kg, s.c.) injections with exposure of the mice to activity test cages for 1 h per day. After administration of saline on the initial day (day 0), D1-M4-KO mice and control littermates were divided into two groups that received either drug (amphetamine or cocaine) or saline for 6 d (days 1–6). After a 13 d period during which mice did not receive any drugs and were not tested, all mice were given amphetamine (2 mg/kg, s.c.) or cocaine (10 mg/kg, s.c.) on day 20 and retested in the locomotor activity cages. On the following day (day 21), all mice were injected with saline and reexposed to the activity cages to examine whether context conditioning had any effect on the observed hyperlocomotor responses.
Drug-induced catalepsy.
D1-M4-KO mice and control littermates were injected with haloperidol (0.3 and 1 mg/kg, i.p.), risperidone (1 and 3 mg/kg, i.p.), or saline (control). Cataleptic activity was assessed 30, 60, and 90 min after injection by lifting the mouse by the tail and placing the front paws on a steel bar and the hind legs on a plane surface. The steel bar (15 cm long, 0.5 mm diameter) was situated horizontally 5.5 cm above the surface level. If the mouse did not stay in this position, the procedure was repeated within 15 s. If the mouse still did not remain in the forced position, the time was registered as 0 s. The time period during which the mice stayed in the unnatural position was recorded (cutoff time: 60 s).
In vivo microdialysis studies.
One hour before surgery, mice were treated with antibiotic [enrofloxacin, 10 mg/kg, s.c. (Baytril, Bayer)] and analgesics [carprofen, 5 mg/kg, s.c. (Rimadyl, Pfizer) and buprenorphine, 0.06 mg/kg, s.c. (Temgesic, Schering-Plough)]. Mice were anesthetized using isoflurane (Baxter) and placed in a stereotaxic frame equipped with a mouse adapter (Kopf). Intracerebral guide cannulae (CMA/7, CMA) were stereotaxically implanted into the brain to allow the positioning of the dialysis probe tip in the nAcc (AP: +1.0 mm; ML: +1.2 mm; DV: −4.2 mm) (Franklin and Paxinos, 2007) and fixed in place with two anchor screws and dental cement. Animals were left to recover for 1 d. The correct placement of the probe was verified histologically at the end of the experiment.
For in vivo microdialysis studies, a Ringer's solution (145 mm NaCl, 3 mm KCl, 1 mm MgCl2, and 1.2 mm CaCl2) was perfused at a rate of 1.0 μl/min through the probe (1 mm CMA/12). Using swivel and bowl (Instech), the animals were able to move freely during the dialysis. Following a 3 h acclimatization period, 30 min samples were collected into a refrigerated fraction collector. Time points were corrected for lag time of the perfusate from the microdialysis site to the probe outlet. Three samples were collected to establish dopamine baseline levels. Subsequently, mice were injected with amphetamine (2 mg/kg, s.c.), and five additional samples were collected. The samples were stored at −80°C until analysis with HPLC coupled to electrochemical detection.
For the measurement of dopamine levels, aliquots of the collected dialysates (20 μl) were injected onto an HPLC column by a refrigerated microsampler system, consisting of a syringe pump, a multicolumn injector, and a temperature regulator. Dopamine was separated by reverse phase liquid chromatography (ESA, ODS 150 × 3.2, 3 μm) using a mobile phase consisting of 90 mm NaH2PO4, 50 mm sodium citrate, 1.7 mm 1-octanesulfonic acid, 50 μm EDTA, and 8% acetonitrile, pH 4.0, at a flow rate of 0.5 ml/min. Electrochemical detection of dopamine was accomplished using a coulometric detector (potential set at E1 = −75 mV and E2 = 350 mV; guard cell = 350 mV; Coulochem II, ESA). Data were collected and analyzed using Chromeleon Clinet 6.30 software (Dionex).
Data analysis.
SHIRPA behavioral scores were compared between genotypes using an unpaired t test. Group differences in the locomotor activity and catalepsy studies were analyzed by two-way ANOVA followed by Bonferroni-corrected pairwise comparisons. Behavioral sensitization and microdialysis data were analyzed using repeated-measures ANOVA with time as within-group factor and genotype and drug treatment as between-group factors as applicable. Significant interactions were followed by within-factor analyses, and significant main effects were further analyzed by Bonferroni-corrected pairwise comparisons. A p value of <0.05 was considered statistically significant.
Results
Generation of mutant mice that lack M4 mAChRs only in D1 receptor-expressing neurons
We initially generated mutant mice that contained a floxed version of the M4 mAChR gene. The general strategy that we used to create these mutant mice is summarized in Figure 1A–C. By using standard gene targeting techniques, we first generated embryonic stem (ES) cells in which the endogenous M4 mAChR gene was replaced with a floxed version of this gene. In the next step, properly targeted ES cells were transfected with a plasmid coding for FLPe recombinase (Schaft et al., 2001), resulting in the deletion of the tk/neo selection cassette. The resulting ES cells were then used to obtain mutant mice in which the M4 mAChR coding sequence was surrounded by loxP sites (Fig. 1A). These mice were then mated with transgenic mice that express Cre recombinase under the control of the D1 dopamine receptor promoter (D1-Cre mice) (Lemberger et al., 2007). In this mouse line, the spatio-temporal pattern of Cre expression closely recapitulates that of the endogenous D1 dopamine receptor gene (Lemberger et al., 2007). This mating scheme eventually yielded mutant mice that lacked M4 mAChRs only in D1 receptor-expressing neurons (M4 fl/fl D1-Cre mice). For the sake of simplicity, we refer to these mice as D1-M4-KO mice throughout the manuscript. Unless stated otherwise, floxed (flanked by loxP) M4 mAChR mice (M4 fl/fl mice) lacking the D1-Cre transgene were used as control animals throughout all experiments. Previous studies have shown that D1-Cre mice are devoid of any neuroanatomical deficits (Lemberger et al., 2007) and do not display any phenotypic changes in various behavioral tests (T. Lemberger, R. Parlato, and G. Schütz, unpublished results). For these reasons, D1-Cre mice were not used as additional control animals.
M4 muscarinic and D1 dopamine receptors are primarily coexpressed by a subset of striatal projection neurons (Bernard et al., 1992; Di Chiara et al., 1994; Ince et al., 1997). Real-time qRT-PCR analysis showed that striatal M4 receptor mRNA expression was nearly abolished in D1-M4-KO mice, as compared to control littermates (Fig. 1D). In addition to the M4 receptor, M1 and M2 mAChRs are also found in the striatum at relatively high levels (Levey, 1993; Wolfe and Yasuda, 1995). qRT-PCR studies demonstrated that the expression levels of the two latter receptor subtypes remained unaffected by the lack of striatal M4 receptors (Fig. 1D).
We obtained complementary results when we examined the striatal expression pattern of the M4 muscarinic and D1 dopamine receptor proteins via immunohistochemical techniques (Fig. 1E). This analysis showed that the two receptors were colocalized in the striatum of control mice, consistent with previous findings (Bernard et al., 1992; Di Chiara et al., 1994; Ince et al., 1997) (Fig. 1E, top). Strikingly, M4 receptor staining was virtually abolished in the striatum of D1-M4-KO mice (Fig. 1E, bottom).
Figure 1F (top) demonstrates the specificity of the M4 receptor antibody used for immunohistochemical studies, as observed with brain sections from wild-type and whole-body M4 receptor KO mice (Gomeza et al., 1999). Consistent with the data presented in Figure 1E, M4 receptor expression was greatly reduced in the striatum of D1-M4-KO mice but was unaffected in areas that do not express significant levels of D1 receptors, such as the corpus callosum (inset) (Fig. 1F, bottom).
To examine whether the lack of striatal M4 receptors affected the expression levels of striatal D1 receptors, we performed saturation binding studies using [3H]-SCH23390, a D1 receptor-selective antagonist, as a radioligand. This analysis indicated that [3H]-SCH23390 labeled a similar number of binding sites in D1-M4-KO and control mice (Bmax in fmol/mg membrane protein: control, 851 ± 6; D1-M4-KO, 850 ± 8; Kd values in nm: control, 0.26 ± 0.01; D1-M4-KO, 0.26 ± 0.01; n = 3).
Muscarinic inhibition of D1 dopamine receptor-mediated cAMP production is abolished in the striatum of D1-M4-KO mice
The M4 muscarinic and the D1 dopamine receptors have opposing effects on the activity of adenylyl cyclase. Whereas M4 receptor activation has an inhibitory effect on adenylyl cyclase (via stimulation of Gi-type G-proteins) (Wess, 1996, Caulfield and Birdsall, 1998), D1 receptor activation leads to increased intracellular cAMP levels mediated by the stimulatory G-proteins Gs or Golf (Zhuang et al., 2000; Holmes et al., 2004). To examine the potential functional interaction among these two receptors in the striatum, we treated striatal membranes prepared from control and D1-M4-KO mice with the D1 receptor agonist, SKF82958. As expected, the D1 receptor agonist triggered concentration-dependent increases in cAMP production (Fig. 2). In control mice, this activity was virtually abolished in the simultaneous presence of carbachol (100 μm), a hydrolytically stable ACh analog (Fig. 2). Strikingly, this inhibitory effect of carbachol was no longer observed in striatal membranes prepared from D1-M4-KO mice (Fig. 2), clearly indicating that M4 receptors inhibit D1 receptor-mediated cAMP production in the striatum.
Figure 2.

Muscarinic inhibition of D1 dopamine receptor-stimulated cAMP accumulation is abolished in the striatum of D1–M4-KO mice. cAMP assays were performed using striatal membranes prepared from D1–M4-KO and control mice by applying increasing concentrations of the D1 receptor agonist, SKF82958, either in the absence or in the presence of 100 μm carbachol (CCh). Data are given as means ± SEM (n = 3).
Basal behavioral testing (primary screen)
We initially subjected naive D1-M4-KO mice and control littermates to a series of basic tests examining reflexes and sensorimotor functions, using the SHIRPA primary screen procedure (Irwin, 1968; Rogers et al., 1997, 1999). We found no significant differences between the two groups of mice in any of the measures investigated (supplemental Table 3, available at www.jneurosci.org as supplemental material).
D1 receptor agonist-, amphetamine-, and cocaine-induced hyperlocomotor responses are increased in D1-M4-KO mice
We next subjected D1-M4-KO mice and control littermates to a series of pharmacological challenges, with a particular focus on behavioral responses that involve the central dopaminergic system. In the first set of experiments, we injected D1-M4-KO and control mice with increasing doses of the D1 receptor agonist, SKF82958 (0.1–1.0 mg/kg, s.c.), and the D2 receptor agonist, quinpirole (3–30 μg/kg, s.c.), followed by locomotor activity measurements. The D1 receptor agonist caused dose-dependent increases in locomotor activity in both groups of mice (Fig. 3A). Interestingly, at the highest dose of SKF82958 used, the D1-M4-KO mice were significantly more active than their control littermates (Fig. 3A).
Figure 3.
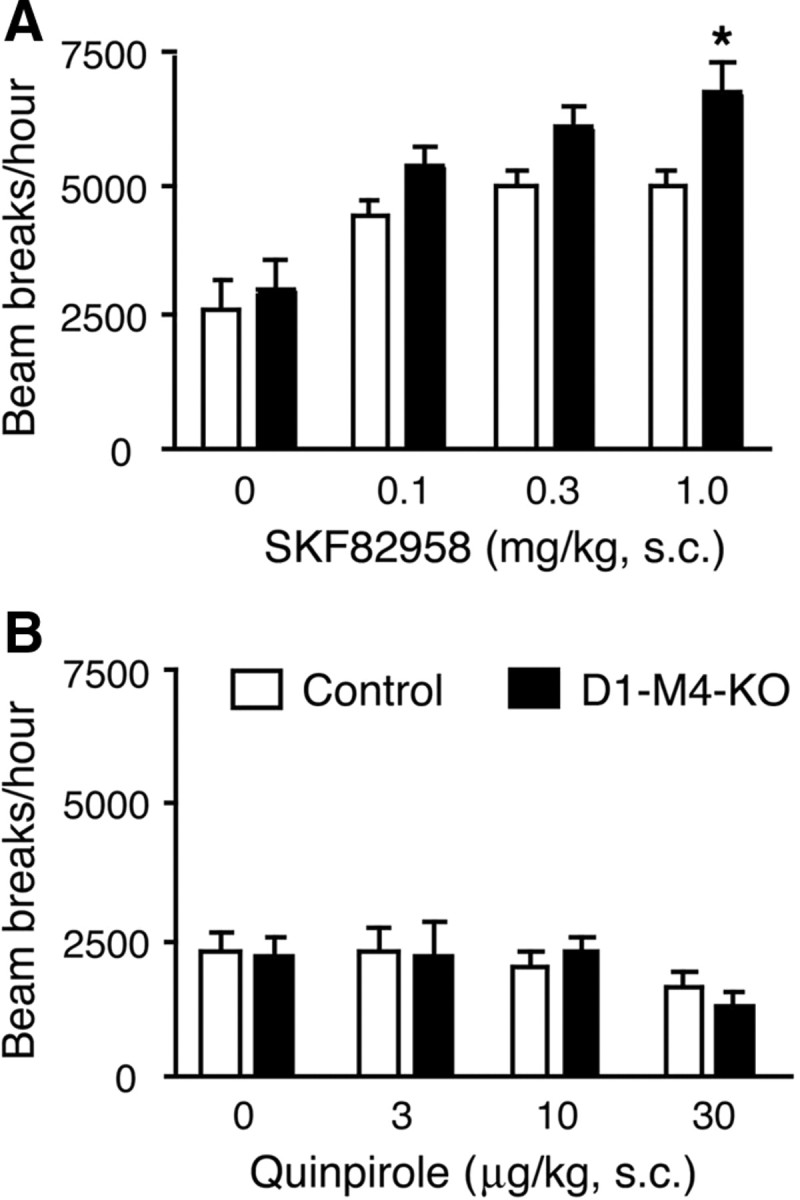
Effects of SKF82958 and quinpirole on locomotor activity in D1–M4-KO and control mice. A, B, D1–M4-KO mice and control littermates were injected subcutaneously with the indicated doses of the D1 receptor agonist, SKF82958 (A), or the D2 receptor agonist, quinpirole (B). Locomotor activity was assessed during a 1 h test period. Data are given as means ± SEM (n = 9–12). *p ≤ 0.05 versus control.
The D2 receptor agonist, quinpirole, only caused a small decrease in locomotor activity in both mouse strains at the highest dose used (30 μg/kg, s.c.) (Fig. 3B). The magnitude of this effect was similar in control and D1-M4-KO mice.
We also performed locomotor activity measurements following administration of amphetamine (1 and 2 mg/kg, s.c.) or cocaine (5 and 10 mg/kg, s.c.), two psychostimulants that increase synaptic dopamine levels by interfering with the activity of the dopamine transporter, in addition to other sites of action. In both control and D1-M4-KO mice, amphetamine and cocaine caused dose-dependent increases in locomotor activity (Fig. 4A,B). We found that the M4 receptor mutant mice were significantly more active than their control littermates at the highest amphetamine and cocaine doses used (Fig. 4A,B).
Figure 4.

Effects of amphetamine and cocaine on locomotor activity in D1–M4-KO and control mice. A, B, D1–M4-KO mice and control littermates were injected subcutaneously with the indicated doses of amphetamine (A) or cocaine (B). Locomotor activity was assessed during a 1 h test period. Data are given as means ± SEM (n = 9–12). **p < 0.01 versus control.
The cataleptic effects of antipsychotic drugs are reduced in D1-M4-KO mice
The clinical use of classical antipsychotic drugs such as haloperidol often leads to severe motor side effects such as parkinsonism. These side effects are thought to involve changes in the balance between dopaminergic and cholinergic neurotransmission in the striatum (Di Chiara et al., 1994). Haloperidol-induced catalepsy is a frequently used animal model to mimic the motor side effects of antipsychotic drugs. In the next set of studies, we therefore examined haloperidol-induced catalepsy in control and D1-M4-KO mice. We injected the mice with two different doses of haloperidol (0.3 and 1 mg/kg, i.p.) and then measured cataleptic activity at three different time points (30, 60, and 90 min after drug administration). Strikingly, the cataleptic responses to haloperidol were significantly decreased in D1-M4-KO mice, independent of time point or haloperidol dose used (Fig. 5A,B).
Figure 5.

Cataleptic responses to haloperidol and risperidone in D1–M4-KO and control mice. A–D, D1–M4-KO mice and control littermates were injected intraperitoneally with the indicated doses of haloperidol (A, B) or risperidone (C, D), and cataleptic responses were assessed 30, 60 and 90 min after drug injection. Significant genotype effects were found at both doses of haloperidol and risperidone, indicating that D1–M4-KO mice showed attenuated cataleptic responses as compared to control mice. Data are given as means ± SEM (n = 8–18). *p < 0.05, ***p < 0.001 versus control.
We obtained similar results when we used an atypical neuroleptic drug, risperidone (1 and 3 mg/kg, i.p.), to induce catalepsy in control and D1-M4-KO mice (Fig. 5C,D). In contrast to haloperidol, risperidone belongs to the group of atypical or second-generation antipsychotic drugs, which usually exhibit reduced motor side effects. As shown in Figure 5, C and D, risperidone-induced cataleptic responses were also significantly attenuated in the D1-M4-KO mice.
Amphetamine-induced behavioral sensitization is enhanced in D1-M4-KO mice
In rodents, repeated injections of amphetamine or other psychostimulants lead to enhanced locomotor stimulation, a phenomenon referred to as behavioral sensitization. Importantly, this effect persists for many weeks or months after the last drug administration, thus mimicking long-term sensitivity to drugs observed in human addicts. To study the role of striatal M4 mAChRs in the behavioral sensitization to amphetamine, we initially injected control and D1-M4-KO mice daily with either saline or amphetamine (2 mg/kg, s.c.) for a 6 d period. Consistent with the results from single amphetamine injections (Fig. 4A), repeated amphetamine injections generally induced higher levels of locomotion in D1-M4-KO than in control mice, reaching significance on days 4 and 5 (Fig. 6A). As expected, repeated saline injections had no stimulatory locomotor effects in either control or M4 receptor mutant mice (Fig. 6A). In D1-M4-KO mice, amphetamine-induced locomotor stimulation was significantly greater on days 4 and 5, as compared to day 1 (day of first amphetamine injection) (Fig. 6A). Amphetamine-injected control mice showed a clear trend toward enhanced locomotor responses on days 2–5, as compared to day 1. However, this increase failed to reach statistical significance, probably due to the moderate amphetamine dose used in these experiments (2 mg/kg, s.c.). The observed differences in locomotor responses between D1-M4-KO and control mice indicate that the mutant mice were clearly more prone to develop amphetamine-induced behavioral sensitization.
Figure 6.

Behavioral sensitization after repeated amphetamine injections in D1–M4-KO mice. A, Locomotor activity during sensitization to amphetamine (2 mg/kg, i.p.). Repeated administration of amphetamine significantly increased the hyperlocomotor response of D1–M4-KO mice. Black arrows indicate treatments that differed from those indicated by the legend. *p < 0.05, **p < 0.01 amphetamine-treated D1–M4-KO versus amphetamine-treated control mice; #p < 0.05, ###p < 0.001 versus amphetamine response on day 1 (same genotype). B, Amphetamine-induced locomotor responses on day 20 (test day). On day 20, all mice, independent of whether they had been treated with saline or amphetamine during the initial 6 d injection period, received a single dose of amphetamine (2 mg/kg, i.p.). For each genotype, data are expressed as percentage increase in amphetamine-induced hyperlocomotor activity observed with amphetamine-pretreated versus saline-pretreated mice. This analysis revealed a significant difference between D1–M4-KO and control mice. *p < 0.05 versus control. Data are given as means ± SEM (n = 11–16).
After the end of the 6 d amphetamine (or saline) injection period (days 1–6), mice were kept test- and drug-free for 13 d. On day 20, all mice, independent of whether they had received saline or amphetamine during the initial 6 d injection period, received a single amphetamine injection (2 mg/kg, s.c.) and were then retested. Strikingly, the D1-M4-KO mice that had received amphetamine during the initial 6 d injection period showed a significantly increased hyperlocomotor response, as compared to amphetamine-pretreated control mice (Fig. 6A). On day 20, the amphetamine-induced locomotor response was 75% higher in D1-M4-KO mice pretreated with amphetamine, as compared to D1-M4-KO mice pretreated with saline (Fig. 6B). In contrast, in control mice, we observed only a 23% increase in response under the same experimental conditions (Fig. 6B), indicative of enhanced behavioral sensitization in the M4 receptor mutant mice.
On day 21, all mice were injected with saline and retested in locomotor activity cages. In this case, all groups of mice showed locomotor responses that were similarly small, independent of genotype and type of pretreatment (Fig. 6A), indicating that the striking sensitizing effects observed with D1-M4-KO mice on day 20 were not due to context conditioning.
We obtained similar results when we performed analogous studies using cocaine (10 mg/kg, s.c.) as a psychostimulant (Fig. 7).
Figure 7.

Behavioral sensitization after repeated cocaine injections in D1–M4-KO mice. A, Locomotor activity during sensitization to cocaine (10 mg/kg, i.p.). Repeated administration of cocaine significantly increased the hyperlocomotor response of D1–M4-KO and control mice. Black arrows indicate when treatment differed from that implied by the legend. Overall analysis revealed a significant genotype effect. ##p < 0.01, ###p < 0.001 versus cocaine response on day 1 (same genotype). B, Cocaine-induced locomotor responses on day 20 (test day). On day 20, all mice, independent of whether they had been treated with saline or cocaine during the initial 6 d injection period, received a single dose of cocaine (10 mg/kg, i.p.). For each genotype, data are expressed as percentage increase in cocaine-induced hyperlocomotor activity observed with cocaine-pretreated versus saline-pretreated mice. This analysis revealed a trend toward increased locomotor activity in D1–M4-KO mice (versus control mice; p = 0.096). Data are given as means ± SEM (n = 11–16).
Dopaminen efflux is enhanced in the nAcc of D1-M4-KO mice
The rewarding effects of all major drugs of abuse involve the release of dopamine in the nAcc (Wise, 1996; Koob et al., 1998), a major component of the ventral striatum that shows a similar cellular architecture as the dorsal striatum and also contains neurons that coexpress M4 muscarinic and D1 dopamine receptors (McGinty, 1999). We therefore used in vivo microdialysis to investigate whether dopamine efflux was altered in the nAcc of D1-M4-KO mice. We found that basal dopamine efflux was increased twofold to threefold in D1-M4-KO mice, as compared to control littermates (Fig. 8A).
Figure 8.

Basal and amphetamine-stimulated dopamine efflux in the nucleus accumbens (nAcc) of D1–M4-KO and control mice. A, Basal dopamine efflux. Basal dopamine levels were determined via in vivo microdialysis. Data are expressed as means ± SEM (n = 6 or 7) of microdialysis samples 1–3 (see Materials and Methods for details). B, Amphetamine-stimulated dopamine efflux. D1–M4-KO mice and control littermates were injected with amphetamine (2 mg/kg, s.c.) at the 90 min time point (arrow). Data are expressed as means ± SEM (n = 6 or 7). *p < 0.05 versus control; #p < 0.05 versus basal dopamine efflux.
We next injected mice with a single dose of amphetamine (2 mg/kg, s.c.) and then measured dopamine efflux in the nAcc over a 150 min period. In both control and D1-M4-KO mice, amphetamine treatment led to significant increases in dopamine release, especially during the first 30 min after drug injection (Fig. 8B). Absolute amphetamine-induced dopamine efflux was significantly enhanced in D1-M4-KO at the 120 min time point, as compared to control mice (Fig. 8B). However, the percentage increases in dopamine efflux above basal levels induced by amphetamine were not significantly different between D1-M4-KO and control mice.
Discussion
A large body of evidence suggests that changes in dopaminergic and muscarinic cholinergic neurotransmission play a critical role in the pathophysiology of various CNS disorders, including Parkinson's disease and schizophrenia (Carlsson, 1988; Gainetdinov et al., 2001; Bymaster et al., 2002; Raedler et al., 2007). The sites of neuronal interactions between dopaminergic and cholinergic signaling pathways that affect motor and behavioral functions are uncertain, primarily because of the involvement of numerous receptor subtypes and many brain regions and neuronal subpopulations. Behavioral and neurochemical analysis of whole-body M4 receptor KO mice suggested that M4 mAChRs play an important role in inhibiting dopaminergic neurotransmission in higher brain regions (Gomeza et al., 1999; Felder et al., 2001; Zhang et al., 2002; Tzavara et al., 2004). The current study provides the first direct demonstration that the M4 mAChR, by acting specifically on a subgroup of D1 receptor-containing striatal projection neurons, mediates dopaminergic/muscarinic cholinergic interactions governing locomotion and various behavioral responses. However, we cannot completely exclude the possibility that other, perhaps minor neuronal subpopulations coexpressing D1 dopamine and M4 muscarinic receptors (that remained undetectable in immunohistochemical studies) might have contributed to the phenotypic changes displayed by the D1-M4-KO mice.
Interestingly, M4 mAChRs are colocalized with D1 dopamine receptors in a subgroup of medium spiny neurons in both the dorsal and ventral striatum (Bernard et al., 1992; Di Chiara et al., 1994; Ince et al., 1997; McGinty, 1999). To test the hypothesis that this subpopulation of M4 mAChRs plays a role in modulating central dopaminergic neurotransmission and dopamine-dependent behavioral processes, we generated mutant mice that selectively lacked M4 mAChRs in D1 receptor-expressing neurons only (D1-M4-KO mice). In vivo phenotyping studies demonstrated that the D1-M4-KO mice displayed several striking behavioral changes. First, we found that the stimulatory locomotor responses following administration of the D1 receptor agonist, SKF82958, amphetamine, or cocaine were significantly enhanced in D1-M4-KO mice. This finding indicates that M4 mAChRs present on D1 receptor neurons inhibit the psychostimulant effects of D1 receptor activation by directly (SKF82958) or indirectly (amphetamine and cocaine) acting dopamine receptor agonists. Since inhibition of amphetamine-induced hyperactivity is frequently used as an animal model to identify drugs with antipsychotic efficacy, our data suggest that M4 mAChRs expressed by D1 receptor neurons play a critical role in mediating the antipsychotic effects of M4 mAChR agonists (allosteric enhancers; see the following paragraph).
Recently, several small molecule ligands that act as allosteric enhancers of M4 mAChR function, including VU0010010 (Shirey et al., 2008) and LY2033298 (Chan et al., 2008), have been identified in large-scale screening programs. These agents selectively increase the affinity and/or efficacy of ACh or other traditional muscarinic agonists at M4 mAChRs (Shirey et al., 2008; Chan et al., 2008; Brady et al., 2008). Importantly, LY2033298 and two centrally active derivatives of VU0010010 displayed pronounced activity in animal models predictive of clinical antipsychotic drug efficacy (Brady et al., 2008; Chan et al., 2008). In a clinical trial, xanomeline, a drug that preferentially activates M1 and M4 mAChRs (Bymaster et al., 2002), showed significant activity in improving both the negative and positive symptoms in schizophrenic patients (Shekhar et al., 2008). Interestingly, xanomeline treatment was superior to that of traditional antipsychotic agents, and the beneficial antipsychotic effects were observed in <1 week after initiation of xanomeline administration (Shekhar et al., 2008). Recent studies with whole-body M4 mAChR KO mice demonstrated that the antipsychotic effects of xanomeline are mediated predominantly by M4 mAChRs (Woolley et al., 2009). Together, these findings strongly suggest that the M4 mAChR subtype represents an attractive new target for the development of novel antipsychotic drugs.
Chronic clinical use of antipsychotic drugs often leads to severe motor side effects such as parkinsonism. These side effects are thought to involve changes in the balance between dopaminergic and muscarinic cholinergic neurotransmission in the striatum (Di Chiara et al., 1994). For drug screening purposes, haloperidol-induced catalepsy is frequently used as an animal model for these unwanted motor side effects. We found that the cataleptic responses to haloperidol, a first-generation antipsychotic drug, were significantly decreased in D1-M4-KO mice. We obtained very similar results when we measured the cataleptic responses to risperidone, a second-generation antipsychotic agent. Although the clinical use of low doses of risperidone is associated with a relatively low incidence of motor side effects, the use of high doses of risperidone leads to a spectrum of motor side effects with a frequency that appears similar to that seen with haloperidol (Tarsy et al., 2002). This observation is in accordance with animal studies demonstrating that high doses of risperidone cause haloperidol-like cataleptic effects in mice (Nakai et al., 2003), as was observed in the present study.
These novel findings strongly suggest that M4 mAChRs present on D1 receptor neurons play a key role in mediating the motor side effects of antipsychotic drugs. These observations also raise the caveat that the potential clinical use of M4 mAChR agonists may increase the likelihood of striatal motor side effects. However, such side effects may be less likely in the case of allosteric enhancers of M4 mAChR function, which require M4 mAChR occupation by endogenous ACh.
Prolonged treatment with amphetamine or other psychostimulants leads to enhanced locomotor stimulation in rodents, a phenomenon referred to as behavioral sensitization. A characteristic feature of behavioral sensitization is that it persists for many weeks or months after termination of the initial drug administration period, thus mimicking long-term sensitivity to drugs observed in human addicts. In the present study, we demonstrated that D1-M4-KO mice showed enhanced behavioral sensitization following repeated amphetamine and, to a lesser degree, cocaine injections. These data support the novel concept that M4 mAChRs present on D1 receptor neurons function to reduce psychostimulant-induced behavioral sensitization. In addition, we demonstrated that basal and amphetamine-induced dopamine efflux were significantly enhanced in the nAcc of D1-M4-KO mice, although the percentage increases in dopamine efflux (above basal levels) induced by amphetamine did not differ between control and D1-M4-KO mice. It is likely that the increase in basal dopamine efflux that we observed with the D1-M4-KO mice involves a multisynaptic mechanism. On the basis of a microdialysis study performed with whole-body (conventional) M4 mAChR KO mice, Tzavara et al. (2004) proposed that activation of M4 receptors expressed by D1 receptor-expressing GABAergic striatal projection neurons leads to decreased GABA release from the nerve terminals. One possibility therefore is that reduced GABA release from these neurons enhances the activity of inhibitory GABAergic interneurons, which are known to contact the cell bodies of midbrain dopamine neurons (Tzavara et al., 2004). This model predicts that the lack of striatal M4 receptors in D1-M4-KO mice results in increased GABA release from striatal projection neurons, triggering enhanced striatal dopamine release from dopaminergic terminals via a multisynaptic mechanism (inhibition of inhibitory GABAergic interneurons). However, this model remains to be tested more rigorously experimentally.
Although D1-M4-KO mice showed a significant increase in basal dopamine efflux in the nucleus accumbens, the mutant mice displayed normal baseline locomotor activity. One possible explanation for this observation is the fact that locomotor behavior is modulated by many neuronal circuits and various other neurotransmitters besides dopamine. It is therefore feasible that D1-M4-KO mice maintained normal basal locomotor activity via activation of compensatory (as yet unknown) pathways.
Similar to amphetamine, most other psychostimulants and drugs of abuse are known to trigger increases in dopamine release in the nAcc (Wise, 1996; Koob et al., 1998). This effect is predicted to be closely associated with the reinforcing effects of these agents (Wise, 1996; Koob et al., 1998). Our findings therefore strongly support the novel concept that centrally active M4 mAChR agonists might be useful in the treatment of drug addiction. This hypothesis needs to be tested more rigorously in future studies.
Several lines of evidence indicate that the D1 dopamine receptor plays a key role in mediating the hyperlocomotor effects of psychostimulants and the behavioral sensitization that can be observed after repeated administration of these agents (Hummel and Unterwald, 2002; Holmes et al., 2004; Le Foll et al., 2009). Moreover, previous work has demonstrated that D1 receptor-mediated activation of the Gs(Golf)/cAMP/PKA signaling cascade, followed by a series of subsequent phosphorylation and downstream signaling events, is critically involved in the manifestation of these psychostimulant-induced behavioral effects (Nestler, 2001; Hummel and Unterwald, 2002; Borgkvist and Fisone, 2007). In the present study, we demonstrated that muscarinic inhibition of D1 receptor-mediated stimulation of cAMP accumulation was abolished in the striatum of D1-M4-KO mice. It is therefore likely that the ability of the Gi-coupled M4 receptor to act as a functional antagonist of cAMP-dependent signaling pathways in D1 receptor-expressing neurons represents the key cellular mechanism underlying the striking behavioral phenotypes displayed by the D1-M4-KO mice.
In conclusion, we demonstrated that a subpopulation of neuronal M4 mAChRs plays a critical role in modulating several important dopamine-dependent behaviors. Our findings provide a rational basis for the development of novel muscarinic drugs for the treatment of various CNS disorders, including Parkinson's disease, schizophrenia, and drug addiction.
Footnotes
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disorders (J.J., Y.C, C.L., C.D., J.W.), the National Institute of Neurological Disorders and Stroke (A.I.L.), the National Institute on Aging and the PhRMA Foundation (A.A.D.), the Lundbeck Foundation, and the Ivan Nielsen Foundation (A.F.). We thank Drs. Nina Wettschureck and Stephan Offermanns (University of Heidelberg, Heidelberg, Germany) for kindly providing the pFlox vector and Dr. Howard Rees (Emory University, Atlanta, GA) for expert technical assistance.
References
- Bernard V, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J Neurosci. 1992;12:3591–3600. doi: 10.1523/JNEUROSCI.12-09-03591.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgkvist A, Fisone G. Psychoactive drugs and regulation of the cAMP/PKA/DARPP-32 cascade in striatal medium spiny neurons. Neurosci Biobehav Rev. 2007;31:79–88. doi: 10.1016/j.neubiorev.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharmacol Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bymaster FP, Felder CC, Ahmed S, McKinzie D. Muscarinic receptors as a target for drugs treating schizophrenia. Curr Drug Targets CNS Neurol Disord. 2002;1:163–181. doi: 10.2174/1568007024606249. [DOI] [PubMed] [Google Scholar]
- Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1988;1:179–186. doi: 10.1016/0893-133x(88)90012-7. [DOI] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJM. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, Christopoulos A, Lazareno S, Birdsall NJ, Bymaster FP, Felder CC. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci U S A. 2008;105:10978–10983. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Everitt BJ. Dopamine receptors in the learning, memory and drug reward circuitry. Semin Cell Dev Biol. 2009;20:403–410. doi: 10.1016/j.semcdb.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Morelli M, Consolo S. Modulatory functions of neurotransmitters in the striatum: ACh/dopamine/NMDA interactions. Trends Neurosci. 1994;17:228–233. doi: 10.1016/0166-2236(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Eglen RM. Muscarinic receptor subtype pharmacology and physiology. Prog Med Chem. 2005;43:105–136. doi: 10.1016/S0079-6468(05)43004-0. [DOI] [PubMed] [Google Scholar]
- Felder CC, Bymaster FP, Ward J, DeLapp N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J Med Chem. 2000;43:4333–4353. doi: 10.1021/jm990607u. [DOI] [PubMed] [Google Scholar]
- Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, Hamilton SE, Gomeza J, Wess J, McKinzie DL. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001;68:2605–2613. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- Franklin KJB, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. New York: Academic; 2007. [Google Scholar]
- Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends Neurosci. 2001;24:527–533. doi: 10.1016/s0166-2236(00)01886-5. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Zhang L, Kostenis E, Felder C, Bymaster F, Brodkin J, Shannon H, Xia B, Deng C, Wess J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M4 muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:10483–10488. doi: 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herroeder S, Reichardt P, Sassmann A, Zimmermann B, Jaeneke D, Hoeckner J, Hollmann MW, Fischer KD, Vogt S, Grosse R, Hogg N, Gunzer M, Offermanns S, Wettschureck N. Guanine nucleotide-binding proteins of the G12 family shape immune functions by controlling CD4+ T cell adhesiveness and motility. Immunity. 2009;30:708–720. doi: 10.1016/j.immuni.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Holmes A, Lachowicz JE, Sibley DR. Phenotypic analysis of dopamine receptor knockout mice; recent insights into the functional specificity of dopamine receptor subtypes. Neuropharmacology. 2004;47:1117–1134. doi: 10.1016/j.neuropharm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- Hummel M, Unterwald EM. D1 dopamine receptor: a putative neurochemical and behavioral link to cocaine action. J Cell Physiol. 2002;191:17–27. doi: 10.1002/jcp.10078. [DOI] [PubMed] [Google Scholar]
- Ince E, Ciliax BJ, Levey AI. Differential expression of D1 and D2 dopamine and m4 muscarinic acetylcholine receptor proteins in identified striatonigral neurons. Synapse. 1997;27:357–366. doi: 10.1002/(SICI)1098-2396(199712)27:4<357::AID-SYN9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Irwin S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioural and physiologic state of the mouse. Psychopharmacologia. 1968;13:222–257. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Gallo A, Le Strat Y, Lu L, Gorwood P. Genetics of dopamine receptors and drug addiction: a comprehensive review. Behav Pharmacol. 2009;20:1–17. doi: 10.1097/FBP.0b013e3283242f05. [DOI] [PubMed] [Google Scholar]
- Lemberger T, Parlato R, Dassesse D, Westphal M, Casanova E, Turiault M, Tronche F, Schiffmann SN, Schütz G. Expression of Cre recombinase in dopaminoceptive neurons. BMC Neurosci. 2007;8:4. doi: 10.1186/1471-2202-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI. Immunological localization of m1–m5 muscarinic acetylcholine receptors in peripheral tissues and brain. Life Sci. 1993;52:441–448. doi: 10.1016/0024-3205(93)90300-r. [DOI] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF. Regulation of neurotransmitter interactions in the ventral striatum. Ann N Y Acad Sci. 1999;877:129–139. doi: 10.1111/j.1749-6632.1999.tb09265.x. [DOI] [PubMed] [Google Scholar]
- Nakai S, Hirose T, Uwahodo Y, Imaoka T, Okazaki H, Miwa T, Nakai M, Yamada S, Dunn B, Burris KD, Molinoff PB, Tottori K, Altar CA, Kikuchi T. Diminished catalepsy and dopamine metabolism distinguish aripiprazole from haloperidol or risperidone. Eur J Pharmacol. 2003;472:89–97. doi: 10.1016/s0014-2999(03)01857-0. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Olianas MC, Maullu C, Onali P. Effects of clozapine on rat striatal muscarinic receptors coupled to inhibition of adenylyl cyclase activity and on the human cloned m4 receptor. Br J Pharmacol. 1997;122:401–408. doi: 10.1038/sj.bjp.0701357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Jones DN, Nelson PR, Jones CM, Quilter CA, Robinson TL, Hagan JJ. Use of SHIRPA and discriminant analysis to characterise marked differences in the behavioural phenotype of six inbred mouse strains. Behav Brain Res. 1999;105:207–217. doi: 10.1016/s0166-4328(99)00072-8. [DOI] [PubMed] [Google Scholar]
- Schaft J, Ashery-Padan R, van der Hoeven F, Gruss P, Stewart AF. Efficient FLP recombination in mouse ES cells and oocytes. Genesis. 2001;31:6–10. doi: 10.1002/gene.1076. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci U S A. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16:23–45. doi: 10.2165/00023210-200216010-00003. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Bymaster FP, Davis RJ, Wade MR, Perry KW, Wess J, McKinzie DL, Felder C, Nomikos GG. M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J. 2004;18:1410–1412. doi: 10.1096/fj.04-1575fje. [DOI] [PubMed] [Google Scholar]
- Vilaró MT, Mengod G, Palacios JM. Advances and limitations of the molecular neuroanatomy of cholinergic receptors: the example of multiple muscarinic receptors. Prog Brain Res. 1993;98:95–101. doi: 10.1016/s0079-6123(08)62385-7. [DOI] [PubMed] [Google Scholar]
- Volpicelli LA, Levey AI. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog Brain Res. 2004;145:59–66. doi: 10.1016/S0079-6123(03)45003-6. [DOI] [PubMed] [Google Scholar]
- Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol. 1996;10:69–99. doi: 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- Wolfe BB, Yasuda RP. Development of selective antisera for muscarinic cholinergic receptor subtypes. Ann N Y Acad Sci. 1995;757:186–193. doi: 10.1111/j.1749-6632.1995.tb17474.x. [DOI] [PubMed] [Google Scholar]
- Woolley ML, Carter HJ, Gartlon JE, Watson JM, Dawson LA. Attenuation of amphetamine-induced activity by the non-selective muscarinic receptor agonist, xanomeline, is absent in muscarinic M4 receptor knockout mice and attenuated in muscarinic M1 receptor knockout mice. Eur J Pharmacol. 2009;603:147–149. doi: 10.1016/j.ejphar.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yamada M, Gomeza J, Basile AS, Wess J. Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1–M5 muscarinic receptor knock-out mice. J Neurosci. 2002;22:6347–6352. doi: 10.1523/JNEUROSCI.22-15-06347.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Belluscio L, Hen R. GOLFα mediates dopamine D1 receptor signaling. J Neurosci. 2000;20:RC91. doi: 10.1523/JNEUROSCI.20-16-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]