

Abstract
AIM
To characterize the formation and urinary elimination of metabolites of S-(+) and R-(−) methamphetamine (MA) in humans.
METHODS
In this 12-subject, six-session, double-blind, placebo-controlled, balanced, crossover design study, the formation of the MA metabolites para hydroxymethamphetamine (pOH-MA) and amphetamine (AMP) were determined in urine after intravenous doses of S-(+)-MA 0.25 and 0.5 mg kg−1, R-(−)-MA 0.25 and 0.5 mg kg−1, racemic MA 0.5 mg kg−1, or placebo. Parent drug and metabolite levels in urine and plasma were measured by gas chromatography-mass spectrometry. Pharmacokinetic parameters were calculated by noncompartmental models using WinNonlin.
RESULTS
An approximately threefold enantioselectivity difference in elimination was observed for AMP, with 7% of the dose converted to S-(+)-AMP vs. 2% to R-(−)-AMP (P < 0.001). Furthermore, less R-(−)-pOH-MA was excreted in the urine compared with S-(+)-pOH-MA (8% vs. 11%, P= 0.02). Correspondingly, S-(+)-MA excretion was less than R-(−)-MA (42% vs. 52%; P= 0.005).
CONCLUSIONS
The metabolism of MA is enantioselective, with formation of AMP having the highest isomer selectivity. A greater percentage of MA is converted to pOH-MA (8–11%) than AMP (2–7%). The formation of pOH-MA was less affected by the MA enantiomer administered, suggesting that urine pOH-MA may be a more stable biomarker of MA metabolism.
Keywords: metabolism, methamphetamine, stereoselectivity
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Methamphetamine (MA) is a chiral compound.
The S-(+) enantiomer is more commonly abused and is more potent in producing central nervous system and cardiovascular effects than the R-(−) enantiomer.
Studies describing the metabolism of MA have mainly been done with the S-(+)-isomer; pharmacokinetic data for R-(−)-MA or racemic MA are very limited.
WHAT THIS STUDY ADDS
Stereoselectivity exists in the metabolism of MA.
Urinary excretion of para hydroxymethamphetamine (pOH-MA) was found to be least affected by stereoselectivity.
It is suggested that pOH-MA may be a more stable biomarker of MA abuse.
Background
Methamphetamine (MA) possesses a chiral centre, giving two enantiomers, S-(+)-MA (dextrorotatory-MA, also called d-MA) and R-(−)-MA (levorotatory-MA, also called l-MA). MA is available in prescription and nonprescription medications and is a significant drug of abuse throughout the world. S-(+)-MA is available by prescription for the treatment of attention deficit disorder, exogenous obesity and narcolepsy [1]. In the USA, R-(−)-MA is the active ingredient of the commonly used nasal decongestant Vick's Vapor Inhaler; Vick's inhalers sold elsewhere do not contain MA. S-(+)-MA is generally associated with more potent physiological and behavioural effects and higher abuse liability, and addicts generally prefer and abuse this enantiomer [2]. Despite easy availability of R-(−)-MA from the over-the-counter Vick's Vapor Inhaler, abuse of this enantiomer is rarely reported. Almost all abused MA contains S-(+)-MA, either as relatively pure S-(+)-MA or as S/R-MA mixture. Isomer differences in the illicit market are probably due to the availability of different precursors, resulting in different final synthetic products. Addicts prefer S-(+)-MA, and the few commercially available precursors yielding this product have become increasingly regulated. There are many precursors with myriad legitimate uses that can serve as starting materials for racemic MA. A likely consequence of decreasing availability of precursors that yield S-(+)-MA is the increased synthesis and availability of racemic MA [2]. Thus, it is important to understand more about the metabolism and patterns of urinary elimination of the MA isomers.
Pharmacological differences between the MA isomers have been recognized, both in animal and human studies, with S-(+)-MA being more potent in producing central nervous system and cardiovascular effects. In humans MA is metabolized by aromatic hydroxylation and N-demethylation, mainly through CYP2D6, with formation of two major metabolites: para-hydroxymethamphetamine (pOH-MA) and amphetamine (AMP), respectively. AMP can be further metabolized into para-hydroxyamphetamine (pOH-AMP), N-hydroxyamphetamine and norephedrine (Figure 1) [3]. Prior studies have estimated that 43% of a MA dose was excreted as parent drug and 4–7% as AMP in the urine [4]. There are several publications describing the metabolism of S-(+)-MA, but few studies are available on the biodisposition of R-(−)-MA and no human studies are available on enantiomeric differences in formation of metabolites [5–8]. The aim of this study was to investigate the stereoselectivity in the disposition of MA in humans.
Figure 1.
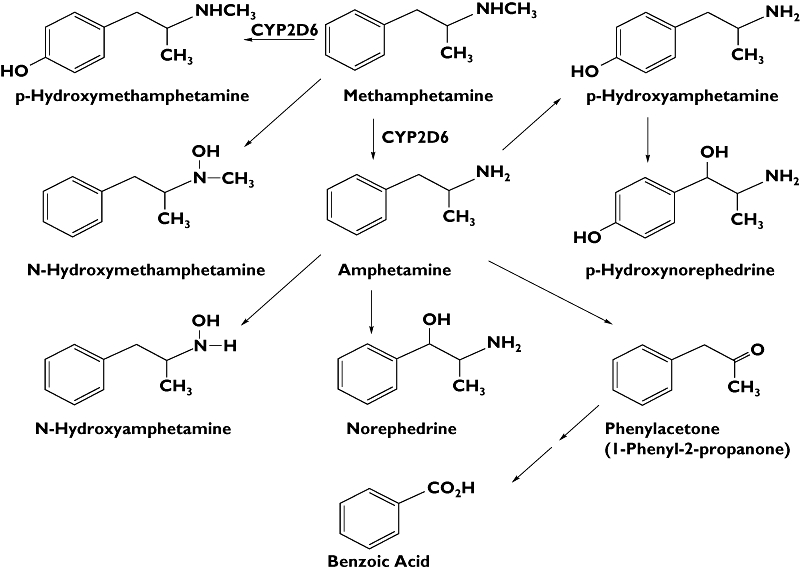
Main metabolic pathways of methamphetamine in humans
Subjects and methods
Subjects
Twelve male intravenous (i.v.) MA users participated in this study. Subjects were between 21 and 45 years old and in good health. They were not dependent on MA, alcohol or other illicit drugs based on criteria in the Diagnostic and Statistical Manual of Mental Disorders, 4th edn. The study protocol was approved by the Committee on Human Research, University of California, San Francisco, and was carried out in accordance with the Declaration of Helsinki.
Study design
A six-session, double-blind, placebo-controlled, Latin-square balanced crossover design was used. Single i.v. doses of S-(+)-MA, R-(−)-MA, racemic MA, or placebo were administered over a period of 1 min via infusion pump control into a forearm vein. The drugs and doses (all i.v.) were as follows:
S-(+)-MA, 0.25 mg kg−1;
S-(+)-MA, 0.5 mg kg−1;
R-(−)-MA, 0.25 mg kg−1;
R-(−)-MA, 0.5 mg kg−1;
racemic MA, 0.5 mg kg−1 (0.25 mg kg−1 S-(+)-MA plus 0.25 mg kg−1 R-(−)-MA); and placebo (0.9% sodium chloride)
Test sessions occurred at approximately weekly intervals.
Blood and urine collection
A plastic catheter was inserted into an arm vein, and 7.5 ml of whole blood was collected before dosing and at 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 30, 36 and 48 h after dosing. Urine was collected at 4, 8, 12, 24 and 48 h after dosing. The volume of urine for each period was measured, and an aliquot from each sample was stored at −80°C until analysis.
Bioassay of urine samples
MA and AMP in urine were extracted and the analytes converted to the trifluoroacetyl derivatives. MA-d9 and AMP–d11, obtained from Cerilliant (Round Rock, TX, USA), were employed as internal standards. pOH-MA and pOH-AMP were extracted from urine and converted to the trifluoroacetyl amide, para-methyl ether derivative. Analogues of each metabolite, deuterium-labelled on the terminal methyl group of the n-propyl side chain, were synthesized in house and employed as the internal standards. Chiral separation was effected by temperature-programmed gas chromatography utilizing a Restek β-DEXcst chiral column. Detection was by isobutane chemical-ionization gas chromatography-mass spectrometry, utilizing the molecular ion species (M + H)+ of each analyte.
The respective limits of quantification (LOQ) of the assay and coefficients of variation at the LOQ were: MA, 2 ng ml−1 (15%); AMP, 0.5 ng ml−1 (11%); pOH-MA, 0.2 ng ml−1 (11%); and pOH-AMP, 0.25 ng ml−1 (9%).
Data analysis
WinNonlin professional software (Version 5.2) was used to obtain pharmacokinetic (PK) estimates. Du (amount recovered) as well as the Pu (percent recovered) for MA and its metabolites were derived from the noncompartment analysis of the urine data (Pu =Du/Dose). AUC0–∞ and CL (total clearance) were derived from the noncompartment analysis of the plasma data. The CLR (renal clearance) of MA was calculated based on CLR=Du/AUC0–∞. The S/R ratio (cumulative amount of S-(+)-isomer divided by that of R-(−)-isomer) at the ending time point of each collection interval after racemate administration was calculated as well. Values are presented as mean ± standard deviation (SD).
Statistical analysis
Statistical analysis for percent recovered in the urine of parent drug as well as its two main metabolites among different sessions was done using a one-way anova test. P-values of ≤0.05 were considered as significant.
Results for MA plasma PKs and pharmacodynamics have been published. This report describes urinary PK of MA and its main metabolites. For more details about the study design and the bioanalysis of plasma samples, please refer to our previously published paper [2].
Results
MA plasma and urinary PKs are summarized in Table 1. The percent urinary recovery of S-(+)-MA was 44.9 ± 17.9%, 43.2 ± 14.8% and 38.3 ± 14.3% for the 0.25 mg kg−1, 0.5 mg kg−1 dose, 0.5 mg kg−1 racemic dose, respectively, while the percent urinary recovery of R-(−)-MA was 55.7 ± 14.1%, 49.1 ± 12.8% and 52.4 ± 12.1%, respectively, under the same dosing conditions. For each isomer, the percent recovery of unchanged MA was not dose dependent (P > 0.05). However, the percent recovery of unchanged MA was isomer dependent with a larger percentage of R-(−)-MA excreted as parent drug (P < 0.01). This difference was the most obvious under the racemic dose condition, where a 14% difference was observed.
Table 1.
Pharmacokinetics of methamphetamine (MA) isomers after intravenous administration of different doses
| 0.25 mg kg−1 | 0.5 mg kg−1 | Racemic (1:1) 0.5 mg kg−1 | ||||
|---|---|---|---|---|---|---|
| S-(+)-MA | R-(−)-MA | S-(+)-MA | R-(−)-MA | S-(+)-MA | R-(−)-MA | |
| AUC0–∞ (ng h−1 ml−1) | 1023 ± 208.9 | 1189.1 ± 271.4 | 1996.5 ± 373.1 | 2367.2 ± 522.3 | 996.3 ± 257.2 | 1394.2 ± 341.4 |
| CL (l h−1 kg−1) | 0.254 ± 0.051 | 0.22 ± 0.048 | 0.257 ± 0.038 | 0.221 ± 0.048 | 0.264 ± 0.057 | 0.189 ± 0.045 |
| CLR (l h−1 kg−1) | 0.114 ± 0.043 | 0.134 ± 0.047 | 0.113 ± 0.034 | 0.118 ± 0.042 | 0.108 ± 0.034 | 0.114 ± 0.043 |
| MA percent recovered in the urine (%) | 44.9 ± 17.9 | 55.7 ± 14.1 | 43.2 ± 14.8 | 49.1 ± 12.8 | 38.3 ± 14.3 | 52.4 ± 12.1 |
| pOH-AMP percent recovered in the urine (%) | 10.3 ± 4.6 | 7.8 ± 4.1 | 11.5 ± 6.0 | 7.1 ± 4.4 | 10.2 ± 4.4 | 9.1 ± 5.4 |
| AMP percent recovered in the urine (%) | 6.8 ± 2.5 | 2.3 ± 0.8 | 6.4 ± 2.3 | 2.1 ± 0.8 | 6.3 ± 2.0 | 2.4 ± 1.0 |
| pOH-AMP percent recovered in the urine (%) | 0.34 ± 0.16 | 0.29 ± 0.21 | 0.34 ± 0.16 | 0.27 ± 0.22 | 0.34 ± 0.17 | 0.38 ± 0.27 |
An approximately threefold difference in the recovery of AMP in the urine was observed with 6–7% of the S-(+)-MA dose converted to S-(+)-AMP compared with 2% of R-(−)-MA to R-(−)-AMP (P < 0.001). This finding is consistent with our published plasma data, where we observed a smaller AUC of R-(−)-AMP compared with that of S-(+)-AMP [2]. In contrast to AMP, the differences in the excretion of pOH-MA between the two isomers were much less, with 11% of S-(+)-MA going to S-(+)-pOH-MA vs. 8% of R-(−)-MA going to R-(−)-pOH-MA. A secondary metabolite of MA, pOH-AMP, was detected in the urine samples but recovery was <0.5% of MA, indicating that this is a minor metabolite.
The CL of S-(+)-MA was greater than that of R-(−)-MA. This was particularly evident with the racemic dose and was described in our previous paper [2]. No difference in the CLR between R-(−)-MA and S-(+)-MA was observed, which implies that enantioselective metabolism rather than enantioselective renal excretion is the possible explanation for the enantioselective PK of MA. The CL of R-(−)-MA after 0.5 mg kg−1 racemic MA was less than the CL of the comparable 0.5 mg kg−1 R-(−)-MA. The fall in clearance was accompanied by an increase in AUC and half-life, suggesting that S-(+)-MA may compete more effectively than R-(−)-MA for biotransformation binding sites in metabolizing enzymes, and the metabolism kinetics of R-(−)-MA is slowed in the presence of S-(+)-MA.
The S/R ratio of MA and its metabolites after 0.5 mg kg−1 racemic MA is illustrated in Figure 2. The S/R ratio for MA was 1:1 at 4 h after dosing and decreased slowly to approximately 0.7:1 at 48 h post dosing. The S/R ratio for pOH-MA was close to 2:1 and its change over time paralleled that of MA, reaching approximately 1.5:1 at 48 h post dosing. The average S/R ratio for AMP was approximately 6:1 at 4 h post dose and decreased to approximately 3:1 at 48 h post dose, with a much steeper decline in the first 24 h compared with the next 24 h.
Figure 2.
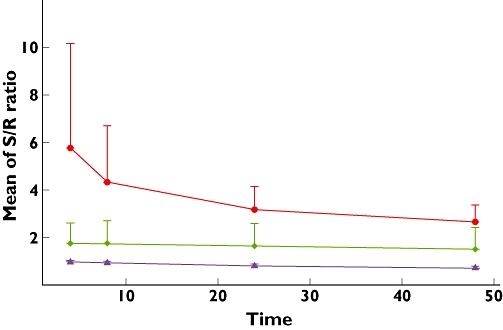
S/R ratio of cumulative amount of methamphetamine (MA) and its metabolites vs. time after 0.5 mg kg−1 racemic MA intake. AMP ( ); MA (
); MA ( ); pOH-MA (
); pOH-MA ( )
)
It is of interest to note that when the data were looked at individually, there was one subject with an extremely low rate of metabolite formation. The percent recovery of parent drug and its two main metabolites in urine of this subject is displayed in Table 2.
Table 2.
Urinary recovery of methamphetamine (MA) and its metabolites of one subject
| 0.25 mg kg−1 | 0.5 mg kg−1 | Racemic (1:1) 0.5 mg kg−1 | ||||
|---|---|---|---|---|---|---|
| S-(+)-MA | R-(−)-MA | S-(+)-MA | R-(−)-MA | S-(+)-MA | R-(−)-MA | |
| MA percent recovered in the urine (%) | 68.8 | 75.4 | 60.4 | 78.6 | 66.9 | 64.9 |
| pOH-MA percent recovered in the urine (%) | 1.21 | 0.26 | 1.63 | 0.26 | 1.23 | 0.31 |
| AMP percent recovered in the urine (%) | 1.89 | 0.91 | 1.48 | 0.81 | 1.33 | 0.76 |
Discussion
This study characterized the urinary excretion of MA enantiomers and their major metabolites following i.v. administration of the individual enantiomers and racemic MA. Due to the relatively long terminal half-life of MA, unchanged MA and its two major metabolites (AMP and pOH-MA) can be detected in the urine for at least 24 h after modest i.v. doses [9]. More MA was metabolized to pOH-MA than AMP and a larger percentage of pOH-MA was excreted in the urine compared with AMP. A small study (only two subjects) using 14C-labelled MA reported results similar to ours. Following an oral dose of 20 mg MA, they found 15% of the dose as pOH-MA and 3% as AMP in urine [8].
The urinary excretion of MA and its two metabolites exhibits stereoselectivity, with AMP exhibiting the largest degree of selectivity. More unchanged R-(−)-MA was excreted than unchanged S-(+)-MA, possibly due to less conversion of R-(−)-MA to R-(−)-AMP and R-(−)-pOH-MA, with the pathway from MA to AMP being most stereoselective. Previous studies have also implied stereoselective metabolism of MA and AMP from the urinary excretion of AMP isomers. However, pOH-MA was not explored in these studies [10, 11]. The main enzyme mediating the metabolism of MA to AMP and pOH-MA is CYP2D6 [3]. CYP2D6 has a greater affinity for the S-enantiomer of the related psychostimulant, 3,4-methylenedioxymethamphetamine (MDMA); in an in vitro enzyme kinetic study greater biotransformation of the S-enantiomer was observed [12]. It is likely that CYP2D6 has similar isomer preference in the metabolism of MA, an assumption that would be consistent with the findings in our study.
The percentage of MA recovered in the urine was independent of dose in our study. With an average weight of 71 ± 5 kg for the 12 subjects in this study, the dose range was from 16.75 mg to 41.95 mg (0.25 mg kg−1 and 0.5 mg kg−1). Our findings are not consistent with the results reported by others, where decreased elimination (from 47.3 ± 19.8% to 31.6 ± 7.2%) was reported following 10 mg and 20 mg oral dose of S-(+)-MA [13]. This author proposed saturated excretion by the kidney as the mechanism of the decrease in urinary elimination of MA. Our lower dose of 0.25 mg kg−1 is comparable to the 20-mg dose in their study. However, the percent recovery of the parent drug in urine is similar to that observed after their 10-mg dose. Although the mean percent drug recovered in the low-dose condition was greater than in the high-dose condition, it was not statistically significant, probably due to the large variation between subjects. It is known that the elimination of MA as well as its metabolite AMP in urine is dependent on the urine pH [5]. Urine pH values were not obtained in our study. Thus, uncontrolled variation in urine pH may contribute to the discrepancy between these two studies. Further study with controlled acidification or alkalization of urine may be needed to verify the dose range for linear kinetics of MA.
The S/R ratio over the time indicates that the AMP exhibits the highest S/R ratio among the three and also shows the greatest decrease over time, implying that AMP formation has the highest stereoselectivity. This stereoselectivity wanes with time with less generation of both metabolites over time. This implies that, compared with R-(−)-MA, S-(+)-MA is metabolized not only more completely but also at a greater rate.
The one subject displaying an extremely low formation of AMP and pOH-MA suggests that this subject may carry a nonfunctional CYP2D6 genotype exhibiting minimal or almost no CYP2D6 activity. We did not genotype subjects in this study. Although the CYP2D6 genotype of this subject is unknown, this outlier raises the interesting possibility that CYP2D6 genotype may have a determining effect on the conversion of MA to AMP and pOH-MA in humans. High intersubject variability in MA concentration after the same single dose has been reported by other studies also [14]. If differences in CYP2D6 genotype account for the population variability in biotransformation of MA, then exploring the genetic effects of CYP2D6 in the PK of MA in humans will be an interesting topic for future studies. In addition, genetic effects of CYP26D in the metabolism of racemic MA can be studied further to compare the degree of stereoselectivity of different CYP2D6 genotypes [15].
Overall, our results clearly indicate that the metabolism of MA is stereoselective. A larger percent of MA proceeds through aromatic hydroxylation rather than through N-demethylation. The aromatic hydroxylation pathway is much less stereoselective, which suggests that pOH-MA may be a more stable biomarker of MA abuse than either urinary MA or AMP, especially considering that stereoselective assay methods are available in only a few laboratories. Interpretation of the drug testing results for MA can be a very challenging and complex task; abuse of racemic MA can make this process even more complex due to the differences in metabolism and excretion between S-(+)-MA and R-(−)-MA [16, 17]. Data from our study may be helpful in the interpretation of urine analytical results in studies of MA abuse in clinical and forensic toxicology.
Competing interests
None to declare.
This study was supported by P50 DA018179, DA 012521 and P30 DA012393. We thank the staff of the Drug Dependence Research Center, General Clinical Research Center, and Investigational Pharmacy at the University of California, San Francisco.
REFERENCES
- 1.Physician's Desk Reference. 54 edn. Montvale, NJ: Medical Economics Company; 2000. [Google Scholar]
- 2.Mendelson J, Uemura N, Harris D, Nath RP, Fernandez E, Jacob P, Everhart T, Jones RT. Human pharmacology of the methamphetamine stereoisomers. Clin Pharmacol Ther. 2006;80:403–20. doi: 10.1016/j.clpt.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Lin LY, Di Stefano EW, Schmitz DA, Hsu L, Ellis SW, Lennard MS, Tucker GT, Cho AK. Oxidation of methamphetamine and methylenedioxymethamphetamine by CYP2D6. Drug Metab Dispos. 1997;25:1059–64. [PubMed] [Google Scholar]
- 4.Baselt R. Disposition of Toxic Drugs and Chemicals in Man. 2nd edn. Foster City, CA: Biomedical Publications; 1978. [Google Scholar]
- 5.Beckett AH, Rowland M. Urinary excretion of methylamphetamine in man. Nature. 1965;206:1260–1. doi: 10.1038/2061260a0. [DOI] [PubMed] [Google Scholar]
- 6.Cook CE, Jeffcoat AR, Hill JM, Pugh DE, Patetta PK, Sadler BM, White WR, Perez-Reyes M. Pharmacokinetics of methamphetamine self-administered to human subjects by smoking S–(+)–methamphetamine hydrochloride. Drug Metab Dispos. 1993;21:717–23. [PubMed] [Google Scholar]
- 7.Cook CE, Jeffcoat AR, Sadler BM, Hill JM, Voyksner RD, Pugh DE, White WR, Perez-Reyes M. Pharmacokinetics of oral methamphetamine and effects of repeated daily dosing in humans. Drug Metab Dispos. 1992;20:856–62. [PubMed] [Google Scholar]
- 8.Caldwell J, Dring LG, Williams RT. Metabolism of (14 C)methamphetamine in man, the guinea pig and the rat. Biochem J. 1972;129:11–22. doi: 10.1042/bj1290011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oyler JM, Cone EJ, Joseph RE, Jr, Moolchan ET, Huestis MA. Duration of detectable methamphetamine and amphetamine excretion in urine after controlled oral administration of methamphetamine to humans. Clin Chem. 2002;48:1703–14. [PubMed] [Google Scholar]
- 10.Gunne LM. The urinary output of d- and l-amphetamine in man. Biochem Pharmacol. 1967;16:863–9. doi: 10.1016/0006-2952(67)90059-7. [DOI] [PubMed] [Google Scholar]
- 11.Beckett AH, Brookes LG, Shenoy EV. Urinary excretion of the drug and its main metabolite in man, after the administration of (plus or minus)-, (plus)- and (minus)-ethylamphetamine. J Pharm Pharmacol. 1969;21(Suppl.):151S+. doi: 10.1111/j.2042-7158.1969.tb08366.x. [DOI] [PubMed] [Google Scholar]
- 12.Meyer MR, Peters FT, Maurer HH. The role of human hepatic cytochrome P450 isozymes in the metabolism of racemic 3,4-methylenedioxy-methamphetamine and its enantiomers. Drug Metab Dispos. 2008;36:2345–54. doi: 10.1124/dmd.108.021543. [DOI] [PubMed] [Google Scholar]
- 13.Kim I, Oyler JM, Moolchan ET, Cone EJ, Huestis MA. Urinary pharmacokinetics of methamphetamine and its metabolite, amphetamine following controlled oral administration to humans. Ther Drug Monit. 2004;26:664–72. doi: 10.1097/00007691-200412000-00013. [DOI] [PubMed] [Google Scholar]
- 14.Huestis MA, Cone EJ. Methamphetamine disposition in oral fluid, plasma, and urine. Ann NY Acad Sci. 2007;1098:104–21. doi: 10.1196/annals.1384.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lind AB, Reis M, Bengtsson F, Jonzier-Perey M, Powell Golay K, Ahlner J, Baumann P, Dahl ML. Steady-state concentrations of mirtazapine, N-desmethylmirtazapine, 8-hydroxymirtazapine and their enantiomers in relation to cytochrome P450 2D6 genotype, age and smoking behaviour. Clin Pharmacokinet. 2009;48:63–70. doi: 10.2165/0003088-200948010-00005. [DOI] [PubMed] [Google Scholar]
- 16.Cody JT, Schwarzhoff R. Interpretation of methamphetamine and amphetamine enantiomer data. J Anal Toxicol. 1993;17:321–26. doi: 10.1093/jat/17.6.321. [DOI] [PubMed] [Google Scholar]
- 17.Musshoff F. Illegal or legitimate use? Precursor compounds to amphetamine and methamphetamine. Drug Metab Rev. 2000;32:15–44. doi: 10.1081/dmr-100100562. [DOI] [PubMed] [Google Scholar]