

Abstract
AIMS
The aim of the study was to determine the effects of oral clotrimazole troches on the pharmacokinetics of oral and intravenous midazolam in the plasma.
METHODS
We conducted a randomized, open-label, four-way crossover study in 10 healthy volunteers. Each volunteer received oral midazolam 2 mg or intravenous midazolam 0.025 mg kg−1 with and without oral clotrimazole troches 10 mg taken three times daily for 5 days. Each study period was separated by 14 days. Serial blood samples were collected up to 24 h after oral midazolam and 6 h after intravenous midazolam. Plasma concentrations for midazolam and its metabolite 1-hydroxymidazolam were measured and fitted to a noncompartmental model to estimate the pharmacokinetic parameters.
RESULTS
Ten healthy volunteers aged 21–26 years provided written informed consent and were enrolled into the study. Clotrimazole decreased the apparent oral clearance of midazolam from 57 ± 13 l h−1[95% confidence interval 48, 66] to 36 ± 9.8 l h−1 (95% confidence interval 29, 43) (P= 0.003). These changes were accompanied by a decrease in the area under the concentration–time curve (mean difference 22 µg h−1 l−1, P= 0.001) and bioavailability (mean difference 0.21, P= NS). There were no significant differences in the systemic clearance of midazolam with or without clotrimazole troches.
CONCLUSIONS
Oral clotrimazole troches decreased the apparent oral clearance of midazolam; no significant differences in the systemic clearance of midazolam were found.
Keywords: clotrimazole, CYP3A4, extraction, drug–drug interaction, pharmacokinetics, ratiomidazolam
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Clotrimazole troches are commonly used as prophylaxis for thrush that may develop in immunocompromised patients.
Conflicting data suggest that clotrimazole may induce or inhibit cytochrome P450 (CYP) 3A4.
WHAT THIS STUDY ADDS?
We have demonstrated that clotrimazole increased the area under the concentration–time curve and decreased the apparent oral clearance of midazolam following oral administration without affecting the pharmacokinetic properties of midazolam after intravenous administration.
These data permit the suggestion that clotrimazole may cause clinically relevant changes in the pharmacokinetics or pharmacodynamics of concurrently administered oral CYP3A substrates that undergo significant first-pass metabolism in this patient population.
Introduction
Clotrimazole is an oral triazole antifungal drug that is effective for the prophylaxis of thrush in immunocompromised patients [1–3]. Drug–drug interactions occur frequently in this patient population and it is known that other triazole antifungal drugs, including itraconazole, fluconazole, posaconazole and voriconazole, may contribute to these known interactions. Itraconazole, fluconazole, posaconazole and voriconazole work by inhibiting the fungal cytochrome P450 14-α-sterol demethylase. Furthermore, it appears that these triazole antifungal drugs inhibit multiple cytochrome P450 (CYP) enzymes, including the most abundant enzyme in the intestines and liver, CYP3A4 [4–13]. However, it is not clear whether clotrimazole troches can inhibit or induce CYP3A in humans, with conflicting data from laboratory and human studies, and whether sufficient amounts of clotrimazole are systemically absorbed following administration of the oral troches to inhibit or induce CYP3A activity in humans [14–19].
A case report described an interaction between clotrimazole and tacrolimus, a calceurin inhibitor that undergoes metabolism by CYP3A4, in a liver transplant recipient [16]. A study was subsequently conducted in renal allograft recipients to determine if clotrimazole increased the steady-state trough concentrations of oral tacrolimus [17]. The mean tacrolimus blood trough concentrations were higher (P < 0.05) and the mean tacrolimus dose was lower (P < 0.05) in patients receiving clotrimazole compared with those receiving nystatin; nystatin is another antifungal drug that is effective for the prophylaxis of thrush. These findings suggested that oral clotrimazole inhibited intestinal CYP3A or P-glycoprotein (P-gp), since oral tacrolimus undergoes transport by intestinal P-gp and presystemic metabolism by intestinal CYP3A [20]. It is assumed that oral clotrimazole troches yield limited systemic concentrations, since this formulation is designed to dissolve in the mouth and treat local infections in the mouth and throat.
Whereas these data suggest that clotrimazole inhibits CYP3A, two laboratory studies conducted in primary human hepatocytes or human colon carcinoma cells suggest that clotrimazole induces CYP3A4. Clotrimazole increased CYP3A4 expression [15, 18] and activated pregnane X receptor [15, 21], an orphan nuclear receptor that influences the transcription of CYP3A4 and other cytochrome P450 isozymes. These data suggest that clotrimazole induces CYP3A4 activity in the laboratory, but inhibits CYP3A in humans. Two additional laboratory studies conducted in human liver microsomes suggest that clotrimazole inhibits CYP3A4 [14, 19]. Although primary human hepatocytes provide a unique cellular system to examine accurately the effects of xenobiotics on cytochrome P450 enzymes [22], the net effect of clotrimazole on CYP3A activity should be examined in humans. Therefore, we determined the effects of oral clotrimazole troches on the pharmacokinetics of midazolam after oral and intravenous (i.v.) administration; midazolam is a common probe drug used as a surrogate measure of CYP3A activity in humans. Of note, one laboratory study to date does indicate that clotrimazole can induce P-gp protein in human colon carcinoma cells; however, other oral triazole antifungal drugs appear to inhibit P-glycoprotein competitively [23–27]. P-gp is not the focus of the current study.
Methods
Study population
Ten healthy volunteers aged 18–55 years were eligible for enrollment. Subjects were required to have normal hepatic, renal and bone marrow function and physical examination. Subjects were excluded for contraindications to clotrimazole or midazolam, including allergy and documented cardiac or pulmonary diseases. Women of childbearing age were excluded if they were pregnant or breastfeeding. All subjects were instructed to use two proven methods of birth control within 2 weeks of starting and completing the study. Smoking, alcohol consumption, caffeine-containing beverages, or medications known to induce or inhibit CYP3A4 were prohibited for at least 1 week before each study visit. Each subject provided written informed consent before any study-related procedures were initiated.
Study design
This randomized, open-label, four-way crossover study was conducted at the Clinical Research Center at the University of Illinois Medical Center, and was approved by the Scientific Advisory Committee for the Clinical Research Center and the Institutional Review Board for the University of Illinois at Chicago. The four study visits included: (i) oral midazolam, without clotrimazole; (ii) oral midazolam, with clotrimazole; (iii) i.v. midazolam, without clotrimazole and (iv) i.v. midazolam, with clotrimazole. Each study visit was separated by a 2-week wash-out.
Oral clotrimazole troches 10 mg three times daily for 5 days were provided before two of the study visits. The subjects were instructed to take the final clotrimazole dose about 1 h before the administration of midazolam. Adherence was assessed by counting the number of pills remaining the morning of the study visit.
For oral administration, each subject received i.v. midazolam solution 2 mg by mouth. The oral midazolam solution was prepared by mixing the i.v. midazolam solution with 50 ml of apple juice [28]. Each subject provided blood at baseline and 0.5, 1, 2, 4, 5, 6, 12 and 24 h after the dose. For i.v. administration, each subject received midazolam 0.025 mg kg−1 as an i.v. bolus. Each subject provided blood at baseline and 0.25, 0.5, 1, 2, 4, 5, 6 h after the dose. Blood was collected in heparin-containing blood collection tubes. The plasma was separated by centrifugation at 4°C for ∼15 min at 1500 g within 30 min of collection and stored at −20°C until analysis.
Determination of midazolam and 1-hydroxymidazolam
All assays were performed in the Clinical Research Laboratory at the University of Illinois, College of Pharmacy. Plasma concentrations for midazolam and hydoxymidazolam were determined using reverse-phase high-performance liquid chromatography [29]. Midazolam was graciously provided by Hoffman-La Roche Inc. (Nutley, NJ, USA). Hydroxymidazolam was purchased from Lipomed Inc. (Cambridge, MA, USA) and the internal standard, flurazepam, was purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
Briefly, subject plasma or pooled human plasma containing known concentrations of midazolam and 1-hydroxymidazolam was mixed with the internal standard flurazepam and 0.1 M sodium acetate buffer, pH 5.0 that contained β-glucuronidase and incubated at 37°C for 24 h. The mixtures were extracted with methyl-tert butyl ether (Fisher Scientific, Fair Lawn, NJ, USA). After centrifugation, the resulting supernatant was evaporated under a stream of nitrogen in a heated water bath. The residue was reconstituted with a mixture of methanol:2-propanol:perchloric acid containing 0.015% perchloric acid. Midazolam and its metabolites were separated on a 5-µm Spherisorb CN 150 mm × 4.6 mm analytical column (Alltech Associates, Inc., Deerfield, IL, USA). The column eluate was monitored at 215 and 245 nm. An isocratic flow rate of 1.5 ml min−1 was maintained for a total run time of 10 min with 1-hydroxymidazolam, midazolam and flurazepam eluting at 3.2, 3.8 and 4.6 min, respectively. The standard curves for midazolam and 1- hydroxymidazolam were linear from 2.5 to 100 µg l−1 and the lower limit of detection was 0.5 µg l−1. The within-day and between-day variability measured for low, medium and high concentrations of the quality control samples were <5% and <10%, respectively, for both midazolam and 1-hydroxymidazolam and the stock solutions for midazolam and 1-hydroxymidazolam were stable at −20°C for 6 months in 100% methanol.
Determination of clotrimazole plasma concentrations
Plasma clotrimazole concentrations were determined using a previously published method with minor modifications [30]. Subject plasma or pooled human plasma containing known concentrations of clotrimazole were mixed with acetonitrile and the internal standard ketoconazole was buffered with methanol–0.05 M sodium hydroxide (40:60 v/v) and extracted with acetonitrile-n-butyl chloride (1:4 v/v). After centrifugation, the solution was evaporated to dryness and clotrimazole was reconstituted in a mixture of water and acetonitrile (50:50, v/v). An aliquot of the reconstituted material was injected into the high-performance liquid chromatography system fitted with a 10-µm Bondapak C18 300 mm × 3.9 mm analytical column (Waters, Milford, MA, USA). The mobile phase was pumped at 1.0 ml min−1 and the column eluate was monitored at 226 nm. The retention times for ketoconazole and clotrimazole were 8.1 and 16.7 min, respectively. The assay was linear from 1 to 20 µg l−1 with a lower limit of detection of 0.05 µg l−1. The within-day and between-day coefficients of variation, which were determined for low, medium and high concentrations of quality control samples included in each analytic run, were <15%. Clotrimazole and the internal standard were purchased from ICN Biomedical, Inc. (Aurora, OH, USA).
Pharmacokinetic analysis
Noncompartmental analysis was used to estimate the pharmacokinetic parameters for midazolam and 1-hydroxymidazolam with the aide of the program WinNonlin® Professional Version 4.0 (Pharsight Corp., Mountain View, CA, USA). The apparent peak plasma concentration (Cmax) and the time to peak concentration (Tmax) for 1-hydroxymidazolam were visually identified from the individual concentration–time profiles. The elimination rate constant was determined by nonlinear least-square regression and the elimination half-life was calculated by dividing this rate constant by the natural logarithm of two. The area under the concentration–time curve (AUC) for midazolam and 1-hydroxymidazolam was calculated using the log-linear trapezoidal rule. The AUC was extrapolated to infinity by dividing the last measured concentration by the terminal elimination rate constant. The extrapolated area for i.v. and oral data was <15% with and without clotrimazole for most volunteers.
Oral bioavailability (F) was determined by the following equation: F= (AUC0→∞ oral/doseoral)/(AUC0→∞ IV/doseIV). The hepatic extraction ratio (EH) was determined by the following equation: EH= CLIV/QH such that QH= 25.4 ml min−1 kg−1× total body weight (kg). The intestinal availability (FG) and intestinal extraction ratio (EG) were determined by the following equations: FG= 1 –[F/(1 –EH)] and EG= 1 –FG[31, 32]. We assumed the liver was the only organ responsible for eliminating i.v. midazolam when determining the hepatic availability (FH= 1 –EH) and that the intestinal absorption of midazolam approaches unity when determining intestinal availability [31–34].
Statistical analysis
All continuous variables are expressed as a mean ± SD and all categorical variables are expressed as number and percentage. Nine subjects were needed to detect a 25% change in the clearance following oral or i.v. administration with 80% power and a level of statistical significance of P= 0.025 (Bonferroni correction). Repeated measures anova was used to assess the effect of clotrimazole and the potential interaction effect between clotrimazole and the manner of clearance (oral vs. systemic). The Proc GLM in the SAS statistical software version 9.1 (SAS Institute Inc., Cary, NC, USA) was used for performing the statistical analysis.
Results
Study population
Ten healthy volunteers, including six men and four women ranging in age from 21 to 26 years, were enrolled and completed the study. The study population included seven Asians, two Whites and one Hispanic. The mean total body weight was 70.4 ± 10.7 kg. No adverse events were reported.
Oral midazolam
The mean apparent oral clearance of midazolam was decreased after clotrimazole administration from 57 ± 13 to 36 ± 9.8 l h−1 (P= 0.003). These changes were accompanied by a statistically significant increase in the mean area under the concentration–time curve (mean difference = 22 µg h−1 l−1, P= 0.001) (Table 1, Figure 1a). The mean bioavailability F increased from 0.56 ± 0.20 (without clotrimazole) to 0.77 ± 0.15 (with clotrimazole), the mean FG increased from 0.75 ± 0.23 to 0.95 ± 0.11 and the mean EG decreased from 0.25 ± 0.23 to 0.06 ± 0.09 after clotrimazole administration; these differences did not reach statistical significance.
Table 1.
Estimates of midazolam and 1-hydroxymidazolam pharmacokinetic parameters determined after oral (2 mg) and intravenous (0.025 mg kg−1) administration of midazolam with and without oral clotrimazole troches 10 mg three times daily for 5 days before the single administration of midazolam
| Parameter | Without clotrimazole | With clotrimazole | P-value |
|---|---|---|---|
| Oral midazolam | |||
| Midazolam | |||
| AUC0→∞ (µg h−1 l−1) | 36 ± 12 | 58 ± 14 | 0.001 |
| CL/F (l h−1) | 57 ± 13 | 36 ± 9.8 | 0.003 |
| t1/2 (h) | 2.1 ± 0.65 | 2.2 ± 1.4 | NS |
| F | 0.56 ± 0.20 | 0.77 ± 0.15 | NS |
| FG | 0.75 ± 0.23 | 0.95 ± 0.11 | NS |
| EG | 0.25 ± 0.23 | 0.06 ± 0.09 | NS |
| Hydroxymidazolam | |||
| Cmax (µg l−1) | 28 ± 7.9 | 19 ± 4.3 | <0.001 |
| AUC0→6 (µg h−1 l−1) | 50 ± 8.9 | 72 ± 12 | <0.0001 |
| Intravenous midazolam | |||
| Midazolam | |||
| Co (µg l−1) | 34 ± 9.3 | 36 ± 7.3 | NS |
| AUC0→∞ (µg h−1 l−1) | 62 ± 25 | 56 ± 13 | NS |
| CL (l h−1) | 31 ± 8.6 | 27 ± 4.6 | NS |
| t1/2 (h) | 1.7 ± 0.49 | 1.8 ± 0.57 | NS |
| EH | 0.30 ± 0.11 | 0.26 ± 0.06 | NS |
| Hydroxymidazolam | |||
| Cmax (µg l−1) | 9.7 ± 5.0 | 9.2 ± 3.5 | NS |
| AUC0→6 (µg h−1 l−1) | 40 ± 18 | 30 ± 14 | NS |
Data are given as mean ± 0.5 standard deviation. AUC, area under the concentration–time curve; CL, systemic clearance; CL/F, apparent oral clearance; Co, initial concentration; Cmax, maximum concentration; EG, intestinal extraction ratio; EH, hepatic extraction ratio; F, bioavailability; FG, intestinal availability; t1/2, terminal elimination half-life; NS, not significant (P > 0.05).
Figure 1.
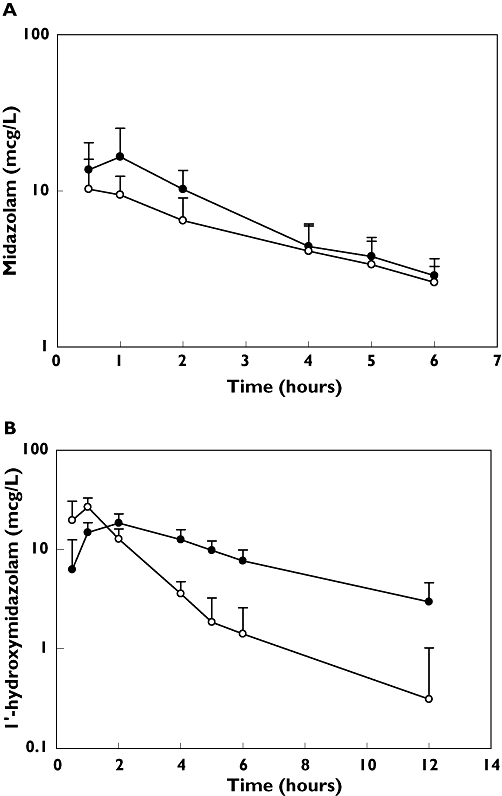
(a) The mean (± standard deviation) plasma concentration–time profile for midazolam in 10 healthy volunteers. Each volunteer received oral midazolam 2 mg with (filled circles) and without (open circles) clotrimazole 10 mg three times daily for 5 days before the single-dose administration of midazolam. (b) The mean (± standard deviation) plasma concentration–time profile for 1-hydroxymidazolam in 10 healthy volunteers. Each volunteer received oral midazolam 2 mg with (filled circles) and without (open circles) clotrimazole 10 mg three times daily for 5 days before the single-dose administration of midazolam. Clotrimazole (—•—); Control (—○—)
The mean Cmax of 1-hydroxymidazolam declined from 28 ± 7.9 to 19 ± 4.3 µg l−1 after clotrimazole administration (P= 0.001). However, the AUC0-6 for 1-hydroxymidazolam increased from 50 ± 8.9 to 72 ± 12 µg h−1 l−1 after receiving clotrimazole (P < 0.001) (Figure 1b).
Intravenous midazolam
There were no significant differences in the systemic clearance of midazolam with or without clotrimazole troches (Table 1, Figure 2a). The hepatic extraction ratio similarly remained unaffected by clotrimazole. Subsequently, the Cmax and AUC0-6 for 1-hydroxymidazolam were unchanged (Table 1, Figure 2b).
Figure 2.
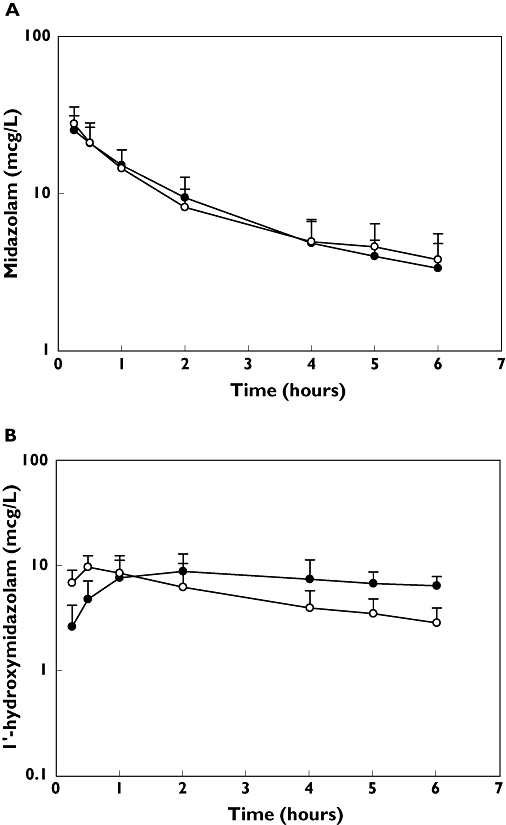
(a) The mean (± standard deviation) plasma concentration–time profile for midazolam in 10 healthy volunteers. Each volunteer received intravenous midazolam 0.025 mg kg−1 with (filled circles) and without (open circles) clotrimazole 10 mg three times daily for 5 days before the single-dose administration of midazolam. (b) The mean (± standard deviation) plasma concentration–time profile for 1-hydroxymidazolam in 10 healthy volunteers. Each volunteer received intravenous midazolam 0.025 mg kg−1 with (filled circles) and without (open circles) clotrimazole 10 mg three times daily for 5 days before the single dose administration of midazolam. Clotrimazole (—•—); Control (—○—)
Clotrimazole concentrations
The subjects achieved mean concentrations of clotrimazole of 2.7 ± 1.8 µg l−1 (7.8 ± 5.2 nm) and 2.4 ± 1.0 µg l−1 (7.0 ± 2.9 nm) 30 min after receiving oral or i.v. midazolam, respectively.
Discussion
The results of this study suggest that oral clotrimazole troches administered for 5 days can reduce the apparent oral clearance of midazolam. We have demonstrated that clotrimazole administered for 5 days before a single oral dose of midazolam 2 mg decreased the apparent oral clearance (∼40%) and increased the AUC (∼40%). Furthermore, oral and intestinal bioavailability were increased, although these differences did not reach statistical significance. The pharmacokinetic properties of midazolam following i.v. administration were not affected by clotrimazole administration. Comparatively, clotrimazole troches caused a 60% decrease in the apparent oral clearance of tacrolimus in kidney transplant recipients [17].
Our findings appear to indicate that clotrimazole reduces the presystemic metabolism of midazolam by intestinal CYP3A4 without affecting the systemic metabolism of midazolam by hepatic CYP3A4. We propose that clotrimazole achieved adequate local concentrations in the intestines to inhibit intestinal CYP3A, but insufficient concentrations in the hepatic circulation to inhibit hepatic CYP3A. Laboratory data in human liver microsomes estimate the Ki to range from 250 pm to 20 nm[14, 19]; our data suggest that the estimate of 20 nm is more accurate, since we noted only inhibition of intestinal CYP3A4 with systemic concentrations of 7 nm. Furthermore, the elimination half-life of clotrimazole may be short and limit the concentrations achieved in the hepatic circulation. The pharmacokinetic properties of clotrimazole following topical administration of oral troches are not available. Alternatively, clotrimazole may have delayed the intestinal dissolution of midazolam following oral administration of the i.v. formulation. The solubility of midazolam is pH dependent and the oral absorption of the parent drug is highly variable following nonstandard oral formulations [35]. Clotrimazole is highly water soluble, whereas midazolam is highly fat soluble. Oral absorption and metabolite production would be delayed, as illustrated in Figure 1a,b.
The effects of clotrimazole on CYP3A activity are consistent with previous reports describing the effects of ketoconazole, posaconazole and fluconazole on midazolam pharmacokinetic parameters in humans [11, 36–39] and on CYP3A4 activity in the laboratory [40], in that these drugs typically inhibit CYP3A activity. These findings support the conclusions generated by the study conducted in renal allograft recipients that oral clotrimazole increased the mean trough concentration for oral tacrolimus by inhibiting CYP3A activity [17]. Since midazolam is not a substrate of P-gp, the findings from these two studies collectively indicate that clotrimazole is a clinically important CYP3A inhibitor, although it was once believed that topical administration of clotrimazole did not lead to systemic absorption. Furthermore, consistent with the results from other studies, it appears that triazole antifungal agents have a profound inhibitory effect on the intestinal metabolism of CYP3A substrates. This implies that inhibiting intestinal CYP3A by these drugs is likely to contribute to the reported drug–drug interactions in immunocompromised patients. However, the consequences of this potential drug–drug interaction depend on multiple factors, including route of administration, schedule of administration, and dose, as well as the correlation between the pharmacokinetics and pharmacodynamic parameters.
The changes in the pharmacokinetic properties of oral midazolam were accompanied by changes in the Cmax and AUC of the 1-hydroxymidazolam metabolite. The Cmax probably decreased secondary to inhibition of CYP3A and the reduced metabolism of the parent drug. However, the AUC0–6 substantially increased. The data appear to suggest that the elimination of this metabolite was suppressed. This metabolite is glucuronidated, then renally eliminated. Since triazole antifungal drugs can inhibit glucuronidation, it is plausible that clotrimazole limited the elimination of this metabolite [41–43]. The Cmax was also delayed following oral administration, suggesting that clotrimazole may be affecting intestinal absorption (Tmax control 0.8 ± 0.5 0.25 h, and clotrimazole 2.0 ± 0.5 0.90 h, P= 0.001); these changes were not observed following i.v. administration.
Laboratory and human data conflict regarding the overall effects of clotrimazole on CYP3A. As stated in the Introduction, two laboratory studies indicate that clotrimazole 2–20 µM substantially induces CYP3A4 protein and activity and activates the pregnane X receptor [15, 18]. These concentrations greatly exceed systemic concentrations in humans and the inhibitory constant reported in the laboratory [33]. Therefore, it is unlikely that induction will occur in humans.
In summary, our data suggest that oral clotrimazole troches inhibit the presystemic metabolism of midazolam without altering systemic metabolism. Therefore, it is plausible that oral clotrimazole troches could reduce the availability of oral drugs that undergo metabolism by intestinal CYP3A4.
Competing interests
The views expressed in this paper reflect the views of the authors and do not reflect official policy. No official endorsement by the FDA is intended or should be inferred.
This study was supported in part by a New Investigator Program grant awarded by the American Association for Colleges of Pharmacy to S.S.S. and the General Clinical Research Center at the University of Illinois at Chicago (NIH grant M01-RR-13987).
REFERENCES
- 1.Gombert ME, duBouchet L, Aulicino TM, Butt KM. A comparative trial of clotrimazole troches and oral nystatin suspension in recipients of renal transplants. Use in prophylaxis of oropharyngeal candidiasis. JAMA. 1987;258:2553–5. [PubMed] [Google Scholar]
- 2.Owens NJ, Nightingale CH, Schweizer RT, Schauer PK, Dekker PT, Quintiliani R. Prophylaxis of oral candidiasis with clotrimazole troches. Arch Intern Med. 1984;144:290–3. [PubMed] [Google Scholar]
- 3.Ruskin JD, Wood RP, Bailey MR, Whitmore CK, Shaw BW. Comparative trial of oral clotrimazole and nystatin for oropharyngeal candidiasis prophylaxis in orthotopic liver transplant patients. Oral Surg Oral Med Oral Pathol. 1992;74:567–71. doi: 10.1016/0030-4220(92)90345-q. [DOI] [PubMed] [Google Scholar]
- 4.Sadaba B, Campanero MA, Quetglas EG, Azanza JR. Clinical relevance of sirolimus drug interactions in transplant patients. Transplant Proc. 2004;36:3226–8. doi: 10.1016/j.transproceed.2004.10.056. [DOI] [PubMed] [Google Scholar]
- 5.Hyland R, Jones BC, Smith DA. Identification of the cytochrome P450 enzymes involved in the N-oxidation of voriconazole. Drug Metab Dispos. 2003;31:540–7. doi: 10.1124/dmd.31.5.540. [DOI] [PubMed] [Google Scholar]
- 6.Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111–80. doi: 10.2165/00003088-200038020-00002. [DOI] [PubMed] [Google Scholar]
- 7.Liu P, Foster G, LaBadie RR, Gutierrez MJ, Sharma A. Pharmacokinetic interaction between voriconazole and efavirenz at steady state in healthy male subjects. J Clin Pharmacol. 2008;48:73–84. doi: 10.1177/0091270007309703. [DOI] [PubMed] [Google Scholar]
- 8.Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805–8. doi: 10.1248/bpb.28.1805. [DOI] [PubMed] [Google Scholar]
- 9.Pea F, Baccarani U, Tavio M, Cojutti P, Adani GL, Londero A, Baraldo M, Franceschi L, Furlanut M, Viale P. Pharmacokinetic interaction between everolimus and antifungal triazoles in a liver transplant patient. Ann Pharmacother. 2008;42:1711–6. doi: 10.1345/aph.1L330. [DOI] [PubMed] [Google Scholar]
- 10.Saari TI, Laine K, Bertilsson L, Neuvonen PJ, Olkkola KT. Voriconazole and fluconazole increase the exposure to oral diazepam. Eur J Clin Pharmacol. 2007;63:941–9. doi: 10.1007/s00228-007-0350-0. [DOI] [PubMed] [Google Scholar]
- 11.Krishna G, Moton A, Ma L, Savant I, Martinho M, Seiberling M, McLeod J. Effects of oral posaconazole on the pharmacokinetic properties of oral and intravenous midazolam: a phase I, randomized, open-label, crossover study in healthy volunteers. Clin Ther. 2009;31:286–98. doi: 10.1016/j.clinthera.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 12.Moton A, Ma L, Krishna G, Martinho M, Seiberling M, McLeod J. Effects of oral posaconazole on the pharmacokinetics of sirolimus. Curr Med Res Opin. 2009;25:701–7. doi: 10.1185/03007990802644209. [DOI] [PubMed] [Google Scholar]
- 13.Wexler D, Courtney R, Richards W, Banfield C, Lim J, Laughlin M. Effect of posaconazole on cytochrome P450 enzymes: a randomized, open-label, two-way crossover study. Eur J Pharm Sci. 2004;21:645–53. doi: 10.1016/j.ejps.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Gibbs MA, Kunze KL, Howald WN, Thummel KE. Effect of inhibitor depletion on inhibitory potency: tight binding inhibition of CYP3A by clotrimazole. Drug Metab Dispos. 1999;27:596–9. [PubMed] [Google Scholar]
- 15.Luo G, Cunningham M, Kim S, Burn T, Lin J, Sinz M, et al. CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab Dispos. 2002;30:795–804. doi: 10.1124/dmd.30.7.795. [DOI] [PubMed] [Google Scholar]
- 16.Mieles L, Venkataramanan R, Yokoyama I, Warty VJ, Starzl TE. Interaction between FK506 and clotrimazole in a liver transplant recipient. Transplantation. 1991;52:1086–7. doi: 10.1097/00007890-199112000-00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasquez E, Pollak R, Benedetti E. Clotrimazole increases tacrolimus blood levels: a drug interaction in kidney transplant patients. Clin Transplant. 2001;15:95–9. doi: 10.1034/j.1399-0012.2001.150203.x. [DOI] [PubMed] [Google Scholar]
- 18.Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49:311–8. [PubMed] [Google Scholar]
- 19.Zhang W, Ramamoorthy Y, Kilicarslan E, Nolte H, Tyndale RF, Sellers EM. Inhibition of cytochrome P450 by antifungal imidazole derivatives. Drug Metab Dispos. 2002;30:314–8. doi: 10.1124/dmd.30.3.314. [DOI] [PubMed] [Google Scholar]
- 20.Hebert MF. Contributions of hepatic and intestinal metabolism and P-glycoprotein to cyclosporine and tacrolimus oral drug delivery. Adv Drug Deliv Rev. 1997;27:201–14. doi: 10.1016/s0169-409x(97)00043-4. [DOI] [PubMed] [Google Scholar]
- 21.Pascussi JM, Drocourt L, Fabre JM, Maurel P, Vilarem MJ. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol Pharmacol. 2000;58:361–72. doi: 10.1124/mol.58.2.361. [DOI] [PubMed] [Google Scholar]
- 22.Gomez-Lechon MJ, Donato T, Ponsoda X, Castell JV. Human hepatic cell cultures: in vitro and in vivo drug metabolism. Altern Lab Anim. 2003;31:257–65. doi: 10.1177/026119290303100307. [DOI] [PubMed] [Google Scholar]
- 23.Heiskanen T, Backman JT, Neuvonen M, Kontinen VK, Neuvonen PJ, Kalso E. Itraconazole, a potent inhibitor of P-glycoprotein, moderately increases plasma concentrations of oral morphine. Acta Anaesthesiol Scand. 2008;52:1319–26. doi: 10.1111/j.1399-6576.2008.01739.x. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of single and multiple doses of itraconazole on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br J Clin Pharmacol. 2006;62:372–6. doi: 10.1111/j.1365-2125.2006.02689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br J Clin Pharmacol. 2006;61:538–44. doi: 10.1111/j.1365-2125.2006.02613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Angirasa AK, Koch AZ. P-glycoprotein as the mediator of itraconazole–digoxin interaction. J Am Podiatr Med Assoc. 2002;92:471–2. doi: 10.7547/87507315-92-8-471. [DOI] [PubMed] [Google Scholar]
- 27.Jalava KM, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin. Ther Drug Monit. 1997;19:609–13. doi: 10.1097/00007691-199712000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Kinirons MT, O'Shea D, Kim RB, Groopman JD, Thummel KE, Wood AJ, Wilkinson GR. Failure of erythromycin breath test to correlate with midazolam clearance as a probe of cytochrome P4503A. Clin Pharmacol Ther. 1999;66:224–31. doi: 10.1016/S0009-9236(99)70029-9. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann B, Boulieu R. Determination of midazolam and its unconjugated 1-hydroxy metabolite in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1995;674:138–42. doi: 10.1016/0378-4347(95)00286-6. [DOI] [PubMed] [Google Scholar]
- 30.de Bruijn P, Kehrer DF, Verweij J, Sparreboom A. Liquid chromatographic determination of ketoconazole, a potent inhibitor of CYP3A4-mediated metabolism. J Chromatogr B Biomed Sci Appl. 2001;753:395–400. doi: 10.1016/s0378-4347(00)00573-9. [DOI] [PubMed] [Google Scholar]
- 31.Lee JI, Chaves-Gnecco D, Amico JA, Kroboth PD, Wilson JW, Frye RF. Application of semisimultaneous midazolam administration for hepatic and intestinal cytochrome P450 3A phenotyping. Clin Pharmacol Ther. 2002;72:718–28. doi: 10.1067/mcp.2002.129068. [DOI] [PubMed] [Google Scholar]
- 32.Gorski JC, Vannaprasaht S, Hamman MA, Ambrosius WT, Bruce MA, Haehner-Daniels B, et al. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin Pharmacol Ther. 2003;74:275–87. doi: 10.1016/S0009-9236(03)00187-5. [DOI] [PubMed] [Google Scholar]
- 33.Gorski JC, Jones DR, Haehner-Daniels BD, Hamman MA, O'Mara EM, Jr, Hall SD. The contribution of intestinal and hepatic CYP3A to the interaction between midazolam and clarithromycin. Clin Pharmacol Ther. 1998;64:133–43. doi: 10.1016/S0009-9236(98)90146-1. [DOI] [PubMed] [Google Scholar]
- 34.Smith MT, Eadie MJ, Brophy TO. The pharmacokinetics of midazolam in man. Eur J Clin Pharmacol. 1981;19:271–8. doi: 10.1007/BF00562804. [DOI] [PubMed] [Google Scholar]
- 35.Cote CJ, Cohen IT, Suresh S, Rabb M, Rose JB, Weldon C, Davis PJ, Bikhazi GB, Karls HW, Hummer KA, Hannallah RS, Khoo KC, Collins P. A comparison of three doses of a commercially prepared oral midazolam syrup in children. Anesth Analg. 2002;94:37–43. doi: 10.1097/00000539-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 36.Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66:461–71. doi: 10.1016/S0009-9236(99)70009-3. [DOI] [PubMed] [Google Scholar]
- 37.Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55:481–5. doi: 10.1038/clpt.1994.60. [DOI] [PubMed] [Google Scholar]
- 38.Ahonen J, Olkkola KT, Neuvonen PJ. Effect of route of administration of fluconazole on the interaction between fluconazole and midazolam. Eur J Clin Pharmacol. 1997;51:415–9. doi: 10.1007/s002280050223. [DOI] [PubMed] [Google Scholar]
- 39.Ahonen J, Olkkola KT, Takala A, Neuvonen PJ. Interaction between fluconazole and midazolam in intensive care patients. Acta Anaesthesiol Scand. 1999;43:509–14. doi: 10.1034/j.1399-6576.1999.430504.x. [DOI] [PubMed] [Google Scholar]
- 40.Gibbs MA, Kunze KL, Howald WN, Thummel KE. Effect of inhibitor depletion on inhibitory potency: tight binding inhibition of CYP3A by clotrimazole. Drug Metab Dispos. 1999;27:596–9. [PubMed] [Google Scholar]
- 41.Sawamura R, Sato H, Kawakami J, Iga T. Inhibitory effect of azole antifungal agents on the glucuronidation of lorazepam using rabbit liver microsomes in vitro. Biol Pharm Bull. 2000;23:669–71. doi: 10.1248/bpb.23.669. [DOI] [PubMed] [Google Scholar]
- 42.Asgari M, Back DJ. Effect of azoles on the glucuronidation of zidovudine by human liver UDP-glucuronosyltransferase. J Infect Dis. 1995;172:1634–6. doi: 10.1093/infdis/172.6.1634. [DOI] [PubMed] [Google Scholar]
- 43.Sampol E, Lacarelle B, Rajaonarison JF, Catalin J, Durand A. Comparative effects of antifungal agents on zidovudine glucuronidation by human liver microsomes. Br J Clin Pharmacol. 1995;40:83–6. doi: 10.1111/j.1365-2125.1995.tb04540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]