

Abstract
AIMS
The pathophysiology of angiotensin-converting enzyme inhibitor (ACEi)-induced angio-oedema remains unclear. We have investigated the impact of ACE insertion/deletion (I/D) polymorphism in combination with serum ACE activity as well as the bradykinin B2 receptor 2/3 and c.C181T polymorphisms.
METHODS
We analysed the ACE I/D as well as bradykinin B2 (2/3 and C181T) receptor polymorphisms in 65 patients with documented episodes of ACEi-induced angio-oedema and 65 patients matched for age and sex being under ACEi treatment without history of angio-oedema. Furthermore, we determined serum ACE activity in 47 of the 65 angio-oedema patients 3 months after the angio-oedema attack and compared these values with 51 healthy individuals (control II).
RESULTS
No risk association was identified between ACE I/D (I-allele: 0.42 vs. 0.41, D-allele: 0.58 vs. 0.59; P= 0.095) or bradykinin B2 receptor polymorphisms and the development of angio-oedema during ACEi treatment. We found a trend of lower serum ACE activity in ACE I/I genotypes in comparison with control II (I/I: 28 ± 4.5 vs. 33 ± 1.8 U l−1; ID: 39 ± 3.3 vs. 41 ± 1 U l−1; DD: 56 ± 6.7 vs. 52 ± 1.8 U l−1; P= 0.9).
CONCLUSIONS
Our data suggest that polymorphism of ACE I/D and the bradykinin B2 receptor polymorphisms are not involved in the development of ACEi-induced angio-oedema when considered individually. Further studies should be carried out to clarify whether a combination of these polymorphisms might be a risk factor for ACEi-induced angio-oedema.
Keywords: ACE I/D polymorphism, angio-oedema, bradykinin, bradykinin B2 receptor polymorphism, serum ACE activity
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Angiotensin-converting enzyme inhibitor (ACEi)-induced angio-oedema is an underestimated clinical life-threatening problem.
The incidence of this non-allergic, bradykinin-induced drug side-effect is 1 : 4000.
Although most ACEi-treated patients probably have an increased bradykinin plasma concentration, only 0.5% of them develop an angio-oedema and nothing is known about potential risk factors.
WHAT THIS STUDY ADDS
In our attempt to elucidate the unpredictable character of ACEi-induced angio-oedema, we investigated bradykinin B2 receptor 2/3 and c.C181T polymorphisms as well as the ACE insertion/deletion polymorphism in combination with serum ACE activity in 65 patients.
ACE insertion/deletion and bradykinin B2 receptor polymorphisms are not involved in the development of ACEi-induced angio-oedema.
Further studies should be carried out to clarify whether a combination of these polymorphisms might be a risk factor for ACEi-induced angio-oedema.
Introduction
Sudden occurrence of subcutaneous or submucosal swelling, so-called angio-oedema, is a well-known side-effect of angiotensin-converting enzyme inhibitors (ACEi), which may become life-threatening if the upper airway is involved [1–3]. More than 6.5 million patients in Germany receive ACEi and, in view of the estimated incidence of 0.5% (1 : 4000) drug-induced angio-oedema, this medication is believed to cause most of the bradykinin-induced angio-oedema [1, 4].
The duration of ACEi treatment at the first manifestation of angio-oedema has been reported as from 1 day to 8 years and it may take up to 10 years until the causal relationship is recognized and the ACEi is discontinued [1]. In our experience, a considerable part of all ACEi-induced angio-oedema occurs at an average of 35.8 ± 5.3 months after initiation of treatment [5]. This substantial delay can be viewed as an important cause for non-identification of late-onset angio-oedema as a drug-related side-effect. Thus, it appears to be of clinical relevance to develop measures that allow to predict ACEi-induced angio-oedema to a certain extent and thereby prevent episodes of this potentially dangerous condition.
At present, ACEi-induced angio-oedema ia believed to result from alterations of bradykinin metabolism pathways causing increased concentrations of bradykinin, a mediator of inflammation, vasodilation and capillary leakage [6–12]. In general, changes of bradykinin steady-state concentration in plasma and tissue can follow either diminished activity of degrading enzymes such as ACE or increased activity of generating enzymes such as kallikrein. So far, it remains unclear why angio-oedema occurs in a small fraction of the many patients treated with ACEi and why many such cases occur only after a considerable time interval of uneventful ACEi medication [5, 13]. Furthermore, several factors other than bradykinin might play a role in the aetiology of ACEi-induced angio-oedema, including substance P and prostaglandin E2[6]. Recently, we reported on significantly increased serum levels of C-reactive protein and fibrinogen in patients with acute ACEi-induced angio-oedema but not in patients with other angio-oedema of unknown origin (idiopathic angio-oedema) [5, 13]. These findings are indicative of an inflammatory pathophysiology of angio-oedema. However, the observation of increased acute-phase proteins during angio-oedema may be an epiphenomenon as well and is currently under further investigation.
ACE catalyses the transformation of angiotensin I to angiotensin II, a key molecule in cardiovascular regulation [14]. In addition, ACE substantially contributes to the inactivation of bradykinin and, therefore, inhibitors of ACE inevitably account for both decreased angiotensin II and increased bradykinin plasma levels [15].
The ACE gene is located on chromosome 17q23 and harbours a functionally relevant polymorphism in intron 16, where a 287-bp alu repeat is either present (I-allele: insertion) or absent (D-allele: deletion) [16, 17]. The ACE D-allele has been found to be associated with higher serum enzyme activity compared with the I-allele, i.e. bradykinin is more (D-allele) or less (I-allele) readily degraded [18–20].
Further determinants that might be involved in genetic predispositions to develop ACEi-induced angio-oedema are bradykinin receptors. In general, nonapeptide bradykinin binds to two distinct G-protein-coupled receptors, the bradykinin receptors B1 and B2[21]. Activation of bradykinin receptors has been implicated in a variety of physiological and pathological processes, including vasodilation, increased vascular permeability, oedema, pain, smooth muscle contraction and cell proliferation [22]. Most of the physiological activities are mediated by the B2 receptor [23, 24]. Hence, genetic variations of the bradykinin B2 receptor may be responsible for triggering ACEi-induced angio-oedema. Two well-defined and frequently occurring polymorphisms have been described for this receptor. One polymorphism located in exon 1 next to the promoter region is a repeat tandem polymorphism consisting of three alleles and starting at nucleotide position 12 after the transcription starting point. DNA sequencing has revealed two (2G) and three (3G) repeat units (GGTGGGGAC), respectively [25]. The second polymorphism, a C→T transition (c.C181T), which leads to an amino acid exchange from arginine to cysteine in the receptor protein at position 14 (p.R14C), is located in exon 2 at nucleotide position 181 [22, 24, 25].
We have investigated the ACE I/D polymorphism in combination with serum ACE activity as well as the bradykinin B2 receptor 2/3 and c.C181T polymorphisms.
Materials and methods
Patients
In a bi-centre setting, 65 consecutive patients presenting with acute ACEi-induced angio-oedema were included, (the characteristics of these patients are summarized in Table 1). Patients with angio-oedema of unknown origin and those with known aetiology other than ACEi-treatment (e.g. C1-esterase inhibitor deficiency type 1/type 2 or urticaria) were excluded. The mean duration of treatment with ACEi was 36.7 ± 4.8 months. The mean duration of the swellings was 60 ± 10 h and the event-free time after diagnosis was 4.3 ± 0.24 years. In 47 (72%) of the 65 angio-oedema patients, we were able to measure serum ACE activity (at least) 3 months after discontinuation of ACEi treatment.
Table 1.
Characteristics of patients with angiotensin-converting enzyme inhibitor (ACEi)-induced angio-oedema (values are given as mean ± SEM)
| Base-line characteristics of the patients | |
|---|---|
| Female (%) | 47 |
| Age (years) | 62 ± 1.6 |
| Blood pressure (mmHg) | |
| Systolic | 148 ± 3 |
| Diastolic | 82 ± 2 |
| Heart rate (frequency min−1) | 85 ± 2 |
| Duration of treatment with ACEi until occurrence of angio-oedema (months) | 36.7 ± 4.8 |
| ACE serum activity (U l−1) | 42.7 ± 3.1 |
| Diseases (n) | |
| Hypertension | 47 |
| Coronary artery disease | 25 |
| Diabetes | 14 |
| Hyperlipidaemia | 7 |
| Chronic renal failure | 1 |
| Laboratory results | |
| Leucocytes (× 1000 µl−1) | 9.1 ± 0.4 |
| Fibrinogen (mg dl−1) | 429 ± 23 |
| C-reactive protein (mg dl−1) | 3.9 ± 0.5 |
| Haemoglobin (g l−1) | 14 ± 0.3 |
The control group consisted of 65 age- and sex-matched patients with similar risk factors and comorbidity also under ACEi medication but without a history of angio-oedema episodes (control I). The mean duration of treatment with ACEi was 49 ± 3.5 months. Furthermore, 51 healthy age- and sex-matched individuals without ACEi medication served as control group for normal serum ACE activity (control II).
The study was approved by the local Ethics Committee. All patients submitted their written consent forms.
ACE I/D genotyping
Genomic DNA was extracted from peripheral blood leucocytes using the QIAmp DNA Blood Mini Kit (Qiagen, Hilden Germany) according to the instructions of the manufacturer. The ACE-I/D polymorphism was analysed by polymerase chain reaction (PCR) amplification of genomic DNA using the following primers (MWG Biotech, Ebersberg, Germany): sense 5′ GCC CTG CAG GTG TCT GCA GCA TGT 3′, antisense 5′ GGA TGG CTC TCC CCG CCT TGT CTC 3′. Thermocycling included an initial denaturation step (5 min at 94°C), and 31 cycles consisting of 30 s at 94°C, 25 s at 64°C and 2 min at 70°C, followed by a final extension step at 70°C for 7 min. Amplification with this primer pair produced products of 597 bp and 310 bp corresponding to the I- and D-alleles, respectively. Since the D-allele is preferentially amplified, mistyping of ID heterozygotes as DD homozygotes is possible; therefore, each sample found to have the DD genotype was subjected to a second independent PCR amplification with an insertion-specific primer pair (MWG Biotech): sense 5′ TGG GAC CAC AGC GCC CGC CAC TAC 3′, antisense 5′ TCG CCA GCC CTC CCA TGC CCA TAA 3′. The reaction yielded a 335-bp amplicon only in the presence of an I-allele.
Serum ACE activity
The activity of ACE was determined with a colorimetric assay (ACE kinetic test®; Buehlmann Laboratories, Schoenenbruch, Switzerland) in serum immediately obtained from venous blood draws. In 47 patients of the 65 (72%) angio-oedema group, we were able to collect a second serum sample 3 months after discontinuation the ACEi medication; here any drug-related alteration of ACE activity could no longer be expected. The control group consisted of healthy sex- and age-matched individuals (n= 51) without ACEi medication.
Bradykinin B2 polymorphisms (exon 1 and exon 2)
The bradykinin B2 receptor exon 1 polymorphism (2/3-polymorphism) was analysed by PCR amplification of genomic DNA using the following primers (MWG Biotech): sense 5′-GCC CTT GAA AGA TGA GCT-3′, antisense 5′-AAC TCC CCA CGA CCA CAG-3′. For analyses of the exon 2 polymorphism (c.T181C-polymorphism), we used the primers: sense 5′-CCA TTT CTC CTC CCT GCT CGGA-3′, antisense GGT GGG CAC GGA GTC CTC TC-3′[24, 26]. Cycling conditions for the PCR of exon 1 were an initial 5 min at 95°C, followed by 40 s at 94°C, 40 s at 53°C and 40 s at 72°C for 29 cycles, and a final extension time of 7 min at 72°C. Cycling conditions for the PCR of exon 2 were an initial 5 min at 95°C, followed by 40 s at 94°C, 40 s at 61°C and 40 s at 72°C for 34 cycles, and a final extension time of 5 min at 72°C. For the exon 1 polymorphism, PCR products were subjected to single-strand conformation polymorphism electrophoresis. A 10-µl portion of the PCR product was denatured at 95°C for 5 min. Electrophoresis was carried out in 1× TBE buffer (89 mM Tris–borate, 2 mM Na2 ethylenediamine tetraaceticacid) at room temperature at constant 50 V for 24 h. For the exon 2 polymorphism, 10 µl of the PCR product was digested with 2 U (0.2 µl) TaqI (Boehringer Mannheim, Mannheim, Germany) at 65°C for 1 h. Electrophoresis separation was done by 2% TBE-gel with 1× TBE buffer containing 2 µl Orange G at 90 V for 120 min.
Statistical analysis
Data were expressed as percentages or means ± SD. For categorical data, Pearson's χ2 test was used. Differences in continuous variables were tested with one-way anova. The expected allele frequencies under the assumption of Hardy–Weinberg equilibrium were compared with those observed in the control group. Furthermore, odds ratios (OR) and their 95% confidence intervals (CI) were determined. Statistical analysis was performed using a standard computer program (Graph Pad Prism®, version 4.0, San Diego, CA, USA). All tests were two-sided and statistical significance was defined as P < 0.05.
Results
ACE-gene I/D polymorphism
Of the 65 patients with ACEi-induced angio-oedema, the ACE I/D genotypes showed the following distribution: 10 I/I (15%), 35 I/D (54%) and 20 D/D (31%) individuals (Table 2). In the group of 65 control I subjects (ACEi-treated patients without angio-oedema), the distribution of the three genotypes were: 12 I/I (18%), 29 I/D (45%) and 24 D/D (37%) individuals (Table 2).
Table 2.
Genotype frequencies of angiotensin-converting enzyme (ACE) I/D, bradykinin B2 receptor exon 1 (2/3) and exon 2 (c.C181T) polymorphism in 65 patients with ACE inhibitor (i)-induced angio-oedema and 65 control subjects under ACEi treatment but without angio-oedema episodes (control I)
| ACEi angio-oedema cases | ACEi controls | |||
|---|---|---|---|---|
| Gene polymorphism | Allele frequency (%) | n | Allele frequency (%) | n |
| ACE | ||||
| I/I | 0.18 | 10 | 0.17 | 12 |
| I/D | 0.24 | 35 | 0.24 | 29 |
| D/D | 0.34 | 20 | 0.35 | 24 |
| B2 receptor exon 1 | ||||
| 2/2 | 0.23 | 16 | 0.18 | 11 |
| 2/3 | 0.25 | 30 | 0.24 | 32 |
| 3/3 | 0.27 | 19 | 0.34 | 22 |
| B2 receptor exon2 | ||||
| C/C | 0.85 | 55 | 0.83 | 54 |
| C/T | 0.07 | 10 | 0.08 | 10 |
| T/T | 0.006 | 0 | 0.008 | 1 |
No significant differences were observed in the allele frequencies of the ACE-gene I/D polymorphisms in the group of angio-oedema patients (I-allele 0.42, D-allele 0.58) and ACEi-treated patients without angio-oedema attack (I-allele 0.41, D-allele 0.59, P= 0.095) (Figure 1). The expected allele frequencies under the assumption of Hardy–Weinberg equilibrium showed no differences in the two groups (Table 2). In addition, no risk association between germ-line heterozygosity (I/D: OR 1.45, 95% CI 0.7, 2.9) or homozygosity (DD: OR 0.76, 95% CI 0.4, 1.5) and the development of an angio-oedema during ACEi treatment could be demonstrated.
Figure 1.
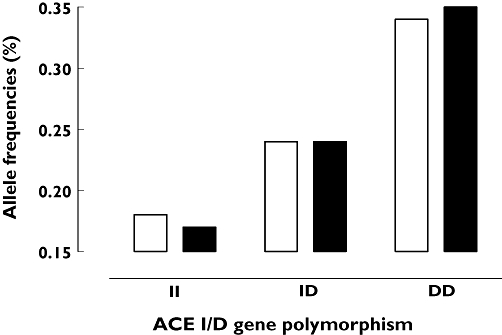
Genotype frequencies of angiotensin-converting enzyme (ACE) I/D polymorphism in 65 patients with ACE inhibitor (i)-induced angio-oedema and 65 control subjects under ACEi treatment but without angio-oedema episodes (control I). ACEi angioedema (n=65) (□); ACEi control I (n=65) ( )
)
Serum ACE activity
Serum ACE activity correlated well with ACE-gene I/D polymorphisms, i.e. the D-allele is associated with increased serum ACE activity (Figure 2). ACE activity after the discontinuation of ACEi medication for (at least) 3 months was not significantly different in any ACE I/D polymorphism subgroups of the 47 angio-oedema patients (I/I 28 ± 4.5 U l−1, ID 39 ± 3.3 U l−1, DD 56 ± 6.7 U l−1; Figure 2) compared with the 51 healthy individuals of control group II (I/I 33 ± 1.8 U l−1, I/D 41 ± 1 U l−1, D/D 52 ± 2.2 U l−1; P= 0.9) (Figure 2). However, the results showed a trend for a decrease of ACE activity in patients with the I/I or I/D polymorphism.
Figure 2.
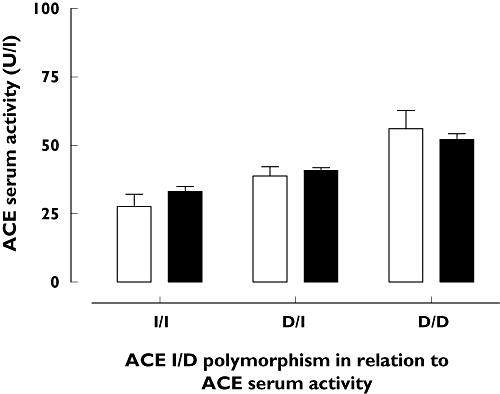
Serum angiotensin-converting enzyme (ACE) activity depending on ACE I/D polymorphism in 47 patients with ACE inhibitor (i)-induced angio-oedema 3 months after discontinuation of ACEi medication and 51 healthy individuals (control II). In both groups of individuals the polymorphism had a significant effect on ACE serum activity (P < 0.0001, two-way anova), but there was no significant difference between angio-oedema patients and controls (P= 0.730, two-way anova). ACEi angiooedema (□); Healthy control 2B ( )
)
Bradykinin B2 receptor polymorphisms
Exon 1 (2/3) polymorphism
Of the 65 patients with ACEi-induced angio-oedema, the exon 1 polymorphisms showed the following distribution: 16 (25%) 2/2, 30 (46%) 2/3, and 19 (29%) 3/3 (Table 2). In control group I (ACEi-treated patients without angio-oedema), the genotypes 2/2, 2/3 and 3/3 were found in 11 (17%) 2/2, 32 (49%) 2/3 and 22 (34%) 3/3 individuals, respectively. No significant differences were observed in the allele frequencies of the bradykinin B2 receptor 2/3 polymorphism (2-allele 0.48 vs. 0.42, 3-allele 0.52 vs. 0.58, P= 0.26; Figure 3), and the same was true for the expected allele frequencies under the assumption of Hardy–Weinberg equilibrium (Table 2). The ORs for development of an ACEi-induced angio-oedema were 1.6 (CI 067, 3.8), 0.88 (CI 0.44, 1.76) and 0.6 (CI 0.26, 1.47) for (2/2), (2/3) and (3/3) individuals, respectively.
Figure 3.
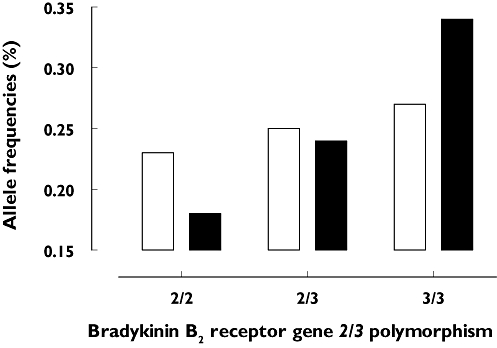
Genotype frequencies of bradykinin B2 receptor exon 1 (2/3) polymorphism in 65 patients with angiotensin-converting enzyme inhibitor (ACEi)-induced angio-oedema and 65 control subjects under ACEi treatment but without angio-oedema episodes (control I). ACEi angioedema (n=65) (□); ACEi control I (n=65) ( )
)
Exon 2 (c.C181T) polymorphism
The group of angio-oedema patients showed only the genotypes C/C 55 (85%) and C/T 10 (15%). Control I subjects showed a similar distribution: C/C 54 (83%), C/T 10 (15%) and T/T 1 (2%). We found no significant differences in the allele frequencies of the bradykinin B2 receptor c.C181T polymorphism in angio-oedema patients (C-allele 0.92, T-allele 0.08) and controls (C-allele 0.91, T-allele 0.09, P= 0.1; see Figure 4). The expected allele frequencies assuming Hardy–Weinberg equilibrium did not differ from those observed in both groups (see Table 2). Finally, the OR for patients to develop an ACEi-induced angio-oedema was 1.12 (CI 0.44, 2.85) in case of homozygote alleles (C/C) and 1.00 (C/T, CI 0.39, 2.59) in case of heterozygote alleles.
Figure 4.
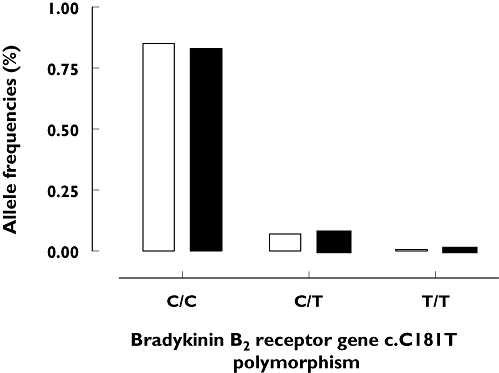
Genotype frequencies of bradykinin B2 receptor exon 2 (c.C181T) polymorphism in 65 patients with angiotensin-converting enzyme inhibitor (ACEi)-induced angio-oedema and 65 control subjects under ACEi treatment but without angio-oedema episodes (control I). ACEi angioedema (n=65) (□); ACEi control I (n=65) ( )
)
Discussion
The clinical importance of uncovering the pathogenic mechanism of ACEi-induced angio-oedema results from the urgent need to identify predictive markers for the development of this potentially life-threatening drug-related side-effect. ACEi inhibits the degradation of bradykinin, and it is therefore not surprising that previous studies have shown increased bradykinin levels during ACEi treatment [10, 11]. Interestingly, a higher risk for the development of ACEi-induced angio-oedema has been described for Black Americans [27, 28], suggesting that a genetic predisposition may modulate the vascular response to bradykinin. Furthermore, it would be expected to find a correlation between occurrence of angio-oedema and the I/I polymorphism, because the highest bradykinin concentrations were found in patients with this particular genotype [29, 30]. In the present study we investigated whether the ACE I/D and two different bradykinin B2 receptor polymorphisms are related to the occurrence of ACEi-induced angio-oedema.
It was shown that the ACE I/D polymorphism is associated with cardiovascular and renal diseases such as myocardial infarction and idiopathic nephrotic syndrome [31, 32]. According to the results of our study there appears to be no independent association between the occurrence of ACEi-induced angio-oedema and ACE I/D polymorphism. However, we have found a trend of decreased serum ACE activity in the angio-oedema group within the subgroups carrying the ACE I/I polymorphism in comparison with healthy individuals (control II). The medication-free interval should preclude a persisting effect of ACEi on the enzyme activity. Theoretically, this particular genotype might be associated with increased plasma and tissue levels of bradykinin, but this has not yet been proven in clinical trials.
ACE-inhibitors act in a competitive manner and their activity is dependent on their plasma concentration. A previous report has shown that the ACE I/D polymorphism might have an effect on ACE activity [33], although the 35% decrease of ACE activity in II vs. DD was not significant. In addition, Rigat et al. have provided data suggesting that there is a 40% decrease of serum ACE concentration in II vs. DD [17]. ACE activity has not been measured. The results of our study extend these previous observations and suggest that ACE activity might be influenced by yet unknown conditions and/or genetic variations different from the I/D polymorphism. We found that control patients and angio-oedema patients with a DD genotype showed a higher serum ACE activity and therefore may have a risk of developing angio-oedema. The ACE D allele has been associated with increased conversion of angiotensin I to angiotensin II and increased degradation of bradykinin in predominantly White populations [33]. The racial difference was most pronounced in subjects heterozygous at the ACE I/D locus in which Blacks had a markedly attenuated response to bradykinin compared with Whites [34]. However, we did not detect a significant increase of the ACE I/I polymorphism in angio-oedema patients, although a trend in this direction could be observed. It is unknown whether this trend may become significant if the number of patients is enhanced. So far, our results suggest that ACE I/D polymorphisms are not involved in the development of ACEi-induced angio-oedema when considered individually.
In some cases there was a time interval of up to 3 years between discontinuation of ACEi and measurement of ACE activity. This might raise some doubts on the diagnosis of ACEi-induced angio-oedema. However, previous investigations have shown that such uneventful periods are not rare [5]. Furthermore, during the mean observation period of approximately 4 years after the study qualifying event and discontinuation of the ACEi, none of the patients experienced another angio-oedema attack. In summary, observations of other groups and those presented in this study suggest that patients with primarily reduced serum ACE activity might have a higher risk of developing angio-oedema in response to ACEi. Further investigations should be performed to clarify (i) whether the low serum ACE activity arises as a result of a reduced concentration or represents a functional disorder at normal ACE plasma concentrations, and (ii) whether a low ACE activity level might be a useful marker to predict the occurrence of ACEi-induced angio-oedema.
Another potential candidate possibly involved in the development of ACEi-induced angio-oedema is the gene for the bradykinin B2 receptor. A noncoding exon 1 polymorphism in which alleles differ by a 9-bp deletion has been suggested to be of clinical significance [29]. The (–) allele determines a splice variant that is more resistant to the action of RNases and appears to confer a higher level of bradykinin B2 receptor expression, because it is always present in the most symptomatic cases of C1 inhibitor deficiency-induced hereditary angio-oedema, i.e. episodes with increased activation of plasma kallikrein and other blood proteases. Hence, the B2 receptor (–) allele may modulate the penetrance of the underlying genetic defect in C1 inhibitor deficiency in a dominant fashion. On the other hand, in a cohort of 37 hereditary angio-oedema patients no correlation was found between clinical status and bradykinin B2 receptor polymorphism in exon 1 [35]. Interestingly, ACEi-induced cough has been reported to be associated with the bradykinin B2 receptor exon 1 polymorphism [36]. In our study, we could find no association between the occurrence of angio-oedema and either of the two investigated bradykinin B2 receptor polymorphisms. However, it should be added that an investigation in a larger group of patients might have revealed a significant influence of the bradykinin B2 receptor exon 1 polymorphism 3/3. Nevertheless, the potential influence of this polymorphism on the incidence of angio-oedema appears to be slight.
In summary, the present study has shown no significant correlation between ACE I/D or bradykinin B2 (2/3; C181T) gene polymorphisms and the occurrence of ACEi-induced angio-oedema. We detected a nonsignificant increase of the ACE I/I polymorphism in angio-oedema patients, but it is unknown whether this trend may become significant if the number of patients is enhanced. Further studies should be carried out to clarify whether a combination of these polymorphisms might be a risk factor for ACEi-induced angio-oedema.
Competing interests
None to declare.
This study was supported by grants from the Medical Faculty of the Heinrich-Heine-University Duesseldorf, Germany (M.B., no. 9772240). The authors are grateful to N. Kuhr and C. Schwand for their excellent technical assistance.
REFERENCES
- 1.Agostoni A, Cicardi M, Cugno M, Zingale LC, Gioffre D, Nussberger J. Angioedema due to angiotensin-converting enzyme inhibitors. Immunopharmacology. 1999;44:21–5. doi: 10.1016/s0162-3109(99)00107-1. [DOI] [PubMed] [Google Scholar]
- 2.Slater EE, Merrill DD, Guess HA, Roylance PJ, Cooper WD, Inman WH, Ewan PW. Clinical profile of angioedema associated with angiotensin converting-enzyme inhibition. JAMA. 1988;260:967–70. [PubMed] [Google Scholar]
- 3.Weiner JM. Failure to recognise the association of life-threatening angio-oedema and angiotensin-converting enzyme inhibitor therapy. Aust NZ J Med. 1995;25:241–2. doi: 10.1111/j.1445-5994.1995.tb01532.x. [DOI] [PubMed] [Google Scholar]
- 4.Messerli FH, Nussberger J. Vasopeptidase inhibition and angio-oedema. Lancet. 2000;356:608–9. doi: 10.1016/S0140-6736(00)02596-4. [DOI] [PubMed] [Google Scholar]
- 5.Bas M, Hoffmann TK, Bier H, Kojda G. Increased C-reactive protein in ACE-inhibitor-induced angioedema. Br J Clin Pharmacol. 2005;59:233–8. doi: 10.1111/j.1365-2125.2004.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vleeming W, van Amsterdam JG, Stricker BH, De Wildt DJ. ACE inhibitor-induced angioedema. Incidence, prevention and management. Drug Saf. 1998;18:171–88. doi: 10.2165/00002018-199818030-00003. [DOI] [PubMed] [Google Scholar]
- 7.Adam A, Cugno M, Molinaro G, Perez M, Lepage Y, Agostoni A. Aminopeptidase P in individuals with a history of angio-oedema on ACE inhibitors. Lancet. 2002;359:2088–9. doi: 10.1016/S0140-6736(02)08914-6. [DOI] [PubMed] [Google Scholar]
- 8.Lefebvre J, Murphey LJ, Hartert TV, Jiao SR, Simmons WH, Brown NJ. Dipeptidyl peptidase IV activity in patients with ACE-inhibitor-associated angioedema. Hypertension. 2002;39:460–4. doi: 10.1161/hy0202.103054. [DOI] [PubMed] [Google Scholar]
- 9.Molinaro G, Cugno M, Perez M, Lepage Y, Gervais N, Agostoni A, Adam A. Angiotensin-converting enzyme inhibitor-associated angioedema is characterized by a slower degradation of des-arginine(9)-bradykinin. J Pharmacol Exp Ther. 2002;303:232–7. doi: 10.1124/jpet.102.038067. [DOI] [PubMed] [Google Scholar]
- 10.Nussberger J, Cugno M, Cicardi M. Bradykinin-mediated angioedema. N Engl J Med. 2002;347:621–2. doi: 10.1056/NEJM200208223470820. [DOI] [PubMed] [Google Scholar]
- 11.Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–7. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 12.Sigler C, Annis K, Cooper K, Haber H, Van deCarr S. Examination of baseline levels of carboxypeptidase N and complement components as potential predictors of angioedema associated with the use of an angiotensin-converting enzyme inhibitor. Arch Dermatol. 1997;133:972–5. [PubMed] [Google Scholar]
- 13.Bas M, Kojda G, Bier H, Hoffmann TK. Durch ACE-Hemmer induziertes Angioödem des Kopf-Hals-Bereichs. Eine Frage der Zeit? HNO. 2004;52:886–90. doi: 10.1007/s00106-003-1017-5. [DOI] [PubMed] [Google Scholar]
- 14.Matsusaka T, Ichikawa I. Biological functions of angiotensin and its receptors. Annu Rev Physiol. 1997;59:395–412. doi: 10.1146/annurev.physiol.59.1.395. [DOI] [PubMed] [Google Scholar]
- 15.Pellacani A, Brunner HR, Nussberger J. Plasma kinins increase after angiotensin-converting enzyme inhibition in human subjects. Clin Sci (Lond) 1994;87:567–74. doi: 10.1042/cs0870567. [DOI] [PubMed] [Google Scholar]
- 16.Soubrier F, henc-Gelas F, Hubert C, Allegrini J, John M, Tregear G, Corvol P. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci USA. 1988;85:9386–90. doi: 10.1073/pnas.85.24.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigat B, Hubert C, Henc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–6. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda S, Elliott HL, Morton JJ, Connell JM. Enhanced pressor response to angiotensin I in normotensive men with the deletion genotype (DD) for angiotensin-converting enzyme. Hypertension. 1995;25:1266–9. doi: 10.1161/01.hyp.25.6.1266. [DOI] [PubMed] [Google Scholar]
- 19.Brown NJ, Vaughan DE. Angiotensin-converting enzyme inhibitors. Circulation. 1998;97:1411–20. doi: 10.1161/01.cir.97.14.1411. [DOI] [PubMed] [Google Scholar]
- 20.Bantis C, Heering PJ, Luther Y, Aker S, Kuhr N, Grabensee B, Ivens K. Influence of cytokine gene polymorphisms on focal segmental glomerulosclerosis. Am J Nephrol. 2004;24:427–31. doi: 10.1159/000080186. [DOI] [PubMed] [Google Scholar]
- 21.Marceau F, Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nat Rev Drug Discov. 2004;3:845–52. doi: 10.1038/nrd1522. [DOI] [PubMed] [Google Scholar]
- 22.Braun A, Maier E, Kammerer S, Muller B, Roscher AA. A novel sequence polymorphism in the promoter region of the human B2-bradykinin receptor gene. Hum Genet. 1996;97:688–9. doi: 10.1007/BF02281884. [DOI] [PubMed] [Google Scholar]
- 23.Burch RM, Kyle DJ. Recent developments in the understanding of bradykinin receptors. Life Sci. 1992;50:829–38. doi: 10.1016/0024-3205(92)90201-y. [DOI] [PubMed] [Google Scholar]
- 24.Braun A, Kammerer S, Bohme E, Muller B, Roscher AA. Identification of polymorphic sites of the human bradykinin B2 receptor gene. Biochem Biophys Res Commun. 1995;211:234–40. doi: 10.1006/bbrc.1995.1801. [DOI] [PubMed] [Google Scholar]
- 25.Braun A, Kammerer S, Maier E, Bohme E, Roscher AA. Polymorphisms in the gene for the human B2-bradykinin receptor. New tools in assessing a genetic risk for bradykinin-associated diseases. Immunopharmacology. 1996;33:32–5. doi: 10.1016/0162-3109(96)00079-3. [DOI] [PubMed] [Google Scholar]
- 26.Kammerer S, Braun A, Arnold N, Roscher AA. The human bradykinin B2 receptor gene: full length cDNA, genomic organization and identification of the regulatory region. Biochem Biophys Res Commun. 1995;211:226–33. doi: 10.1006/bbrc.1995.1800. [DOI] [PubMed] [Google Scholar]
- 27.Brown NJ, Ray WA, Snowden M, Griffin MR. Black Americans have an increased rate of angiotensin converting enzyme inhibitor-associated angioedema. Clin Pharmacol Ther. 1996;60:8–13. doi: 10.1016/S0009-9236(96)90161-7. [DOI] [PubMed] [Google Scholar]
- 28.Gibbs CR, Lip GY, Beevers DG. Angioedema due to ACE inhibitors: increased risk in patients of African origin. Br J Clin Pharmacol. 1999;48:861–5. doi: 10.1046/j.1365-2125.1999.00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lung CC, Chan EK, Zuraw BL. Analysis of an exon 1 polymorphism of the B2 bradykinin receptor gene and its transcript in normal subjects and patients with C1 inhibitor deficiency. J Allergy Clin Immunol. 1997;99:134–46. doi: 10.1016/s0091-6749(97)70310-5. [DOI] [PubMed] [Google Scholar]
- 30.van Dijk MA, Kroon I, Kamper AM, Boomsma F, Danser AH, Chang PC. The angiotensin-converting enzyme gene polymorphism and responses to angiotensins and bradykinin in the human forearm. J Cardiovasc Pharmacol. 2000;35:484–90. doi: 10.1097/00005344-200003000-00020. [DOI] [PubMed] [Google Scholar]
- 31.Seckin D, Ilhan N, Ilhan N, Ozbay Y. The relationship between ACE insertion/deletion polymorphism and coronary artery disease with or without myocardial infarction. Clin Biochem. 2006;39:50–4. doi: 10.1016/j.clinbiochem.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Tsai IJ, Yang YH, Lin YH, Wu VC, Tsau YK, Hsieh FJ. Angiotensin-converting enzyme gene polymorphism in children with idiopathic nephrotic syndrome. Am J Nephrol. 2006;26:157–62. doi: 10.1159/000092982. [DOI] [PubMed] [Google Scholar]
- 33.Brown NJ, Blais C, Jr, Gandhi SK, Adam A. ACE insertion/deletion genotype affects bradykinin metabolism. J Cardiovasc Pharmacol. 1998;32:373–7. doi: 10.1097/00005344-199809000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Gainer JV, Nadeau JH, Ryder D, Brown NJ. Increased sensitivity to bradykinin among African Americans. J Allergy Clin Immunol. 1996;98:283–7. doi: 10.1016/s0091-6749(96)70151-3. [DOI] [PubMed] [Google Scholar]
- 35.Freiberger T, Vyskocilova M, Kolarova L, Kuklinek P, Krystufkova O, Lahodna M, Hanzlikova J, Litzman J. Exon 1 polymorphism of the B2BKR gene does not influence the clinical status of patients with hereditary angioedema. Hum Immunol. 2002;63:492–4. doi: 10.1016/s0198-8859(02)00397-x. [DOI] [PubMed] [Google Scholar]
- 36.Mukae S, Itoh S, Aoki S, Iwata T, Nishio K, Sato R, Katagiri T. Association of polymorphisms of the renin–angiotensin system and bradykinin B2 receptor with ACE-inhibitor-related cough. J Hum Hypertens. 2002;16:857–63. doi: 10.1038/sj.jhh.1001486. [DOI] [PubMed] [Google Scholar]