

Abstract
Given their essential function in aerobic metabolism, mitochondria are intuitively of interest in regard to the pathophysiology of diabetes. Qualitative, quantitative, and functional perturbations in mitochondria have been identified and affect the cause and complications of diabetes. Moreover, as a consequence of fuel oxidation, mitochondria generate considerable reactive oxygen species (ROS). Evidence is accumulating that these radicals per se are important in the pathophysiology of diabetes and its complications. In this review, we first present basic concepts underlying mitochondrial physiology. We then address mitochondrial function and ROS as related to diabetes. We consider different forms of diabetes and address both insulin secretion and insulin sensitivity. We also address the role of mitochondrial uncoupling and coenzyme Q. Finally, we address the potential for targeting mitochondria in the therapy of diabetes. Antioxid. Redox Signal. 12, 537–577.
I. Introduction
Approximately 7% of the U.S. population has diabetes, and the prevalence is increasing. Most (∼90–95%) represents type 2 diabetes. The prevalence of type 2 diabetes is higher in certain ethnic populations, and the incidence is increasing worldwide, particularly in developing nations. Thus, diabetes and the related problems of obesity and vascular disease represent major global health-care issues.
Type 2 diabetes is associated with both impaired insulin action at target tissues and impaired insulin release. Defects at both levels are evident early in the course of the disorder, and evidence suggests that mitochondria play a role in both processes. In contrast, type 1 diabetes results primarily from autoimmune β-cell destruction. Nonetheless, mitochondria also are important in type 1 diabetes, if not in pathogenesis, then in treatment of the disorder and in prevention of long-term consequences. Both type 1 and type 2 diabetes, and less common forms as well, are associated with similar long-term complications that, at least in part, appear to result from pathogenic processes at the mitochondrial level.
Mitochondrial function has different implications for diabetes in different cells and tissues. This review emphasizes the major cell types responsible for insulin secretion (pancreatic β cells) and insulin action (skeletal and cardiac myocytes and hepatocytes), as well as target organs for the major complications of diabetes (renal, retinal, neural, and vascular cells).
The objectives of this review are initially to provide a basic overview of mitochondrial physiology; to discuss qualitative, quantitative, and functional perturbations in mitochondria as related to diabetes; to discuss the implications of mitochondrial reactive oxygen species (ROS) for diabetes; and to conclude by addressing therapeutic implications. In the course of this review, we also address the role of mitochondrial uncoupling and coenzyme Q.
We address several controversial topics. These include the following: whether diabetes results from perturbed mitochondria or vice versa; the importance of mitochondrial dysfunction versus altered numbers of mitochondria; sites of mitochondrial ROS production; the role of ROS in diabetes and its complications; the role of membrane potential in regulating ROS; and the merits of mitochondria-directed therapy.
Sections II and III address basic mitochondrial physiology and methods for study. This information should be helpful to readers with a background in diabetes but less familiar with mitochondrial work per se. Readers well versed in mitochondrial physiology may wish to skip ahead to Section IV.
II. Basic Physiology
Mitochondria generate energy as electrons are passed from donors at lower to acceptors at higher redox potential through various protein complexes. Along with this process, protons are pumped from the matrix outward, generating a potential difference across the inner membrane. The resulting potential energy is transferred to ATP or dissipated as heat as protons leak back toward the matrix. Although most electrons are eventually passed to molecular oxygen, a small portion are leaked during transport. This results in one-electron reduction of oxygen to superoxide, which subsequently is converted to additional radical species (Section II.B). Although the ROS so generated may be destructive, these radicals also serve metabolic purposes such as induction of mitochondrial uncoupling (92, 93) and cell signaling (further discussed later).
A. Electron transport
Figure 1 is a schematic diagram of electron transport and its relation to ROS production and uncoupling protein activity. The term electron transport “chain” has been criticized as misleading, because it implies a linear progression along a single pathway. In actuality, electrons enter the electron-transport system (ETS) or branched electron-transport chain (ETC) at four separate sites that are convergent, in that all eventuate in the reduction of coenzyme Q (Fig. 1). Electrons donated by NADH enter at complexes I (NADH ubiquinone reductase), whereas succinate conversion to fumarate generates electrons at complex II (succinate dehydrogenase). Electrons derived from FADH2 may enter the convergent pathway through the electron-transport flavoprotein (ETF). Electrons from glycerol 3-phosphate enter by way of a mitochondrial form of glycerol 3-phosphate dehydrogenase (GAPDH), located on the outer face of the inner membrane (Fig. 1).
FIG. 1.

Mitochondrial electron transport and ROS production. The schematic illustration depicts the convergent nature of electron donation at one of four sites: complex I (NADH ubiquinone reductase), complex II (succinate dehydrogenase), the electron-transfer flavoprotein (ETF), or a mitochondrial form of GAPDH. Reduced ubiquinone is processed through the Q-cycle in complex III, where protons are pumped and electrons passed to mobile cytochrome c and then cytochrome oxidase. ATP formation through ATP synthase (not shown) is coupled to mitochondrial potential generated by proton pumping at complexes I, III, and IV and offset by proton transfer in the opposite direction (proton leak), mediated in part by uncoupling proteins (UCPs). Superoxide (O2·−) produced at complex III in the Q-cycle results from electron leaks generated by the reactive semiquinone intermediate, O2·− (295). Superoxide is also produced at complex I (see text, section II.B), where it is released to the matrix. Note that superoxide is shown (dotted lines) to activate proton transfer by UCP. Red arrows, Electron transport. Blue arrows, H+ movement either away from (proton pumping) or back toward (proton leak) the matrix. Black arrows, Electron leaks leading to one-electron reduction of oxygen to O2·−.
Electron flow from entry sites is directed through the mobile intermediate ubiquinone, followed by oxidation of ubiquinol by complex III (ubiquinol–cytochrome c reductase) (284). Electrons are then transferred to another mobile intermediate, cytochrome c, which directs flow to complex IV (cytochrome c oxidase). ATP synthase (F0·F1-ATPase), also called complex V, consists of a joined membrane-bound F0-ATPase and apparently attached rotatory F1-ATPase. The complex is capable of “coupling” proton flow to conversion of ADP to ATP in an intricate manner that still remains incompletely understood (16).
Substrates for the TCA cycle enter the mitochondrial matrix through pyruvate dehydrogenase, carrier proteins, or one of multiple shuttle mechanisms. Fatty acyl-CoAs enter through the carnitine palmitoyl transferase system (CPT-I and CPT-II) for β-oxidation. Metabolism of different substrates results in electron donation to specific complexes or sites. For example, oxidation of glutamate, malate, and pyruvate provide NADH for electron entry at complex I, whereas succinate is used as substrate at complex II. Fatty acyl-CoAs can be used as substrates if carnitine is added to enable transport into mitochondria. β-Oxidation of fatty acyl-CoAs generates electrons for entry at complex I or complex II by way of acetyl-CoA metabolism through the TCA cycle. In addition, fatty acyl-CoA metabolism provides electrons through the ETF via FADH2, which is a product of β-oxidation, independent of the TCA cycle.
When isolated mitochondria are incubated in vitro, these substrates can be used to study electron flow specifically through that site/complex. For example, in the presence of complex I substrates, flux through the entire TCA cycle is limited by shuttle systems that do not allow a fully operational (closed) cycle. Hence, one can examine respiration or other mitochondrial functions (like ATP or ROS production) as affected by fuel use specifically at complex I. In studies using fatty acyl-CoAs as substrate, malate can be added to maintain the TCA cycle by replenishing oxaloacetate for reaction with acetyl-CoA at the citrate synthase step.
Mitochondrial respiratory states were originally defined by Chance and Williams (58). Respiration with substrate added in excess and during ADP conversion to ATP is referred to as state 3. In the absence of ADP (with excess substrate), for example, by using oligomycin to block ATP synthase, or after consumption of all added ADP, respiration is referred to as state 4. Chance and Williams further defined respiration with no ADP or substrate as state 1, with added ADP and before endogenous substrate exhaustion as state 2, and anaerobic respiration after exhaustion of oxygen as state 5. Although originally defined thus, the term “state 2 respiration” also has be used to imply respiration in the presence of substrate but without added ADP (34).
Mitochondrial membrane potential is generated by proton pumping at complexes I, III, and IV and offset by proton transfer in the opposite direction, referred to as proton leak. Although this process can occur in less-defined ways, apparently independent of known enzymes or carriers (117), much of the proton leak is a catalytic property of specific molecules termed uncoupling proteins (UCPs). The best characterized of these are UCP1, UCP2, and UCP3. UCP1, initially termed “thermogenin,” was the first of these to be described and is responsible for converting mitochondrial membrane potential to heat production in brown adipose tissue (233). UCP3, which also is expressed in BAT, is the major form present in skeletal muscle and heart. UCP2, the more ubiquitous form, is expressed in many tissues, most prominently spleen, lung, kidney, and, important with regard to diabetes, in insulin-producing pancreatic islet β cells. The likely functions of UCP2 and UCP3 (further addressed in Section VI) do not appear to include thermogenesis. Rather, these involve mitigation of ROS generation, export of fatty acids outward from mitochondria (UCP3), and regulation of insulin release (UCP2).
B. Reactive oxygen species and mitochondria
The ETS generates substantial superoxide derived from electron leaks as substrates are metabolized (294). Hence, mitochondrial oxygen use is associated with a cost in terms of generation of oxygen radicals and consequent oxidative damage. Biologically important ROS include the superoxide radical, O2·−, hydrogen peroxide, H2O2, and the hydroxyl radical, OH·. At physiologic pH, superoxide self-reacts (dismutates) or, more efficiently, is catalyzed by superoxide dismutase to form H2O2 (107). Although, superoxide per se is not thought to be particularly destructive, its impact arises from generation of the hydroxyl radical through a series of steps dependent on the presence of redox metals, such as iron or copper. This occurs as follows:
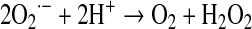 |
Summing these reactions yields
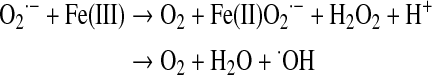 |
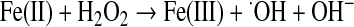 |
Mitochondria are considered the major intracellular site of superoxide production (57, 117, 263). However, exact quantification is difficult, and the mitochondrial contribution varies with the respiratory state, being greater near state 4 when membrane potential is less mitigated by ATP synthesis (118). The major sites of superoxide production within mitochondria have been controversial, but evidence indicates that most derive from complex I and III (263). Complex I superoxide is released nearly exclusively to the matrix side of the inner membrane, whereas complex III likely generates superoxide to both the matrix and outward to the intermembrane and extramitochondrial space (128, 299).
Complex I superoxide arises from bound flavin reduced to FMNH2 by NADH. Eventually, electrons are passed to membrane-embedded ubiquinone. In this process, electrons may be passed by FMNH2 or the partially reduced flavosemiquinone to oxygen-generating superoxide (282). Complex III superoxide is generated during the Q cycle, wherein coenzyme Q undergoes redox cycling through a reactive semiquinone species (Fig. 2) (295). As is evident in Fig. 2, the half-life of the semiquinone is highly dependent on potential and thus can be regulated by uncoupling.
FIG. 2.

Q-cycle at complex III. Mitochondrial inner membrane is depicted with (+) outside and (−) inside charge. bL and bH represent low- and high-potential cytochrome heme content. FeSIII represents non-heme iron–sulfur cluster of complex III. Electron (e−) flow follows along arrows, as depicted. Oxidation of CoQH2 directs electrons either to the iron–sulfur cluster and cytochrome c (Cyt C) or to generate the semiquinone form of CoQ, which passes electrons back through bL and bH to complete the cycle. Accompanying this process, two hydrogen ions are pumped outward from the negatively charged matrix.
Mitochondrial ROS may be generated at other sites as well, such as α-ketoglutarate dehydrogenase (6) and the iron–sulfur centers in the aconitase protein, where conversion of superoxide to the hydroxyl radical results in inactivation of the enzyme (110, 319).
Although this review is concerned with mitochondrial ROS, it should be recognized that considerable ROS derives from outside this organelle, including oxygen radicals from peroxisomal β-oxidation of fatty acids (24), NAD(P)H oxidase (53), xanthine oxidase, arachidonic acid metabolism, microsomal P-450 enzymes (75), and the prooxidant heme molecule (226).
Mitochondria have the capacity to invoke adaptive mechanisms that have evolved to defend against oxidative stress (117). One putative mechanism is a physiologic or “mild” uncoupling of oxidative phosphorylation, which would reduce superoxide generation by reducing mitochondrial membrane potential (295). This is detailed in Section VI. In addition, several enzymatic means may protect against ROS in mitochondria. These include conversion of superoxide to H2O2 by manganese superoxide dismutase (MnSOD) and scavenging of H2O2 by catalase, glutathione peroxidase (GPX), or peroxiredoxin III (117). Although catalase is highly efficient in scavenging hydrogen peroxide, the enzyme has classically been considered a cytoplasmic rather than mitochondrial protein. Conversely, recent studies document the expression and antioxidant activity in liver mitochondria (195, 280), and reports exist of catalase in heart mitochondria (262, 313).
C. Mitochondrial nitric oxide
Nitric oxide (NO) can interact with proteins containing heme or thiols. In mitochondria, this radical can decrease respiration and potentially trigger apoptosis or cell necrosis or both by inhibiting cytochrome oxidase (41). Moreover, superoxide is known to react very rapidly with NO (107), generating a damaging peroxynitrite (ONOO−) radical (98, 144, 246), which can impair proteins involved in electron transport (261). In endothelial mitochondria, superoxide consumption of NO could be especially problematic if it were to interfere with NO signaling, a process that regulates vasodilation through release of NO to vascular smooth muscle.
Although this implies that NO interacts with mitochondrial proteins, a lack of confirmed evidence exists to suggest that mitochondria contain a nitric oxide synthase (NOS) and are capable of actually producing the radical. Although several earlier studies suggested the presence of a mitochondrial NOS, other reports question this and raise technical concerns, as described in a recent review (183). Venkatakrishnan et al. (320) recently carried out detailed studies of highly purified liver mitochondria and submitochondrial particles by using HPLC–mass spectroscopy and found no evidence of NOS-derived peptides, calmodulin (needed for NOS activity), or NOS activity measured as arginine-to-citrulline conversion.
D. Role of calcium and the mitochondrial permeability transition pore
Calcium is taken up by mitochondria both through a uniporter (thus, independent of other ion or molecule) and through a pulsed or rapid mode, dependent on pre-pulse calcium and inhibited by calcium concentration >100–150 nM (123). Separate molecular structures responsible for the uniporter and rapid-mode transport have not been identified, and the two processes may derive from similar modes of operation. The conserved nature of these mechanisms suggests important functional consequences. The major roles of calcium in mitochondria appear to be to stimulation of ATP production and induction of the mitochondrial permeability transition (124). Both Na+-dependent and -independent processes for calcium efflux exist (125), which, of course, must match the amount taken in as a result of influx pulses.
Calcium, ROS, high pH, low membrane potential, and oxidized pyridine nucleotides all may lead to opening of the mitochondrial permeability transition pore (MPTP) (284). In its dramatic form, the MPTP results in a marked increase in mitochondrial inner-membrane permeability, which will decrease ΔΨ and lead to mitochondrial swelling, release of cytochrome c, cell damage, and apoptosis. Conversely, transient opening may be physiologic, being involved in functions such as voltage, redox or pH sensing, divalent cation sensing, or regulation of adenine nucleotide concentrations (178). It is believed that the MPTP is mediated by the reversible opening of a protein pore in the inner membrane (257). The composition of the MPTP is still unclear, but creation of the pore may involve the interaction of several proteins. This includes adenine nucleotide translocase (ANT), which imports ADP and exports ATP through binding of these nucleotides on opposite sides and directing the carrier opening toward or away from the mitochondrial matrix (284). Cyclosporin A, which interacts with the matrix protein cyclophilin D, inhibits the MPTP. The pore requires calcium for opening. So, under the conditions in which we and others have studied respiratory coupling in isolated mitochondria, the MPTP is inhibited by lack of calcium in the medium.
III. Assessing Mitochondrial Function
A. Respiration and potential
With a simple oxygen electrode and small-volume chamber, which can be less <1 ml, it is easy to measure oxygen consumption by isolated mitochondria over time, a process that is generally linear over several minutes, even as the chamber oxygen content decreases. In isolated mitochondria, particular energy substrates will initiate respiration through action at a specific complex or entry point (Section II.A). Direct addition of NADH to isolated mitochondria would not be expected to initiate respiration unless the outer membrane is disrupted, which is not uncommon, because preparation of mitochondria may not produce perfectly intact organelles.
Oxygen consumption can also be assessed in whole cells by measuring the decrease in O2 content in the medium. Another way to do this, which avoids changes in cell oxygen tension, is to perfuse cells by using small oxygen electrodes proximal and distal to the cell preparation. This has been described even for cells perfused under microscopy (149) or, as recently described by our laboratory (104), on bovine aortic endothelial cells grown on glass beads and perfused on columns. It is necessary to calibrate precisely the proximal and distal electrodes to each other, which can be done by using a shunt pathway around the cell preparation. By adding various inhibitors of mitochondrial respiration or ATP synthase or both, it is possible to use this type of system to study mitochondrial function in intact cells (235).
Mitochondrial membrane potential can be estimated with a number of potential-sensitive fluorescent compounds both in isolated mitochondria and in intact cells, although it is difficult to quantify in this fashion. Quantitatively to assess potential, we and others measured the concentration of mitochondria-targeted cations, such as methyltriphenylphosphonium (TPMP) or tetraphenylphosphonium (TPP) inside and outside the mitochondrial matrix (154). Potential can then be calculated by using the Nernst equation. The probe concentration can be determined either by using an electrode sensitive to the cation (154) or by using the radiolabeled compound.
B. ATP production and the proton leak
Mitochondrial membrane potential (charge differential across the inner membrane), often referred to as ΔΨ, is generated as protons are pumped outward from the matrix, a process that depends on substrate utilization and electron transport. Loss of membrane potential will result from any process wherein protons move back toward the matrix (for example, the proton leak as induced by the catalytic action of specific uncoupling proteins). Loss of potential also may result from nonspecific proton leaks, opening of the MPTP (Section II.D), interactions of fatty acids with mitochondrial proteins (136, 163, 289), drug or chemical action (56), or general disruption of the inner membrane. According to the chemiosmotic theory (219), the extent of ADP conversion to ATP is determined by membrane potential per se; in other words, the net effect of all processes contributing to or detracting from potential.
ATP production by isolated mitochondria incubated in vitro can be determined by removing medium at different time points and measuring the ATP content (34). The ADP/O ratio (ADP converted to ATP per unit oxygen consumed) provides an index of the efficiency of ATP production. This can be determined in isolated mitochondria by adding ADP and observing the rate of oxygen consumption, which will increase immediately on addition of ADP and usually will decrease sharply at the point where ADP is completely consumed. The ADP/O ratio is sometimes used as an index of uncoupling activity or the proton leak. However, the ADP/O ratio also is affected by ATP synthase and the efficiency of electron transfer, as well as the proton leak. Typical values for the ADP/O ratio range up to just above 3, dependent on the processes (see earlier) that control membrane potential.
The respiratory control ratio (RCR or ratio of state 3 to state 4 respiration) also provides an index of the efficiency of ATP formation. State 4 respiration will increase, and the RCR will decrease in mitochondria subject to uncoupling, as discharge of potential will be compensated by increased respiration. However, state 4 respiration is not the best estimate of the proton leak because it is influenced not just by uncoupling but also by substrate oxidation and the efficiency of electron transport.
A more direct and probably the best assessment of the proton leak in isolated mitochondria can be accomplished by measurement of inner membrane potential simultaneous with respiration under conditions set so that oxygen consumption is proportional to proton pumping (39). Under these conditions, it is possible to assess the relation of hydrogen transfer to potential (the driving force behind ATP synthesis) and to calculate the proton conductance (in other words, proton transfer per unit potential expressed in units of nmole H/min/mg mitochondrial protein/mV). In comparing different experimental states, the proton leak will manifest as greater proton conductance. Assessing proton conductance under differing degrees of electron transfer, adjusted by using inhibitors, enables us to perform kinetic analysis (39). An increase in the proton leak between two conditions will manifest as a shift in the curve of oxygen use versus potential upward and to the left, indicating greater oxygen consumption (proportional to hydrogen transfer) at any given membrane potential. An example is shown in Fig. 3, which depicts proton-leak kinetics in brown fat mitochondria of mice fed low-fat and high-fat diets (102). Leak kinetics was assessed in the presence or absence of the uncoupling protein 1 inhibitor GDP. The mitochondria from the high-fat–fed mice manifested greater GDP inhibitable proton conductance, reflecting the UCP1-mediated proton leak.
FIG. 3.

Kinetics of the proton leak in mitochondria isolated from brown adipose tissue mitochondria of C57Bl/6 mice fed low-fat (LF) or high-fat (HF) diets. Leak kinetics were assessed in the presence or absence of the uncoupling protein 1 inhibitor GDP. Mitochondria from the high-fat–fed mice manifest greater GDP-inhibitable proton conductance, reflecting the UCP1-mediated proton leak. For both panels, the increased proton leak in the absence of GDP compared with the presence of GDP is evident as a shift in the curve of oxygen use versus potential upward and to the left.
Proton conductance is more difficult to measure in intact cells, but it has been accomplished (259). Coupling of respiration and TCA activity, which should decrease with uncoupling, has been estimated in muscle in vivo by using magnetic resonance spectroscopy to assess ATP production and 13C enrichment of glutamate by acetate to assess TCA activity (64).
C. ROS production by isolated mitochondria
ROS production is maximal during state 4 respiration, wherein radical formation is enhanced as electron flow leads to high potential, unmitigated by ATP generation (31). Although mitochondria in vivo are rarely, if ever, in this unmitigated state, it is theorized that mitochondria in diabetes, when exposed to high glucose and fatty acid concentrations, may be driven toward greater oxygen use and higher potential, thereby forming more ROS (87, 237, 335). Under state 4 conditions in liver or heart mitochondria, ROS production has been estimated to account for as much as 2% of oxygen consumed (57).
ROS produced by isolated mitochondria can be detected with a variety of techniques not reviewed here. Fluorescent measurements are often used; but with any method, it is very important to use all possible caution to avoid measuring nonspecific signals. This can arise from probe interaction with a wide variety of substances, including substrates, test substances, and components of mitochondria or cells per se. Signal intensity in the presence and absence of superoxide dismutase can help validate specificity for superoxide. Likewise, H2O2 can be determined in the presence or absence of catalase. To the extent that these enzymes or mimetics penetrate into the compartment under study, and assuming that the enzymes themselves do not alter fluorescence, catalase or superoxide should reduce fluorescence to near the detectable limit. In studies of isolated mitochondria, it also is important that fluorescence be determined under no-substrate conditions and that this be considered with respect to substrate-induced ROS.
A specific but somewhat cumbersome way to assess oxygen radical formation by isolated mitochondria is through EPR spectroscopy. This can be done by detecting specific signals resulting from free radical interactions with added compounds, as spin traps (105, 241). We used the spin trap, 5,5-dimethyl-l-pyrroline-N-oxide (DMPO) to detect superoxide generating a specific signal representing either this compound or the hydroxy radical (see Section III.D). These two possibilities can be separated by adding SOD, which should abolish the signal generated by superoxide.
D. Site specificity of mitochondrial superoxide production
We suggested a way to measure superoxide from isolated mitochondria in a manner that imparts a degree of specificity for matrix ROS compared with superoxide released external to the organelles. Fluorescent H2O2 probes such as 10-acetyl-3,7-dihydroxyphenoxazine (DHPA) and EPR spectroscopy measure mitochondrial superoxide in a different fashion. DHPA detects superoxide indirectly. When added to isolated mitochondria, the probe detects H2O2 generated from superoxide by matrix MnSOD. H2O2 so generated diffuses outward from mitochondria and reacts with horseradish peroxidase in the incubation medium to trigger fluorescence. H2O2 produced in this way derives largely from superoxide generated at complex I and released to the matrix (299). In contrast, the EPR spin trap, DMPO, detects superoxide directly after efflux outward from mitochondria. Superoxide produced in this way derives largely from the Q cycle at complex III (299). Some superoxide also is released to the matrix. However, because DMPO will not easily penetrate mitochondria and because matrix superoxide is rapidly converted to H2O2, the spin trap should detect very little matrix superoxide.
We carried out substrate and inhibitor studies of ROS production by isolated bovine aortic endothelial (BAE) cell mitochondria, which support these contentions (241). ROS was detected as H2O2 by DHPA fluorescence (Fig. 4) and directly as superoxide by EPR (Fig. 5). These experiments showed (241) that the complex I inhibitor, rotenone, markedly decreased succinate-driven ROS production (reverse transport through complex I), as detected with fluorescence, but had no effect on succinate-driven superoxide production with EPR spectroscopy. Moreover, antimycin (active in complex III to increase ROS by its effect on the Q-cycle to prolong the half-life of the semiquinone) markedly increased succinate-driven superoxide by EPR, but decreased ROS by fluorescent detection. Further, stigmatellin, which blocks electron entry to the complex III Q-cycle, markedly reduced the EPR signal. Thus, we suggest that, in combination, EPR spectroscopy and fluorescence assessment of H2O2 release from mitochondria impart at least a degree of specificity for complex I (largely matrix) or III (externally released as well as matrix) superoxide. Figure 6 depicts the origin of mitochondrial superoxide production assessed in this way. We applied these methods to assess superoxide release by mitochondria isolated from tissues of diabetic rodents (Section IV.C.7) and to assess the effects of a mitochondria-targeted coenzyme Q derivative in BAE cells (Section VIII.C).
FIG. 4.

H2O2 production by mitochondria isolated from mouse hindlimb muscle (A) or bovine aortic endothelial cells (B and C), measured as 10-acetyl-3,7-dihydroxyphenoxazine (DHPA) fluorescence. Mitochondria were incubated in the presence of the substrates or inhibitors shown (or both), including 5 mM succinate (succ), 5 mM glutamate + 1 mM malate (glut + mal), 5 μM rotenone (rot), or 1 μM antimycin A (ant A). n = 4 to 6 mitochondrial preparations for each data point. *p < 0.01, †p < 0.05 by one-way ANOVA compared with succinate condition.
FIG. 5.

EPR spectra generated by using the spin-trap DMPO, which is specific for superoxide or the hydroxyl radical. Spectra were determined in the presence of bovine aortic endothelial cell mitochondria incubated in respiratory buffer plus the substrates or compounds indicated or both. Spectral signals were abolished by addition of MnSOD, demonstrating specificity for superoxide. Note the scale difference for the succinate + antimycin A condition. Additions consisted of 5 mM succinate, 5 μM rotenone, 1 μM antimycin A, or 200 μg/ml manganese superoxide dismutase (SOD).
FIG. 6.

Schematic diagram depicting electron transport, the action of substrates and inhibitors, and proposed sites of ROS production, as detected with H2O2 fluorescence and EPR spectroscopy. Straight lines with arrows, Forward electron transport. Dashed lines, Reverse transport. Dashed-dotted lines, Diffusion of Q compounds in complex III or of H2O2 out from the matrix, as shown. Dotted boxes, Complexes I, II, and III. X, Sites of inhibition by rotenone (ROT), stigmatellin (STIG), or antimycin A (ANT A). Dark arrows, Direction of superoxide release or conversion to H2O2.
Theoretically, it is possible to assess complex III superoxide released to the cytoplasmic side of isolated mitochondria simply by measuring H2O2 production (for example, as DHPA fluorescence in the presence and absence of added SOD). SOD should increase fluorescence to the extent that it would then include the contribution of externally released superoxide to the H2O2 pool. Superoxide production has been effectively assessed in this way in studies of the topology of muscle, heart, and liver mitochondria (299), although that required mathematical correction for fluorescent interference.
E. Mitochondrial ROS production in intact cells
Several studies measured intact-cell total ROS production as H2O2 by using fluorescent probes such as carboxydichlorodihydrofluorescein (with more or less attention to radical specificity). However, most intact-cell studies do not separate mitochondrial from cytoplasmic ROS. A degree of specificity for intact-cell mitochondrial superoxide, as opposed to cytoplasmic, can be detected by using mitochondria- targeted dihydroethidine (DHE) or “MitoSOX”. MitoSOX is a DHE derivative conjugated to the cation triphenylphosphonium, resulting in potential-dependent accumulation of the probe in the mitochondrial matrix. The accumulation in the matrix should be very large, as cationic triphenylphosphonium-conjugated molecules accumulate by many fold (230). The difference in fluorescence between untargeted DHE and MitoSOX may provide a semiquantitative index of relative cytoplasmic and mitochondrial superoxide. A concern, however, is the degree to which MitoSOX could undergo oxidation in the cytoplasm, which is difficult to ascertain. Because DHE and MitoSOX do not measure H2O2, treatment with an SOD mimetic should decrease fluorescence and may serve as a means of validation that superoxide is being measured. Another important consideration with respect to mitochondria-targeted DHE is that the probe is dependent on mitochondrial membrane potential to enter the organelles. Resolution of this requires that potential be monitored and that an appropriate correction be applied. Although difficult, this has been accomplished by using tetramethylrhodamine methyl ester (TMRM) to measure fluorescence in cerebellar granule neurons (149).
DHE has been criticized as nonspecific, and some advocate analysis of the oxidation products with high-pressure liquid chromatography (HPLC) to document specificity for superoxide as opposed to H2O2 (188). In using rhodamine derivatives like TMRM, attention has to be paid to the capacity for these compounds themselves to be a source of ROS (255).
F. Oxidative damage to mitochondria in intact cells
Distinct from assessing ongoing ROS production, several techniques are available to assess oxidative damage done to mitochondria and intact cells and have been used in diabetes-related studies. These include markers of oxidative damage to proteins, lipids, and DNA. Chronic radical production is also compensated by a variety of enzymatic and other mechanisms that can be assessed as evidence of oxidative stress.
Oxidative damage to DNA can cause structural modifications of the nucleotide bases or by cross-linking. This can lead to gene mutation and cell damage. A common way to detect oxidative damage to DNA is to measure 8-hydroxy-2′-deoxyguanosine (8-OHdG), a compound formed by oxidation of deoxyguanosine in blood or urine, which can be analyzed by various analytic methods, including HPLC, gas chromatography–mass spectrometry, and enzyme-linked immunosorbent assay (ELISA) (258).
Because of the key role of lipids in biomembranes, lipid peroxidation is of great concern in regard to disease states like diabetes. Moreover, in obesity and diabetes, lipids have been observed to accumulate near mitochondria, and products of lipid oxidation can interfere with cell signaling (see Section IV.C.5). Also of concern is that lipid peroxidation can occur as a chain reaction that can self-perpetuate, thereby amplifying an initial oxygen radical insult by severalfold. The occurrence of this self-sustaining reaction has therapeutic implications (Section VIII.C).
Unstable lipid peroxides derived from polyunsaturated fatty acid breakdown to several compounds amenable to biochemical assay. For example, isoprostanes are often measured as the marker, 8-isoprostane, formed by peroxidation of arachidonic acid (217). Isoprostanes have adverse vascular effects, including mitogenesis and altered vascular reactivity (217). Other markers of lipid peroxidation are alkanals, which can be measured as 4-hydroxy-2-nonenal (4-HNE), malondialdehyde (MDA), and acrolein (258).
Oxidative damage also can modify amino acids and, therefore, change structure and function and lead to cross-linking or protein breakdown (258). Peroxynitrite, which results from oxygen radical interaction with nitric oxide, can cause nitration of tyrosine, resulting in a formation of the marker compound, nitrotyrosine (216).
Certain methods can be used to measure oxidative damage specifically within mitochondria. One method is to determine the activity of the aconitase enzyme, a protein that is highly sensitive to oxidative damage (110). Mitochondrial protein also can be evaluated for 4-HNE protein adducts with immunoblotting antibody (34), although specificity can be questioned.
IV. Mitochondrial Metabolism and Diabetes
This section focuses on the relation of mitochondrial metabolism to the clinical problem of diabetes. After a brief general discussion, we address mitochondrial diabetes and then consider the role of mitochondria in the more common forms, classified as type 1 and type 2. We first discuss issues related to morphology, numbers of mitochondria, and mitochondrial biogenesis. After this, we address the role of mitochondria in insulin sensitivity and insulin secretion and the role of respiratory uncoupling. We next address the relation of mitochondria to diabetic complications in non–insulin-sensitive tissues and, finally, the importance of fuel selectivity and its relation to the important problem of diabetic cardiomyopathy.
A. General considerations
As the major sites for energy disposition, it should not be surprising that mitochondria appear to be important in multiple aspects of this disorder, including cause, complications, management, and prevention. In the unusual case, genetic mutations in mitochondrial DNA lead to “mitochondrial diabetes” (see later). As opposed to mitochondrial diabetes, the large majority of cases fall into the broad classifications of type 1 and type 2 diabetes (10).
It is widely accepted that the etiology of type 2 diabetes involves both pancreatic β-cell dysfunction and insulin resistance in insulin-sensitive tissues, including hepatocytes, myocytes, and adipocytes. Moreover, type 2 diabetes is well known to be a progressive disorder (120), characterized by deteriorating capacity for both insulin release and insulin action. Both defects can be identified early and are present even in nondiabetic offspring of patients with type 2 diabetes (112, 122, 249). However, general consensus is found that insulin sensitivity is substantially impaired early in the course, whereas worsening of hyperglycemia over time is related to β-cell dysfunction, with diminished ability of insulin secretion to keep up with the demand imposed by insulin resistance.
In contrast, type 1 diabetes has a completely different etiology: autoimmune destruction of pancreatic β cells. Once under way, this process evolves over months or years to a point at which insulin secretion is low enough to induce hyperglycemic symptoms. From that point, the course continues downward to complete insulin deficiency and, in the absence of insulin therapy, a ketotis-prone and life-threatening state.
Next we begin with an overview of mitochondrial diabetes, followed by mitochondrial aspects related to the much more common forms of diabetes, classified as type 1 or type 2.
B. Mitochondrial diabetes
Mitochondrial diabetes usually is first seen at middle age, is maternally transmitted (the mode of inheritance of mitochondrial DNA), and often is associated with hearing loss, particularly for high tones. The most common mutation leading to mitochondrial diabetes is the A3243G mutation in the mitochondrial encoded tRNA (Leu, UUR) gene (205, 206). The defect in tRNA leads to impaired synthesis of multiple mitochondrial proteins and overall mitochondrial dysfunction. Although the phenotype will often look like type 2 diabetes, treatment with the commonly used drug, metformin, should be avoided because of a propensity to lactic acidosis, a well-recognized but otherwise rare adverse effect of this drug.
The A3243G form of mitochondrial diabetes is characterized by decreased glucose-induced insulin release but not insulin resistance, suggesting that the major pathology occurs within mitochondria of pancreatic β cells. Conversely, recent studies also provide evidence for hepatic dysfunction (305) and decreased skeletal muscle glucose uptake (200) associated with the A3243G mutation.
Interestingly, this syndrome is initially mild and worsens over time. One possible reason may be mitochondrial dysfunction compounded by hyperglycemia-induced ROS, oxidative damage, and worsening hyperglycemia (117). Pancreatic β cells may be particularly prone to oxidative damage. β Cells exposed to hyperglycemia, and consequent increased intracellular calcium, are prone to high levels of reducing equivalents and consumption of ADP (327), resulting in higher membrane potential and, therefore, more ROS production (see Section V.A.1). Moreover, β cells have relatively low levels of expression of antioxidant enzymes (190), and LDH levels are low (290), so glucose is driven to mitochondrial substrates generating higher membrane potential.
Other possible reasons for the progression of mitochondrial diabetes have been proposed. Decreased numbers of functioning β cells will decrease insulin responsiveness to glucose and perhaps reset glucose-induced insulin release to a higher level of glycemia. In addition, generalized respiratory depression will result in decreased ATP formation, which will impair glucose-induced insulin release.
Cells contain numerous mitochondria, which can number in the hundreds or thousands, and each has multiple copies of mitochondrial DNA (mtDNA). However, mtDNA mutations are present in only a portion of the mtDNA, a situation referred to as heteroplasty. In mitochondria of cells with the A3243G mutant mtDNA, the extent of heteroplasty is related to mitochondrial oxygen consumption, which seems to decrease sharply when heteroplasty reaches ∼70% (205). Conversely, it is not clear that heteroplasty worsens with age, so this is apparently not an explanation for the late onset and progression of mitochondrial diabetes.
Clinically, mitochondrial diabetes must be differentiated from other unusual single-gene defects, such as MODY diabetes, due to defects in glucokinase or hepatic transcription factors. These disorders often are first seen earlier in life (111). The A3243AG mutation also is seen in the MELAS (mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes) syndrome (115).
C. Type 1 and type 2 diabetes
Several studies have demonstrated perturbations in mitochondria in both insulin-deficient and insulin-resistant states and in the related condition of obesity. Although the phrase “mitochondrial dysfunction” is often used in this respect, it must be remembered that beyond dysfunction, evidence exists for defects in mitochondrial biogenesis, number, morphology, and dynamics, including fusion and fission. It is a matter of debate whether the insulin resistance seen in type 2 diabetes is related to mitochondrial function as opposed to number or other characteristic(s) or a combination of these.
1. Mitochondrial number and morphology
Biopsies of skeletal muscle from subjects with type 2 diabetes and obesity reveal mitochondria of smaller size and number per unit volume (density) compared with those in lean controls (161). Size appears to correlate with whole-body insulin sensitivity (161). Moreover, mitochondria of offspring of diabetic subjects are lower in density compared with those of controls (225). Mitochondrial subtype selectivity to morphologic alterations also may be noted. Skeletal myocytes and cardiomyocytes contain two populations of mitochondria; subsarcolemmal (SLM) and intermyofibrillar (IMFM). The SLM are larger, lamellar shaped, and located below the sarcolemma (169, 268). The IMFM are smaller and located between contractile elements. It is thought that the SLM contribute energy for membrane and transport processes, whereas the IMFM contribute more to contractile function. Studies with transmission electron microscopy revealed reduced numbers of SLM in skeletal muscle of type 2 diabetes and obese subjects associated with reduced electron-transport activity per unit mitochondrial DNA, suggesting functional impairment as well (268).
Insulin deficiency, as seen in type 1 diabetes, is also associated with alterations in mitochondrial morphology. Skeletal muscle mitochondria of insulin-deficient rats made diabetic with the β-cell toxin streptozotocin, thereby mimicking type 1 diabetes, showed a loss of cristae and an increase in electron-dense granules along with lipid droplets around the mitochondria (59). Studies using alloxan to damage β cells and induce diabetes in rats revealed a decrease in mitochondrial number in liver and heart, with mitochondrial swelling and damage to mitochondrial membranes and cristae (209). An increase in mitochondrial area seen with transmission electron microscopy was reported in endothelial cells of women with type 1 diabetes (54). By using isotopic techniques to measure volumes of [3H]H2O and [14C]sucrose, we found no change in matrix volumes of gastrocnemius, heart, and liver mitochondria isolated from severely hyperglycemic streptozotocin (STZ)-diabetic rats compared with controls (134).
A recent study using electron microscopy to examine heart mitochondria of insulin-deficient Akita mice, who develop diabetes as a result of a mutation in the insulin gene, revealed reduced crista density and greater mean area but unchanged mitochondrial numbers in affected compared with wild-type (WT) mice (42). Conversely, observations by the same group (34) revealed that mitochondrial number was increased and that mitochondria were smaller in cardiac muscle of db/db obese diabetic mice compared with WT. db/db mice have a defect in the leptin receptor, leading to obesity and hyperinsulinemia (as opposed to the insulin-defective Akita mice) and manifest steatosis in cardiomyocytes. Possibly, the contrast between these models relates to the extent of lipid accumulation, which was marked in cardiomyocytes of the obese model.
In summary, it is clear that morphologic changes in mitochondria occur in diabetic states, although the data are not completely consistent over different studies. One difficulty is that different methods have been used for assessment. Moreover, once mitochondria are isolated and removed from the in vivo environment or fixed within tissue preparations, factors such as osmotic forces, tissue turgor, or membrane integrity may be altered and could easily affect mitochondrial size and morphology.
2. Fission/fusion
Besides mitochondrial number and morphology, mitochondrial metabolism also depends on the dynamic movement and distribution of the organelles, which tend to localize to intracellular sites where ATP production is most essential (196). As mitochondria move within cells, they undergo both fission, needed for distribution and networking, and fusion, needed for mixing of the mitochondrial genome. These processes depend on certain proteins, including two isoforms of mitofusin (MFN) involved in docking to initiate fusion and the presenillin-associated rhomboid-like (PARL) protein important for morphologic integrity (63). Evidence now indicates that obesity in both humans and rodents is associated with reduced MFN (19). Moreover, polymorphisms of PARL in humans are associated with insulin resistance (325).
3. Mitochondrial biogenesis
Perturbed mitochondrial biogenesis has been suggested as the cause for reduced mitochondrial number as well as reduced capacity for oxidative phosphorylation in diabetes. An important factor driving mitochondrial biogenesis at the molecular level is the peroxisome proliferator–activated receptor gamma (PPARγ) coactivator or PGC-1α. PGC-1α represents a coactivator of nuclear transcription factors (NRFs) 1 and 2 and mitochondrial transcription factor A, as well as PPARγ and PPARα (89, 334). These factors regulate the expression of genes involved in mitochondrial replication as well as oxidative phosphorylation (334). PGC-1α also serves to coactivate transcription factors for several other genes involved in energy homeostasis (100).
The regulation of PGC-1α is an evolving issue. As might be expected, given its importance in bioenergetics, PGC-1α transcription or activity or both are enhanced by two important enzymes viewed as metabolic sensors: AMP-activated protein kinase (AMPK) and the mammalian counterpart of silent information regulator 2 (SIRT1). These enzymes alter PGC-1α through phosphorylation or deacetylation, respectively. States of energy depletion, such as reduced caloric intake and exercise, result in an increase in the ratio of AMP to ATP, thereby activating AMPK (41, 52, 266). In turn, this increases PGC-1α transcription and directly activates PGC-1α through phosphorylation. Caloric restriction or exercise also increases tissue NAD+ content relative to NADH and, thereby, activates the NAD+-dependent histone deacetylase, SIRT1. SIRT1 enhances PGC-1α by deacetylation at specific lysine residues (272).
Nitric oxide also appears important as a regulator of biogenesis (238). As indicated in Section II.C, nitric oxide acutely inhibits mitochondrial respiration by binding to cytochrome c oxidase (41). Conversely, smaller and prolonged increases in NO induce mitochondrial biogenesis, as observed in several cell types (239). Moreover, NO-stimulated biogenesis was observed in brown adipose, muscle, and heart tissues and appeared dependent on PGC-1α as well as cyclic GMP (239).
Overexpression of PGC-1α in cultured muscle cells increased β-oxidation of fatty acids and reduced acylcarnitine levels, which reflect partial breakdown products of β-oxidation (168). Overexpression of PGC-1α in mouse skeletal muscle increased muscle glucose uptake as well as the expression of proteins involved in fat oxidation and glucose transport (27). Consistent with this finding, mice deficient in PGC-1α were found to have defective contractility of skeletal and heart muscle (15, 89). Moreover, PGC-1α enhanced oxidative phosphorylation and appeared a major factor regulating the muscle fiber type favoring the generation of oxidative type 1 muscle fibers (198).
Considering this, PGC-1α might be important in the pathogenesis of insulin-resistant states, and defects in PGC-1α expression or activity might result in reduced insulin sensitivity. Muscle-biopsy studies showed that PGC-1α is reduced in patients with type 2 diabetes (215, 222, 247) as well as in family members of individuals with type 2 diabetes (247). Moreover, PGC-1α expression could be restored in type 2 diabetes by the insulin-sensitizing drug, rosiglitazone (215).
Conversely, mice deficient in muscle PGC-1α have normal peripheral insulin sensitivity (129), and globally deficient mice are resistant to diet-induced obesity, although associated with many other systemic problems (191, 199). Moreover, reduced PGC-1α or reductions in down-line transcription factors were not seen in muscle biopsy specimens from family members of subjects with type 2 diabetes, even though these subjects had increased intramyocellular lipid and decreased numbers of mitochondria (225). Thus, factors other than PGC-1α appear involved in the relations between mitochondria and insulin resistance.
Liver also represents an important insulin-sensitive tissue wherein mitochondrial biogenesis may be critical to the pathogenesis of diabetes. In hepatocytes, PGC-1α is important in the regulation of both gluconeogenesis and fat oxidation (271). The NAD+-dependent histone deacetylase, SIRT1 increases gluconeogenesis in liver cells through its effects on PGC-1α (272). Consistent with this, mice deficient in PGC-1α develop hepatic steatosis and are prone to hypoglycemia (191, 199), among several other multisystem abnormalities.
4. Mitochondrial function in type 2 diabetes and insulin-resistant states
As discussed earlier, type 2 diabetes is a progressive disorder worsening over time. The reasons(s) underlying the onset and worsening of this condition are still unresolved. However, considerable suspicion falls on mitochondrial function, in regard to both insulin resistance and insulin secretion.
Studies of human subjects and rodents provide evidence for impaired oxidative phosphorylation in muscle mitochondria in insulin-resistant states. Kelley et al. (161) studied mitochondria isolated from human muscle biopsy specimens obtained from type 2 diabetes, obese, and lean individuals. These investigators demonstrated reduced NADH oxidoreductase and reduced citrate synthase activity in the mitochondria of the diabetes and obese subjects compared with lean subjects. Citrate synthase governs the condensation of acetyl-CoA with oxaloacetate generating citrate and is important in setting the rate of the TCA cycle. Mitochondrial oxidative phosphorylation has been also been assessed in human muscle in vivo by using 13C nuclear magnetic resonance (NMR) to assess TCA flux rates along with 31P NMR to assess phosphorylation of ADP. These studies showed that skeletal muscle oxidative phosphorylation was impaired in insulin-resistant offspring of individuals with type 2 diabetes, associated with increased intramyocellular lipid (253). Similar findings were reported in muscle of elderly subjects with insulin resistance compared with young controls (252). In a further study, type 2 diabetes was characterized by increased lipid content in myocytes, as well as by a relative decrease in the proportion of enzymes regulating oxidative as opposed to glycolytic metabolism (131).
Consistent with this, evidence exists of decreased mRNA expression of several genes associated with oxidative phosphorylation, including genes coordinately regulated by PGC-1α and nuclear respiratory factors (222, 247, 334). This has been observed not only in subjects with type 2 diabetes but also in first-degree relatives. Evidence at the protein level suggests that muscle of subjects with type 2 diabetes manifests impaired ATP production, suggested by reduced ATP synthase and creatine kinase B (139).
Although this is consistent with impaired mitochondrial function in type 2 diabetes per unit muscle tissue, controversy exists regarding whether mitochondria per se are defective or whether the problem is restricted to mitochondrial number. Boushel et al. (35) examined mitochondrial function in permeabilized skeletal muscle fibers of 11 subjects with type 2 diabetes. These investigators found reduced oxygen use under conditions of ADP stimulation (coupled respiration) and maximal uncoupling by carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone (FCCP) in the diabetic subjects compared with nondiabetic controls. However, the differences were resolved when the data were normalized to mitochondrial DNA or to citrate synthase activity. If these parameters actually reflect mitochondrial numbers, the implication is that the decreased respiration in type 2 diabetes can be attributed to reduced muscle mitochondrial content.
Studies of mitochondrial function also have been carried out in rodent models of type 2 diabetes. Boudina et al. (34) examined heart mitochondrial function in saponin-permeabilized heart muscle fibers isolated from insulin-resistant, diabetic, leptin receptor–deficient db/db mice compared with lean controls. These investigators reported decreased respiration on complex I substrates and palmitoyl-carnitine, associated with proportionately reduced ATP production and therefore no change in ADP/O ratios. These investigators also reported decreased content of the F1 α-subunit of ATP synthase and an increase in fatty acid–induced proton conductance based on proton-leak kinetics. These findings were associated with reduced cardiac muscle function in the db/db mice.
High-fat feeding is associated with insulin resistance (286) and may lead to oversupply of fatty acids to mitochondria. In humans, high-fat feeding results in downregulation of several genes associated with oxidative phosphorylation and mitochondrial biogenesis (298). Recent metabolomic studies in rodents suggest that enhanced fat metabolism seen with high-fat feeding overloads mitochondria with β-oxidation products in a way that restricts their ability to metabolize these products completely to CO2 and restricts their capacity to switch from fat to glucose oxidation (228). Koves et al. (170) showed that high-fat feeding increased muscle even chain acylcarnitines and acid-soluble metabolites of labeled fatty acids; these compounds representing products of incomplete β oxidation. This was associated with decreased TCA intermediates and an inability of mitochondria to switch from using fat-derived substrates to using the glucose-derived metabolite, pyruvate. These perturbations could be prevented by restricting mitochondrial entry of fatty acids by KO of malonyl-CoA decarboxylase (MCD). MCD breaks down malonyl-CoA, thereby relieving inhibition of carnitine palmitoyltransferase 1 (170). Similar findings were observed by silencing of MCD in muscle cells (36). Hence these data imply that mitochondria of high-fat–fed rodents are exposed to increased rather than decreased rates of β oxidation but become impaired and unable to handle the high rate of flux.
In liver, the forkhead transcription factor Foxa2 activates transcription of genes regulating lipid metabolism and ketogenesis. Wolfrum et al. (330) showed that in insulin-resistant or hyperinsulinemic mice, Foxa2 is inactive and confined to the cytoplasm of hepatocytes. The inactivity of this factor promotes lipid accumulation at the expense of oxidation in the liver and impairs insulin sensitivity, encouraging export of fat, ketones, and glucose. Indeed, degradation of malonyl Co-A in liver (through overexpression the degrading enzyme malonyl-CoA-decarboxylase) favors mitochondrial fat oxidation and reduces circulating free fatty acids and ketones (β-hydroxybutyrate), improving insulin sensitivity in both muscle and liver (12). Although this may appear to differ from the effect of inhibiting malonyl-CoA in muscle, it should be appreciated that liver is far more capable than muscle of packaging and exporting fatty acids. Hence, it has been suggested that muscle mitochondria, as opposed to liver, are more vulnerable to energy overload (170). Another contrast between the phenotypic results of malonyl-CoA manipulation in liver and muscle is evident in studies of mice deficient in acetyl-CoA carboxylase 2 (ACC2). These mice demonstrated reduced malonyl-CoA levels and a higher rate of fatty acid oxidation and resisted diet-induced obesity and diabetes (5). At first thought, this differs from the effects of MCD KO (previous paragraph). However, this can be explained because the ACC2 KO mice showed minor adjustments in muscle lipid metabolism but marked changes in liver, including protection against hepatic steatosis. Again, this underscores basic differences in lipid handling between muscle and liver with packaging and export in liver compared with the more singular role of lipid oxidation in muscle.
Evidence also exists for altered mitochondrial function of adipocytes in type 2 diabetes. Mitochondrial function, as indicated by respiration and fatty acid oxidation, were reported to be decreased in db/db mice, a leptin receptor–deficient obese model of type 2 diabetes (61). Mitochondrial numbers, as indicated by mtDNA and histologic tagging with the marker “MitoTracker,” also were reduced. To the extent that these findings might be applicable to human type 2 diabetes, it is possible that reduced adipose mitochondrial function may result in a lack of suppression of lipolysis. Because fatty acids impair muscle and liver insulin sensitivity (177), the consequent increase in fatty acid release due to adipose mitochondrial dysfunction could contribute to the insulin resistance of type 2 diabetes. Further, this could be compounded by adipocyte release of inflammatory cytokines associated with increased fat mass. The insulin-sensitizing thiazolidinedione drugs seem to improve adipose mitochondrial function (30), possibly a mechanism for improved whole-body insulin sensitivity.
5. Is mitochondrial impairment a cause of insulin resistance?
As discussed in the preceding section, insulin-resistant states are associated with mitochondrial dysfunction or decreased mitochondrial content or both. However, whether mitochondrial dysfunction is the cause of insulin resistance and type 2 diabetes or is a consequence of this disorder remains controversial. Insulin action results from a cascade of events after insulin interaction with α-subunits of the insulin receptor (IR), which extends outward from the external surface of the cell membrane (251, 350). In response to an insulin-induced conformational change in the internal or β subunits of the receptor, tyrosine residues undergo autophosphorylation, and the IR acquires tyrosine kinase activity, phosphorylating the intracellular insulin receptor substrate (IRS) family of molecules. This results in downstream activation of phosphatidylinositol 3-kinase (PI3K) and activation of AKT. In muscle and fat, which express the insulin-sensitive glucose transporter type 4 isoform (GLUT4), AKT induces translocation of GLUT4 to the cell membrane, resulting in increased glucose uptake. In liver, insulin activate enzymes that impair gluconeogenesis and reduce hepatic glucose output. In endothelial cells, insulin activates endothelial nitric oxide synthase, resulting in vasodilation (227, 251). Apart from these effects, insulin signaling also triggers protein–protein interactions, which activate the mitogen-activated protein kinase pathway, favoring cell growth and mitogenesis.
As depicted in Fig. 7 for muscle cells, a rationale exists whereby mitochondrial dysfunction (or reduced mitochondrial density) might impair insulin signaling (177). Mitochondrial dysfunction should lead to impaired fatty acid oxidation, resulting in increased intracellular fatty acyl-CoA and diacylglycerol content, with consequent activation of protein kinase C (1, 204). This, in turn, triggers a serine kinase cascade, ultimately resulting in serine phosphorylation of insulin-receptor substrate type 1 (IRS-1). This has the consequence of blocking the tyrosine kinase activity of the IR on IRS-1, thereby blocking the insulin signaling pathway (Fig. 7).
FIG. 7.

Effect of mitochondrial dysfunction to inhibit insulin signaling in GLU4-expressing muscle cells. The schematic diagram depicts major steps in insulin signal transduction as well as the consequences of excess fatty acyl-CoA and ROS production on insulin signaling. Insulin interacts with α-subunits of its receptor (IR), which extends outward from the cell membrane. In response to an induced conformational change in the internal or β-subunits of the receptor, tyrosine residues undergo autophosphorylation, and the IR acquires tyrosine kinase activity. This leads to phosphorylation of insulin-receptor substrate-1 (IRS-1), triggering a downstream cascade leading to activation of Akt and translocation of the glucose transporter type 4 (GLUT4) to the cell membrane. GLUT-4 fusion with the membrane results in glucose uptake by facilitated diffusion. Mitochondrial dysfunction is depicted to oppose insulin signaling in two ways: first, by interfering with oxidation of fatty acyl-CoA and consequent accumulation of intracellular lipid and diacylglycerol, and second, through generation of ROS. Both processes activate serine kinase reactions, leading to serine phosphorylation of IRS-1 and interference with insulin signal transduction. IRS-1, insulin receptor substrate-1; GLUT4, glucose transporter 4; FA, fatty acid; FATPs, various transport proteins that have been described as active in fatty acid uptake.
An additional way that mitochondrial dysfunction may result in insulin resistance might follow from excess production of ROS, a topic discussed later (Section V.A.2) in more detail. In support of a role for ROS, Houstis et al. (142) reported that 3T3-L1 adipocytes, treated with either tumor necrosis factor-α (TNF-α) or glucocorticoids, generated more hydrogen peroxide, expressed genes associated with oxidative stress, and exhibited higher levels of protein carbonylation; the latter representing a marker of cumulative oxidative stress. These authors suggested mitochondrial involvement because TNF-α and dexamethasone can induce mitochondrial ROS through ceramide formation. Moreover, manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) prevented the effect of TNF-α or dexamethasone to reduce serine phosphorylation of AKT (an important parameter of insulin signaling) and prevented threonine phosphorylation of c-Jun N-terminal kinase (JNK), a kinase linked to insulin resistance (137). MnTBAP has catalytic effects similar to those of the antioxidant enzymes superoxide dismutase and catalase (142).
Clinical evidence indicates that defects in mitochondrial function may be a primary cause of insulin resistance. Family members of persons with insulin-resistant type 2 diabetes (247) demonstrate reduced expression of the PGC-1α and PGC-1β, coactivators of NRF-1 and PPARγ-dependent transcription involved in the expression of multiple genes associated with oxidative phosphorylation. In addition, persons with impaired glucose tolerance or “prediabetes” also show evidence of reduced expression of oxidative phosphorylation genes (222). Further, women with polycystic ovarian syndrome (PCO), and therefore at risk for type 2 diabetes, show a reduction in nuclear genes involved with mitochondrial oxidative metabolism (293).
Conversely, such clinical evidence for a primary role of mitochondria in insulin resistance is debatable. This is because insulin resistance is already present in persons with impaired glucose tolerance and in family members of individuals with type 2 diabetes. Moreover, women with PCO are often already insulin resistant, a common trait associated with PCO. Hence it is difficult clearly to ascertain whether defects in mitochondria occur before or after the onset on insulin resistance. Evidence also suggests that insulin signaling per se may be required for intact mitochondrial function. Prolonged insulin infusion increases mitochondrial ATP production (301) in vastus lateralis muscle and ATP synthesis (254, 303) in healthy controls, whereas subjects with type 2 diabetes were resistant to this effect of insulin.
Elegant studies of relatives of persons with type 2 diabetes with magnetic resonance spectroscopy revealed impaired oxidative phosphorylation (253); impaired stimulated ATP synthesis (254); increased intramyocellular lipid with decreased glucose uptake and reduced mitochondrial density (225); and decreased TCA-cycle substrate oxidation (26). Hence these studies might argue for a genetic predisposition to type 2 diabetes involving genes important in mitochondrial function. However, it is important that, even though diabetes was not present at the time of investigation, individuals in these studies had insulin resistance at the time of study, so again, it is difficult to conclude that mitochondrial dysfunction caused insulin resistance.
It might be expected that a transcription factor that induced mitochondrial biogenesis might be important in the pathogenesis of insulin-resistant states. However, as stated earlier, mice deficient in muscle PGC-1α have normal peripheral insulin sensitivity (129). Moreover, globally deficient mice are resistant to diet-induced obesity. The meaning of this is not clear, because these animals have a number of problems affecting multiple organs including the central nervous system (191, 199). In addition, muscles of mice deficient in certain oxidative phosphorylation genes do not show reduced insulin-responsive glucose utilization in vitro. These gene defects include deletion of the mitochondrial flavoprotein apoptosis-inducing factor, which can initiate progressive dysfunction of oxidative phosphorylation (260, 316), and deletion of mitochondrial transcription factor A, which results in mice with myopathy and progressively deteriorating respiratory-chain function (332, 333).
PGC-1α also plays a critical role in regulating nutrient flux in the liver. Low-glucose and low-nutrient conditions (possibly through increased NAD+ or NAD+/NADH) activate the NAD+ dependent histone deacetylase, SIRT1, which, as discussed in Section IV.C.3, deacetylates and activates PGC-1α (271). In turn, PGC-1α activates transcription factors including FoxO1 and HNF4α, thereby inducing metabolic genes important for gluconeogenesis and fat oxidation. However, any defect in hepatic PGC-1α that might reduce gluconeogenesis would decrease rather than increase circulating glucose, so it does not seem that such a defect would be causative of insulin resistance. In fact, knockdown of PGC-1α and SIRT1 in mice can reduce hepatic glucose output, reduce glycemia, increase insulin sensitivity, and also lead to increased hepatic free fatty acid content (273).
To summarize this, the pathophysiology of insulin resistance is, at least in part, related to the inability of liver and skeletal muscle effectively to oxidize fatty acids at the mitochondrial level. It is quite reasonable to think that mitochondria play an important role in regard to insulin resistance. However, whether defects in mitochondria are primary or secondary to the process remains uncertain. In any case, it is important to realize that even if mitochondrial dysfunction were secondary to insulin resistance, mitochondrial defects could add to hyperglycemia once the insulin resistance is in place and lead to progressive worsening of the diabetic state.
6. Mitochondrial respiratory coupling and insulin release
In addition to their effects on insulin sensitivity in insulin-target tissues, mitochondria may play an important role in modulating pancreatic islet β cells insulin secretion, also a critical element in the pathogenesis of type 2 diabetes (Fig. 8). As suggested by Fig. 8, any component of mitochondrial function that could alter ATP production should have major impact on the capacity of glucose to trigger insulin secretion.
FIG. 8.

Role of mitochondria in regulating insulin secretion. As shown, glucose sensing and glucose-induced insulin release is dependent on mitochondrial ATP generation and affected by both mitochondrial ROS and UCP2. ATP is essential for opening of potassium ATP channels and, therefore, for entry of calcium and insulin release from storage granules. Under conditions of hyperglycemia, it is possible that excess ROS may lead to oxidative damage, gradually impairing insulin secretion over time, with worsening of the diabetic state. +, Positive effect. Dash, Negative effect. VDCC, voltage dependent calcium channel; GK, glucokinase.
In particular, uncoupling protein 2 (UCP2), the UCP subtype expressed in islets, would be suspect, given its effect to reduce ATP production at any given level of fuel oxidation. Indeed, Zhang et al. (346) reported that mice genetically deficient in UCP2 manifest higher islet ATP levels and increased glucose-stimulated insulin release. The role of UCP2 was further examined in leptin-deficient obese ob/ob mice (346). These mice are characterized by impaired first-phase insulin release; in other words, acute release occurring over ∼10 min after an exposure to a rapid increase in glucose. UCP2 knockout (KO) in these mice restored first-phase insulin release (346). In addition, UCP2 KO reduced blood glucose and increased insulin when measured in the fed state in ob/ob mice observed over a period of 15 weeks. Interestingly, a kinetic analysis (9) revealed that the ATP/ADP ratio was much more regulated by mitochondria in islet β cells (modeled by INS-1E insulinoma cells) than by mitochondria of skeletal muscle, supporting the importance of mitochondria in regulating islet insulin secretion. Further, evidence indicates that the impaired insulin secretion observed during treatment of isolated islets with high glucose can be mitigated by UCP2 knockdown (176) and that UCP2 KO protects mice from fatty acid–induced impairment in glucose-induced insulin release (152, 153).
As opposed to UCP knockdown, overexpression of UCP2 inhibits glucose-induced insulin release, as demonstrated by our laboratory with INS-1 cells (140) and by Chan et al. (55) in cultured pancreatic islets. In contrast, Wang et al. (326) found that adenoviral expression of UCP2 in islets isolated from ZDF rats increased proinsulin and improved glucose-induced insulin secretion. However, this discrepancy may be explained by the effect of UCP on fat oxidation in islets from these rats, because ZDF islets are known to contain large amounts of fat, inducing a lipotoxic state. The in vivo depletion of islet fat as a result of troglitazone treatment of these animals induced UCP2 expression, reduced islet fat, and improved insulin secretion (291). Additional work by Krauss et al. (176) showed that induction of UCP2 by endogenous superoxide impaired insulin secretion from isolated islets in WT but not UCP2 KO mice.
In past years, the major factors regulating glucose-induced insulin secretion have been considered to be glycolysis and glucokinase (the hexokinase predominantly expressed in β cells) (211). However, these considerations now direct attention to mitochondria with a major role for UCP2 in modulating mitochondrial respiration and membrane potential, ATP production, and therefore insulin release (8).
Nonesterified fatty acids associated with obesity and type 2 diabetes may impair islet function by several mechanisms, as recently reviewed (194). These include fatty acid–induced apoptosis, accumulation of malonyl-CoA, and consequent accumulation of cytoplasmic fatty acyl-CoA molecules, decreased insulin gene transcription, and induction of ROS. The role of ROS in islet function is discussed in Section V.A.1. Conversely, Moore et al. (221) found that fatty acid inhibition of insulin secretion in cultured pancreatic islets was not associated with increased peroxide or nitric oxide and was not prevented by antioxidants. However, the effect could be reproduced by diacylglycerol, suggesting some effect downstream of this metabolite.
At present, little is known about how mitochondria affect islet function in human type 2 diabetes. Nonetheless, from our discussion of “mitochondrial diabetes” (Section IV.B), it is clear that mitochondria do affect insulin secretion in humans.
7. Mitochondrial function in insulin-deficient diabetes
Reports of mitochondrial function in insulin-deficient diabetic states date back to the 1950s. Early studies revealed decreased respiration on complex I or complex II substrates in liver and muscle mitochondria isolated from rodents or cats made diabetic by pancreatectomy, alloxan, or streptozotocin (38, 119, 127, 322). Other more recent reports describe mitochondrial bioenergetics in mitochondria from different tissues obtained from insulin-deficient diabetic rats. Overall, the results appear somewhat variable, suggesting that respiration is impaired in mitochondria isolated from heart and brain mitochondria (101, 223) but actually increased in kidney mitochondria respiring on succinate or complex I substrates (159). More recent studies of heart and kidney mitochondria from STZ diabetic rats revealed no significant change in respiration (223).
Recently Bugger et al. (42) studied heart mitochondria of insulin-deficient Akita mice which develop diabetes because of a defect in the insulin gene, Ins2 (42). These authors reported decreased state 3 respiration on complex I substrates pyruvate and glutamate but not on palmitoyl carnitine. Coupling of oxidation and phosphorylation was not altered on any substrate, as assessed with ADP/O ratios or with proton-leak kinetics. The ATP production rate was reduced on the complex I substrates but in proportion to reduced oxygen consumption, explaining the lack of change in the indices of respiratory coupling. We recently reported that heart and gastrocnemius muscle mitochondria isolated from STZ-diabetic rats manifest reduced maximally uncoupled respiration on FCCP but without a significant difference in state 3 respiration, ADP/O ratio, or ATP-production rate (134). These findings were more pronounced in 8-week than in 2-week diabetic rats. Based on the kinetic relation between hydrogen transfer and membrane potential, our data indicated that the proton conductance of muscle mitochondria of the diabetic rats was reduced compared with nondiabetic controls (curve shifted down and to the right; that is, opposite to what would be expected if diabetic muscle mitochondria were uncoupled). We do not know the reason we observed more efficient coupling without altered ATP production, but this could have resulted from a defect in a protein involved in oxidative phosphorylation or in ATP. Such defects were reported in the previously noted study of heart mitochondria of Akita mice (42). We also examined liver mitochondria of the STZ-diabetic rats and observed no significant perturbations in respiration, respiratory coupling, or proton-leak kinetics, although a strong trend was noted toward reduced respiration. In a recent study of heart mitochondria isolated from hyperglycemic ketotic and hyperglycemic nonketotic streptozotocin diabetic rats, Lashin and Romani (186) reported reduced state 3 respiration but only in the ketotic animals associated with an increase in state 4 respiration. These authors suggested that insulin was an important factor regulating mitochondrial function, because the concentrations of this hormone were much lower in the ketotic animals.
A recent study of muscle mitochondria isolated from humans with type 1 diabetes showed that brief discontinuation of insulin therapy resulted in a decrease in ATP production by the isolated organelles (157). Whole-body oxygen consumption was increased in these subjects. Although these authors considered that respiratory uncoupling might explain their results, they were careful to point out that no data to support this. The authors suggested that the known increased splanchnic oxygen consumption that characterizes type 1 diabetes might account for their results. Human studies are limited by tissue availability and by difficulty studying the untreated disease state, especially in type 1 diabetes which would require insulin discontinuation.
Although these studies have not generated consistent results, taken together, they seem to suggest that respiration and perhaps ATP production in muscle and heart mitochondria are impaired under conditions of insulin deficiency, at least when mitochondria are isolated and studied ex vivo. Respiratory uncoupling in heart and skeletal muscle is not evident, perhaps because of proportional reduction in respiration and ATP synthesis.
Interestingly, UCP3 expression is increased in insulin-deficient diabetic rodents but with no associated increase (42) or an actual decrease (134) in the proton conductance (decrease in the proton leak) of isolated mitochondria incubated in vitro. This resembles the discordance in UCP3 expression and proton-leak activity seen in rodents subject to starvation (44) or lipopolysaccharide-induced free fatty acid release (344). The reasons for this discordance are not clear, although we suggest that this might result from a reduction in coenzyme Q content (Section VII) or in superoxide production (Section V.A.2), which activates UCP3 (94).
The mitochondrial membrane permeability transition induced by inorganic phosphate, uncouplers, or prooxidants results in calcium-induced ROS production (321). This has been attributed to structural changes in the of the inner mitochondrial membrane lipids, resulting in disorganization of proteins of the respiratory chain. Oliveria et al. (244) reported that heart mitochondria isolated from 21-day STZ-diabetic rats with severe hyperglycemia demonstrated increased sensitivity to calcium-triggered reduction in membrane potential. Prevention of this with cyclosporin suggested that this was due to greater susceptibility of these mitochondria to opening of the MPTP (244). Conversely, milder hyperglycemia observed in the Goto-Kakizaki diabetic rat model was associated with upregulation of antiapoptotic proteins (116), a phenomenon that might represent an adaptive change to a milder condition. Taken together, these studies suggest that diabetes-induced changes in calcium transport and ROS may evolve over time but eventually result in more permanent or irreversible damage (or both) and apoptosis.
8. Diabetes and mitochondrial function in non–insulin-sensitive tissues
Glucose uptake by many cell types occurs by facilitated diffusion independent of circulating insulin but highly affected by the blood glucose concentration. In particular, this is the case for the nervous system, vascular endothelium, retina, and kidney, tissues most susceptible to the long-term complications of diabetes. Hyperglycemia in these insulin-independent tissues appears to generate increased mitochondrial substrates for the ETS and to increase the propensity for ROS production (87, 237), Therefore, much work attempting to link mitochondria to the long-term complications of diabetes has focused on the role of ROS. This is discussed in more detail in Section V.B.
Consistent with excess ROS production, retinal capillary cells exposed to hyperglycemia become dysfunctional and manifest proapoptotic BAX translocation (171). Mitochondria and cytosol isolated from retinal endothelial cells and pericytes of rats with STZ-induced diabetes revealed increased cytochrome c release to the cytosol and translocation of the proapoptotic protein BAX to mitochondria (172). Cui et al. (76) reported that UCP2 expression was increased in cultured retinal capillary cells exposed to high glucose. This was postulated to be an attempt to adapt to high mitochondrial membrane potential and to protect against resulting ROS. Another study of retinal mitochondria from STZ diabetic rats reported a reduction in the NAD+/NADH ratio, consistent with hypoxia (242).
Diabetes-induced mitochondria changes also have been reported in renal cells. Recent immunohistochemical studies revealed an increase in proximal tubular UCP2 expression in STZ diabetic rats (108). This was reversible by insulin, suggesting that hyperglycemia and not STZ toxicity was the cause. Isolated mitochondria from kidneys of these diabetic rats manifest increased glutamate-stimulated oxygen consumption, which was blocked by GDP (which inhibits uncoupling protein activity). The authors interpreted this as evidence for increased uncoupling associated with the increase in UCP2. These results appear at odds with another recent report showing increased mitochondrial membrane potential in kidney mitochondria of STZ diabetic rats and reduced UCP2 by immunoblotting of mitochondrial protein (79). Interestingly, these changes were prevented with an angiotensin-receptor blocker (which also reduced H2O2 production). The differences between these two reports could reflect marked differences in methods, with the former study focusing on proximal tubular UCP2 rather than whole-kidney mitochondria. These studies also examined different degrees of severity and duration of STZ diabetes; the former report examined a short (2-week) duration of diabetes with a lower dose of STZ.
Recent studies of brain mitochondria isolated from streptozotocin-treated rats revealed decreased respiration during state 3 and state 4, with no change in the ADP/O ratio (223), raising the possibility of impaired electron transport or ATP synthase. These changes were not observed in kidney mitochondria.
9. Mitochondria and cell-fuel selectivity
The ability of cells to switch between substrate utilization is essential to survival, as is obvious when fat stores are used by muscle during starvation. Moreover, the importance of fuel selectivity is underscored by studies indicating that “metabolic inflexibility” or impaired capacity to switch between nutrient utilization, in particular between fatty acid and glucose oxidation, has a pathogenic role in the insulin resistance commonly seen in type 2 diabetes and obesity (228). Metabolic inflexibility may also affect the vascular complications of diabetes, because a switch to glucose utilization represents an important cardiac adaptation to stress (33).
According to the classic Randle hypothesis, fat and glucose metabolism compete and undergo regulation based on the acetyl-CoA/CoA ratio and citrate concentrations, with consequent effects on enzymes regulating glucose and fat metabolism. Later work did not verify this, but placed emphasis on intracellular signaling pathways (270). More recent metabolomic studies now suggest that enhanced fat metabolism seen with high-fat feeding leads to high levels of β-oxidation products that alter mitochondria in a way that restrict the ability to switch to glucose oxidation (228). Insulin deficiency, like high-fat feeding, is also a state associated with increased circulating fatty acids and accelerated delivery to mitochondria. Therefore, it seems plausible that mitochondria might be altered in similar fashion in insulin-deficient states and lose the capacity to switch from fat to glucose metabolism.
Of course, many factors regulate nutrient selectivity. Prominent among these are AMPK, which can stimulate both glucose and fat oxidation through activation of myriad targets; the pyruvate dehydrogenase complex (PDC), essential for pyruvate entry into mitochondria; CPT-I and CPT-II, essential for fatty acyl-CoA entry; malonyl-CoA, which inhibits CPT; AKT, which activates glucose transport by GLUT4 (and many other steps); and phosphofructokinase (PFK), an important regulator of glycolysis.
We recently reported data showing that a mitochondria-targeted coenzyme Q analogue, designed as an antioxidant, altered intact-cell fuel selectivity, favoring glucose use over fatty acid oxidation. This is discussed later (Section VIII.D).
10. Diabetic cardiomyopathy and mitochondrial function
Diabetes is associated with ischemia and hypertension, both of which contribute to cardiomyopathy. Moreover, circulating FFA concentrations are increased because of insulin resistance or decreased circulating insulin or both. When the cardiac supply of FFAs exceeds oxidative capacity, intramyocardial triglyceride accumulation and lipotoxicity compound the problem of cardiomyopathy. Hypertension and triglyceride accumulation are associated with diastolic dysfunction (267, 317), a major characteristic of diabetic cardiomyopathy.
As mentioned in the previous section, “metabolic flexibility” is particularly important to cardiac tissue under stress conditions. Given the need for large amounts of ATP to match cardiac work requirements, it is not surprising that the heart has evolved to use fatty acids efficiently for energy; this fuel type provides optimal ATP production per molecule metabolized (317). By comparison, glucose oxidation provides less ATP per molecule oxidized. However, glucose oxidation requires less oxygen per ATP generated. Hence, glucose use becomes advantageous when the oxygen supply relative to work is limited, as applies in diabetic cardiomyopathy, especially in the face of enhanced work demand.
Unfortunately, insulin resistance or insulin deficiency or both, which represent the fundamental characteristics of diabetes, impair the ability to switch to carbohydrate metabolism. This is compounded by intracellular events resulting from lipotoxicity (Fig. 7). Moreover, accelerated fat metabolism will generate ROS, which will lead to mitochondrial respiratory uncoupling, which will further decrease ATP production per unit O2 consumed. These downward-spiraling events result in a scenario wherein the myocardium is faced with a need to use more glucose but, at the same time, an impaired capacity to do so (Fig. 9). To counter this progressive scenario, treatments are needed that encourage metabolic flexibility, in particular, the capacity to switch to glucose oxidation. This is further discussed in Section VIII.
FIG. 9.

Events associated with diabetic cardiomyopathy contributing to the incongruity between glucose need and glucose oxidation (rectangular boxes). Ischemia, pressure load (often related to hypertension), and lipid overload all contribute to greater oxygen demand. This is compounded by ROS and fatty acid–induced uncoupling, which increases the oxygen requirement for a given degree of ADP conversion to ATP. These events might be offset if the myocardium could more efficiently use glucose rather than fat, because the former requires less oxygen per molecule ATP produced. Unfortunately, insulin resistance or defective insulin secretion or both, as well as excess lipid supply, all decrease the capacity of the heart to use glucose when it is needed most.
Evidence implicates impaired mitochondrial function very early in the course of events leading to diabetes and diabetic cardiomyopathy. Katakam et al. (158) documented abnormalities in heart mitochondria of insulin-resistant Zucker obese rats, compared with Zucker lean, at age 10–12 weeks; a time at which the animals were euglycemic and, thus, in the prediabetic stage. The abnormalities included decreased mitochondrial numbers, increased MnSOD, and dysmorphic features of swelling, disorganized cristae, and vacuolation. These findings were associated with impairment of effective ischemic preconditioning, a property dependent on mitochondrial ATP-activated potassium channels.
11. Summary
Salient points are as follows. As opposed to specific mutations that affect mitochondrial proteins (mitochondrial diabetes), the role of mitochondria in the pathophysiology of type I and type II diabetes is much more diffuse and involves both insulin sensitivity and secretion. Defects in mitochondrial morphology, fission, fusion, biogenesis, and oxidative phosphorylation are all associated with insulin resistance. Work is needed the better to define and prioritize these defects and to determine whether mitochondrial dysfunction is a cause or consequence of insulin resistance. With respect to insulin secretion, any perturbation affecting mitochondrial ATP function will alter insulin release; in particular, mitochondrial uncoupling has attracted recent interest. Controversy exists regarding the capacity for insulin-deficient diabetic muscle and heart mitochondria to generate ATP, with discordant findings over different models.
Diabetes is characterized by excess glucose and fatty acid flux to non–insulin-sensitive tissues, which may enhance mitochondrial substrate supply and ROS production. Excess fatty acid flux to muscle and heart alters nutrient selectivity and impairs metabolic flexibility. An important consequence is impaired capacity of the heart to use glucose under stress conditions.
V. Mitochondrial ROS and Diabetes
The previous section was directed primarily at mitochondrial metabolism as related to diabetes. Here we focus on a critical related aspect: that of mitochondrial ROS production. Reasons exist to believe that mitochondrial ROS are involved in both the pathogenesis and long-term complications of diabetes. This follows from evidence that elevated glucose or free fatty acids or both drive the formation of ROS (87, 237, 335), impairing both β-cell insulin release and insulin sensitivity and contributing to the complications of diabetes (31, 87, 117). Exactly how exposing cells to high glucose or fatty acid concentrations or both leads to increased mitochondrial ROS production is a question that, we believe, requires more detailed resolution. The supposition is that metabolism of these nutrients generates high levels of substrate flux to mitochondria and, consequently, greater overall electron-transport activity and more electron leak.
Plasma levels of markers of lipid peroxides, such as 8-iso-prostaglandin F2α (138), conjugated dienes, and lipid hydroperoxides (210), are elevated early in the course of type 1 diabetes, whereas antioxidant capacity assayed as total plasma antioxidant capacity (TRAP) is reduced (210). Moreover, DNA damage is detectable in circulating lymphocytes of subjects with insulin-dependent diabetes and correlates to the extent of glucose elevation (65). Further, the extent of urinary 8-OHdG excretion, a marker of DNA damage, correlates with the extent of renal damage in subjects with type 2 diabetes (155).
Although we believe it is too simplistic to invoke ROS as the entire or unifying factor explaining diabetes and its complications, ROS are of major concern. Figure 10 depicts a simplistic scheme wherein ROS may be involved in a vicious self-perpetuating process favoring the development and worsening of the diabetic state. Details are discussed later.
FIG. 10.

Self-perpetuating vicious cycle wherein excess nutrient supply to islet cells and insulin-sensitive myocytes leads to worsening of insulin secretion and insulin action. Concurrently worsening glycemia and elevated free fatty acids (FFAs) lead to worsening diabetic complications.
A. ROS production and the cause of diabetes
Mitochondrial ROS appear important in the pathogenesis of impaired islet β-cell insulin secretion as seen in both type 1 and type 2 diabetes, as well as in the insulin resistance that characterizes type 2 diabetes. These two issues are addressed in the following sections: first with regard to islet β-cell function and then with regard to insulin sensitivity in insulin-responsive tissues.
1. Oxidative damage and pancreatic islet β cells
Type 1 diabetes is widely believed to result from autoimmune destruction of islet β cells. Although this appears the primary process, ROS production induced by inflammation may account for a significant part of the damage. The capacity for ROS to destroy pancreatic β cells is evident in the toxicity imposed by alloxan or streptozotocin, which are commonly used to induce diabetes for experimental purposes. These agents are known to cause free radical damage to islets (279, 312). Evidence indicates that treatment by overexpressing superoxide dismutase (179) or glutathione peroxidase (231) mitigates radical-induced islet damage due to alloxan or streptozotocin. It is also noteworthy that antioxidant protective enzymes, including SOD, catalase, and GPX, are expressed at lower levels in mouse islets compared with liver, kidney, brain, lung, skeletal muscle, heart, adrenal gland, and pituitary gland (190), accounting for the particular sensitivity of pancreatic β cells to cytotoxic damage by these diabetogenic compounds.
Prooxidant heme may have a role in the islet pathology of diabetes. Induction of heme oxygenase-1 (HO-1) with cobalt protoporphyrin (CoPP) in nonobese diabetic (NOD) mice decreased the blood glucose and increased antiapoptotic proteins in the pancreas (193). Heme oxygenase (HO) catalyzes the rate-limiting step in heme degradation, converting heme to biliverdin while consuming oxygen and generating Fe2+ and carbon monoxide (3). This may affect mitochondrial function, at least based on studies in renal mitochondria of diabetic rats. These studies showed that CoPP increased the expression of the carnitine, citrate, deoxynucleotide, dicarboxylate, and ADP/ATP carriers associated with an increase in cytochrome c oxidase activity and phosphorylation of the antiapoptotic proteins AKT and BcL-XL (82).
Oxidative damage to islet β cells also has been observed in human type 2 diabetes by nitrotyrosine staining of islets obtained at autopsy (130). Islets from rats exposed to high fat in the form of oleate infused in vivo demonstrated impaired glucose-stimulated insulin release. This could be inhibited by the antioxidants taurine or N-acetylcysteine, which increase glutathione (245). When incubated ex vivo, the islets that had been exposed to oleate demonstrated increased H2O2 production, again preventable by the antioxidant compounds or by the SOD mimetic Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy).
UCP2 may be important in ROS-mediated islet toxicity. Emre et al. (96) found that mice deficient in UCP2 were more sensitive to diabetes induced by multiple low doses of streptozotocin compared with wild-type mice. This was accompanied by evidence for increased damage due to ROS and nitric oxide radicals, along with greater intraislet lymphocytic infiltration. Hence, these changes were thought to be related to decreased mitochondrial oxidant protection normally mediated by UCP2. Conversely, these findings are somewhat opposite to the findings mentioned earlier (Section IV.C.6) wherein UCP2-deficient mice had higher islet ATP levels and increased glucose-stimulated insulin secretion (346). In this case, the protection was thought to be related to higher islet ATP or to the ATP/ADP ratio consequent on reduced UCP2. Hence, this poses a difficult question: Does ROS protection come at a cost (that is, increased superoxide induced uncoupling and reduced ATP). That would mean that any UCP2-mediated reduction in superoxide production would reduce ATP generation with negative rather than positive effects on glucose-induced insulin secretion. Conversely, it can be speculated that even at a cost of decreased ATP, UCP2-mediated reduction in superoxide might prevent irreversible damage to islet cells, whereas reduced ATP would, at most, induce a temporary functional loss of glucose-induced insulin release. These concepts are depicted in Fig. 8.
Nitric oxide (NO) may be important in the pathophysiology of autoimmune diabetes by a complex mechanism, in part involving mitochondria (175). NO has a positive effect on insulin secretion except when present at high levels, wherein the radical can become cytotoxic to β cells. The NO supply is regulated by inducible nitric oxide, which generates NO from l-arginine. At the same time, glutamine drives the formation of the protective antioxidant glutathione (GSH). These reactions appear to be involved in the β-cell destruction seen in early type 1 diabetes (175). Inflammation may result in glutamate consumption, decreasing the availability of GSH. Although poor nutrition or acidosis associated with insulin deficiency may compromise l-arginine production by the kidney, this reduction tends to be compensated by diversion of glutamate-induced GSH to glutamate-mediated production of l-arginine through the urea cycle (175). Although this would increase l-arginine (and NO), it would also decrease GSH-mediated protection. This situation is further compounded because β cells express low amounts of NADPH-dependent GSSH reductase.
2. ROS and oxidative damage in insulin-sensitive target tissues
It has been theorized that mitochondrial dysfunction in insulin-sensitive tissues, including muscle, heart, and liver, could contribute to deterioration in the diabetic state over time. A unifying explanation for both the insulin resistance and impaired insulin secretion that characterizes type 2 diabetes is ongoing damage to mitochondria of insulin-sensitive peripheral cells (288), along with progressive impairment in mitochondria of islet β cells (117).
Muscle represents the major peripheral tissue that transports and uses glucose in response to insulin. Given the dependency of glucose transport on insulin, mitochondria within the myocyte interior are not subject to the ROS driving force of glycemia in the same way as are non–insulin-sensitive tissues such as neural or endothelial cells. However, this is not the case for fatty acids, which circulate in higher concentrations in both type 1 and type 2 diabetes in the untreated state. Intramyocellular lipid content is elevated in humans with obesity, diabetes, and insulin resistance, and much of this lipid is actually localized near mitochondria (286) and is potentially sensitive to ROS-induced peroxidation. Skeletal muscle lipid peroxides are elevated in muscles of subjects with obesity and insulin resistance (277). These products induce UCP3-mediated uncoupling, which, in theory (see Section VI), may be beneficial by reducing ROS and enhancing export of toxic fatty acids (136). Conversely, this might be a purely compensatory response that could conceivably have a cost in terms of an uncoupling-induced decrease in ATP production.
Boudina et al. (34) reported an increase in ROS production, a decrease in ATP production, and an increase in a marker of oxidative damage (4-HNE) in heart mitochondria of insulin-resistant, obese, and leptin receptor–deficient db/db mice, a model of extreme obesity associated with diabetes. Conversely, this group noted a decrease in ROS production from heart mitochondria of an insulin-deficient model, the Akita mouse, which more closely resembles human type 1 diabetes (42). Hence, these findings suggest fundamental differences in the mechanisms underlying ROS production and ROS protection between heart mitochondria of insulin-deficient mice and mitochondria isolated from an obese, insulin-resistant strain.
We also noted no increase (or an actual decrease) in ROS production measured both as fluorescent H2O2 release and as superoxide with EPR from mitochondria of heart, gastrocnemius muscle, and liver of insulin-deficient rats made diabetic with streptozotocin (134). Our findings were associated with an upregulation of MnSOD and UCP3, as well as cytoplasmic catalase in heart and muscle and an increase in glutathione peroxidase in liver mitochondria. Our results are in agreement with the previously mentioned studies of insulin-deficient Akita mice, in that both reports show that insulin deficiency does not cause an intrinsic increase in mitochondrial superoxide production. However, the upregulation of antioxidant protection does suggest that islets isolated from insulin-deficient mice had been exposed to antecedent in vivo oxidative stress.
With respect to insulin-resistant states, in data as yet unpublished, we examined superoxide production both as H2O2 fluorescence and with EPR spectroscopy in mitochondria isolated from muscle and heart of rats subject to high-fat feeding along with a low dose of streptozotocin. These treatments led to a state resembling very mild human type 2 diabetes or “prediabetes,” defined as an increase in the fasting blood glucose to >100 mg/dl but not >125 mg/dl (10). Our results did not show excess superoxide production, indicating that the mitochondria, incubated in vitro, were not intrinsically altered to generate excess ROS.
Impaired aldehyde dehydrogenase (ALDH) has been implicated in diabetic complications. As opposed to excess generation of ROS and products of oxidative damage, ALDH is important in detoxification. Impaired ALDH will increase levels of lipid peroxidation products such as 4-HNE, a reactive aldehyde that modifies proteins. An example is the FAD-containing subunit of succinate dehydrogenase, which is so modified in hearts of STZ-diabetic rats associated with defective mitochondrial respiration (187). Evidence suggests that glycoxidation or hyperglycemic pseudohypoxia or both impair ALDH and lead to accumulation of lipid peroxides in liver of insulin-deficient diabetic rats (311). Hyperglycemic pseudohypoxia refers to the increased NADH-to-NAD+ ratio observed in insulin-deficient diabetes without a decrease in tissue po2 (328).
GSH content also is reduced in liver mitochondria of insulin-deficient STZ-diabetic rats, as we and others observed (21, 134). In our work, the reduction in GSH was associated with an increase in GPx expression, apparently in compensation for oxidative stress. Interestingly, mitochondria isolated from fatty liver of obese mice demonstrated opposite alterations, showing increased GSH and a reduction in GPx enzyme activity (336). This likely exemplifies the different pathophysiology of insulin deficiency compared with obesity and insulin resistance.
B. ROS and the complications of diabetes
High glucose or fatty acid flux or both to mitochondria and consequent production of mitochondrial oxygen radicals may be a major factor underlying the complications of diabetes. Generation of mitochondrial oxygen radicals by endothelial cells exposed to a high circulating glucose level has been proposed as a mechanism for glycemic damage to the vasculature in diabetes mellitus (237). Glycemic effects of this nature have been reported for mitochondria of diverse cell types, including bovine endothelial cells (86, 237), retinal endothelial cells (87), renal mesangial cells (165), cardiomyocytes (338), and epineural blood vessels (70). Moreover, diabetes is associated with increased fatty acid oxidation and increased intracellular fat accumulation, both of which have been implicated in mitochondrial ROS generation (288, 335).
Multiple mechanisms exist by which ROS could lead to the complications of diabetes. As indicated in Section II.C, superoxide reacts with nitric oxide to form peroxynitrite. This will induce lipid peroxidation and consume nitric oxide, which can impair endothelium-mediated vasodilation. Superoxide can also damage iron–sulfur centers, reducing catalysis by enzymes such as aconitase (319). Moreover, hydrogen peroxide, produced from superoxide by MnSOD, can react with iron to form the very reactive hydroxyl molecule. Thus, mitochondrial superoxide generates other radicals, thereby imparting diffuse damage to protein, DNA, RNA, and lipids. Moreover, mitochondrial damage and consequent dysfunction will disrupt calcium transit and can induce the mitochondrial permeability transition, leading to apoptosis (146).
Based on studies in BAE cells, it has been posited that hyperglycemia-induced mitochondrial ROS lead to diabetic complications through at least three separate metabolic pathways (237), including generation of advanced glycosylation end products, protein kinase C (PKC) activation, and polyol formation. In the first case, glucose-induced ROS increase levels of methylglyoxal, which is known to induce the formation of advanced glycosylation end products. In addition, ROS-activated PKC can lead to diabetic complications by triggering the production of several proteins. Examples are renal mesangial matrix proteins, leading to glomerular damage (74) or platelet-derived growth factor and the vasoconstrictive endothelelin-1, which are associated with diabetic retinopathy (340). Moreover, antioxidant administration decreases sorbitol accumulation in BAE cells exposed to high glucose. This implies that ROS increase glucose-driven polyol formation through the aldose reductase pathway, a mechanism linked to diabetic complications (237).
The next two sections address ROS damage; first to the non–insulin-sensitive tissues classically affected by diabetes (eye, kidney, and nervous tissue) and then to the vasculature per se.
1. Non–insulin-sensitive tissues (retina, renal, neural cells)
As opposed to tissues such as muscle, liver, and heart, which depend on insulin for glucose transport and utilization, most tissues take up glucose by facilitated diffusion, independent of insulin (250). Hence, glucose influx is increased by high circulating glucose per se, thus rendering cells susceptible to consequences of excess supply. This applies to the classic sites of diabetic complications, including the retina, kidney, neurons, and vascular endothelial cells. It has been suggested that the excess glucose load results in increased substrate flow to mitochondria and consequent enhanced ROS production (236).
In transformed retinal cells (rMC-1s) and bovine retinal endothelial cells (BRECs), incubation in 25 mM, as opposed to 5 mM, glucose increased superoxide production (87). This was thought to be primarily from mitochondria, because inhibition of the mitochondrial electron-transport chain complex II normalized superoxide production, whereas inhibition of NADPH oxidase or nitric oxide synthase had little or no effect. In contrast, Busik et al. (43) showed that 25 mM glucose did not increase ROS production in human retinal endothelial cells. The increased glucose concentration did not actually increase glucose utilization in these cells. In contrast to the lack of effect of glucose, these authors (43) found that stimulation by interleukin-1β or TNF-α did induce ROS production in human retinal endothelial cells, suggesting that diabetes-related endothelial injury may be related more to cytokine production than to excess glucose. This study used specific spin traps to verify intracellular production of superoxide by electron paramagnetic resonance spectroscopy. In this work, a mitochondrial source of ROS was suggested, based on the accumulation of a probe, MitoTracker Red, which is dependent on potential to enter mitochondria.
Kanwar et al. (156) reported that superoxide production, measured as lucigenin fluorescence, was increased in retinal tissue isolated from streptozotocin-diabetic mice with blood glucose concentrations ∼400 mg/dl. This was prevented by overexpression of MnSOD in the diabetic mice before isolation of the retinal tissue. These authors also reported that diabetes decreased mitochondrial content of reduced glutathione. Cui et al. (76) used a confocal microscopy approach and reported that high glucose in the culture medium increased ROS production in bovine retinal capillary endothelial cells and pericytes associated with apoptosis. These authors also noted increased uncoupling protein expression and MnSOD, suggesting mitochondrial compensation for ROS. Oddly, the induced UCPs included UCP1, generally expressed only in brown fat. Consistent with these studies, Koluru et al. (172) showed that retinal mitochondria from rats after 8 months (but not 2 months) of STZ-induced diabetes are characterized by leakage of markers of apoptosis (cytochrome c and the BAX protein). In another report, this group showed that MnSOD overexpression in transgenic mice inhibited oxidative damage to the retina, manifest as 8-OHdG and nitrotyrosine (173).
Evidence suggests that mitochondrial ROS contribute to diabetic nephropathy. Friederich et al. (108) showed that diabetic rats express increased mitochondrial UCP-2 in proximal tubular cells associated with increased oxygen use and suggested that the increase in UCP2 was protective against oxidative stress. Another report showed that UCP2 was negatively associated with H2O2 production in kidney mitochondria of diabetic rats (79). Manabe et al. (208) reported that high glucose increased ROS fluorescence in human mesangial cells associated with potentially harmful cytokine expression, an effect that was blocked by astaxanthin, a carotenoid that accumulated in mitochondria. High glucose also reportedly increased H2O2 production by dichlorodihydrofluorescein fluorescence in human mesangial cells (165). This was suppressed by reduction in membrane potential by chemical inhibition or by UCP1 overexpression, but, curiously, also suppressed by MnSOD, which should actually increase H2O2 production from superoxide.
Coughlan et al. (72) demonstrated renal mitochondrial oxidative damage in 32-week streptozotocin diabetic rats manifest as lucigenin luminescence in kidney slices, an effect that was reduced by alagebrium, a crosslink inhibitor of advanced glycosylation end product (AGE) accumulation. Interestingly, renal carboxymethyl lysine, an AGE marker of glycoxidation and lipid peroxidation, also was inhibited, linking oxidative damage to protein glycosylation. In another report, methylglyoxal formation (a precursor to AGEs) accompanied an increase in superoxide production by renal cortical mitochondria of 12-month STZ-diabetic rats (274). Mitochondrial ROS also were implicated in renal pathology in the Goto-Kakizaki rat, a rodent model of type 2 diabetes (275). This study showed a reduction in tissue aconitase activity, a mitochondrial enzyme susceptible to inactivation by reactive oxygen, along with an increase in lipid peroxides.
Moreira et al. (223) reported no increase in H2O2 production by brain mitochondria isolated from 12-week streptozotocin diabetic rats. However, that study did show increased H2O2 production accompanied by upregulation of glutathione peroxidase in kidney mitochondria of the diabetic rats.
2. ROS and vascular cells
Diabetes increases the risk of cardiovascular events by two- to fourfold (http://www.diabetes.org/diabetes-statistics.jsp). In part, this could be due to impaired vascular function, because both endothelial and smooth muscle cell–mediated vascular reactivity are impaired by diabetes (20, 292). Therefore, mitochondrial function, as affected by diabetes, is particularly important with respect to vascular cells.
The three major factors produced by the endothelium that contribute to the regulation of vascular relaxation are NO, prostacyclin, and the as-yet-unidentified factor referred to as endothelium-derived hyperpolarizing factor (EDHF). Impaired endothelium-dependent vasodilation has been demonstrated in various vascular beds of animal models of diabetes and humans with type 1 and type 2 diabetes (80). One of the mechanisms attributed to diabetes-induced endothelium dysfunction is increased oxidative stress. Hyperglycemia-induced production of superoxide by mitochondria of endothelial cells has been suggested as a common link for mechanisms of diabetes-induced vascular dysfunction (237). Studies in our laboratory of epineural arterioles of the sciatic nerve derived from diabetic rats have provided evidence that the generation of oxidative stress through the production of superoxide and peroxynitrite impairs vascular function and endothelium-dependent vascular relaxation (67–69, 342). These studies provided results suggesting that complex I of the mitochondrial electron-transport chain was responsible for the increase in superoxide formation observed with epineurial arterioles from the sciatic nerve of diabetic rats (70). It was shown that pretreating epineurial arterioles from diabetic rats with rotenone reduced superoxide formation (70). Further, treating diabetic rats with three different types of antioxidants prevented the diabetes-induced increase in superoxide and peroxynitrite formation in aorta and epineurial arterioles of the sciatic nerve and diabetes-induced vascular and neural dysfunction, thereby providing evidence that increased oxidative stress contributes to diabetes-induced vascular and neural disease (67–69).
Studies from other laboratories provided further evidence that antioxidants may prevent vascular complications in diabetes. Treating diabetic rats with Tempol, a stable superoxide dismutase mimetic, abolished the diabetes-induced increase in vascular superoxide, malondialdehyde, and 8-epi-prostaglandin F(2α) and also the impairment in relaxation of aortic rings to acetylcholine (232). Cameron and colleagues (46, 48–50, 145) demonstrated that treating diabetic rats with α-lipoic acid or the metal chelators, hydroxyethyl starch deferoxamine or trientine, prevented the diabetes-induced impairment in vascular relaxation associated with hyperalgesia and neurovascular deficits. In addition, Keegan et al. (160) demonstrated that treating diabetic rats with α-lipoic acid improved endothelium-dependent vascular relaxation of corpus cavernosum smooth muscle. These studies imply that increased superoxide formation via the mitochondrial electron-transport chain and perhaps NAD(P)H oxidase are partially responsible for reduced vascular reactivity observed in epineurial arterioles of the sciatic nerve from diabetic rats (70). Because metal chelators and hydroxyl radical scavengers have also been demonstrated to be effective in preventing diabetes-induced vascular and neural dysfunction, it is likely that the formation of hydroxyl radicals also contributes to impairment of vascular reactivity and nerve function in diabetes (46–51, 145).
Finally, heme oxygenase reportedly protects the vasculature in diabetes. Biliverdin, a product of HO-1 catalysis, has antioxidant properties, whereas another product, carbon monoxide, has vasodilatory, antiinflammatory, and antiproliferative effects (226). The inducible subtype HO-1 is present in many tissues and is upregulated by several stimuli, including growth factors, inflammatory cytokines, hypoxia, peroxynitrite, and nitric oxide. HO-1 improves endothelial dysfunction in diabetes (314) and has angiogenic properties (88). Further, treatment of genetically obese mice with CoPP ameliorated obesity, visceral, and subcutaneous fat, increased adiponectin, and improved insulin sensitivity (192).
C. Summary
ROS contribute to defects in both insulin secretion and insulin action and to the long-term complications of diabetes. Inflammatory damage that characterizes type 1 diabetes is mediated at least in part through islet ROS. In persons with type 2 diabetes, the high nutrient flux and consequent ROS production appear to mediate loss of β-cell function. In insulin-sensitive tissues, including liver, muscle, and heart, high fatty-acid flux leads to oxidative damage. At the same time, non–insulin-sensitive tissues, including the eye, kidney, nervous system, and vasculature, are exposed to both high circulating glucose and fatty acid levels and, consequently, ROS-induced diabetic complications. An important, yet still open question is whether oxidative damage to islets and to insulin-sensitive target tissues is responsible for the progressive nature of human type 2 diabetes.
VI. Mitochondrial Membrane Potential and Diabetes
The last two sections addressed mitochondrial metabolism and mitochondria-induced oxidative damage as related to diabetes. In this section, we focus on a still-controversial issue critical to both metabolism and ROS production: the importance of mitochondrial membrane potential and its regulation by uncoupling proteins.
A. Role of uncoupling proteins
Given their effects on mitochondrial bioenergetics, the question arises as to whether uncoupling proteins are involved in the pathogenesis or pathophysiology or both of diabetes. Until the 1980s, poor coupling of respiration to ATP formation was often considered a property of poorly prepared mitochondria, so it was initially surprising that a “proton leak” would prove to be a catalyzed activity of a specific mitochondrial protein. However, it is now widely accepted that, in brown adipose tissue (BAT), UCP1 does function in this way. UCP1 is a 32-kDa protein encoded by a nuclear rather than a mitochondrial gene, is localized to the inner mitochondrial membrane, and is abundantly expressed in rodent brown adipose tissue (300). Consistent with physiologic energy demands, UCP1 is expressed at higher levels during cold exposure and adrenergic stimulation (300). However, since humans, other than newborns, had not until very recently been found to express BAT or UCP1, there has been doubt that the protein has any role in human diabetes. This now needs to be reconsidered since very recent findings indicate that humans do express BAT in the neck region (78, 278, 318, 323) and the amount of BAT correlates inversely with body-mass index (78).
UCP1 homologues including UCP2 and the long and short forms of UCP3 and two brain mitochondrial proteins termed brain mitochondrial carrier-1 and UCP4 have been identified (31). UCPs 2 and 3 each have considerable homology to UCP1 and considerable interest has been generated regarding their relation to diabetes. UCP2 is expressed in a variety of tissues, including adipose tissue, muscle, heart, liver, and pancreatic islets, and is responsive to nutritional regulation (31, 106) and, as discussed (Section IV.C.6), may be important in regulating insulin release. UCP3 is 73% homologous to UCP2 in humans and is predominantly expressed in human and rodent skeletal muscle and in rodent BAT (32). UCP3 is known to be upregulated under conditions of increased free fatty acid delivery to mitochondria, including fasting (44), insulin-deficient diabetes (134), and high-fat feeding (135). However, mitochondria isolated from animals exposed to these states do not show altered proton-leak kinetics, suggesting that the major role for UCP3 is not to enhance the basal proton conductance (hydrogen transfer in the absence of any physiologic inducer such as superoxide).
UCP2 and UCP3 KO mice are not obese or diabetic. Moreover, mitochondria isolated from UCP3 (45, 91) or UCP2 KO mice (73, 176) do not show a decrease in basal proton conductance. In addition, skeletal muscle mitochondria from a human with dysfunctional UCP3 showed no change in respiratory coupling (62). However, proton conductance of UCP2 and UCP3 can be activated by matrix superoxide or 4-hydroxy-2-nonenal (4-HNE), a byproduct of lipid peroxidation (45, 91, 93). The mechanism appears to involve catalysis of hydrogen transfer from fatty acid to UCPs by coenzyme Q and coupling to fatty acids (94). Superoxide production by mitochondria may be sensitive to membrane potential, and “mild uncoupling” has been proposed as a means of defense against mitochondrial oxidative stress (295). This has led to the idea that superoxide may act in feedback fashion to induce uncoupling and thereby control its own production (92, 306). This induced proton-leak activity can be inhibited by GDP, further supporting physiologic relevance. Importantly, GDP-sensitive proton conductance triggered by superoxide or 4-HNE is not present in mitochondria from the muscle of UCP3 knockout mice (91) or from the kidney of UCP2-deficient mice (176).
How might UCP2 and UCP3 be related to the pathophysiology of diabetes? One obvious and important role might be to protect against oxidative damage to pancreatic islets (see Section V.A.1). Moreover, reduction in ROS production due to decreased membrane potential might also mitigate damage to non–insulin-sensitive target tissues, including retinal, renal, neural, and vascular endothelial cells (Section V.B). In this regard, this superoxide-inducible proton leak and feedback protection from further ROS may come into play. In this scenario, superoxide or products of oxidative damage trigger uncoupling, which would then limit further superoxide production and limit further damage (Fig. 11).
FIG. 11.

Antioxidant role of UCP3. By reducing membrane potential in the face of high substrate flux to mitochondria and consequent ROS production, UCP3 may act to effect a type of feedback inhibition. As shown, ROS would activate UCP3, thereby triggering UCP3-mediated uncoupling and close the loop.
In addition, evidence indicates that an important role for UCP3 may be to export fatty acids (136, 289). This might have a protective role in diabetes, mitigating the high flux of β-oxidation products to mitochondria. Such an increase in flux might occur both in insulin-deficient type 1 diabetes, because of rapid fat mobilization, and in insulin-resistant states or in overfeeding, which increase circulating free fatty acids.
A proposed mechanism for UCP3 both to export fatty acids and to discharge inner-membrane potential involves fatty-acid export (Fig. 12). This could occur in two ways (136, 289). One is through a “flip-flop” mechanism wherein the lipophilic portion of the fatty-acid chain inserts into the inner membrane, with subsequent flip of the polar head to the matrix inside (136). The negative matrix would then encourage dissociation of a proton and the trapped fatty-acid anion would be exported by UCP3. Hence, the process transfers a proton from outside the inner membrane to the matrix, accounting for UCP3-mediated discharge of membrane potential or uncoupling. Further, fatty-acid export would protect against lipid peroxidation of the trapped anion.
FIG. 12.

Possible role of UCP3 to export excess mitochondrial fatty acids. Under conditions of high availability, fatty acid anions might accumulate in the mitochondrial matrix in two ways. One is through the flip-flop phenomenon (136). The other is through the carnitine palmitoyl transferase (CPT) system, followed by conversion of excess fatty acyl-CoAs to fatty acid anions and coenzyme A by mitochondrial thioesterase-1 (MTE-1) (289). In both cases, UCP3 would function to export the resultant trapped fatty acid anion protecting mitochondria from excess accumulation of fat and lipid peroxides. FAO, fatty acid oxidation.
A second way in which UCP3 might respond to high fatty acid exposure involves the action of mitochondrial thioesterase-1 (MTE-1) on fatty acyl-CoAs that enter through the carnitine palmitoyl transferase system (289) (Fig. 12). Excess fatty acyl-CoAs are broken down to coenzyme A and anion, with export of the anion. As depicted in Fig. 12, UCP3 is again postulated to act as a channel across the inner mitochondrial membrane amenable to export of these fatty-acid anions. Again the result would be the uptake of the proton associated with the exported anion. Interestingly, however, the investigators who described this latter pathway recently found that UCP3 is not actually essential for fatty acid export (289). This was based on studies of fatty acid oxidation and export in skeletal muscle mitochondria of wild-type and UCP3-deficient mice. Although fatty acid export did occur in mitochondria of both the wild-type and KO mice, this occurred equally for both groups. Conversely, UCP3 was necessary for enhancement of fatty acid oxidation during fasting.
Clinical studies indicate a role for UCP3 in the pathogenesis of diabetes. UCP3 expression is reportedly reduced by approximately half in type 2 diabetes (287). Consistent with this finding, type 2 diabetes also is characterized by decreased oxidative phosphorylation activity (161), altered mitochondrial morphology (161), and damage to mitochondrial DNA (197). In addition, humans with a splice-donor mutation in the UCP3 gene have decreased UCP3 levels and diminished fat oxidation (17). Moreover, oxidative gene expression is, in general, reduced in subjects with type 2 diabetes (222, 247).
B. Does membrane potential actually protect against superoxide production?
Although this concept is often cited in regard to uncoupling and ROS, controversy still remains. This is particularly true when extending the argument to the intact-cell environment.
Superoxide production by mitochondria may be sensitive to membrane potential and, as suggested earlier, “mild uncoupling” has been proposed as a means of defense against mitochondrial stress (295). Membrane potential also can be reduced by active ATP synthesis wherein potential energy is diverted to the ATP synthase reaction. This would also be expected to reduce ROS production (167), although little is known about this process in the intact cell. In whole cells, mitochondria generally function in a state between maximally active ATP synthesis (state 3 respiration) and respiration in the absence of ATP formation (state 4).
As mentioned earlier, ROS generation is particularly high under conditions wherein the complex II substrate, succinate, is metabolized, generating reverse transport of electrons back to complex I (185, 241, 324). This process is known to be sensitive to potential, so it is thought that “mild uncoupling” may protect against ROS generated in this way. Moreover, in the Q-cycle (Fig. 2), a high ΔΨ (positive outside-membrane charge) would impair conversion of low-potential cytochrome bL to bH, prolonging the half-life of CoQ·−. Visualized this way, reduction in potential will decrease the CoQ·− lifetime and protect against its oxidative capacity (294). However, in this regard, it should be acknowledged that the actual mechanism of superoxide production in complex III may be more complex than illustrated in Fig. 2, because recent evidence suggests that superoxide may actually be produced in a reverse reaction through oxidation of reduced heme bL (85).
Notwithstanding the preceding, the concept of “mild uncoupling” as a defense against ROS generation has been criticized. Nicholls et al. (151, 234) point out that this may not be the case in intact cells where mitochondria predominantly oxidize NADH-linked substrates at complex I. These authors also point out that, because complex I is thermodynamically weak, relative to the other proton-pumping complexes, bypassing complex I (an artificial state created in isolated mitochondria fueled by succinate) allows mitochondria to generate another 5–10 mV of potential across the inner membrane. It is over this increase or hyperpolarization of the membrane that considerable superoxide is generated. Recent work by this group (151) showed that a small, chemically induced 10-mV decrease in mitochondrial membrane potential in intact cells (rat cerebellar cells) did not affect mitochondrial superoxide production and actually increased cytoplasmic superoxide. Moreover, they point out that the relation of mitochondrial respiration to potential is not particularly conducive to a controlled degree of “mild uncoupling.” This is because mitochondria respond to uncoupling by increasing respiration. So, in actuality, membrane potential decreases very little until the capacity to increase respiration is exceeded, a process that is not inherently efficient (151, 234). Work reported several years ago suggested that UCP1 overexpression in BAE cells protected against ROS (240). However, this overexpression is clearly not physiologic (300). Moreover, in past work in our laboratory (105), we were unable to show any effect of overexpression of UCPs (either UCP1 or UCP2) in BAE cells on ROS. That work was limited to ROS detected by EPR, which we believe represents superoxide generated mostly at complex III.
To summarize Section VI.C, the issue of reduced mitochondrial potential as a means to control ROS is still somewhat controversial, especially in intact cells, and the question requires further study.
C. Summary
UCP2 in pancreatic tissue and UCP3 in muscle and heart are important in the pathogenesis of diabetes. These proteins are induced by and appear protective against superoxide-induced damage. Conversely, this occurs at a cost of reduced ATP, which could result in loss of insulin secretory capacity by islet β cells or loss of function in heart and in muscle. UCP2 and UCP3 are upregulated under conditions of excess fatty acid flux and counter lipotoxicity. Reasons exist to believe that mild uncoupling reduces ROS production through reduced potential; however, the issue is still controversial.
VII. Coenzyme Q and Diabetes
This section addresses the relation of coenzyme Q or ubiquinone to diabetes. Obviously, CoQ is a fundamental life molecule without which all electron transport and energy production would cease. Hence, it seems a bit odd to these authors that more attention has not been directed to CoQ and its biosynthesis, as related to diabetes.
CoQ is fat soluble and localizes to hydrophobic regions within mitochondrial membranes wherein it is mobile and functions as an electron carrier. About one half of total body CoQ is synthesized endogenously, whereas the remainder derives from the diet (113). CoQ side-chain synthesis is dependent on 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase and proceeds through steps common to cholesterol biosynthesis to the intermediate compound farnesyl pyrophosphate which then diverges either to cholesterol, cis-prenylation to dolichol, or trans-prenylation to the side chain of ubiquinone. Synthesis of the benzoquinone ring of CoQ and the long side-chain converge when the two are linked by the COQ2 gene product. At that point CoQ synthesis proceeds within or adjacent to the mitochondrial inner membrane, where at least 10 different gene products are involved in the final generation of CoQ.
Ubiquinone, in final form, consists of a 50-carbon side chain attached to the quinone moiety. The carbon chain consists of ten 5-carbon prenyl units; hence the name CoQ10. In rats and mice, unlike humans, the predominant form contains nine 5-carbon units and is designated CoQ9 (2, 297). The completed CoQ with its long carbon side chain is very mobile within lipid membranes and, thus, able to serve its essential role as an electron carrier. In addition to mitochondrial membranes, CoQ also is present in Golgi vesicles and lysosomes and is present in other membrane structures as well (315).
The antioxidant properties of CoQ are not due to direct scavenging of superoxide but appear to be mediated through regeneration of active ascorbic acid and tocopherol, the reduced from of vitamin E (66). Moreover, CoQ in the semiquinone form also acts as a chain-breaking agent, thereby protecting against lipid peroxidation (147). Although often thought of (and marketed as) an antioxidant, it is important to point out that CoQ also has prooxidant properties. As depicted in Figs. 1 and 2, the semiquinone form of CoQ leaks electrons to form superoxide during electron transport through the Q-cycle in complex III. Moreover, at least suggestive evidence exists for involvement of the semiquinone form of CoQ in electron transport at complex I (40, 90, 185). In this regard, CoQ may actually be needed for superoxide formation, because the radical may have important signaling properties, for example, to induce UCP activity, as discussed earlier, or to enhance the expression/activity of regulatory proteins like AMPK (60, 141). Moreover, it is worth noting that superoxide may not be harmful if followed by detoxification to H2O2 by SOD and breakdown of H2O2 by catalase.
Coenzyme Q concentrations appear to be reduced in diabetic states. An early study of liver mitochondria, which used cytochrome c oxidation to measure CoQ, reported increased concentrations in mitochondria of pancreatectomized diabetic rats (37). However, later studies with HPLC reported a reduction in liver (180) and kidney (180, 223) mitochondrial CoQ content and an ∼75% reduction in heart mitochondria of 8-week STZ-diabetic rats (180). Unpublished studies in our laboratory show a substantial depletion of skeletal muscle mitochondrial CoQ in insulin-deficient diabetic rats. Evidence also exists for reduced CoQ in plasma of humans with diabetes (113).
It is possible that coenzyme Q may be particularly important when diabetes patients are treated with HMG-CoA reductase inhibitors (commonly referred to as “statins”), widely used in patients with diabetes to reduce cholesterol. HMG-CoA blockade is more potent at inhibiting cholesterol biosynthesis than CoQ biosynthesis because of differing Km values for trans-prenyltransferase (leads to CoQ) and squalene synthetase (leads to cholesterol) at the farnesyl-PP branch point (315). Conversely, it is possible that CoQ depletion in diabetes might engender more sensitivity to adverse effects of these drugs, in particular, myopathy. We believe this issue needs further study directed at mechanisms underlying the reductions in CoQ seen in diabetes. Severe myopathy is rare, but milder muscle pain remains a major reason that some patients do not tolerate statins. Moreover, in diabetes, this is of particular importance, given the need to control concomitant cholesterol elevation.
To summarize, CoQ is related to diabetes through its fundamental action on electron transport, direct involvement in both the generation of ROS and antioxidant protection, and as an important regulator of mitochondrial uncoupling. Important therapeutic implications exist, as discussed in the next section.
VIII. Therapeutic Implications
Therapy targeting mitochondria may provide novel ways to treat diabetes or to minimize the complications of diabetes or both. Here we consider ways to alter mitochondrial function in general, as well as mitochondrial ROS production in particular.
A. Improving mitochondrial metabolism
1. Lifestyle modification
Lifestyle modification, including exercise and diet, decreases the risk for developing type 2 diabetes (166), whereas physical activity improves glucose tolerance (132). Moreover, mitochondrial dysfunction may underlie features of the metabolic syndrome, including obesity, hyperlipidemia, hypertension, and vascular disease. In this regard, exercise offers several benefits, including increased electron-transport activity in muscle, stimulation of mitochondrial biogenesis through effects on PGC-1α, and improved sensitivity to insulin (266, 269). Exercise also activates AMPK, which improves both glucose and fat oxidation (266).
Calorie restriction is known to prolong the lifetime of rodents, nematodes, and maybe humans (265). How this is related to mitochondrial function is not clear, but evidence suggests that calorie restriction favors mitochondrial biogenesis, oxygen use and ATP formation, and expression of SIRT1, which activates PGC1-α (121, 203). Calorie restriction also has been shown partially or completely to prevent the effect of aging to decrease the expression of genes involved in mitochondrial biogenesis and function (189).
2. Pharmacologic intervention
Attempts to improve mitochondrial function go back to the 1930s, when attempts were made to treat human obesity with the mitochondrial chemical uncoupler dinitrophenol (77). This was quite effective, thereby providing proof of concept. However, this treatment had to be quickly abandoned because of an unacceptable risk of fulminant liver failure. Conversely, recent research has uncovered several molecular targets that may prove amenable to therapies directed at mitochondrial function (Fig. 13).
FIG. 13.

Examples representing molecular targets for mitochondrial therapy directed at diabetes, insulin resistance, and obesity.
Although not targeted to weight reduction, certain drugs commonly used for patients with type 2 diabetes may improve mitochondrial function. The thiazolidinedione class of insulin sensitizers (ligands for PPARγ) are used to improve insulin resistance and reduce blood glucose levels. The thiazolidinedione, pioglitazone, was found to induce mitochondrial biogenesis in adipose tissue as well as expression of PGC-1α and genes in the fatty acid–oxidation pathway (30). Interestingly, however, the use of thiazolidinediones is limited by a tendency for weight gain in part because of increased subcutaneous, but not intraabdominal, fat (337) and, in part, because of fluid retention by an uncertain mechanism (29).
Metformin is most often the initial pharmacologic agent used in type 2 diabetes. Metformin has mitigating effects on ROS production, activates AMPK, and favors mitochondrial proliferation (181, 353). In clinical use, metformin, unlike insulin or insulin secretagogues, is not associated with weight gain. Another group of drugs that improve insulin sensitivity and enhance mitochondrial biogenesis are the angiotensin-receptor blockers or inhibitors of angiotensin-converting enzyme. These agents also reduce oxidative stress, although the mechanisms still require clarification (164).
Newer pharmacologic approaches to improving mitochondrial function may be on the horizon. Resveratrol, an ingredient in red wines, is a polyphenolic SIRT1 activator that, like calorie restriction, has anti-aging effects in lower organisms (22, 143, 331), reduces signs of aging in mice (248), and extends survival (22). In mice, resveratrol improves insulin resistance, protects against diet-induced obesity, induces genes for oxidative phosphorylation, and activates PGC-1α (22, 184). Other related small molecules are more potent than resveratrol to enhance the action of SIRT1 on substrates for deacetylation (218). These compounds also improve insulin sensitivity in obese rodents (218). Pharmacologic activation of AMPK, like resveratrol, increases PGC-1α, favoring mitochondrial biogenesis (28, 329).
As discussed in Section IV.C.4, high fatty acyl-CoA flux may result in mitochondrial overload with adverse consequences on carbohydrate metabolism. In this regard, it may be possible to improve glucose utilization through measures that inhibit mitochondrial uptake of long-chain acyl-CoA molecules. For example, lipid suppression of glucose utilization is mitigated by etomoxir, an inhibitor of carnitine palmitoyltransferase 1, or by knockdown of malonyl-CoA decarboxylase, an enzyme that promotes mitochondrial β-oxidation by preventing malonyl-CoA–induced inhibition of CPT-I (36, 170). A seemingly opposite approach would be pharmaceutic blockade of acetyl-CoA carboxylase 2 (ACC2), leading to decreased malonyl-CoA. As indicated in Section IV.C.4, ACC2 KO mice demonstrate enhanced rates of fatty acid oxidation in liver and muscle (4, 99). This might be envisioned to increase muscle fatty acid oxidation and therefore to decrease glucose utilization. However, the predominant effect appears to be in liver, which would decrease hepatic packaging and export of lipids and decrease the lipid supply to muscle. Consistent with this concept, glucose oxidation, as well as fat oxidation, was increased in cardiac muscle of ACC2 KO mice (99).
Other targets potentially amenable to pharmacologic manipulation might include AMPK, which enhances both glucose and fat oxidation (109, 276), pyruvate dehydrogenase (213), or the various shuttle mechanisms regulating uptake of TCA intermediates (114). Finally, manipulating mitochondrial reactive oxygen should also have metabolic consequences. H2O2 appears to activate AMPK through an increase in the AMP/ATP ratio and by threonine phosphorylation (60). Moreover, as we recently showed (104), mitochondria-targeted antioxidants may alter intact-cell fuel selectivity. This is discussed in the next section.
B. Controlling ROS production and oxidative damage
Various vitamins and chemical compounds with antioxidant properties and effects on mitochondria have been used in attempts to prevent, control, or reduce the complications of diabetes. These include coenzyme Q, vitamin E, α-lipoic acid, N-acetylcysteine (NAC), vitamin C, inducers of heme oxygenase, and the SOD mimetic MnTRAP.
As the major mobile electron carrier, coenzyme Q has long been of interest as a therapy for obesity and general health and to improve diabetic states. However, the therapeutic use of CoQ10 and other antioxidants in vivo, particularly in human studies directed at vascular events, has been disappointing (341, 345). This may be due to concerns about toxicity and consequent inadequate dosing or the inability to deliver agents to target sites of ROS production. Attempts to treat diabetes, either in rodents (220, 264) or in humans (13, 133, 220, 302), have met with variable results, showing either a beneficial effect on markers of oxidative damage (182, 302) or glycemia (220), no effect (13, 133), or mixed results depending on tissue and markers of oxidative damage (264). CoQ effectively reduced elevated insulin concentrations, improved endothelial function, and reduced markers of oxidative damage when administered through the drinking water to variants of the spontaneously hypertensive rat (SHR), that have features of the metabolic syndrome (182). In another report, oral decylQ (a CoQ analogue) reduced plasma malondialdehyde, blood pressure, and cholesterol in an SHR rat model prone to stroke (229). Further, intraperitoneal CoQ treatment for 7 weeks reportedly improved the respiratory-control ratio and the ADP/O ratio in brain mitochondria and slightly reduced glucose in diabetic Goto-Kakizaki rats treated with a neurotoxin (224). Although human studies are limited, evidence suggests a net benefit of CoQ supplements to humans with neurodegenerative disease or genetic defects in mitochondrial function (126). However, a problem is that the effectiveness of CoQ delivery to mitochondria was not determined in these studies.
Ubiquinol, the reduced from of CoQ, acts as an antioxidant in mitochondria, both by regeneration of vitamin E and by directly reacting with peroxyl radicals. Thus, CoQ acts in mitochondria both as an antioxidant and as a mobile electron carrier (97, 207). However, in our experience, CoQ10 in either the ubiquinol or ubiquinone redox state does not appear to have direct effects on mitochondrial ROS (104, 241) and may not easily enter mitochondria. As indicated earlier (Section VII), the final convergent steps in the biosynthetic pathways generating coenzyme Q occur within or along the inner mitochondrial membrane, so endogenous biosynthesis may be critical to proper localization of mitochondrial coenzyme Q. The antioxidant properties of vitamin E are thought to be based on its oxidation to the tocopheroxyl radical, which allows this lipophilic molecule to inhibit lipid peroxidation (207). Evidence exists for a correlation between mitochondrial lipid peroxidation and vitamin E concentration (310). The tocopheroxyl radical is recycled to vitamin E by water-soluble agents, such as ascorbate and glutathione. Moreover, electron-transport activity within submitochondrial membrane particles can also recycle vitamin E (207). However, despite this, vitamin E did not improve cardiovascular outcomes in a large multicenter trial and actually increased congestive heart failure (201). Vitamin E also did not prevent the progression of carotid intima-media thickness in high-risk patients with diabetes (202). Moreover, despite reports that short-term vitamin E may have beneficial effects on endothelial function in diabetes, prolonged vitamin E supplementation offered no benefit or even had vasoconstrictive effects on endothelial function (23, 95). Thus, it is disturbing that a compound with antioxidant effects on mitochondrial lipids is not effective or is even harmful in the clinical research setting.
Water-soluble ascorbic acid (vitamin C) is widely marketed for its antioxidant properties (212, 296) and, as stated earlier, appears to regenerate reduced vitamin E. Although these compounds may have general effects to improve the overall cell redox state, no evidence supports a role in the management of diabetes (11).
Other antioxidant molecules of potential therapeutic importance include α-lipoic acid and N-acetylcysteine (NAC). In vivo, α-lipoic acid is enzymatically reduced to dihydrolipoic acid and, as such, is an effective scavenger of superoxide (343). In this form, the compound regenerates other antioxidants including glutathione, vitamin C, and vitamin E. In STZ-diabetic rats, α-lipoic acid mitigated the diabetes-induced decrease in retinal mitochondrial and cytosolic NAD+/NADH ratio (243). This compound also prevented lipid peroxidation when administered to rats (83) and improved β-cell function in apolipoprotein E–deficient mice given STZ (339). α-Lipoic acid also protected the retinal microvasculature in diabetic rats by reducing nitrotyrosine and oxidized DNA (174). In human studies, α-lipoic acid has been administered intravenously and improved diabetic peripheral neuropathy (352). Oral α-lipoic acid also improved peripheral neuropathy but caused nausea, vomiting, and vertigo (351).
NAC interacts with multiple radical species, forming NAC disulfide as the end product (71). This agent penetrates cells where the thiol group confers antioxidant activity, effectively removing H2O2 by transfer of electrons from the SH group. With respect to oxygen radicals, NAC rapidly reduces ·OH and HOCl. Interaction with H2O2 is slow, and interaction with O·− is minor at most. Nonetheless, by scavenging the hydroxyl radical, NAC is effective in preventing a damaging radical formed consequent to H2O2 and O·− (Section II.B). In vitro evidence indicates that NAC may exert antioxidant effects through increasing GSH, facilitating the action of GPx. NAC counters ROS in nonalcoholic steatohepatitis, a disorder often associated with type 2 diabetes and characterized by mitochondrial ROS production (214). NAC also is used to mitigate contrast-induced nephropathy in diabetes patients undergoing coronary angiography (150).
As indicated in Section V, inducers of heme oxygenase mitigated islet damage, improved glucose in diabetic mice (193), and improved obesity and insulin sensitivity in genetically obese mice (192). However, these findings have not yet been translated to human studies.
Besides these antioxidant approaches, efforts are now under way to develop antioxidant compounds specifically targeted to mitochondria. Examples are described in the following section.
C. Mitochondria-targeted antioxidants
The likely role of mitochondrial ROS in human disease has led to efforts to develop effective antioxidant compounds targeted to mitochondria. One approach to this involves the synthesis of compounds linking agents such as redox forms of quinone (ubiquinol and ubiquinone) or vitamin E to form alkylated triphenylphosphonium compounds. These are lipophilic cations avidly taken up into the relatively negative mitochondrial matrix (162). As discussed in Section III. A, we and several others used the triphenylmethylphosphonium or tetraphenylphosphonium cations to measure mitochondrial membrane potential (103, 105).
Two such compounds (alkyltriphenylphosphonium cations) incorporating ubiquinone or vitamin E, termed mitoQ and mitoVit E, respectively, have been synthesized (162). By virtue of their delocalized positive charge, these agents accumulate several hundredfold in mitochondria (230). MitoQ (mitoquinone or mitoquinol or a mixture of the two redox cycling compounds) is currently under development as a therapeutic agent in humans for neurodegenerative disorders including Parkinson disease and hepatitis (http://www.antipodeanpharma.com/). Although mitoQ appears to have protective effects in certain cell types, the mechanism of action is not well defined and appears complex. A major action may be to decrease lipid peroxidation by virtue of the quinol moiety acting as a chain-breaking antioxidant (147). Figure 14 illustrates the structures of ubiquinone (CoQ10 named for the ten 5-carbon prenyl units composing the long side arm), mitochondrial-targeted CoQ, and related compounds used as controls in studies of targeted CoQ.
FIG. 14.

Structures of ubiquinone (CoQ10), mitochondrial targeted CoQ (mitoQ), and related compounds.
Oral administration of mitoQ (500 μM in drinking water administered ad libitum) to normal male rats protected heart muscle function, prevented myocardial cell death, and improved the respiratory-control ratio (state 3 to state 4 respiration) in rats subject to ischemia/reperfusion injury (7). Mitochondrial-targeted antioxidants protected Friedreich ataxia fibroblasts, in which glutathione synthesis was blocked, from oxidative stress (148), and mitoQ reduced telomere shortening in fibroblasts exposed to oxidative stress (281). In BAE cells, mitoQ reduced oxidative damage in cells stressed by 25 mM glucose and glucose oxidase (81). Moreover, mitoQ also reduced ROS and reduced activation of the mitogen-activated protein kinase, p42ERK2, in endothelial cells after hypoxic stress (283).
D. Metabolic effects of mitochondria-targeted antioxidants
Given the mitochondrial action of mitoQ, an important issue arises as to whether treatment with such agents might have consequences beyond antioxidant effects. We recently reported that, in BAE cell mitochondria, mitoQ (either as mitoquinone or mitoquinol) had prooxidant as well as antioxidant effects (241) and metabolic effects (104). These compounds markedly increased or decreased superoxide production, when assayed as DHPA fluorescence, depending on substrate provided for fuel (241). MitoQ markedly increased superoxide production during forward electron transport in mitochondria respiring on complex I substrates. Conversely, mitoQ inhibited superoxide generated by BAE mitochondria respiring on the complex II substrate, succinate; a condition wherein ROS production occurs through reverse electron transport or backflow of electrons to complex I originating from complex II. This was likely due to a subtle “mild uncoupling” effect of mitoQ. During respiration on complex I substrates, superoxide appears to result from redox cycling of endogenous CoQ10 or exogenous analogues at Q-binding sites within complex I (84, 185, 241).
As opposed to DHPA fluorescence, we found that mitoquinone had essentially no effect on superoxide production with EPR spectroscopy (241). Based on the considerations depicted in Fig. 6 (Section III.D), DHPA fluorescence measures matrix superoxide largely from complex I, whereas EPR detects superoxide released external to mitochondria, largely from complex III. Hence the effect of mitoquinone to increase DHPA fluorescence, but not the EPR signal, suggests action within complex I. Subsequently, we found that mitoquinone increased respiration by isolated mitochondria, but only on complex I substrates (104).
These considerations led us to think that if mitoquinone increased respiration selectively on particular substrates, then perhaps mitoquinone could alter intact cell-nutrient selectivity. We observed a substantial increase in glucose use and a decrease in fat oxidation in BAE cells (104) as well as C2C12 mouse myocytes (Fig. 15) exposed to mitoQ. However, we do not know the mechanism underlying this effect. Several possible ways exist by which an agent that affects mitochondrial function might alter nutrient selectivity (Section IV.C.9), so this will require extensive additional study. Speculative possibilities include mitochondrial redox effects, which could affect the state of cytoplasmic reducing equivalents, with consequent effects on myriad enzyme systems and kinases. These could then alter fuel selectivity at notable steps, such as AMPK kinase, pyruvate dehydrogenase, phosphofructokinase, or others. Moreover, it is possible that mitoquinone might have direct effects on mitochondrial proteins such as the pyruvate dehydrogenase complex or the electron-transport flavoprotein:ubiquinone oxidoreductase.
FIG. 15.

Dose-dependent effect of mitoquinone on fuel selectivity in bovine endothelial cells. BAE cells were incubated overnight (16 h) in the presence of culture medium with 5.5 mM glucose. 1-[14C]oleic acid or d-[14C(U)]glucose was added along with 50 μM oleate, and cells were incubated for 120 min before trapping of CO2. Data are expressed relative to incubation in the presence of vehicle alone (0 point on x-axis, otherwise depicted on log scale). Data represent mean ± SEM (n = 4 for each determination). *p < 0.05 or **p < 0.01 compared with vehicle by one-way ANOVA with repeated measures. The absolute values for glucose and oleate oxidation in the presence of vehicle were 6.21 ± 0.54 nmol/well and 71 ± 18 pmol/well, respectively.
The effects we observed of mitoQ on nutrient selectivity by intact cells do not appear to be a response to cell toxicity. We found no evidence for this in cytotoxic assays (104). Also, dose/response studies demonstrated effects of mitoquinone extending an order of magnitude or more downward from the 1 μM (or higher) dose used in other reported studies of the cellular actions of this compound (25, 81, 283), some of which reported a mitoQ-induced resistance to apoptosis (25, 81). Moreover, decylTPP (which differs from mitoQ only in the absence of the Q moiety) (Fig. 14), did not affect fuel selectivity despite a greater reduction in membrane potential than that with mitoquinone (104).
E. Mitochondria-targeted antioxidant peptides
In addition to the previously mentioned triphenylphosphonium cationic molecules, other approaches to mitochondrial antioxidant therapy are being considered. One involves synthetic peptides with antioxidant properties designed to target mitochondria. These have been shown to penetrate mitochondria targeting the inner membrane by a poorly understood mechanism (304). Peptides containing tyrosine residues have been found effectively to scavenge oxygen radicals and peroxynitrite and to inhibit lipid peroxidation (304, 349). Such peptides reduce intracellular ROS and cell death in neuronal (256) and pancreatic islet cells (309) and recently were reported to reduce skeletal myocyte H2O2 production and to preserve insulin sensitivity in rats fed a high-fat diet (14). Unfortunately, these peptides also possess potent opioid-receptor affinity and activity (285, 347, 348). This activity may potentially limit their clinical effectiveness. Novel agents are needed to retain the antioxidant activity while reducing opioid-receptor activity.
F. Targeting superoxide
Tempol is an SOD mimetic that has been administered to rodents, apparently without toxicity (308). The agent is not specific for mitochondrial as opposed to total cellular superoxide, but effects do appear to include mitochondria. Tang et al. (307) studied the effect of hyperglycemia on islet β-cell function in rats after antecedent exposure to in vivo hyperglycemia with or without coadministration of antioxidants. Tempol both prevented β-cell dysfunction and increased superoxide induced by hyperglycemia. Tempol was observed to prevent excess total as well as mitochondrial superoxide, as evidenced by the effect of this agent on DHE and mitochondria-targeted DHE fluorescence (see Section III.E). In another study of rats infused with oleate for 48 h, co-infusion of Tempol improved insulin secretion and mitigated oxidative stress in islets subsequently studied ex vivo (245).
Tempol also may have beneficial effects on diabetic complications. The agent reportedly reduced renal mesangial expansion and decreased transforming growth factor β in diabetic rats (18). Conversely, the Tempol-induced conversion of superoxide to H2O2 appeared to be followed by increased hypochlorite production from H2O2 (18). In another report, 8 weeks of subcutaneous Tempol treatment was found to mitigate endothelial dysfunction as well as oxidative damage to vascular cells in rats with STZ-diabetes (232).
IX. Summary
Mitochondria, by virtue of numbers or functional properties or both, are critically involved in the pathophysiology of diabetes. At the islet β-cell level, acute insulin release is regulated by mitochondrial ATP production and mitochondrial ROS may contribute to the long-term deterioration of insulin secretory capacity seen in type 2 diabetes. Mitochondrial function also appears a critical determinant of insulin sensitivity within muscle, liver, and adipose tissue. Moreover, ROS appear important in the autoimmune destruction that characterizes type 1 diabetes, as well as in the pathophysiology of the long-term complications that characterize both classes of diabetes. Mitochondria also play a primary role in the etiology of genetic forms of “mitochondrial” diabetes. New diabetes-treatment strategies are needed to address both mitochondrial function and ROS production. Pharmacologic interventions must focus on mechanisms regulating mitochondrial biogenesis, ROS, and respiration. At the functional level, effective pharmacologic agents are needed that can be safely delivered to targeted sites within cells and within mitochondria.
Abbreviations Used
- 4-HNE
4-hydroxy-2-nonenal
- 8-OHdG
8-hydroxy-2′deoxyguanosine
- ACC2
acetyl-CoA carboxylase 2
- AGE
glycosylation end product
- AKT
serine/threonine kinase protein kinase B
- ALDH
aldehyde dehydrogenase
- AMPK
AMP-activated protein kinase
- ANT
adenine nucleotide translocase
- BAE
bovine aortic endothelial
- BAT
brown adipose tissue
- BRECs
bovine retinal endothelial cells
- CoPP
cobalt protoporphyrin
- CoQ
coenzyme Q
- CoQ9
coenzyme Q containing nine prenyl subunits
- CoQ10
coenzyme Q containing 10 prenyl subunits
- CPT
carnitine palmitoyl transferase
- CPT-I
carnitine palmitoyl transferase I
- CPT-II
carnitine palmitoyl transferase II
- DHE
dihydroethidine
- DHPA
10-acetyl-3,7-dihydroxyphenoxazine
- DMPO
5,5-dimethyl-l-pyrroline-N-oxide
- EDHF
endothelium-derived hyperpolarizing factor
- ELISA
enzyme-linked immunosorbent assay
- EPR
electron paramagnetic resonance
- ETF
electron-transport flavoprotein
- ETS
electron-transport system
- FCCP
carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone
- F0F1-ATPase
ATP synthase
- GAPDH
glycerol 3-phosphate dehydrogenase
- GLUT4
glucose transporter type 4
- GPX
glutathione peroxidase
- GSH
glutathione
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- HO-1
heme oxygenase-1
- HPLC
high-pressure liquid chromatography
- IMFM
intermyofibrillar mitochondria
- IR
insulin receptor
- IRS
insulin-receptor substrate
- IRS-1
insulin-receptor substrate type 1
- JNK
c-Jun N-terminal kinase
- KO
knock-out
- LDH
lactate dehydrogenase
- MCD
malonyl-CoA decarboxylase
- MDA
malondialdehyde
- MFN
mitofusin
- MnSOD
manganese superoxide dismutase
- MnTBAP
manganese (III) tetrakis (4-benzoic acid) porphyrin
- MPTP
mitochondrial permeability transition pore
- mtDNA
mitochondrial DNA
- MTE-1
mitochondrial thioesterase-1
- NAC
N-acetylcysteine
- NMR
nuclear magnetic resonance
- NO
nitric oxide
- NOS
mitochondrial nitric oxide synthase
- NRF-1
nuclear transcription factor 1
- NRFs
nuclear transcription factors
- PARL
presenillin-associated rhomboid-like
- PCO
polycystic ovarian syndrome
- PDC
pyruvate dehydrogenase complex
- PFK
phosphofructokinase
- PGC-1α or β
peroxisome proliferator–activated receptor gamma coactivator type 1 alpha or 1 beta
- PI3K
phosphatidylinositol 3-kinase
- PKC
protein kinase C
- PPARα
peroxisome proliferator–activated receptor alpha
- PPARγ
peroxisome proliferator–activated receptor gamma
- rMC-1
transformed retinal cells
- ROS
reactive oxygen species
- SHR
spontaneously hypertensive rat
- SIRT1
mammalian counterpart silent information regulator 2
- SLM
subsarcolemmal mitochondria
- SOD
superoxide dismutase
- STZ
streptozotocin
- TMRM
tetramethylrhodamine methyl ester
- TNF-α
tumor necrosis factor-alpha
- TPMP
methyltriphenylphosphonium
- TPP
tetraphenylphosphonium
- TRAP
plasma antioxidant capacity
- UCP
uncoupling protein
- UCP1,2,3,4
uncoupling proteins 1, 2, 3, or 4
- Vo2max
maximal oxygen consumption
- WT
wild type
Footnotes
Reviewing Editors: Nader Abraham, David Busija, Anonymous, Alexander Galkin, Thomas Kietzmann, Renu Kowluru, Matthew Picklo, and Yuichiro J. Suzuki
Acknowledgments
This work was supported by Veterans Affairs Research Funds and by NIH grants DK25295 and DK073990.
References
- 1.Abdul-Ghani MA. DeFronzo RA. Mitochondrial dysfunction, insulin resistance, and type 2 diabetes mellitus. Curr Diabetis Rep. 2008;8:173–178. doi: 10.1007/s11892-008-0030-1. [DOI] [PubMed] [Google Scholar]
- 2.Aberg F. Appelkvist EL. Dallner G. Ernster L. Distribution and redox state of ubiquinones in rat and human tissues. Arch Biochem Biophys. 1992;295:230–234. doi: 10.1016/0003-9861(92)90511-t. [DOI] [PubMed] [Google Scholar]
- 3.Abraham NG. Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 4.Abu-Elheiga L. Matzuk MM. Abo-Hashema KA. Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 5.Abu-Elheiga L. Oh W. Kordari P. Wakil SJ. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc Natl Acad Sci U S A. 2003;100:10207–10212. doi: 10.1073/pnas.1733877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal. 2005;7:1140–1149. doi: 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- 7.Adlam VJ. Harrison JC. Porteous CM. James AM. Smith RA. Murphy MP. Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 8.Affourtit C. Br MD. On the role of uncoupling protein-2 in pancreatic beta cells. Biochim Biophys Acta. 2008;1777:973–979. doi: 10.1016/j.bbabio.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 9.Affourtit C. Br MD. Stronger control of ATP/ADP by proton leak in pancreatic beta-cells than skeletal muscle mitochondria. Biochem J. 2006;393:151–159. doi: 10.1042/BJ20051280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009;32(suppl 1):S62–S67. doi: 10.2337/dc09-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.American Diabetes Association. Standards of medical care in diabetes. Diabetes Care. 2009;32(suppl 1):S13–S61. doi: 10.2337/dc09-S013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An J. Muoio DM. Shiota M. Fujimoto Y. Cline GW. Shulman GI. Koves TR. Stevens R. Millington D. Newgard CB. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med. 2004;10:268–274. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- 13.Andersen CB. Henriksen JE. Hother-Nielsen O. Vaag A. Mortensen SA. Beck-Nielsen H. The effect of coenzyme Q10 on blood glucose and insulin requirement in patients with insulin dependent diabetes mellitus. Mol Aspects Med. 1997;18(suppl):S307–S309. doi: 10.1016/s0098-2997(97)00010-1. [DOI] [PubMed] [Google Scholar]
- 14.Anderson EJ. Lustig ME. Boyle KE. Woodlief TL. Kane DA. Lin CT. Price JW., 3rd Kang L. Rabinovitch PS. Szeto HH. Houmard JA. Cortright RN. Wasserman DH. Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009 doi: 10.1172/JCI37048. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arany Z. He H. Lin J. Hoyer K. Handschin C. Toka O. Ahmad F. Matsui T. Chin S. Wu PH. Rybkin II. Shelton JM. Manieri M. Cinti S. Schoen FJ. Bassel-Duby R. Rosenzweig A. Ingwall JS. Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Arechaga I. Ledesma A. Rial E. The mitochondrial uncoupling protein UCP1: a gated pore. IUBMB Life. 2001;52:165–173. doi: 10.1080/15216540152845966. [DOI] [PubMed] [Google Scholar]
- 17.Argyropoulos G. Brown AM. Willi SM. Zhu J. He Y. Reitman M. Gevao SM. Spruill I. Garvey WT. Effects of mutations in the human uncoupling protein 3 gene on the respiratory quotient and fat oxidation in severe obesity and type 2 diabetes. J Clin Invest. 1998;102:1345–1351. doi: 10.1172/JCI4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asaba K. Tojo A. Onozato ML. Goto A. Fujita T. Double-edged action of SOD mimetic in diabetic nephropathy. J Cardiovasc Pharmacol. 2007;49:13–19. doi: 10.1097/FJC.0b013e31802b6530. [DOI] [PubMed] [Google Scholar]
- 19.Bach D. Pich S. Soriano FX. Vega N. Baumgartner B. Oriola J. Daugaard JR. Lloberas J. Camps M. Zierath JR. Rabasa-Lhoret R. Wallberg-Henriksson H. Laville M. Palacin M. Vidal H. Rivera F. Brand M. Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism: a novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 20.Baron AD. Insulin resistance and vascular function. J Diabetes Complications. 2002;16:92–102. doi: 10.1016/s1056-8727(01)00209-4. [DOI] [PubMed] [Google Scholar]
- 21.Bastar I. Seckin S. Uysal M. Aykac-Toker G. Effect of streptozotocin on glutathione and lipid peroxide levels in various tissues of rats. Res Commun Mol Pathol Pharmacol. 1998;102:265–272. [PubMed] [Google Scholar]
- 22.Baur JA. Pearson KJ. Price NL. Jamieson HA. Lerin C. Kalra A. Prabhu VV. Allard JS. Lopez-Lluch G. Lewis K. Pistell PJ. Poosala S. Becker KG. Boss O. Gwinn D. Wang M. Ramaswamy S. Fishbein KW. Spencer RG. Lakatta EG. Le Couteur D. Shaw RJ. Navas P. Puigserver P. Ingram DK. de Cabo R. Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beckman JA. Goldfine AB. Gordon MB. Garrett LA. Keaney JF., Jr Creager MA. Oral antioxidant therapy improves endothelial function in Type 1 but not Type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol. 2003;285:H2392–H2398. doi: 10.1152/ajpheart.00403.2003. [DOI] [PubMed] [Google Scholar]
- 24.Beckman KB. Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 25.Bedogni B. Pani G. Colavitti R. Riccio A. Borrello S. Murphy M. Smith R. Eboli ML. Galeotti T. Redox regulation of cAMP-responsive element-binding protein and induction of manganous superoxide dismutase in nerve growth factor-dependent cell survival. J Biol Chem. 2003;278:16510–16519. doi: 10.1074/jbc.M301089200. [DOI] [PubMed] [Google Scholar]
- 26.Befroy DE. Petersen KF. Dufour S. Mason GF. de Graaf RA. Rothman DL. Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benton CR. Nickerson JG. Lally J. Han XX. Holloway GP. Glatz JF. Luiken JJ. Graham TE. Heikkila JJ. Bonen A. Modest PGC-1alpha overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. J Biol Chem. 2008;283:4228–4240. doi: 10.1074/jbc.M704332200. [DOI] [PubMed] [Google Scholar]
- 28.Bergeron R. Ren JM. Cadman KS. Moore IK. Perret P. Pypaert M. Young LH. Semenkovich CF. Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 29.Berlie HD. Kalus JS. Jaber LA. Thiazolidinediones and the risk of edema: a meta-analysis. Diabetes Res Clin Pract. 2007;76:279–289. doi: 10.1016/j.diabres.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Bogacka I. Xie H. Bray GA. Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54:1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 31.Boss O. Hagen T. Lowell BB. Uncoupling proteins 2 and 3: potential regulators of mitochondrial energy metabolism. Diabetes. 2000;49:143–156. doi: 10.2337/diabetes.49.2.143. [DOI] [PubMed] [Google Scholar]
- 32.Boss O. Samec S. Paoloni-Giacobino A. Rossier C. Dulloo A. Seydoux J. Muzzin P. Giacobino JP. Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997;408:39–42. doi: 10.1016/s0014-5793(97)00384-0. [DOI] [PubMed] [Google Scholar]
- 33.Boudina S. Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 34.Boudina S. Sena S. Theobald H. Sheng X. Wright JJ. Hu XX. Aziz S. Johnson JI. Bugger H. Zaha VG. Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 35.Boushel R. Gnaiger E. Schjerling P. Skovbro M. Kraunsoe R. Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouzakri K. Austin R. Rune A. Lassman ME. Garcia-Roves PM. Berger JP. Krook A. Chibalin AV. Zhang BB. Zierath JR. Malonyl CoenzymeA decarboxylase regulates lipid and glucose metabolism in human skeletal muscle. Diabetes. 2008;57:1508–1516. doi: 10.2337/db07-0583. [DOI] [PubMed] [Google Scholar]
- 37.Boveris A. Oshino N. Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boveris AA. Cattaneo de Peralta R. Stoppani AO. Foglia VG. Phosphorylation, oxidation, and ubiquinone content in diabetic mitochondria. Proc Soc Exp Biol Med. 1969;132:171–174. doi: 10.3181/00379727-132-34174. [DOI] [PubMed] [Google Scholar]
- 39.Brand MD. Brindle KM. Buckingham JA. Harper JA. Rolfe DF. Stuart JA. The significance and mechanism of mitochondrial proton conductance. Int J Obes Rel Metab Dis, J Int Assoc Study Obes. 1999;23:S4–S11. doi: 10.1038/sj.ijo.0800936. [DOI] [PubMed] [Google Scholar]
- 40.Brandt U. Energy converting NADH:quinone oxidoreductase (complex I) Annu Rev Biochem. 2006;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- 41.Brown GC. Borutaite V. Nitric oxide, mitochondria, and cell death. IUBMB Life. 2001;52:189–195. doi: 10.1080/15216540152845993. [DOI] [PubMed] [Google Scholar]
- 42.Bugger H. Boudina S. Hu XX. Tuinei J. Zaha VG. Theobald HA. Yun UJ. McQueen AP. Wayment B. Litwin SE. Abel ED. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008;57:2924–2932. doi: 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Busik JV. Mohr S. Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57:1952–1965. doi: 10.2337/db07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cadenas S. Buckingham JA. Samec S. Seydoux J. Din N. Dulloo AG. Br MD. UCP2 and UCP3 rise in starved rat skeletal muscle but mitochondrial proton conductance is unchanged. FEBS Lett. 1999;462:257–260. doi: 10.1016/s0014-5793(99)01540-9. [DOI] [PubMed] [Google Scholar]
- 45.Cadenas S. Echtay KS. Harper JA. Jekabsons MB. Buckingham JA. Grau E. Abuin A. Chapman H. Clapham JC. Br MD. The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein-3. J Biol Chem. 2002;277:2773–2778. doi: 10.1074/jbc.M109736200. [DOI] [PubMed] [Google Scholar]
- 46.Cameron NE. Cotter MA. Effects of an extracellular metal chelator on neurovascular function in diabetic rats. Diabetologia. 2001;44:621–628. doi: 10.1007/s001250051669. [DOI] [PubMed] [Google Scholar]
- 47.Cameron NE. Cotter MA. Metabolic and vascular factors in the pathogenesis of diabetic neuropathy. Diabetes. 1997;46(suppl 2):S31–S37. doi: 10.2337/diab.46.2.s31. [DOI] [PubMed] [Google Scholar]
- 48.Cameron NE. Cotter MA. Neurovascular dysfunction in diabetic rats: potential contribution of autoxidation and free radicals examined using transition metal chelating agents. J Clin Invest. 1995;96:1159–1163. doi: 10.1172/JCI118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cameron NE. Cotter MA. Maxfield EK. Anti-oxidant treatment prevents the development of peripheral nerve dysfunction in streptozotocin-diabetic rats. Diabetologia. 1993;36:299–304. doi: 10.1007/BF00400231. [DOI] [PubMed] [Google Scholar]
- 50.Cameron NE. Jack AM. Cotter MA. Effect of alpha-lipoic acid on vascular responses and nociception in diabetic rats. Free Radic Biol Med. 2001;31:125–135. doi: 10.1016/s0891-5849(01)00564-0. [DOI] [PubMed] [Google Scholar]
- 51.Cameron NE. Tuck Z. McCabe L. Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44:1161–1169. doi: 10.1007/s001250100626. [DOI] [PubMed] [Google Scholar]
- 52.Canto C. Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ceriello A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care. 2003;26:1589–1596. doi: 10.2337/diacare.26.5.1589. [DOI] [PubMed] [Google Scholar]
- 54.Cester N. Rabini RA. Salvolini E. Staffolani R. Curatola A. Pugnaloni A. Brunelli MA. Biagini G. Mazzanti L. Activation of endothelial cells during insulin-dependent diabetes mellitus: a biochemical and morphological study. Eur J Clin Invest. 1996;26:569–573. doi: 10.1046/j.1365-2362.1996.1750526.x. [DOI] [PubMed] [Google Scholar]
- 55.Chan CB. MacDonald PE. Saleh MC. Johns DC. Marban E. Wheeler MB. Overexpression of uncoupling protein 2 inhibits glucose-stimulated insulin secretion from rat islets. Diabetes. 1999;48:1482–1486. doi: 10.2337/diabetes.48.7.1482. [DOI] [PubMed] [Google Scholar]
- 56.Chan K. Truong D. Shangari N. O'Brien PJ. Drug-induced mitochondrial toxicity. Expert Opin Drug Metab Toxicol. 2005;1:655–669. doi: 10.1517/17425255.1.4.655. [DOI] [PubMed] [Google Scholar]
- 57.Chance B. Sies H. Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 58.Chance B. Williams GR. Respiratory enzymes in oxidative phosphorylation, III: the steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- 59.Chao TT. Ianuzzo CD. Armstrong RB. Albright JT. Anapolle SE. Ultrastructural alterations in skeletal muscle fibers of streptozotocin-diabetic rats. Cell Tissue Res. 1976;168:239–246. doi: 10.1007/BF00215880. [DOI] [PubMed] [Google Scholar]
- 60.Choi SL. Kim SJ. Lee KT. Kim J. Mu J. Birnbaum MJ. Soo Kim S. Ha J. The regulation of AMP-activated protein kinase by H(2)O(2) Biochem Biophys Res Commun. 2001;287:92–97. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 61.Choo HJ. Kim JH. Kwon OB. Lee CS. Mun JY. Han SS. Yoon YS. Yoon G. Choi KM. Ko YG. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia. 2006;49:784–791. doi: 10.1007/s00125-006-0170-2. [DOI] [PubMed] [Google Scholar]
- 62.Chung WK. Luke A. Cooper RS. Rotini C. Vidal-Puig A. Rosenbaum M. Chua M. Solanes G. Zheng M. Zhao L. LeDuc C. Eisberg A. Chu F. Murphy E. Schreier M. Aronne L. Caprio S. Kahle B. Gordon D. Leal SM. Goldsmith R. Andreu AL. Bruno C. DiMauro S. Heo M. Lowe WL. Lowell BB. Allison DB. Leibel RL. Genetic and physiologic analysis of the role of uncoupling protein 3 in human energy homeostasis. Diabetes. 1999;48:1890–1895. doi: 10.2337/diabetes.48.9.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Civitarese AE. Ravussin E. Minireview: mitochondrial energetics and insulin resistance. Endocrinology. 2008;149:950–954. doi: 10.1210/en.2007-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cline GW. Vidal-Puig AJ. Dufour S. Cadman KS. Lowell BB. Shulman GI. In vivo effects of uncoupling protein-3 gene disruption on mitochondrial energy metabolism. J Biol Chem. 2001;276:20240–20244. doi: 10.1074/jbc.M102540200. [DOI] [PubMed] [Google Scholar]
- 65.Collins AR. Raslova K. Somorovska M. Petrovska H. Ondrusova A. Vohnout B. Fabry R. Dusinska M. DNA damage in diabetes: correlation with a clinical marker. Free Radic Biol Med. 1998;25:373–377. doi: 10.1016/s0891-5849(98)00053-7. [DOI] [PubMed] [Google Scholar]
- 66.Constantinescu A. Maguire JJ. Packer L. Interactions between ubiquinones and vitamins in membranes and cells. Mol Aspects Med. 1994;15(suppl):s57–s65. doi: 10.1016/0098-2997(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 67.Coppey LJ. Gellett JS. Davidson EP. Dunlap JA. Lund DD. Salvemini D. Yorek MA. Effect of M40403 treatment of diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Br J Pharmacol. 2001;134:21–29. doi: 10.1038/sj.bjp.0704216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coppey LJ. Gellett JS. Davidson EP. Dunlap JA. Lund DD. Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- 69.Coppey LJ. Gellett JS. Davidson EP. Dunlap JA. Yorek MA. Effect of treating streptozotocin-induced diabetic rats with sorbinil, myo-inositol or aminoguanidine on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Int J Exp Diabetes Res. 2002;3:21–36. doi: 10.1080/15604280212525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coppey LJ. Gellett JS. Davidson EP. Yorek MA. Preventing superoxide formation in epineurial arterioles of the sciatic nerve from diabetic rats restores endothelium-dependent vasodilation. Free Radic Res. 2003;37:33–40. doi: 10.1080/1071576021000028442. [DOI] [PubMed] [Google Scholar]
- 71.Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- 72.Coughlan MT. Thallas-Bonke V. Pete J. Long DM. Gasser A. Tong DC. Arnstein M. Thorpe SR. Cooper ME. Forbes JM. Combination therapy with the advanced glycation end product cross-link breaker, alagebrium, and angiotensin converting enzyme inhibitors in diabetes: synergy or redundancy? Endocrinology. 2007;148:886–895. doi: 10.1210/en.2006-1300. [DOI] [PubMed] [Google Scholar]
- 73.Couplan E. del Mar Gonzalez-Barroso M. Alves-Guerra MC. Ricquier D. Goubern M. Bouillaud F. No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J Biol Chem. 2002;277:26268–26275. doi: 10.1074/jbc.M202535200. [DOI] [PubMed] [Google Scholar]
- 74.Craven PA. Studer RK. Negrete H. DeRubertis FR. Protein kinase C in diabetic nephropathy. J Diabetes Complications. 1995;9:241–245. doi: 10.1016/1056-8727(95)80012-4. [DOI] [PubMed] [Google Scholar]
- 75.Cross AR. Jones OT. Enzymic mechanisms of superoxide production. Biochim Biophys Acta. 1991;1057:281–298. doi: 10.1016/s0005-2728(05)80140-9. [DOI] [PubMed] [Google Scholar]
- 76.Cui Y. Xu X. Bi H. Zhu Q. Wu J. Xia X. Qiushi R. Ho PC. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: the role of reactive oxygen species in diabetic retinopathy. Exp Eye Res. 2006;83:807–816. doi: 10.1016/j.exer.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 77.Cutting WC. Mehrtens HG. Tainter ML. Actions and uses of dinitrophenol. JAMA. 1933;101:193–195. [Google Scholar]
- 78.Cypess AM. Lehman S. Williams G. Tal I. Rodman D. Goldfine AB. Kuo FC. Palmer EL. Tseng YH. Doria A. Kolodny GM. Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Cavanagh EM. Ferder L. Toblli JE. Piotrkowski B. Stella I. Fraga CG. Inserra F. Renal mitochondrial impairment is attenuated by AT1 blockade in experimental Type I diabetes. Am J Physiol Heart Circ Physiol. 2008;294:H456–H465. doi: 10.1152/ajpheart.00926.2007. [DOI] [PubMed] [Google Scholar]
- 80.De Vriese AS. Verbeuren TJ. Van de Voorde J. Lameire NH. Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dhanasekaran A. Kotamraju S. Kalivendi SV. Matsunaga T. Shang T. Keszler A. Joseph J. Kalyanaraman B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J Biol Chem. 2004;279:37575–37587. doi: 10.1074/jbc.M404003200. [DOI] [PubMed] [Google Scholar]
- 82.Di Noia MA. Van Driesche S. Palmieri F. Yang LM. Quan S. Goodman AI. Abraham NG. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome C oxidase activity in experimental diabetes. J Biol Chem. 2006;281:15687–15693. doi: 10.1074/jbc.M510595200. [DOI] [PubMed] [Google Scholar]
- 83.Dincer Y. Telci A. Kayali R. Yilmaz IA. Cakatay U. Akcay T. Effect of alpha-lipoic acid on lipid peroxidation and anti-oxidant enzyme activities in diabetic rats. Clin Exp Pharmacol Physiol. 2002;29:281–284. doi: 10.1046/j.1440-1681.2002.03642.x. [DOI] [PubMed] [Google Scholar]
- 84.Doughan AK. Dikalov SI. Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid Redox Signal. 2007;9:1825–1836. doi: 10.1089/ars.2007.1693. [DOI] [PubMed] [Google Scholar]
- 85.Drose S. Brandt U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J Biol Chem. 2008;283:21649–21654. doi: 10.1074/jbc.M803236200. [DOI] [PubMed] [Google Scholar]
- 86.Du XL. Edelstein D. Rossetti L. Fantus IG. Goldberg H. Ziyadeh F. Wu J. Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Du Y. Miller CM. Kern TS. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic Biol Med. 2003;35:1491–1499. doi: 10.1016/j.freeradbiomed.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 88.Dulak J. Deshane J. Jozkowicz A. Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008;117:231–241. doi: 10.1161/CIRCULATIONAHA.107.698316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Duncan JG. Fong JL. Medeiros DM. Finck BN. Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dutton PL. Moser CC. Sled VD. Daldal F. Ohnishi T. A reductant-induced oxidation mechanism for complex I. Biochim Biophys Acta. 1998;1364:245–257. doi: 10.1016/s0005-2728(98)00031-0. [DOI] [PubMed] [Google Scholar]
- 91.Echtay KS. Esteves TC. Pakay JL. Jekabsons MB. Lambert AJ. Portero-Otin M. Pamplona R. Vidal-Puig AJ. Wang S. Roebuck SJ. Br MD. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Echtay KS. Murphy MP. Smith RA. Talbot DA. Br MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side: studies using targeted antioxidants. J Biol Chem. 2002;277:47129–47135. doi: 10.1074/jbc.M208262200. [DOI] [PubMed] [Google Scholar]
- 93.Echtay KS. Roussel D. St-Pierre J. Jekabsons MB. Cadenas S. Stuart JA. Harper JA. Roebuck SJ. Morrison A. Pickering S. Clapham JC. Br MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 94.Echtay KS. Winkler E. Frischmuth K. Klingenberg M. Uncoupling proteins 2 and 3 are highly active H(+) transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone) Proc Natl Acad Sci U S A. 2001;98:1416–1421. doi: 10.1073/pnas.98.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Economides PA. Khaodhiar L. Caselli A. Caballero AE. Keenan H. Bursell SE. King GL. Johnstone MT. Horton ES. Veves A. The effect of vitamin E on endothelial function of micro- and macrocirculation and left ventricular function in type 1 and type 2 diabetic patients. Diabetes. 2005;54:204–211. doi: 10.2337/diabetes.54.1.204. [DOI] [PubMed] [Google Scholar]
- 96.Emre Y. Hurtaud C. Karaca M. Nubel T. Zavala F. Ricquier D. Role of uncoupling protein UCP2 in cell-mediated immunity: how macrophage-mediated insulitis is accelerated in a model of autoimmune diabetes. Proc Natl Acad Sci U S A. 2007;104:19085–19090. doi: 10.1073/pnas.0709557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ernster L. Forsmark P. Nordenbr K. The mode of action of lipid-soluble antioxidants in biological membranes: relationship between the effects of ubiquinol and vitamin E as inhibitors of lipid peroxidation in submitochondrial particles. Biofactors. 1992;3:241–248. [PubMed] [Google Scholar]
- 98.Erusalimsky JD. Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–2531. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- 99.Essop MF. Camp HS. Choi CS. Sharma S. Fryer RM. Reinhart GA. Guthrie PH. Bentebibel A. Gu Z. Shulman GI. Taegtmeyer H. Wakil SJ. Abu-Elheiga L. Reduced heart size and increased myocardial fuel substrate oxidation in ACC2 mutant mice. Am J Physiol Heart Circ Physiol. 2008;295:H256–H265. doi: 10.1152/ajpheart.91489.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Feige JN. Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17:292–301. doi: 10.1016/j.tcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 101.Ferko M. Gvozdjakova A. Kucharska J. Mujkosova J. Waczulikova I. Styk J. Ravingerova T. Ziegelhoffer-Mihalovicova B. Ziegelhoffer A. Functional remodeling of heart mitochondria in acute diabetes: interrelationships between damage, endogenous protection and adaptation. Gen Physiol Biophys. 2006;25:397–413. [PubMed] [Google Scholar]
- 102.Fink BD. Herlein JA. Almind K. Cinti S. Kahn CR. Sivitz WI. The mitochondrial proton leak in obesity-resistant and obesity-prone mice. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1773–R1780. doi: 10.1152/ajpregu.00478.2007. [DOI] [PubMed] [Google Scholar]
- 103.Fink BD. Hong YS. Mathahs MM. Scholz TD. Dillon JS. Sivitz WI. UCP2-dependent proton leak in isolated mammalian mitochondria. J Biol Chem. 2002;277:3918–3925. doi: 10.1074/jbc.M107955200. [DOI] [PubMed] [Google Scholar]
- 104.Fink BD. O'Malley Y. Dake BL. Ross NC. Prisinzano TE. Sivitz WI. Mitochondrial targeted coenzyme Q, superoxide, and fuel selectivity in endothelial cells. PLoS ONE. 2009;4:e4250. doi: 10.1371/journal.pone.0004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fink BD. Reszka KJ. Herlein JA. Mathahs MM. Sivitz WI. Respiratory uncoupling by UCP1 and UCP2 and superoxide generation in endothelial cell mitochondria. Am J Physiol, Endocrinol Metab. 2005;288:E71–E79. doi: 10.1152/ajpendo.00332.2004. [DOI] [PubMed] [Google Scholar]
- 106.Fleury C. Neverova M. Collins S. Raimbault S. Champigny O. Levi-Meyrueis C. Bouillaud F. Seldin MF. Surwit RS. Ricquier D. Warden CH. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet. 1997;15:269–272. doi: 10.1038/ng0397-269. [DOI] [PubMed] [Google Scholar]
- 107.Fridovich I. Superoxide anion radical (O2−·), superoxide dismutases, and related matters. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 108.Friederich M. Fasching A. Hansell P. Nordquist L. Palm F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta. 2008;1777:935–940. doi: 10.1016/j.bbabio.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 109.Fujii N. Jessen N. Goodyear LJ. AMP-activated protein kinase and the regulation of glucose transport. Am J Physiol Endocrinol Metab. 2006;291:E867–E877. doi: 10.1152/ajpendo.00207.2006. [DOI] [PubMed] [Google Scholar]
- 110.Gardner PR. Superoxide-driven aconitase FE-S center cycling. Biosci Rep. 1997;17:33–42. doi: 10.1023/a:1027383100936. [DOI] [PubMed] [Google Scholar]
- 111.Gat-Yablonski G. Shalitin S. Phillip M. Maturity onset diabetes of the young: review. Pediatr Endocrinol Rev. 2006;3(suppl 3):514–520. [PubMed] [Google Scholar]
- 112.Gautier JF. Wilson C. Weyer C. Mott D. Knowler WC. Cavaghan M. Polonsky KS. Bogardus C. Pratley RE. Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes. 2001;50:1828–1833. doi: 10.2337/diabetes.50.8.1828. [DOI] [PubMed] [Google Scholar]
- 113.Ghirlanda G. Oradei A. Manto A. Lippa S. Uccioli L. Caputo S. Greco AV. Littarru GP. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol. 1993;33:226–229. doi: 10.1002/j.1552-4604.1993.tb03948.x. [DOI] [PubMed] [Google Scholar]
- 114.Gnaiger E. Mitochondrial pathways and respiratory control. Innsbruck: OROBOROS MiPNet Publications; 2008. pp. 7–15. [Google Scholar]
- 115.Goto Y. Nonaka I. Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 116.Goto Y. Oliveira PJ. Rolo AP. Seica R. Palmeira CM. Santos MS. Moreno AJ. Decreased susceptibility of heart mitochondria from diabetic GK rats to mitochondrial permeability transition induced by calcium phosphate. Biosci Rep. 2001;21:45–53. doi: 10.1023/a:1010482017540. [DOI] [PubMed] [Google Scholar]
- 117.Green K. Brand MD. Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes. 2004;53:S110–S118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- 118.Grivennikova VG. Vinogradov AD. Generation of superoxide by the mitochondrial complex I. Biochim Biophys Acta. 2006;1757:553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 119.Gross MD. Harris S. Beyer RE. The effect of streptozotocin-induced diabetes on oxidative phosphorylation and related reactions in skeletal muscle mitochondria. Horm Metab Res. 1972;4:1–7. doi: 10.1055/s-0028-1094106. [DOI] [PubMed] [Google Scholar]
- 120.Group US. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33): UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 121.Guarente L. Mitochondria: a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gulli G. Ferrannini E. Stern M. Haffner S. DeFronzo RA. The metabolic profile of NIDDM is fully established in glucose-tolerant offspring of two Mexican-American NIDDM parents. Diabetes. 1992;41:1575–1586. doi: 10.2337/diab.41.12.1575. [DOI] [PubMed] [Google Scholar]
- 123.Gunter TE. Buntinas L. Sparagna G. Eliseev R. Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 2000;28:285–296. doi: 10.1054/ceca.2000.0168. [DOI] [PubMed] [Google Scholar]
- 124.Gunter TE. Gunter KK. Uptake of calcium by mitochondria: transport and possible function. IUBMB Life. 2001;52:197–204. doi: 10.1080/15216540152846000. [DOI] [PubMed] [Google Scholar]
- 125.Gunter TE. Yule DI. Gunter KK. Eliseev RA. Salter JD. Calcium and mitochondria. FEBS Lett. 2004;567:96–102. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- 126.Haas RH. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion. 2007;7(suppl):S136–S145. doi: 10.1016/j.mito.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 127.Hall JC. Sordahl LA. Stefko PL. The effect of insulin on oxidative phosphorylation in normal and diabetic mitochondria. J Biol Chem. 1960;235:1536–1539. [PubMed] [Google Scholar]
- 128.Han DWE. Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Handschin C. Choi CS. Chin S. Kim S. Kawamori D. Kurpad AJ. Neubauer N. Hu J. Mootha VK. Kim YB. Kulkarni RN. Shulman GI. Spiegelman BM. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J Clin Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hayden MR. Sowers JR. Isletopathy in type 2 diabetes mellitus: implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antioxid Redox Signal. 2007;9:891–910. doi: 10.1089/ars.2007.1610. [DOI] [PubMed] [Google Scholar]
- 131.He J. Watkins S. Kelley DE. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes. 2001;50:817–823. doi: 10.2337/diabetes.50.4.817. [DOI] [PubMed] [Google Scholar]
- 132.Henriksen EJ. Invited review: effects of acute exercise and exercise training on insulin resistance. J Appl Physiol. 2002;93:788–796. doi: 10.1152/japplphysiol.01219.2001. [DOI] [PubMed] [Google Scholar]
- 133.Henriksen JE. Andersen CB. Hother-Nielsen O. Vaag A. Mortensen SA. Beck-Nielsen H. Impact of ubiquinone (coenzyme Q10) treatment on glycaemic control, insulin requirement and well-being in patients with type 1 diabetes mellitus. Diabetes Med. 1999;16:312–318. doi: 10.1046/j.1464-5491.1999.00064.x. [DOI] [PubMed] [Google Scholar]
- 134.Herlein JA. Fink BD. O'Malley Y. Sivitz WI. Superoxide and respiratory coupling in mitochondria of insulin-deficient diabetic rats. Endocrinology. 2009;150:46–55. doi: 10.1210/en.2008-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hesselink MK. Greenhaff PL. Constantin-Teodosiu D. Hultman E. Saris WH. Nieuwlaat R. Schaart G. Kornips E. Schrauwen P. Increased uncoupling protein 3 content does not affect mitochondrial function in human skeletal muscle in vivo. J Clin Invest. 2003;111:479–486. doi: 10.1172/JCI16653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hesselink MKC. Mensink M. Schrauwen P. Human uncoupling protein-3 and obesity: an update. Obesity Res. 2003;11:1429–1443. doi: 10.1038/oby.2003.192. [DOI] [PubMed] [Google Scholar]
- 137.Hirosumi J. Tuncman G. Chang L. Gorgun CZ. Uysal KT. Maeda K. Karin M. Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 138.Hoeldtke RD. Bryner KD. McNeill DR. Warehime SS. Van Dyke K. Hobbs G. Oxidative stress and insulin requirements in patients with recent-onset type 1 diabetes. J Clin Endocrinol Metab. 2003;88:1624–1628. doi: 10.1210/jc.2002-021525. [DOI] [PubMed] [Google Scholar]
- 139.Hojlund K. Wrzesinski K. Larsen PM. Fey SJ. Roepstorff P. Handberg A. Dela F. Vinten J. McCormack JG. Reynet C. Beck-Nielsen H. Proteome analysis reveals phosphorylation of ATP synthase beta-subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J Biol Chem. 2003;278:10436–10442. doi: 10.1074/jbc.M212881200. [DOI] [PubMed] [Google Scholar]
- 140.Hong Y. Fink BD. Dillon JS. Sivitz WI. Effects of adenoviral overexpression of uncoupling protein-2 and -3 on mitochondrial respiration in insulinoma cells. Endocrinology. 2001;142:249–256. doi: 10.1210/endo.142.1.7889. [DOI] [PubMed] [Google Scholar]
- 141.Horie T. Ono K. Nagao K. Nishi H. Kinoshita M. Kawamura T. Wada H. Shimatsu A. Kita T. Hasegawa K. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J Cell Physiol. 2008;215:733–742. doi: 10.1002/jcp.21353. [DOI] [PubMed] [Google Scholar]
- 142.Houstis N. Rosen ED. Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 143.Howitz KT. Bitterman KJ. Cohen HY. Lamming DW. Lavu S. Wood JG. Zipkin RE. Chung P. Kisielewski A. Zhang LL. Scherer B. Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 144.Huie RE. Padmaja S. The reaction of no with superoxide. Free Radic Res Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 145.Inkster ME. Cotter MA. Cameron NE. Effects of trientine, a metal chelator, on defective endothelium-dependent relaxation in the mesenteric vasculature of diabetic rats. Free Radic Res. 2002;36:1091–1099. doi: 10.1080/1071576021000028325. [DOI] [PubMed] [Google Scholar]
- 146.James AM. Murphy MP. How mitochondrial damage affects cell function. J Biomed Sci. 2002;9:475–487. doi: 10.1159/000064721. [DOI] [PubMed] [Google Scholar]
- 147.James AM. Smith RA. Murphy MP. Antioxidant and prooxidant properties of mitochondrial coenzyme Q. Arch Biochem Biophys. 2004;423:47–56. doi: 10.1016/j.abb.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 148.Jauslin ML. Meier T. Smith RA. Murphy MP. Mitochondria-targeted antioxidants protect Friedreich ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 149.Jekabsons MB. Nicholls DG. In situ respiration and bioenergetic status of mitochondria in primary cerebellar granule neuronal cultures exposed continuously to glutamate. J Biol Chem. 2004;279:32989–33000. doi: 10.1074/jbc.M401540200. [DOI] [PubMed] [Google Scholar]
- 150.Jo SH. Koo BK. Park JS. Kang HJ. Kim YJ. Kim HL. Chae IH. Choi DJ. Sohn DW. Oh BH. Park YB. Choi YS. Kim HS. N-acetylcysteine versus ascorbic acid for preventing contrast-induced nephropathy in patients with renal insufficiency undergoing coronary angiography NASPI study: a prospective randomized controlled trial. Am Heart J. 2009;157:576–583. doi: 10.1016/j.ahj.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 151.Johnson-Cadwell LI. Jekabsons MB. Wang A. Polster BM. Nicholls DG. “Mild uncoupling” does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- 152.Joseph JW. Koshkin V. Saleh MC. Sivitz WI. Zhang CY. Lowell BB. Chan CB. Wheeler MB. Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004;279:51049–51056. doi: 10.1074/jbc.M409189200. [DOI] [PubMed] [Google Scholar]
- 153.Joseph JW. Koshkin V. Zhang CY. Wang J. Lowell BB. Chan CB. Wheeler MB. Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes. 2002;51:3211–3219. doi: 10.2337/diabetes.51.11.3211. [DOI] [PubMed] [Google Scholar]
- 154.Kamo N. Muratsugu M. Hongoh R. Kobatake Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J Membr Biol. 1979;49:105–121. doi: 10.1007/BF01868720. [DOI] [PubMed] [Google Scholar]
- 155.Kanauchi M. Nishioka H. Hashimoto T. Oxidative DNA damage and tubulointerstitial injury in diabetic nephropathy. Nephron. 2002;91:327–329. doi: 10.1159/000058412. [DOI] [PubMed] [Google Scholar]
- 156.Kanwar M. Chan PS. Kern TS. Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007;48:3805–3811. doi: 10.1167/iovs.06-1280. [DOI] [PubMed] [Google Scholar]
- 157.Karakelides H. Asmann YW. Bigelow ML. Short KR. Dhatariya K. Coenen-Schimke J. Kahl J. Mukhopadhyay D. Nair KS. Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes. 2007;56:2683–2689. doi: 10.2337/db07-0378. [DOI] [PubMed] [Google Scholar]
- 158.Katakam PV. Jordan JE. Snipes JA. Tulbert CD. Miller AW. Busija DW. Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007;292:R920–R926. doi: 10.1152/ajpregu.00520.2006. [DOI] [PubMed] [Google Scholar]
- 159.Katyare SS. Satav JG. Effect of streptozotocin-induced diabetes on oxidative energy metabolism in rat kidney mitochondria: a comparative study of early and late effects. Diabetes Obes Metab. 2005;7:555–562. doi: 10.1111/j.1463-1326.2004.00429.x. [DOI] [PubMed] [Google Scholar]
- 160.Keegan A. Cotter MA. Cameron NE. Effects of diabetes and treatment with the antioxidant alpha-lipoic acid on endothelial and neurogenic responses of corpus cavernosum in rats. Diabetologia. 1999;42:343–350. doi: 10.1007/s001250051161. [DOI] [PubMed] [Google Scholar]
- 161.Kelley DE. He J. Menshikova EV. Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 162.Kelso GF. Porteous CM. Coulter CV. Hughes G. Porteous WK. Ledgerwood EC. Smith RA. Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- 163.Khailova LS. Prikhodko EA. Dedukhova VI. Mokhova EN. Popov VN. Skulachev VP. Participation of ATP/ADP antiporter in oleate- and oleate hydroperoxide-induced uncoupling suppressed by GDP and carboxyatractylate. Biochim Biophys Acta. 2006;1757:1324–1329. doi: 10.1016/j.bbabio.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 164.Kim JA. Wei Y. Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kiritoshi S. Nishikawa T. Sonoda K. Kukidome D. Senokuchi T. Matsuo T. Matsumura T. Tokunaga H. Brownlee M. Araki E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: potential role in diabetic nephropathy. Diabetes. 2003;52:2570–2577. doi: 10.2337/diabetes.52.10.2570. [DOI] [PubMed] [Google Scholar]
- 166.Knowler WC. Barrett-Connor E. Fowler SE. Hamman RF. Lachin JM. Walker EA. Nathan DM. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Korshunov SS. Skulachev VP. Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 168.Koves TR. Li P. An J. Akimoto T. Slentz D. Ilkayeva O. Dohm GL. Yan Z. Newgard CB. Muoio DM. Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem. 2005;280:33588–33598. doi: 10.1074/jbc.M507621200. [DOI] [PubMed] [Google Scholar]
- 169.Koves TR. Noland RC. Bates AL. Henes ST. Muoio DM. Cortright RN. Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am J Physiol Cell Physiol. 2005;288:C1074–C1082. doi: 10.1152/ajpcell.00391.2004. [DOI] [PubMed] [Google Scholar]
- 170.Koves TR. Ussher JR. Noland RC. Slentz D. Mosedale M. Ilkayeva O. Bain J. Stevens R. Dyck JR. Newgard CB. Lopaschuk GD. Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 171.Kowluru RA. Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal. 2005;7:1581–1587. doi: 10.1089/ars.2005.7.1581. [DOI] [PubMed] [Google Scholar]
- 172.Kowluru RA. Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003;44:5327–5334. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- 173.Kowluru RA. Kowluru V. Xiong Y. Ho YS. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006;41:1191–1196. doi: 10.1016/j.freeradbiomed.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 174.Kowluru RA. Odenbach S. Effect of long-term administration of alpha-lipoic acid on retinal capillary cell death and the development of retinopathy in diabetic rats. Diabetes. 2004;53:3233–3238. doi: 10.2337/diabetes.53.12.3233. [DOI] [PubMed] [Google Scholar]
- 175.Krause Mda S. de Bittencourt PI., Jr Type 1 diabetes: can exercise impair the autoimmune event: the L-arginine/glutamine coupling hypothesis. Cell Biochem Funct. 2008;26:406–433. doi: 10.1002/cbf.1470. [DOI] [PubMed] [Google Scholar]
- 176.Krauss S. Zhang CY. Scorrano L. Dalgaard LT. St-Pierre J. Grey ST. Lowell BB. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction [see comment] J Clin Invest. 2003;112:1831–1842. doi: 10.1172/JCI19774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Krebs M. Roden M. Molecular mechanisms of lipid-induced insulin resistance in muscle, liver and vasculature. Diabetes Obes Metab. 2005;7:621–632. doi: 10.1111/j.1463-1326.2004.00439.x. [DOI] [PubMed] [Google Scholar]
- 178.Kroemer G. Zamzami N. Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 179.Kubisch HM. Wang J. Bray TM. Phillips JP. Targeted overexpression of Cu/Zn superoxide dismutase protects pancreatic beta-cells against oxidative stress. Diabetes. 1997;46:1563–1566. doi: 10.2337/diabetes.46.10.1563. [DOI] [PubMed] [Google Scholar]
- 180.Kucharska J. Braunova Z. Ulicna O. Zlatos L. Gvozdjakova A. Deficit of coenzyme Q in heart and liver mitochondria of rats with streptozotocin-induced diabetes. Physiol Res. 2000;49:411–418. [PubMed] [Google Scholar]
- 181.Kukidome D. Nishikawa T. Sonoda K. Imoto K. Fujisawa K. Yano M. Motoshima H. Taguchi T. Matsumura T. Araki E. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55:120–127. [PubMed] [Google Scholar]
- 182.Kunitomo M. Yamaguchi Y. Kagota S. Otsubo K. Beneficial effect of coenzyme Q10 on increased oxidative and nitrative stress and inflammation and individual metabolic components developing in a rat model of metabolic syndrome. J Pharmacol Sci. 2008;107:128–137. doi: 10.1254/jphs.fp0072365. [DOI] [PubMed] [Google Scholar]
- 183.Lacza Z. Pankotai E. Busija DW. Mitochondrial nitric oxide synthase: current concepts and controversies. Front Biosci. 2009;14:4436–4443. doi: 10.2741/3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lagouge M. Argmann C. Gerhart-Hines Z. Meziane H. Lerin C. Daussin F. Messadeq N. Milne J. Lambert P. Elliott P. Geny B. Laakso M. Puigserver P. Auwerx J. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 185.Lambert AJ. Br MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 186.Lashin O. Romani A. Hyperglycemia does not alter state 3 respiration in cardiac mitochondria from type-I diabetic rats. Mol Cell Biochem. 2004;267:31–37. doi: 10.1023/b:mcbi.0000049360.75392.89. [DOI] [PubMed] [Google Scholar]
- 187.Lashin OM. Szweda PA. Szweda LI. Romani AM. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic Biol Med. 2006;40:886–896. doi: 10.1016/j.freeradbiomed.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 188.Laurindo FR. Fernandes DC. Santos CX. Assessment of superoxide production and NADPH oxidase activity by HPLC analysis of dihydroethidium oxidation products. Methods Enzymol. 2008;441:237–260. doi: 10.1016/S0076-6879(08)01213-5. [DOI] [PubMed] [Google Scholar]
- 189.Lee CK. Klopp RG. Weindruch R. Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 190.Lenzen S. Drinkgern J. Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- 191.Leone TC. Lehman JJ. Finck BN. Schaeffer PJ. Wende AR. Boudina S. Courtois M. Wozniak DF. Sambandam N. Bernal-Mizrachi C. Chen Z. Holloszy JO. Medeiros DM. Schmidt RE. Saffitz JE. Abel ED. Semenkovich CF. Kelly DP. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li M. Kim DH. Tsenovoy PL. Peterson SJ. Rezzani R. Rodella LF. Aronow WS. Ikehara S. Abraham NG. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526–1535. doi: 10.2337/db07-1764. [DOI] [PubMed] [Google Scholar]
- 193.Li M. Peterson S. Husney D. Inaba M. Guo K. Terada E. Morita T. Patil K. Kappas A. Ikehara S. Abraham NG. Interdiction of the diabetic state in NOD mice by sustained induction of heme oxygenase: possible role of carbon monoxide and bilirubin. Antioxid Redox Signal. 2007;9:855–863. doi: 10.1089/ars.2007.1568. [DOI] [PubMed] [Google Scholar]
- 194.Li N. Frigerio F. Maechler P. The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem Soc Trans. 2008;36:930–934. doi: 10.1042/BST0360930. [DOI] [PubMed] [Google Scholar]
- 195.Li X. May JM. Catalase-dependent measurement of H2O2 in intact mitochondria. Mitochondrion. 2002;1:447–453. doi: 10.1016/s1567-7249(02)00010-7. [DOI] [PubMed] [Google Scholar]
- 196.Li Z. Okamoto K. Hayashi Y. Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 197.Liang P. Hughes V. Fukagawa NK. Increased prevalence of mitochondrial DNA deletions in skeletal muscle of older individuals with impaired glucose tolerance: possible marker of glycemic stress. Diabetes. 1997;46:920–923. doi: 10.2337/diab.46.5.920. [DOI] [PubMed] [Google Scholar]
- 198.Lin J. Wu H. Tarr PT. Zhang CY. Wu Z. Boss O. Michael LF. Puigserver P. Isotani E. Olson EN. Lowell BB. Bassel-Duby R. Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 199.Lin J. Wu PH. Tarr PT. Lindenberg KS. St-Pierre J. Zhang CY. Mootha VK. Jager S. Vianna CR. Reznick RM. Cui L. Manieri M. Donovan MX. Wu Z. Cooper MP. Fan MC. Rohas LM. Zavacki AM. Cinti S. Shulman GI. Lowell BB. Krainc D. Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 200.Lindroos MM. Majamaa K. Tura A. Mari A. Kalliokoski KK. Taittonen MT. Iozzo P. Nuutila P. The m.3243A>G mutation in mitochondrial DNA leads to decreased insulin sensitivity in skeletal muscle and to progressive {beta}-cell dysfunction. Diabetes. 2008;58:543–549. doi: 10.2337/db08-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lonn E. Bosch J. Yusuf S. Sheridan P. Pogue J. Arnold JM. Ross C. Arnold A. Sleight P. Probstfield J. Dagenais GR. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 202.Lonn E. Yusuf S. Dzavik V. Doris C. Yi Q. Smith S. Moore-Cox A. Bosch J. Riley W. Teo K. Effects of ramipril and vitamin E on atherosclerosis: the study to evaluate carotid ultrasound changes in patients treated with ramipril and vitamin E (SECURE) Circulation. 2001;103:919–925. doi: 10.1161/01.cir.103.7.919. [DOI] [PubMed] [Google Scholar]
- 203.Lopez-Lluch G. Hunt N. Jones B. Zhu M. Jamieson H. Hilmer S. Cascajo MV. Allard J. Ingram DK. Navas P. de Cabo R. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lowell BB. Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 205.Maassen JA. LM TH. Van Essen E. Heine RJ. Nijpels G. Jahangir Tafrechi RS. Raap AK. Janssen GM. Lemkes HH. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53(suppl 1):S103–S109. doi: 10.2337/diabetes.53.2007.s103. [DOI] [PubMed] [Google Scholar]
- 206.Maassen JA. Hart LM. Janssen GM. Reiling E. Romijn JA. Lemkes HH. Mitochondrial diabetes and its lessons for common type 2 diabetes. Biochem Soc Trans. 2006;34:819–823. doi: 10.1042/BST0340819. [DOI] [PubMed] [Google Scholar]
- 207.Maguire JJ. Wilson DS. Packer L. Mitochondrial electron transport-linked tocopheroxyl radical reduction. J Biol Chem. 1989;264:21462–21465. [PubMed] [Google Scholar]
- 208.Manabe E. Handa O. Naito Y. Mizushima K. Akagiri S. Adachi S. Takagi T. Kokura S. Maoka T. Yoshikawa T. Astaxanthin protects mesangial cells from hyperglycemia-induced oxidative signaling. J Cell Biochem. 2008;103:1925–1937. doi: 10.1002/jcb.21583. [DOI] [PubMed] [Google Scholar]
- 209.Marinari UM. Monacelli R. Cottalasso D. Novelli A. Effects of alloxan diabetes and insulin on morphology and certain functional activities of mitochondria of the rat liver and heart. Acta Diabetol Lat. 1974;11:296–314. doi: 10.1007/BF02581234. [DOI] [PubMed] [Google Scholar]
- 210.Marra G. Cotroneo P. Pitocco D. Manto A. Di Leo MA. Ruotolo V. Caputo S. Giardina B. Ghirlanda G. Santini SA. Early increase of oxidative stress and reduced antioxidant defenses in patients with uncomplicated type 1 diabetes: a case for gender difference. Diabetes Care. 2002;25:370–375. doi: 10.2337/diacare.25.2.370. [DOI] [PubMed] [Google Scholar]
- 211.Matschinsky FM. Glaser B. Magnuson MA. Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes. 1998;47:307–315. doi: 10.2337/diabetes.47.3.307. [DOI] [PubMed] [Google Scholar]
- 212.May JM. Qu ZC. Li X. Ascorbic acid blunts oxidant stress due to menadione in endothelial cells. Arch Biochem Biophys. 2003;411:136–144. doi: 10.1016/s0003-9861(02)00715-4. [DOI] [PubMed] [Google Scholar]
- 213.Mayers RM. Leighton B. Kilgour E. PDH kinase inhibitors: a novel therapy for type II diabetes? Biochem Soc Trans. 2005;33:367–370. doi: 10.1042/BST0330367. [DOI] [PubMed] [Google Scholar]
- 214.Mehta K. Van Thiel DH. Shah N. Mobarhan S. Nonalcoholic fatty liver disease: pathogenesis and the role of antioxidants. Nutr Rev. 2002;60:289–293. doi: 10.1301/002966402320387224. [DOI] [PubMed] [Google Scholar]
- 215.Mensink M. Hesselink MK. Russell AP. Schaart G. Sels JP. Schrauwen P. Improved skeletal muscle oxidative enzyme activity and restoration of PGC-1 alpha and PPAR beta/delta gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int J Obes (Lond) 2007;31:1302–1310. doi: 10.1038/sj.ijo.0803567. [DOI] [PubMed] [Google Scholar]
- 216.Mercuri F. Quagliaro L. Ceriello A. Oxidative stress evaluation in diabetes. Diabetes Technol Ther. 2000;2:589–600. doi: 10.1089/15209150050502014. [DOI] [PubMed] [Google Scholar]
- 217.Mezzetti A. Cipollone F. Cuccurullo F. Oxidative stress and cardiovascular complications in diabetes: isoprostanes as new markers on an old paradigm. Cardiovasc Res. 2000;47:475–488. doi: 10.1016/s0008-6363(00)00118-8. [DOI] [PubMed] [Google Scholar]
- 218.Milne JC. Lambert PD. Schenk S. Carney DP. Smith JJ. Gagne DJ. Jin L. Boss O. Perni RB. Vu CB. Bemis JE. Xie R. Disch JS. Ng PY. Nunes JJ. Lynch AV. Yang H. Galonek H. Israelian K. Choy W. Iffland A. Lavu S. Medvedik O. Sinclair DA. Olefsky JM. Jirousek MR. Elliott PJ. Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 220.Modi K. Santani DD. Goyal RK. Bhatt PA. Effect of coenzyme Q10 on catalase activity and other antioxidant parameters in streptozotocin-induced diabetic rats. Biol Trace Elem Res. 2006;109:25–34. doi: 10.1385/BTER:109:1:025. [DOI] [PubMed] [Google Scholar]
- 221.Moore PC. Ugas MA. Hagman DK. Parazzoli SD. Poitout V. Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes. 2004;53:2610–2616. doi: 10.2337/diabetes.53.10.2610. [DOI] [PubMed] [Google Scholar]
- 222.Mootha VK. Lindgren CM. Eriksson KF. Subramanian A. Sihag S. Lehar J. Puigserver P. Carlsson E. Ridderstrale M. Laurila E. Houstis N. Daly MJ. Patterson N. Mesirov JP. Golub TR. Tamayo P. Spiegelman B. Lander ES. Hirschhorn JN. Altshuler D. Groop LC. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 223.Moreira PI. Rolo AP. Sena C. Seica R. Oliveira CR. Santos MS. Insulin attenuates diabetes-related mitochondrial alterations: a comparative study. Med Chem. 2006;2:299–308. doi: 10.2174/157340606776930754. [DOI] [PubMed] [Google Scholar]
- 224.Moreira PI. Santos MS. Sena C. Nunes E. Seica R. Oliveira CR. CoQ10 therapy attenuates amyloid beta-peptide toxicity in brain mitochondria isolated from aged diabetic rats. Exp Neurol. 2005;196:112–119. doi: 10.1016/j.expneurol.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 225.Morino K. Petersen KF. Dufour S. Befroy D. Frattini J. Shatzkes N. Neschen S. White MF. Bilz S. Sono S. Pypaert M. Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Morita T. Heme oxygenase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1786–1795. doi: 10.1161/01.ATV.0000178169.95781.49. [DOI] [PubMed] [Google Scholar]
- 227.Muniyappa R. Montagnani M. Koh KK. Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 228.Muoio DM. Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 229.Murad LB. Guimaraes MR. Vianna LM. Effects of decylubiquinone (coenzyme Q10 analog) supplementation on SHRSP. Biofactors. 2007;30:13–18. doi: 10.1002/biof.5520300102. [DOI] [PubMed] [Google Scholar]
- 230.Murphy MP. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997;15:326–330. doi: 10.1016/S0167-7799(97)01068-8. [DOI] [PubMed] [Google Scholar]
- 231.Mysore TB. Shinkel TA. Collins J. Salvaris EJ. Fisicaro N. Murray-Segal LJ. Johnson LE. Lepore DA. Walters SN. Stokes R. Chandra AP. O'Connell PJ. d'Apice AJ. Cowan PJ. Overexpression of glutathione peroxidase with two isoforms of superoxide dismutase protects mouse islets from oxidative injury and improves islet graft function. Diabetes. 2005;54:2109–2116. doi: 10.2337/diabetes.54.7.2109. [DOI] [PubMed] [Google Scholar]
- 232.Nassar T. Kadery B. Lotan C. Da'as N. Kleinman Y. Haj-Yehia A. Effects of the superoxide dismutase-mimetic compound tempol on endothelial dysfunction in streptozotocin-induced diabetic rats. Eur J Pharmacol. 2002;436:111–118. doi: 10.1016/s0014-2999(01)01566-7. [DOI] [PubMed] [Google Scholar]
- 233.Nicholls DG. A history of UCP1. Biochem Soc Trans. 2001;29:751–755. doi: 10.1042/bst0290751. [DOI] [PubMed] [Google Scholar]
- 234.Nicholls DG. Johnson-Cadwell L. Vesce S. Jekabsons M. Yadava N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res. 2007;85:3206–3212. doi: 10.1002/jnr.21290. [DOI] [PubMed] [Google Scholar]
- 235.Nicholls DG. Johnson-Cadwell L. Vesce S. Jekabsons M. Yadava N. Johnson-Cadwell LI. Jekabsons MB. Wang A. Polster BM. Nicholls DG. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity: “mild uncoupling” does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurosci Res. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- 236.Nishikawa T. Edelstein D. Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int Suppl. 2000;77:S26–S30. doi: 10.1046/j.1523-1755.2000.07705.x. [DOI] [PubMed] [Google Scholar]
- 237.Nishikawa T. Edelstein D. Du XL. Yamagishi S. Matsumura T. Kaneda Y. Yorek MA. Beebe D. Oates PJ. Hammes HP. Giardino I. Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 238.Nisoli E. Carruba MO. Nitric oxide and mitochondrial biogenesis. J Cell Sci. 2006;119:2855–2862. doi: 10.1242/jcs.03062. [DOI] [PubMed] [Google Scholar]
- 239.Nisoli E. Clementi E. Paolucci C. Cozzi V. Tonello C. Sciorati C. Bracale R. Valerio A. Francolini M. Moncada S. Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 240.Novials A. Vidal J. Franco C. Ribera F. Sener A. Malaisse WJ. Gomis R. Mutation in the calcium-binding domain of the mitochondrial glycerophosphate dehydrogenase gene in a family of diabetic subjects. Biochem Biophys Res Commun. 1997;231:570–572. doi: 10.1006/bbrc.1997.6147. [DOI] [PubMed] [Google Scholar]
- 241.O'Malley Y. Fink BD. Ross NC. Prisinzano TE. Sivitz WI. Reactive oxygen and targeted antioxidant administration in endothelial cell mitochondria. J Biol Chem. 2006;281:39766–39775. doi: 10.1074/jbc.M608268200. [DOI] [PubMed] [Google Scholar]
- 242.Obrosova IG. Drel VR. Kumagai AK. Szabo C. Pacher P. Stevens MJ. Early diabetes-induced biochemical changes in the retina: comparison of rat and mouse models. Diabetologia. 2006;49:2525–2533. doi: 10.1007/s00125-006-0356-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Obrosova IG. Stevens MJ. Lang HJ. Diabetes-induced changes in retinal NAD-redox status: pharmacological modulation and implications for pathogenesis of diabetic retinopathy. Pharmacology. 2001;62:172–180. doi: 10.1159/000056091. [DOI] [PubMed] [Google Scholar]
- 244.Oliveira PJ. Seica R. Coxito PM. Rolo AP. Palmeira CM. Santos MS. Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003;554:511–514. doi: 10.1016/s0014-5793(03)01233-x. [DOI] [PubMed] [Google Scholar]
- 245.Oprescu AI. Bikopoulos G. Naassan A. Allister EM. Tang C. Park E. Uchino H. Lewis GF. Fantus IG. Rozakis-Adcock M. Wheeler MB. Giacca A. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56:2927–2937. doi: 10.2337/db07-0075. [DOI] [PubMed] [Google Scholar]
- 246.Padmaja S. Huie RE. The reaction of nitric oxide with organic peroxyl radicals. Biochem Biophys Res Commun. 1993;195:539–544. doi: 10.1006/bbrc.1993.2079. [DOI] [PubMed] [Google Scholar]
- 247.Patti ME. Butte AJ. Crunkhorn S. Cusi K. Berria R. Kashyap S. Miyazaki Y. Kohane I. Costello M. Saccone R. Landaker EJ. Goldfine AB. Mun E. DeFronzo R. Finlayson J. Kahn CR. Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Pearson KJ. Baur JA. Lewis KN. Peshkin L. Price NL. Labinskyy N. Swindell WR. Kamara D. Minor RK. Perez E. Jamieson HA. Zhang Y. Dunn SR. Sharma K. Pleshko N. Woollett LA. Csiszar A. Ikeno Y. Le Couteur D. Elliott PJ. Becker KG. Navas P. Ingram DK. Wolf NS. Ungvari Z. Sinclair DA. de Cabo R. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Perseghin G. Ghosh S. Gerow K. Shulman GI. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46:1001–1009. doi: 10.2337/diab.46.6.1001. [DOI] [PubMed] [Google Scholar]
- 250.Pessin JE. Richardson JM. Sivitz WI. Regulation of the glucose transporter in animal models of diabetes. Adv Exp Med Biol. 1991;293:249–262. doi: 10.1007/978-1-4684-5949-4_23. [DOI] [PubMed] [Google Scholar]
- 251.Pessin JE. Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Petersen KF. Befroy D. Dufour S. Dziura J. Ariyan C. Rothman DL. DiPietro L. Cline GW. Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Petersen KF. Dufour S. Befroy D. Garcia R. Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. [see comment] N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Petersen KF. Dufour S. Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Petrat F. Pindiur S. Kirsch M. de Groot H. NAD(P)H, a primary target of 1O2 in mitochondria of intact cells. J Biol Chem. 2003;278:3298–3307. doi: 10.1074/jbc.M204230200. [DOI] [PubMed] [Google Scholar]
- 256.Petri S. Kiaei M. Damiano M. Hiller A. Wille E. Manfredi G. Calingasan NY. Szeto HH. Beal MF. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98:1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- 257.Pfeiffer DR. Gunter TE. Eliseev R. Broekemeier KM. Gunter KK. Release of Ca2+ from mitochondria via the saturable mechanisms and the permeability transition. IUBMB Life. 2001;52:205–212. doi: 10.1080/15216540152846019. [DOI] [PubMed] [Google Scholar]
- 258.Piconi L. Quagliaro L. Ceriello A. Oxidative stress in diabetes. Clin Chem Lab Med. 2003;41:1144–1149. doi: 10.1515/CCLM.2003.177. [DOI] [PubMed] [Google Scholar]
- 259.Porter RK. Br MD. Mitochondrial proton conductance and H+/O ratio are independent of electron transport rate in isolated hepatocytes. Biochem J. 1995;310:379–382. doi: 10.1042/bj3100379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Pospisilik JA. Knauf C. Joza N. Benit P. Orthofer M. Cani PD. Ebersberger I. Nakashima T. Sarao R. Neely G. Esterbauer H. Kozlov A. Kahn CR. Kroemer G. Rustin P. Burcelin R. Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 261.Radi R. Rodriguez M. Castro L. Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- 262.Radi R. Turrens JF. Chang LY. Bush KM. Crapo JD. Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991;266:22028–22034. [PubMed] [Google Scholar]
- 263.Raha S. Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:222–230. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 264.Rauscher FM. Sanders RA. Watkins JB., 3rd Effects of coenzyme Q10 treatment on antioxidant pathways in normal and streptozotocin-induced diabetic rats. J Biochem Mol Toxicol. 2001;15:41–46. doi: 10.1002/1099-0461(2001)15:1<41::aid-jbt5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 265.Redman LM. Ravussin E. Endocrine alterations in response to calorie restriction in humans. Mol Cell Endocrinol. 2008;299:129–136. doi: 10.1016/j.mce.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Reznick RM. Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–39. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rijzewijk LJ. van der Meer RW. Smit JW. Diamant M. Bax JJ. Hammer S. Romijn JA. de Roos A. Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52:1793–1799. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 268.Ritov VB. Menshikova EV. He J. Ferrell RE. Goodpaster BH. Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 269.Rockl KS. Witczak CA. Goodyear LJ. Signaling mechanisms in skeletal muscle: acute responses and chronic adaptations to exercise. IUBMB Life. 2008;60:145–153. doi: 10.1002/iub.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Roden M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol Sci. 2004;19:92–96. doi: 10.1152/nips.01459.2003. [DOI] [PubMed] [Google Scholar]
- 271.Rodgers JT. Lerin C. Gerhart-Hines Z. Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Rodgers JT. Lerin C. Haas W. Gygi SP. Spiegelman BM. Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 273.Rodgers JT. Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Rosca MG. Mustata TG. Kinter MT. Ozdemir AM. Kern TS. Szweda LI. Brownlee M. Monnier VM. Weiss MF. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol. 2005;289:F420–F430. doi: 10.1152/ajprenal.00415.2004. [DOI] [PubMed] [Google Scholar]
- 275.Rosen P. Wiernsperger NF. Metformin delays the manifestation of diabetes and vascular dysfunction in Goto-Kakizaki rats by reduction of mitochondrial oxidative stress. Diabetes Metab Res Rev. 2006;22:323–330. doi: 10.1002/dmrr.623. [DOI] [PubMed] [Google Scholar]
- 276.Ruderman NB. Saha AK. Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology. 2003;144:5166–5171. doi: 10.1210/en.2003-0849. [DOI] [PubMed] [Google Scholar]
- 277.Russell AP. Gastaldi G. Bobbioni-Harsch E. Arboit P. Gobelet C. Deriaz O. Golay A. Witztum JL. Giacobino JP. Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: a case of good vs. bad lipids? FEBS Lett. 2003;551:104–106. doi: 10.1016/s0014-5793(03)00875-5. [DOI] [PubMed] [Google Scholar]
- 278.Saito M. Okamatsu-Ogura Y. Matsushita M. Watanabe K. Yoneshiro T. Nio-Kobayashi J. Iwanaga T. Miyagawa M. Kameya T. Nakada K. Kawai Y. Tsujisaki M. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Sakurai K. Katoh M. Someno K. Fujimoto Y. Apoptosis and mitochondrial damage in INS-1 cells treated with alloxan. Biol Pharm Bull. 2001;24:876–882. doi: 10.1248/bpb.24.876. [DOI] [PubMed] [Google Scholar]
- 280.Salvi M. Battaglia V. Brunati AM. La Rocca N. Tibaldi E. Pietrangeli P. Marcocci L. Mondovi B. Rossi CA. Toninello A. Catalase takes part in rat liver mitochondria oxidative stress defense. J Biol Chem. 2007;282:24407–24415. doi: 10.1074/jbc.M701589200. [DOI] [PubMed] [Google Scholar]
- 281.Saretzki G. Murphy MP. von Zglinicki T. MitoQ counteracts telomere shortening and elongates lifespan of fibroblasts under mild oxidative stress. Aging Cell. 2003;2:141–143. doi: 10.1046/j.1474-9728.2003.00040.x. [DOI] [PubMed] [Google Scholar]
- 282.Sazanov LA. Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry. 2007;46:2275–2288. doi: 10.1021/bi602508x. [DOI] [PubMed] [Google Scholar]
- 283.Schafer M. Schafer C. Ewald N. Piper HM. Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ Res. 2003;92:1010–1015. doi: 10.1161/01.RES.0000070882.81508.FC. [DOI] [PubMed] [Google Scholar]
- 284.Scheffler I. Mitochondria. New York: Wiley-Liss; 1999. Mitochondrial electron transport and oxidative phosphorylation; pp. 141–245. [Google Scholar]
- 285.Schiller PW. Nguyen TM. Berezowska I. Dupuis S. Weltrowska G. Chung NN. Lemieux C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur J Med Chem. 2000;35:895–901. doi: 10.1016/s0223-5234(00)01171-5. [DOI] [PubMed] [Google Scholar]
- 286.Schrauwen P. High-fat diet, muscular lipotoxicity and insulin resistance. Proc Nutr Soc. 2007;66:33–41. doi: 10.1017/S0029665107005277. [DOI] [PubMed] [Google Scholar]
- 287.Schrauwen P. Hesselink MK. Blaak EE. Borghouts LB. Schaart G. Saris WH. Keizer HA. Uncoupling protein 3 content is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2001;50:2870–2873. doi: 10.2337/diabetes.50.12.2870. [DOI] [PubMed] [Google Scholar]
- 288.Schrauwen P. Hesselink MC. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes. 2004;53:1412–1417. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- 289.Seifert EL. Bezaire V. Estey C. Harper ME. Essential role for uncoupling protein-3 in mitochondrial adaptation to fasting but not in fatty acid oxidation or fatty acid anion export. J Biol Chem. 2008;283:25124–25131. doi: 10.1074/jbc.M803871200. [DOI] [PubMed] [Google Scholar]
- 290.Sekine N. Cirulli V. Regazzi R. Brown LJ. Gine E. Tamarit-Rodriguez J. Girotti M. Marie S. MacDonald MJ. Wollheim CB, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells: potential role in nutrient sensing. J Biol Chem. 1994;269:4895–4902. [PubMed] [Google Scholar]
- 291.Shimabukuro M. Zhou YT. Lee Y. Unger RH. Troglitazone lowers islet fat and restores beta cell function of Zucker diabetic fatty rats. J Biol Chem. 1998;273:3547–3550. doi: 10.1074/jbc.273.6.3547. [DOI] [PubMed] [Google Scholar]
- 292.Sivitz WI. Wayson SM. Bayless ML. Sinkey CA. Haynes WG. Obesity impairs vascular relaxation in human subjects: hyperglycemia exaggerates adrenergic vasoconstriction arterial dysfunction in obesity and diabetes. J Diabetes Complications. 2007;21:149–157. doi: 10.1016/j.jdiacomp.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 293.Skov V. Glintborg D. Knudsen S. Jensen T. Kruse TA. Tan Q. Brusgaard K. Beck-Nielsen H. Hojlund K. Reduced expression of nuclear-encoded genes involved in mitochondrial oxidative metabolism in skeletal muscle of insulin-resistant women with polycystic ovary syndrome. Diabetes. 2007;56:2349–2355. doi: 10.2337/db07-0275. [DOI] [PubMed] [Google Scholar]
- 294.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- 295.Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta. 1998;1363:100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- 296.Smith AR. Visioli F. Hagen TM. Vitamin C matters: increased oxidative stress in cultured human aortic endothelial cells without supplemental ascorbic acid. FASEB J. 2002;16:NIL_125–NIL_144. doi: 10.1096/fj.01-0825fje. [DOI] [PubMed] [Google Scholar]
- 297.Sohal RS. Forster MJ. Coenzyme Q, oxidative stress and aging. Mitochondrion. 2007;7(suppl):S103–S111. doi: 10.1016/j.mito.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Sparks LM. Xie H. Koza RA. Mynatt R. Hulver MW. Bray GA. Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 299.St-Pierre J. Buckingham JA. Roebuck SJ. Br MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;4277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 300.Stuart JA. Cadenas S. Jekabsons MB. Roussel D. Br MD. Mitochondrial proton leak and the uncoupling protein 1 homologues. Biochim Biophys Acta. 2001;1504:144–158. doi: 10.1016/s0005-2728(00)00243-7. [DOI] [PubMed] [Google Scholar]
- 301.Stump CS. Short KR. Bigelow ML. Schimke JM. Nair KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci U S A. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Suzuki S. Hinokio Y. Ohtomo M. Hirai M. Hirai A. Chiba M. Kasuga S. Satoh Y. Akai H. Toyota T. The effects of coenzyme Q10 treatment on maternally inherited diabetes mellitus and deafness, and mitochondrial DNA 3243 (A to G) mutation. Diabetologia. 1998;41:584–588. doi: 10.1007/s001250050950. [DOI] [PubMed] [Google Scholar]
- 303.Szendroedi J. Schmid AI. Chmelik M. Toth C. Brehm A. Krssak M. Nowotny P. Wolzt M. Waldhausl W. Roden M. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007;4:e154. doi: 10.1371/journal.pmed.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Szeto HH. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann N Y Acad Sci. 2008;1147:112–121. doi: 10.1196/annals.1427.013. [DOI] [PubMed] [Google Scholar]
- 305.Takahashi Y. Iida K. Takeno R. Kitazawa R. Kitazawa S. Kitamura H. Fujioka Y. Yamada H. Kanda F. Ohta S. Nishimaki K. Fujimoto M. Kondo T. Iguchi G. Takahashi K. Kaji H. Okimura Y. Chihara K. Hepatic failure and enhanced oxidative stress in mitochondrial diabetes. Endocrinol J. 2008;55:509–514. doi: 10.1507/endocrj.k07e-091. [DOI] [PubMed] [Google Scholar]
- 306.Talbot DA. Lambert AJ. Br MD. Production of endogenous matrix superoxide from mitochondrial complex I leads to activation of uncoupling protein 3. FEBS Lett. 2004;556:111–115. doi: 10.1016/s0014-5793(03)01386-3. [DOI] [PubMed] [Google Scholar]
- 307.Tang C. Han P. Oprescu AI. Lee SC. Gyulkhandanyan AV. Chan GN. Wheeler MB. Giacca A. Evidence for a role of superoxide generation in glucose-induced beta-cell dysfunction in vivo. Diabetes. 2007;56:2722–2731. doi: 10.2337/db07-0279. [DOI] [PubMed] [Google Scholar]
- 308.Thiemermann C. Membrane-permeable radical scavengers (tempol) for shock, ischemia-reperfusion injury, and inflammation. Crit Care Med. 2003;31:S76–S84. doi: 10.1097/00003246-200301001-00011. [DOI] [PubMed] [Google Scholar]
- 309.Thomas DA. Stauffer C. Zhao K. Yang H. Sharma VK. Szeto HH. Suthanthiran M. Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J Am Soc Nephrol. 2007;18:213–222. doi: 10.1681/ASN.2006080825. [DOI] [PubMed] [Google Scholar]
- 310.Thomas SM. Gebicki JM. Dean RT. Radical initiated alpha-tocopherol depletion and lipid peroxidation in mitochondrial membranes. Biochim Biophys Acta. 1989;1002:189–197. doi: 10.1016/0005-2760(89)90286-5. [DOI] [PubMed] [Google Scholar]
- 311.Traverso N. Menini S. Odetti P. Pronzato MA. Cottalasso D. Marinari UM. Diabetes impairs the enzymatic disposal of 4-hydroxynonenal in rat liver. Free Radic Biol Med. 2002;32:350–359. doi: 10.1016/s0891-5849(01)00811-5. [DOI] [PubMed] [Google Scholar]
- 312.Turk J. Corbett JA. Ramanadham S. Bohrer A. McDaniel ML. Biochemical evidence for nitric oxide formation from streptozotocin in isolated pancreatic islets. Biochem Biophys Res Commun. 1993;197:1458–1464. doi: 10.1006/bbrc.1993.2641. [DOI] [PubMed] [Google Scholar]
- 313.Turko IV. Murad F. Quantitative protein profiling in heart mitochondria from diabetic rats. J Biol Chem. 2003;278:35844–35849. doi: 10.1074/jbc.M303139200. [DOI] [PubMed] [Google Scholar]
- 314.Turkseven S. Kruger A. Mingone CJ. Kaminski P. Inaba M. Rodella LF. Ikehara S. Wolin MS. Abraham NG. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am J Physiol Heart Circ Physiol. 2005;289:H701–H707. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- 315.Turunen M. Olsson J. Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta. 2004;1660:171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 316.Vahsen N. Cande C. Briere JJ. Benit P. Joza N. Larochette N. Mastroberardino PG. Pequignot MO. Casares N. Lazar V. Feraud O. Debili N. Wissing S. Engelhardt S. Madeo F. Piacentini M. Penninger JM. Schagger H. Rustin P. Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.van der Meer RW. Rijzewijk LJ. de Jong HW. Lamb HJ. Lubberink M. Romijn JA. Bax JJ. de Roos A. Kamp O. Paulus WJ. Heine RJ. Lammertsma AA. Smit JW. Diamant M. Pioglitazone improves cardiac function and alters myocardial substrate metabolism without affecting cardiac triglyceride accumulation and high-energy phosphate metabolism in patients with well-controlled type 2 diabetes mellitus. Circulation. 2009;119:2069–2077. doi: 10.1161/CIRCULATIONAHA.108.803916. [DOI] [PubMed] [Google Scholar]
- 318.van Marken Lichtenbelt WD. Vanhommerig JW. Smulders NM. Drossaerts JM. Kemerink GJ. Bouvy ND. Schrauwen P. Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 319.Vasquez-Vivar J. Kalyanaraman B. Kennedy MC. Mitochondrial aconitase is a source of hydroxyl radical: an electron spin resonance investigation. J Biol Chem. 2000;284:19843–19855. doi: 10.1074/jbc.275.19.14064. 275: 14064–14069. [DOI] [PubMed] [Google Scholar]
- 320.Venkatakrishnan P. Nakayasu ES. Almeida IC. Miller RT. Absence of nitric oxide synthase in sequentially purified rat liver mitochondria. J Biol Chem. 2009;284:19843–19855. doi: 10.1074/jbc.M109.003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Vercesi AE. Kowaltowski AJ. Grijalba MT. Meinicke AR. Castilho RF. The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep. 1997;17:43–52. doi: 10.1023/a:1027335217774. [DOI] [PubMed] [Google Scholar]
- 322.Vester JW. Stadie WC. Studies of oxidative phosphorylation by hepatic mitochondria from the diabetic cat. J Biol Chem. 1957;227:669–676. [PubMed] [Google Scholar]
- 323.Virtanen KA. Lidell ME. Orava J. Heglind M. Westergren R. Niemi T. Taittonen M. Laine J. Savisto NJ. Enerback S. Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 324.Votyakova TV. Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 325.Walder K. Kerr-Bayles L. Civitarese A. Jowett J. Curran J. Elliott K. Trevaskis J. Bishara N. Zimmet P. Mandarino L. Ravussin E. Blangero J. Kissebah A. Collier GR. The mitochondrial rhomboid protease PSARL is a new candidate gene for type 2 diabetes. Diabetologia. 2005;48:459–468. doi: 10.1007/s00125-005-1675-9. [DOI] [PubMed] [Google Scholar]
- 326.Wang MY. Shimabukuro M. Lee Y. Trinh KY. Chen JL. Newgard CB. Unger RH. Adenovirus-mediated overexpression of uncoupling protein-2 in pancreatic islets of Zucker diabetic rats increases oxidative activity and improves beta-cell function. Diabetes. 1999;48:1020–1025. doi: 10.2337/diabetes.48.5.1020. [DOI] [PubMed] [Google Scholar]
- 327.Wiederkehr A. Wollheim CB. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta-cell. Cell Calcium. 2008;44:64–76. doi: 10.1016/j.ceca.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 328.Williamson JR. Chang K. Frangos M. Hasan KS. Ido Y. Kawamura T. Nyengaard JR. van den Enden M. Kilo C. Tilton RG. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–813. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- 329.Winder WW. Holmes BF. Rubink DS. Jensen EB. Chen M. Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- 330.Wolfrum C. Asilmaz E. Luca E. Friedman JM. Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004;432:1027–1032. doi: 10.1038/nature03047. [DOI] [PubMed] [Google Scholar]
- 331.Wood JG. Rogina B. Lavu S. Howitz K. Helfand SL. Tatar M. Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 332.Wredenberg A. Freyer C. Sandstrom ME. Katz A. Wibom R. Westerblad H. Larsson NG. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun. 2006;350:202–207. doi: 10.1016/j.bbrc.2006.09.029. [DOI] [PubMed] [Google Scholar]
- 333.Wredenberg A. Wibom R. Wilhelmsson H. Graff C. Wiener HH. Burden SJ. Oldfors A. Westerblad H. Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Wu Z. Puigserver P. Andersson U. Zhang C. Adelmant G. Mootha V. Troy A. Cinti S. Lowell B. Scarpulla RC. Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 335.Yamagishi SI. Edelstein D. Du XL. Kaneda Y. Guzman M. Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;276:25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- 336.Yang S. Zhu H. Li Y. Lin H. Gabrielson K. Trush MA. Diehl AM. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys. 2000;378:259–268. doi: 10.1006/abbi.2000.1829. [DOI] [PubMed] [Google Scholar]
- 337.Yang X. Smith U. Adipose tissue distribution and risk of metabolic disease: does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia. 2007;50:1127–1139. doi: 10.1007/s00125-007-0640-1. [DOI] [PubMed] [Google Scholar]
- 338.Ye G. Metreveli NS. Donthi RV. Xia S. Xu M. Carlson EC. Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes. 2004;53:1336–1343. doi: 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- 339.Yi X. Maeda N. alpha-Lipoic acid prevents the increase in atherosclerosis induced by diabetes in apolipoprotein E-deficient mice fed high-fat/low-cholesterol diet. Diabetes. 2006;55:2238–2244. doi: 10.2337/db06-0251. [DOI] [PubMed] [Google Scholar]
- 340.Yokota T. Ma RC. Park JY. Isshiki K. Sotiropoulos KB. Rauniyar RK. Bornfeldt KE. King GL. Role of protein kinase C on the expression of platelet-derived growth factor and endothelin-1 in the retina of diabetic rats and cultured retinal capillary pericytes. Diabetes. 2003;52:838–845. doi: 10.2337/diabetes.52.3.838. [DOI] [PubMed] [Google Scholar]
- 341.Yorek MA. The role of oxidative stress in diabetic vascular and neural disease. Free Radic Res. 2003;37:471–480. doi: 10.1080/1071576031000083161. [DOI] [PubMed] [Google Scholar]
- 342.Yorek MA. Coppey LJ. Gellett JS. Davidson EP. Bing X. Lund DD. Dillon JS. Effect of treatment of diabetic rats with dehydroepiandrosterone on vascular and neural function. Am J Physiol Endocrinol Metab. 2002;283:E1067–E1075. doi: 10.1152/ajpendo.00173.2002. [DOI] [PubMed] [Google Scholar]
- 343.Yorek MA. Coppey LJ. Gellett JS. Davidson EP. Lund DD. Effect of fidarestat and alpha-lipoic acid on diabetes-induced epineurial arteriole vascular dysfunction. Exp Diabesity Res. 2004;5:123–135. doi: 10.1080/15438600490277824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Yu XX. Barger JL. Boyer BB. Brand MD. Pan G. Adams SH. Impact of endotoxin on UCP homolog mRNA abundance, thermoregulation, and mitochondrial proton leak kinetics. Am J Physiol Endocrinol Metab. 2000;279:E433–E446. doi: 10.1152/ajpendo.2000.279.2.E433. [DOI] [PubMed] [Google Scholar]
- 345.Yusuf S. Dagenais G. Pogue J. Bosch J. Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients: the Heart Outcomes Prevention Evaluation Study investigators. N Engl J Med. 2000;342:154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 346.Zhang CY. Baffy G. Perret P. Krauss S. Peroni O. Grujic D. Hagen T. Vidal-Puig AJ. Boss O. Kim YB. Zheng XX. Wheeler MB. Shulman GI. Chan CB. Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes [comment] Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- 347.Zhao GM. Qian X. Schiller PW. Szeto HH. Comparison of [Dmt1]DALDA and DAMGO in binding and G protein activation at μ, d, and κ opioid receptors. J Pharmacol Exp Ther. 2003;307:947–954. doi: 10.1124/jpet.103.054775. [DOI] [PubMed] [Google Scholar]
- 348.Zhao K. Luo G. Zhao GM. Schiller PW. Szeto HH. Transcellular transport of a highly polar 3+ net charge opioid tetrapeptide. J Pharmacol Exp Ther. 2003;304:425–432. doi: 10.1124/jpet.102.040147. [DOI] [PubMed] [Google Scholar]
- 349.Zhao K. Zhao GM. Wu D. Soong Y. Birk AV. Schiller PW. Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 350.Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE. 2005;2005:pe4. doi: 10.1126/stke.2682005pe4. [DOI] [PubMed] [Google Scholar]
- 351.Ziegler D. Ametov A. Barinov A. Dyck PJ. Gurieva I. Low PA. Munzel U. Yakhno N. Raz I. Novosadova M. Maus J. Samigullin R. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care. 2006;29:2365–2370. doi: 10.2337/dc06-1216. [DOI] [PubMed] [Google Scholar]
- 352.Ziegler D. Hanefeld M. Ruhnau KJ. Meissner HP. Lobisch M. Schutte K. Gries FA. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid: a 3-week multicentre randomized controlled trial (ALADIN Study) Diabetologia. 1995;38:1425–1433. doi: 10.1007/BF00400603. [DOI] [PubMed] [Google Scholar]
- 353.Zou MH. Kirkpatrick SS. Davis BJ. Nelson JS. Wiles WGt. Schlattner U. Neumann D. Brownlee M. Freeman MB. Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo: role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–43951. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]