

Abstract
RSK2, an ERK downstream kinase, is a novel mediator of skeletal muscle cell differentiation through its regulation of NFAT3 activity. We found that the N-terminal (amino acids (aa) 1–68) and C-terminal (aa 416–674) kinase domains of RSK2 directly interacted with nuclear localization signal 1, the Ser/Pro repeat, and the polyproline domains (aa 261–365) of NFAT3. Upon A23187 stimulation, RSK2 induced nuclear localization of NFAT3. RSK2 phosphorylated NFAT3 in vitro (Km = 3.559 μm), and activation of NFAT3 by RSK2 enhanced the promoter activity of NFAT3 downstream target genes in vivo. Furthermore, nuclear accumulation of NFAT3 was attenuated markedly in RSK2−/− cells compared with wild-type RSK2+/+ cells. Notably, RSK2 and NFAT3 induced a significant differentiation of C2C12 myoblasts to multinucleated myotubes. Multinucleated myotube differentiation was inhibited by small interfering RNA against RSK2, ERK1/2, or NFAT3. These results demonstrate that RSK2 is an important kinase for NFAT3 in mediating myotube differentiation.
Myoblast cell differentiation to muscle fibers depends on myogenic transcription factors, particularly myogenin (1). Many studies addressing the involvement of the mitogen-activated protein kinase (MAPK)2 pathways are controversial because results showed both positive and negative effects of this signaling pathway on myogenesis. The MAPK cascades are implicated in the regulation of cell proliferation, survival, growth, and motility (2, 3) as well as tumorigenesis (4). Extracellular signal-regulated kinase (ERK)-1 and ERK2 (5, 6) mediate the 90-kDa ribosomal S6 kinases (RSKs), which are a family of broadly expressed serine/threonine kinases that respond to many growth factors, peptide hormones, and neurotransmitters (7, 8). When activated, RSK2 translocates to the nucleus, where it can phosphorylate various nuclear proteins, including c-Fos, Elk-1, histones, and cAMP-responsive element-binding protein (CREB) (9-13); activating transcription factor-4 (14); and p53 (15). Moreover, because RSKs have broad substrate specificity, RSK2 may be able to bind and/or phosphorylate a number of diverse substrates that regulate cell proliferation or differentiation or the cell cycle, depending on the specific situation.
The nuclear factor of activated T cell (NFAT) family of transcription factors has been characterized primarily in immune cells (16). However, accumulating evidence has demonstrated that NFAT transcription factors are present in a wide range of cell types and tissues (17–22). NFAT3 and NFAT4 have an ∼48% amino acid homology in the whole protein and exhibit an especially high degree of similarity (i.e. ∼80%) in the Rel homology domain, which contains the DNA-binding motif. Moreover, the N-terminal domains (amino acids (aa) 1–360) of NFAT3 and NFAT4 have only a 30% amino acid similarity. Therefore, we proposed that the N-terminal domains of NFAT3 and NFAT4 might bind to different proteins, which might also phosphorylate key residues. c-Jun N-terminal kinase (JNK)-2 phosphorylates NFAT4, but not NFAT3, at serines 163 and 165, which correspond to serines 168 and 170 of NFAT3 (23). The phosphorylation of NFAT4 at serines 163 and 165 by JNK2 inhibits nuclear localization, and mutation of these serines to alanine induces nuclear localization (24). Moreover, p38 was reported to phosphorylate serines 168 and 170 of NFAT3 and shows the same phenomenon for subcellular distribution as described for NFAT4 (25). However, in experiments with dominant-negative Ras, c-Raf, or ERK2 or chemical inhibitors of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK)-1, the Ras-regulated pathway was shown to stimulate NFAT activity (26). Furthermore, the RSK2-NFAT3 complex was shown to induce transcriptional activity for adipogenesis by forming a transcription activation complex (27). Taken together, these results strongly suggest that the ERK-RSK2 signaling pathway might be involved in the positive regulation of NFAT3 activity.
In this study, we used the mammalian two-hybrid system to identify novel binding protein(s) of RSK2 and found that NFAT3 interacts very strongly with RSK2. We further demonstrated that RSK2 phosphorylates multiple serine residues of NFAT3, resulting in NFAT3 activation and nuclear localization. Notably, cotransfection of RSK2 and NFAT3 markedly enhanced multinucleated myotube differentiation of C2C12 myoblasts. In addition, RSK2 mutation or knockdown dramatically decreased NFAT3 activity and promoter activity of NFAT3 target genes as well as myotube differentiation. These results suggest that NFAT3 is critical for myotube differentiation. Moreover, RSK2 is shown to be a key protein kinase that phosphorylates NFAT3, which is critical in the process of differentiation.
Experimental Procedures
Reagents and Antibodies
Some antibodies for immunoblotting and immunoprecipitation analysis and Tris, NaCl, and SDS for molecular biology and buffer preparation were purchased from Sigma. Some antibodies were also obtained from BD Biosciences or Upstate Biotechnology, Inc. (Charlottesville, VA). Restriction enzymes and some modifying enzymes were acquired from Roche Diagnostics. Cell culture media, other supplements, and SuperScript II RNase H− reverse transcriptase was from Invitrogen, and Taq DNA polymerase was from Qiagen Inc. (Valencia, CA). The DNA ligation kit (Version 2.0) was from Takara Bio, Inc. (Otsu, Shiga, Japan). The Checkmate mammalian two-hybrid system, including expression vectors and the reporter luciferase vector, was obtained from Promega Corp. (Madison, WI).
Cell Culture and Transfections
293, RSK2+/+, and RSK2−/− mouse embryonic fibroblast (MEF) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C in a 5% CO2 incubator. C2C12 myoblasts were cultured in DMEM supplemented with 10% FBS and 4 mm l-glutamine. The cells were maintained by splitting at 80–90% confluence, and media were changed every 3 days. When cells reached 50% confluence, transfection of the expression vectors was performed using jetPEI (Qbiogene, Inc., Montreal, Quebec, Canada) following the manufacturer's suggested protocol.
Reverse Transcription-PCR Amplification of Transcription Factors and RSK2
The transcription factor (pACT-TF), including NFAT3 and other known substrates of RSK2, and RSK2 (pBIND-RSK2FL) cDNAs were cloned by PCR-based amplification as described previously (15). The pACT-TF and pBIND-RSK2FL constructs were confirmed by restriction mapping and DNA sequencing.
Construction of Deletion Mutants for RSK2 and NFAT3
Deletion mutants of RSK2 were constructed as described previously (15). Wild-type (wt) NFAT3 and deletion mutants were generous gifts from Dr. C. W. Chow (Department of Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY) (25). To construct glutathione S-transferase (GST)-NFAT3-(261-365), the cDNA fragment was amplified with sense primer 5′-GCA GGA TCC TGC GGC CTG CCT CTC CAT GTG GCA-3′ (with the BamHI site underlined) and antisense primer 5′-GCT CTA GAG GAG GAG CCA CAG ACT CCT CTG-3′ (with the XbaI site underlined). The cDNA fragment was introduced into the BamHI/XbaI sites of pGEX-5X-C (28) and was designated pGST-wtNFAT3D4. All constructs were confirmed by restriction mapping and DNA sequencing.
Construction of the Green Fluorescent Protein (GFP) Fusion Protein
The GFP fusion proteins for RSK2 and NFAT3 were constructed with PCR-amplified open reading frame DNA fragments and the pEGFP-C1 vector (BD Biosciences). GFP-fused NFAT3 deletion mutants were also constructed by PCR-based amplification using specific primers. The reading frame of each plasmid construct was confirmed by restriction mapping and DNA sequencing.
Mammalian Two-hybrid Assay
To screen for protein-binding partners, we used the mammalian two-hybrid assay following the Promega Checkmate mammalian two-hybrid system protocols. 293 cells were maintained in 10% FBS-containing DMEM, seeded into 48-well plates (2.0 × 104), and finally incubated with 10% FBS-containing DMEM for 18 h before transfection. The pACT-wtNFAT3, pBIND-RSK2, and pG5-luciferase DNAs were combined at the same molar ratio (1:1:1), and the total amount of DNA was not more than 100 ng/well. Transfections were performed using jetPEI following the manufacturer's recommended protocols. For the luciferase assay, the cells were disrupted by the addition of lysis buffer and gentle shaking for 30 min at room temperature. Following the addition of 100 μl of substrate buffer, luminescence was measured automatically using a computer-programmed Luminoskan Ascent machine (MTX Lab Systems, Inc., Vienna, VA). The relative luciferase activity was calculated using the pG5-luciferase basal control and normalized against Renilla luciferase activity, which included the pBIND vector.
In Vitro Kination Assay
GST-wtNFAT3D4 and point mutant proteins were used for the in vitro kination assay using active RSK2 (Upstate Biotechnology, Inc). Reactions were carried out at 30 °C for 30 min in a mixture containing 50 μm unlabeled ATP and 10 μCi of [γ-32P]ATP and then stopped by the addition of 6× SDS sample buffer. Samples were boiled, separated by 12% SDS-PAGE, and visualized by autoradiography, Western blotting, or Coomassie Blue staining.
Immunocytofluorescence Assay
293 (7.5 × 104), C2C12, RSK2+/+, or RSK2−/− cells were seeded in two-chamber slides and mock-transfected or transfected with pEGFP-NFAT3, pEGFP-RSK2, or pEGFP-NFAT3/pcDNA4-HisG-RSK2 depending on the experiment. Transfection was followed by a 12-h incubation and A23187 stimulation for 36 h. At each time point, the cells were washed and fixed in 4% formalin, and the GFP fusion protein was visualized under a fluorescence microscope (magnification ×200).
NFAT3 Activity Assay
293 cells (2.0 × 104) were seeded into 48-well plates and incubated with 10% FBS-containing DMEM for 18 h before transfection. The 3 × NFAT-luciferase reporter plasmid, which consists of the minimal interleukin-2 promoter containing three NFAT-binding consensus sequences (29), was transfected with pcDNA3-FLAG-NFAT3, pcDNA4-RSK2, or both vectors into 293 cells. The cells were cultured for 12 h and were or were not stimulated with 1 μm A23187 and then cultured for an additional 24 h. At each time point, cells were disrupted and analyzed for firefly luciferase activity. The 3× NFAT-luciferase activity was normalized against Renilla luciferase activity (pRL-SV40).
NFAT3 Protein Domain Analysis
Data for analyzing the NFAT3 protein domains were downloaded from the ExPASy proteomics server (NiceProt view of Swiss-Prot entry Q14934). Putative phosphorylation sites were predicted by the NetPhos 2.0 server (www.cbs.dtu.dk/services/NetPhos).
Western Blotting
Proteins were extracted with Nonidet P-40 cell lysis buffer by freezing/thawing, and the concentration was measured. The same amount of protein was resolved by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes, which were then blocked in 5% skim milk for 1 h at room temperature. Antibody hybridization was performed overnight at 4°C, and horseradish peroxidase (HRP)-conjugated secondary antibody binding was conducted for 1 h at room temperature. The membrane was washed, and proteins were visualized using an ECL detection kit (Amersham Biosciences).
Nuclear Localization of NFAT3
To separate the nuclear and cytosolic fractions of C2C12 cells during differentiation, C2C12 cells were plated into 100-mm dishes and cultured to 90% confluence, at which time differentiation was induced with 2% horse serum (HS). The cells were harvested at 24-h intervals up to 96 h, and the nuclear and cytosolic fractions were extracted using NE-PER® nuclear and cytoplasmic extraction reagent (Pierce) following the manufacturer's suggested protocol.
Myotube Differentiation
Each indicated vector in Figs. 6 and 7 was transfected into C2C12 skeletal myoblasts. These cells were cultured for 36 h in a 5% CO2 incubator, and myotube differentiation was induced with 2% HS. At 24-h intervals, the 2% HS differentiation medium was replaced with fresh 2% HS, and differentiation termination was accomplished at 96 h post-induction. The cells were fixed with 4% formalin and permeabilized with 0.5% Triton X-100 and 1× PBS for 10 min. Cells were hybridized with anti-myosin heavy chain antibody MF20 as a primary antibody and Texas Red- or HRP-conjugated goat anti-mouse IgG as a secondary antibody. Differentiation of C2C12 cells to myotubes was observed by fluorescence microscopy. Scale bars indicate 50 μm at a magnification of ×200. The fusion index was obtained by the following formula: % = (numbers of nuclear fused/numbers of total nuclear) × 100. The average length of each myotube was calculated according to the longest axis.
Figure 6. RSK2 induces NFAT3-mediated myotube differentiation of C2C12 myoblasts.

A, endogenous NFAT3 protein levels in C2C12 myoblasts. C2C12 myoblasts were harvested, and proteins were extracted in Nonidet P-40cell lysis buffer. Proteins (50 μg) were resolved by SDS-PAGE, transferred onto PVDF membranes, and hybridized with anti-total NFAT3 or anti-phospho-NFAT3 (Ser168/170) (p-NFAT3) antibody. The bands were visualized using the ECL detection kit. B, localization of the NFAT3 and RSK2 proteins during myotube differentiation of C2C12 myoblasts. To analyze the localization of the NFAT3 and RSK2 proteins during myotube differentiation, pGFP-wtNFAT3FLor pGFP-RSK2FLwas transfected into C2C12 myoblasts. After 36 h, cells were induced to myotube differentiation with 2% HS and then fixed with 4% formalin at the indicated time points. GFP-NFAT3 and GFP-RSK2 proteins were visualized under a fluorescence microscope using fluorescein isothiocyanate. Scale bars = 50 μm at a magnification of ×200. C, protein expression profile during C2C12 differentiation. C2C12 myoblasts were induced to myotube differentiation as described for B. At each time point, the cells were harvested and disrupted in Nonidet P-40 cell lysis buffer. The proteins (50 μg) were resolved by SDS-PAGE, transferred onto PVDF membranes, hybridized with antibodies as indicated, and visualized using the ECL detection kit. β-Actin was used as an internal control to verify equal protein loading. D, RSK2-NFAT3 binding is increased in the early stage of differentiation. C2C12 myoblasts were induced to myotube differentiation as described for B, and proteins were extracted as described for C. Total protein (500 μg) was used for immunoprecipitation (IP) with anti-RSK2 antibody and agarose A/G beads overnight at 4 °C. Proteins were resolved by SDS-PAGE, transferred onto PVDF membranes, and hybridized with anti-NFAT3 antibody. Protein bands were visualized using the ECL detection kit. IB, immunoblot; IgG H.C., IgG heavy chain. E, nuclear localization of NFAT3 and RSK2 during C2C12 differentiation. C2C12 myoblasts were induced to myotube differentiation as described for B, and then nuclear and cytosolic proteins were isolated at each indicated time point. Total nuclear proteins (25 μg) were resolved by SDS-PAGE, transferred onto PVDF membranes, hybridized with anti-NFAT3 or anti-RSK2 antibody, and visualized using the ECL detection kit. Lamin was used for an internal control as a nuclear marker and to monitor equal protein loading. F, Western blotting for marker proteins of muscle cell differentiation. An identical experimental set was conducted to assess marker proteins of muscle cell differentiation in C2C12 cells stimulated with 2% HS. Cells from various time points were harvested and disrupted with Nonidet P-40 cell lysis buffer combined with freezing/thawing. Each marker protein was visualized by Western blotting with specific antibodies against myogenin, MyoD, and myosin heavy chain (MyHC). β-Actin was used as an internal control to verify equal protein loading. G, RSK2 markedly enhances myotube differentiation mediated by NFAT3 in C2C12 cells. The expression vectors pcDNA4 (mock), pcDNA3-FLAG-NFAT3, pcDNA4-HisG-RSK2, and pcDNA3-FLAG-NFAT3/pcDNA4-HisG-RSK2 were transfected into C2C12 myoblasts. Following 36 h of culture, myotube differentiation was conducted as described under “Experimental Procedures.” The cells were hybridized with anti-myosin heavy chain antibody MF20 as a primary antibody and HRP-conjugated goat anti-mouse IgG as a secondary antibody and visualized by the addition FAST 3,3′-diaminobenzidine peroxidase substrate. Nuclear staining was performed using Hoechst (0.5 μg/ml),and myotube differentiation was observed under a fluorescence microscope. Scale bars = 50 μm at a magnification of ×200. Each bar indicates the mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using Student's t test (*, p < 0.05).
Figure 7. The MEK1-ERK1-RSK2-NFAT3 axis is a key signaling pathway for muscle cell differentiation.

A, MEK1 inhibition suppresses C2C12 differentiation to multinucleated myotubes. C2C12 cells were stimulated with 2% HS with or without 10 μm PD98059 (a MEK1 inhibitor). Myotube differentiation was conducted as described under “Experimental Procedures” and visualized using HRP-conjugated goat anti-mouse IgG by the addition of FAST 3,3′-diaminobenzidine peroxidase substrate under a light microscope. Scale bars = 50 μm at a magnification of ×200. B, knockdown of RSK2, ERK1,or ERK2 has varying effects on the expression of marker proteins of muscle cell differentiation. C2C12 cells were transfected with pU6pro-mock, psi-RSK2, psi-ERK1, or psi-ERK2. At 48 h after induction with 2% HS, cells were harvested, and proteins were extracted by freezing/thawing. Protein levels were analyzed by Western blotting using specific antibodies. β-actin was used as an internal control to verify equal protein loading. MyHC, myosin heavy chain. C, C2C12 cells were transfected with pU6pro-mock (si-mock), pcDNA4-RSK2, psi-RSK2, psi-ERK1, psi-ERK1/pcDNA4-RSK2, or psi-ERK2/pcDNA4-RSK2 to knock down respective target protein expression. Following 36 h of culture, myotube differentiation was conducted as described under “Experimental Procedures.” Scale bars = 50 μm at a magnification of ×200. Each bar represents the mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using Student's t test (*, p < 0.05). D, knockdown of endogenous NFAT3. C2C12 cells were transfected with pU6pro-mock or psi-NFAT3 and cultured for 36 h in a 5% CO2 incubator. Proteins (100 μg) were resolved, transferred onto PVDF membranes, hybridized with anti-total NFAT3 antibody, and visualized using the ECL detection kit. The band intensity of NFAT3 siRNA (siNFAT3) was compared with that of mock siRNA (100%) using the Scion Image Version Beta 4.0.3 computer program and quantified as shown in the right panel. E, NFAT3 siRNA efficiently suppresses RSK2-induced myotube differentiation. C2C12 cells were transfected with pU6pro-mock, pcDNA4-RSK2, psi-NFAT3, or psi-NFAT3/pcDNA4-RSK2 to knock down respective target protein expression. Following 36 h of culture, myotube differentiation was conducted as described under “Experimental Procedures.” Scale bars = 50 μm at a magnification of ×200. Each bar represents the mean ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using Student's t test (*, p < 0.05). For B-E, we used a control siRNA vector for GFP using primers 5′-TTTGGGTTATGTACAGGAACGCATTCAAGAGATGCGTTCCTGTACATAACCTTTTT-3′ (sense) and 5′-CTAGAAAAAGGTTATGTACAGGAACGCATCTCTTGAATGCGTTTCTGTACATAACC-3′ (antisense), and this was designated as si-mock.
Results
NFAT3 Is a Novel RSK2-interacting Protein—To identify novel substrates of RSK2, we introduced RSK2 cDNA, including the open reading frame, into the pBIND mammalian two-hybrid system vector (pBIND-RSK2) as bait. The open reading frame of each transcription factor was amplified by PCR and then introduced into the pACT mammalian two-hybrid system vector (pACT-TF) (15). The individual pACT-TF plasmids and the pG5-luciferase reporter plasmid were cotransfected into 293 cells with pBIND-RSK2. Each interaction activity was compared against pG5-luciferase/pBIND-RSK2 as the basal level (Fig. 1A, first bar). c-Fos, activating transcription factor-4 (CREB2), CREB1, CREB3, and p53 were used as positive controls (Fig. 1A, second through sixth bars), and E2F6 and c-Jun were used as negative controls (ninth and tenth bars). NFAT3 (NFATc4) showed an ∼62-fold interacting activity with pBIND-RSK2 compared with the pG5-luciferase/pBIND-RSK2 control (Fig. 1A, eighth bar versus first bar). Therefore, we selected NFAT3 for further study.
Figure 1. NFAT3 is a novel RSK2-interacting protein.
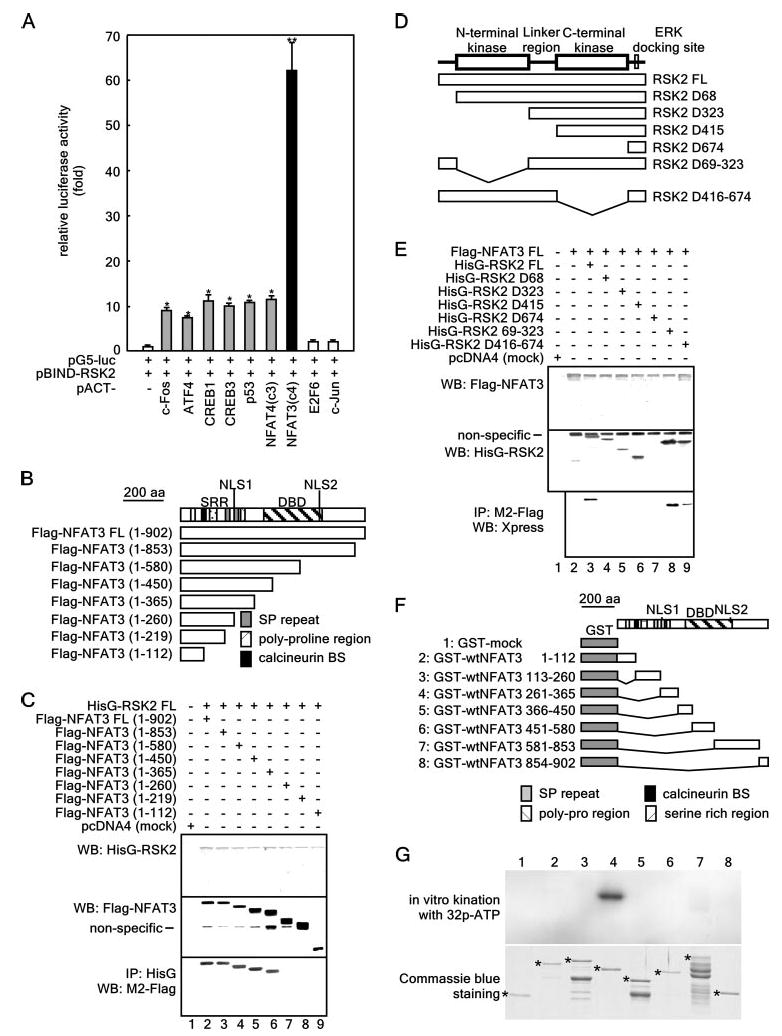
A, assessment of the in vivo protein-protein interactions of pBIND-RSK2 and pACT-TF plasmids by the mammalian two-hybrid assay. Activity is expressed as relative luminescence units normalized to a negative control (value for cells transfected with only pG5-luciferase (luc)/pBIND-RSK2 = 1.0). Data are expressed as the means ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using Student's t test (*, p < 0.01;**, p < 0.0001). ATF4, activating transcription factor-4. B, structure and schematic diagrams of NFAT3 deletion mutant constructs. SRR, serine-rich region; DBD, DNA-binding domain; SP repeat, Ser/Pro repeat; BS, binding site. C, identification of the NFAT3 domain that binds RSK2. To identify the domain of NFAT3 to which RSK2 binds, eight pcDNA3-FLAG-NFAT3 deletion constructs were individually cotransfected with pcDNA4-HisG-RSK2FL into 293 cells. After culturing for 48 h, cells were disrupted with Nonidet P-40 cell lysis buffer and immunoprecipitated (IP) with anti-HisG monoclonal antibody. NFAT3 was detected by Western blotting (WB) using HRP-conjugated anti-FLAG antibody M2. D, structure and schematic diagrams of RSK2 deletion mutant constructs. E, identification of the domain of RSK2 that binds NFAT3. To identify the domain of RSK2 to which NFAT3 binds, seven pcDNA4-RSK2 deletion constructs were individually cotransfected with pcDNA3-FLAG-NFAT3FL into 293 cells. After culturing for 48 h, cells were disrupted with Nonidet P-40 cell lysis buffer and immunoprecipitated with anti-FLAG monoclonal antibody M2. RSK2 was detected by Western blotting using HRP-conjugated anti-Xpress antibody. F, structure and schematic diagrams of GST-NFAT3 fusion constructs. G, target domain mapping of NFAT3. To identify the target domain of NFAT3 for RSK2, each GST-NFAT3 fusion protein was partially purified, directly subjected to an in vitro phosphorylation assay with active RSK2, and then visualized by autoradiography. Each asterisk indicates the respective GST fusion protein.
To identify the NFAT3 domain involved in the RSK2-NFAT3 interaction, we conducted immunoprecipitation experiments with pcDNA4-HisG-RSK2FL and individual pcDNA3-FLAG-NFAT3 deletion mutants (Fig. 1B) that were transfected into 293 cells. The results from the immunoprecipitation experiment with anti-HisG monoclonal antibody showed that deletion mutants from the C terminus to aa 365 were coprecipitated with RSK2 at the same density (Fig. 1C, lower panel, lanes 2–6). When aa 365 to 260 were deleted, the coprecipitated NFAT3 protein disappeared (Fig. 1C, lower panel, lane 7), and any mutants with deletions to aa 219 or 112 did not coprecipitate with the RSK2 protein (lower panel, lanes 8 and 9). These results demonstrate that nuclear localization signal (NLS)-1, the Ser/Pro repeat, and the polyproline domains spanning aa 261-365 of NFAT3 play a key role in the interaction with RSK2. At the same time, identical experiments to test the interaction of FLAG-tagged full-length (FL) NFAT3 and HisG-RSK2 deletion mutants (Fig. 1D) were conducted. The binding of RSK2FL to NFAT3FL was significantly reduced in the mutant with a deleted N-terminal domain (aa 1–68) (Fig. 1E, lower panel, lane 4), and none of the serial mutants of RSK2 were able to interact with NFAT3FL (lower panel, lanes 5–7). Furthermore, the RSK2D416–674 (C-terminal kinase) deletion mutant also displayed reduced binding activity with NFAT3FL (Fig. 1E, lower panel, lane 9), indicating that the C-terminal kinase domain may also be important in the RSK2 interaction with NFAT3. To identify the target domain of NFAT3 against RSK2, we constructed and partially purified seven GST fusion proteins (Fig. 1F) and conducted in vitro phosphorylation assays with active RSK2 (Fig. 1G). The results showed 32P-labeled radioisotope labeling of the GST-wtNFAT3 protein (aa 261–365), demonstrating that this fragment of the NFAT3 protein contains one or more phosphorylation targets of RSK2.
RSK2 Phosphorylates Multiple Serine Sites of NFAT3
To identify potential phosphorylation site(s), we compared the amino acid similarity between activating transcription factor-4 (14) and NFAT3 (Fig. 2A). We found that NFAT3 contains two SPXXS motifs in a reverse repeat within aa 261–365 (Fig. 2B). A search using the NetPhos 2.0 server suggested that these motifs contain putative phosphorylation sites. To confirm the possibility that RSK2 phosphorylates serine residues in this region, we constructed and purified several GST-NFAT3 point mutants and then directly subjected the mutants to an in vitro phosphorylation assay (Fig. 2C). In vitro phosphorylation of the mutants indicated that serines 281 and 285 of the NFAT3 protein might be target amino acids of RSK2 phosphorylation (Fig. 2C, lanes 8–11). However, the GST-NFAT3(S274A/S278A/S281A/S285A) mutant still showed some 32P labeling (Fig. 2C, lane 11), suggesting the presence of additional phosphorylation sites in this region. To identify additional phosphorylation sites, we used an LTQ Orbitrap hybrid mass spectrometer (MS), which provides low parts/million (<2) mass accuracy, and an LTQ linear ion trap, which allows for MSn fragmentation (where n ranges from 2 to 10 and is the number of times a fragmentation can be isolated and fragmented again). In these experiments, the ions generated in the collision-induced dissociation tandem mass spectrometry ion trap scan were used to identify potential phosphopeptides by looking for the characteristic neutral losses of H3PO4 and H3PO4 + H2O from the precursor ions during collision-induced dissociation fragmentation. The results from GST-NFAT3-(261–365) showed tandem mass spectra from three different [M + 2H]2+ precursor ions, each with the most abundant fragment ion corresponding to the loss of 49 from the precursor. In each case, the tandem mass spectrometry fragmentation was adequate enough to identify the phosphopeptide (Fig. 2D). The tandem and multiple mass spectra were manually interpreted to verify that the diagnostic PO3 site ions were among the abundant fragment ions present (supplemental Fig. 1). The results indicated that RSK2 phosphorylated two additional sites at Ser289 (peptide 2) and Ser344 (peptide 3) (Fig. 2E). Peptide 1, which contains serines 281 and 285 phosphorylated by RSK2 (Fig. 2C), overlaps the nuclear localization signal (KRR) and also contains the serine-rich Ser/Pro repeat (Fig. 2F). Therefore, we propose that this region may be important for the nuclear localization and activity of NFAT3. To further characterize the RSK2 interaction with NFAT3, we measured the enzyme kinetics of RSK2 phosphorylation of the NFAT3-(261–365) protein. The results showed that RSK2 has a very high affinity for NFAT3 with Km= 3.559 μm and Vmax = 11.5 μmol of ATP/min/μg of protein (Fig. 2G).
Figure 2. RSK2 phosphorylates NFAT3 at multiple serine sites.
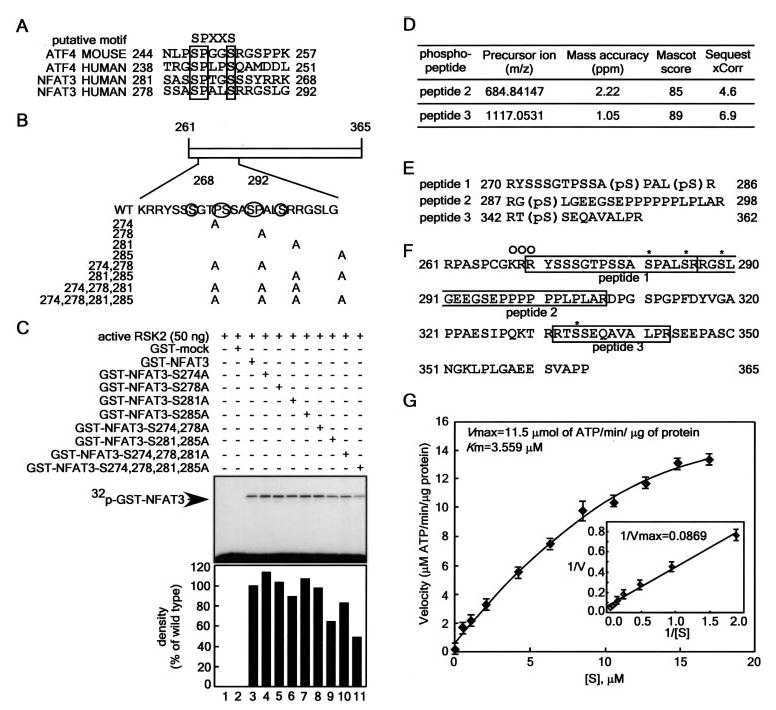
A, amino acid similarity of activating transcription factor-4 (ATF4) and NFAT3. B, schematic diagram of the construction of GST-NFAT3-(261–365) point mutants. C, effect of point mutation of GST-NFAT3 on its phosphorylation by RSK2. Active RSK2 was combined with each GST-NFAT3 mutant protein and 50 μm unlabeled ATP plus 10 μCi of [γ-32P]ATP. Reactions were conducted as described under “Experimental Procedures.” The band intensity of each mutant was compared with that of active wtRSK2/GST-NFAT3-(261–365) (100%) using the Scion Image Version Beta 4.0.3 computer program and quantified as shown in the lower panel. D, identification of phosphorylated peptides. The GST-NFAT3-(261–365) phosphorylation sites were determined using an LTQ Orbitrap hybrid mass spectrometer. The ions generated in the collision-induced dissociation tandem mass spectrometry ion trap scan were used to identify potential phosphopeptides by looking for the characteristic neutral losses of H3PO4 and H3PO4 + H2O from precursor ions during collision-induced dissociation fragmentation. In each case, the tandem mass spectrometry fragmentation was adequate enough to identify the phosphopeptide. The phosphopeptides were scored from both a Mascot and Sequest search of all species in the Swiss Protein Database (March 3, 2006), allowing only full tryptic peptides, with the variable modification allowing for phosphorylation of serine, threonine, and tyrosine. The precursor ion masses were taken from the Orbitrap Fourier transform full scan. E, target amino acid sequences of NFAT3-(261–365) containing phosphorylation site(s). F, amino acid alignment of NFAT3-(261–365). The peptides identified are boxed, and the vertical line indicates a trypsin digestion site. Asterisks indicate sites phosphorylated by RSK2, and open circles indicate the NLS. G, enzyme kinetics of RSK2 phosphorylation of NFAT3-(261–365). To analyze the kinetics of RSK2 phosphorylation of NFAT3, 5 ng of active RSK2 were reacted for 20 min with the indicated concentrations of purified NFAT3-(261–365) in 25 μl of reaction mixture containing 10 μCi of [γ-32P]ATP. Enzyme activity is represented by Lineweaver-Burk plots. Data points are represented as the means ± S.D. of three determinations obtained in one experiment.
RSK2 Induces NFAT3 Activity
We hypothesized that RSK2, which is an ERK downstream serine/threonine protein kinase, may play a key role in NFAT3 activity because the Ras-MEK-ERK pathway positively regulates NFAT3 activity (26). To examine this hypothesis, the 3× NFAT-luciferase reporter gene (consisting of a minimal interleukin-2 promoter containing three NFAT-binding consensus sequences) assay was conducted. The results showed that 3× NFAT-luciferase activity was increased in a dose-dependent manner by the presence of RSK2 (Fig. 3A) and that 3× NFAT-luciferase activity was also induced dose-dependently by pcDNA3-FLAG-NFAT3FL transfection (Fig. 3B). Moreover, after treatment with the calcium ionophore A23187, NFAT3-dependent 3× NFAT-luciferase activity increased considerably (Fig. 3B). Notably, the A23187-induced 3× NFAT-luciferase activity was more enhanced by cotransfection of NFAT3 and increasing amounts of RSK2 (Fig. 3C). These results demonstrate that RSK2 mediates NFAT3 activity.
Figure 3. RSK2 induces NFAT3 activation.
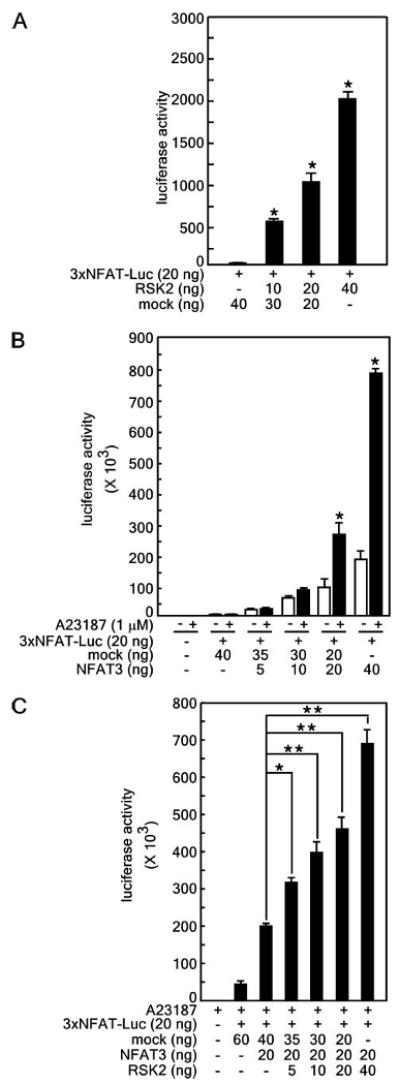
A, RSK2 induces NFAT3 activity in a dose-dependent manner. The 3× NFAT-luciferase (Luc) reporter gene was cotransfected with pcDNA4-RSK2 into 293 cells. Following 48 h of culture, cells were disrupted with lysis buffer and analyzed for firefly luciferase activity. B, NFAT3 activity is induced by the calcium ionophore A23187. To confirm NFAT3 stimulation by A23187, the 3× NFAT-luciferase reporter plasmid was cotransfected with pcDNA3-FLAG-NFAT3 into 293 cells. Following 12 h of culture, cells were stimulated with 1 μm A23187 for 24 h and analyzed for firefly luciferase activity. C, RSK2 induces increased NFAT3 activity stimulated by A23187. To confirm that RSK2 mediates NFAT3 activity, the pcDNA3-FLAG-NFAT3 construct (20 ng) was cotransfected into 293 cells with various amounts of the pcDNA4-RSK2 construct (0, 5,10, 20, and 40 ng). Following 12 h of culture, cells were stimulated with 1 μm A23187 for 24 h and analyzed for firefly luciferase activity. For all experiments (A–C), the 3× NFAT-luciferase activity was normalized against Renilla luciferase activity (pRL-SV40), and data are expressed as the means ± S.D. of values from triplicate experiments. Also for A–C, significant differences were evaluated using Student's t test (A and B,*, p < 0.001; C,*, p < 0.05 and **, p < 0.001).
RSK2 Induces the Nuclear Localization of NFAT3
To begin to study the localization of NFAT3 and RSK2, we first transfected the pGFP-RSK2FL expression vector into 293 cells and found that the GFP-RSK2FL protein was localized in both the nucleus and cytoplasm (Fig. 4A). Then, to examine whether GFP-wtNFAT3FL localization was mediated by RSK2, we introduced pGFP-wtNFAT3FL or pGFP-wtNFAT3FL/pcDNA4-RSK2FL into cells, followed by stimulation with A23187. The results indicated that RSK2 caused the GFP-wtNFAT3FL protein to accumulate in the nucleus (Fig. 4B), whereas cotransfection of pGFP-wtNFAT3FL with pcDNA3-FLAG-JNK1, which has been shown to inhibit NFAT4 nuclear localization (24), resulted in no nuclear accumulation of NFAT3 with or without A23187 stimulation (Fig. 4B). Furthermore, we found that 3× NFAT3-luciferase reporter activity was increased by A23187 stimulation by ∼5.5-fold in RSK2+/+ (but not RSK2−/−) MEF cells (Fig. 4, C and D). These results support our hypothesis that RSK2 induces the nuclear localization and activation of NFAT3. This conclusion was supported by another experiment performed to determine the localization of GFP-wtNFAT3FL in RSK2+/+ and RSK2−/− MEF cells (Fig. 4E). In RSK2+/+ cells, GFP-wtNFAT3FL was observed to accumulate in the nucleus at 60 min after A23187 treatment and continued to accumulate until 240 min (Fig. 4 E, left panels, arrowheads). However, in RSK2−/− cells, accumulation of GFP-wtNFAT3FL was attenuated and observed only at 240 min after A23187 treatment (Fig. 4E, right panels, arrowheads). Nuclear GFP-wtNFAT3FL was redistributed into the cytoplasm at 480 min after A23187 treatment in both cell types (Fig. 4E). Upon A23187 stimulation, the nuclear protein level of endogenous NFAT3 was increased at 120 and 240 min in RSK2+/+ MEF cells. However, RSK2−/− cells showed an increase only at 240 min (Fig. 4F). These results support the idea that RSK2 mediates NFAT3 localization.
Figure 4. RSK2 induces nuclear localization of NFAT3.
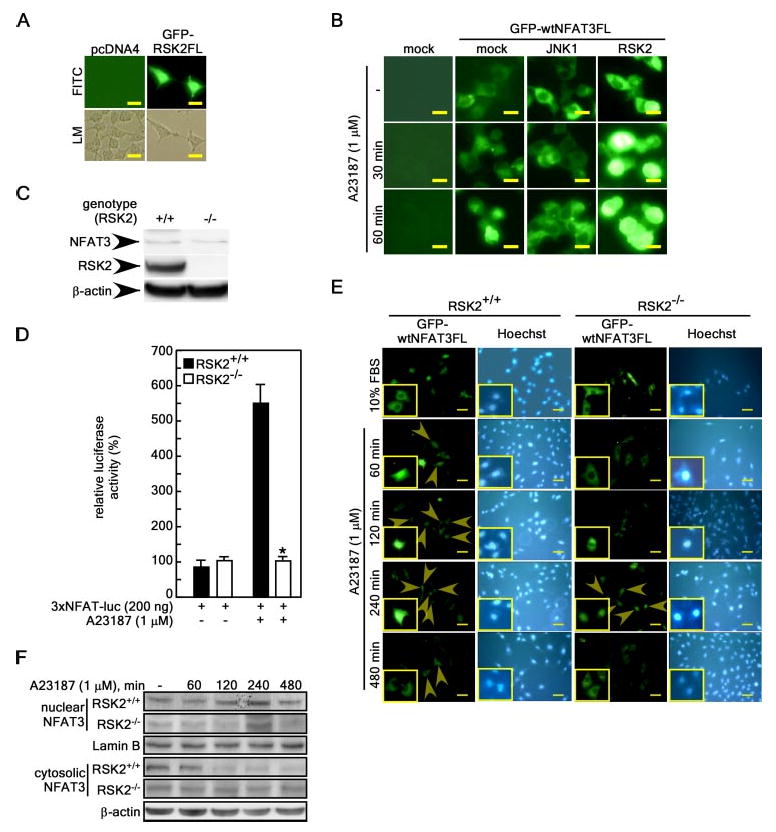
A, cellular localization of RSK2. 293 cells were transfected with a mock vector (pcDNA4) or pEGFP-RSK2FL. At 36 h after transfection,ectopically expressed proteins were visualized using fluorescein isothiocyanate (FITC) under a fluorescence microscope. The fluorescein isothiocyanate and light microscopy (LM) images represent exactly the same region of cells for each. B, effects of RSK2 expression on the nuclear translocation of NFAT3 induced by A23187 stimulation. 293 cells were transfected with pEGFP-wtNFAT3FLwith or without pcDNA3-FLAG-JNK1 or pcDNA4-RSK2. After fixation, the distribution of the NFAT3 protein was visualized using fluorescein isothiocyanate under a fluorescence microscope at the indicated time points. For A and B, scale bars = 10 μm at a magnification of ×200. C, RSK2 expression in RSK2+/+ and RSK2−/− embryonic fibroblast cells. Cells were cultured in 10% FBS-containing DMEM and harvested, and proteins were extracted in Nonidet P-40 cell lysis buffer. Proteins were resolved by SDS-PAGE, transferred onto PVDF membranes, hybridized with anti-RSK2 or anti-NFAT3 antibody, and then visualized using the ECL detection kit. D, RSK2 deficiency suppresses 3× NFAT-luciferase activity. 3× NFAT-luciferase (luc) reporter plasmids were transfected into RSK2+/+ or RSK2−/− MEF cells and stimulated with 1 μm A23187 for 24 h, and then firefly luciferase activity was analyzed. Firefly luciferase activity was normalized against Renilla luciferase activity (pRL-SV40). Data are expressed as the means ± S.D. of values obtained from triplicate experiments. Significant differences were evaluated using Student's t test (*, p < 0.001). E, nuclear accumulation of NFAT3 is attenuated in RSK2−/− MEFs. RSK2+/+ or RSK2−/− MEF cells were transfected with pEGFP-wtNFAT3FL. At 36 h after transfection, the GFP-NFAT3 protein was visualized using fluorescein isothiocyanate under a fluorescence microscope at various time points following A23187 treatment. Scale bars = 10 μm at a magnification of ×200. Arrowheads indicate nuclear accumulated GFP-NFAT3. F, endogenous nuclear NFAT3 protein level. RSK2+/+ and RSK2−/− cells were stimulated with A23187 (1 μm), and nuclear and cytosolic proteins were extracted at the indicated time points. Proteins (30 μg) were resolved by SDS-PAGE, transferred onto PVDF membranes, hybridized with anti-NFAT3 antibody, and visualized using the ECL detection kit.
NLS1, the Ser/Pro Repeat, and the Polyproline Regions Spanning aa 261–365 Play a Key Role in the Nuclear Localization and Activation of NFAT3
To determine whether NFAT3-(261–365) can be phosphorylated under physiological conditions, pEGFP-C1-NFAT3-(261–365) was transfected into RSK2+/+ and RSK2−/− MEFs. The serine phosphorylation levels were then analyzed by immunoprecipitation and Western blotting with a phosphoserine-specific antibody. The results indicated that the phosphorylation level of the NFAT3-(261– 365) protein was decreased in RSK2−/− MEF cells (Fig. 5A). To examine whether RSK2 mediates transactivation, we constructed a Gal4-NFAT3-(1–360) fusion construct and analyzed transactivation activity in RSK2+/+ and RSK2−/− MEF cells using the p5×Gal4-luciferase reporter gene assay (Fig. 5B). The results indicated that RSK2-mediated transactivation activity was suppressed in RSK2−/− MEF cells. Moreover, the results of the mammalian two-hybrid assay indicated that mutation of NFAT3 at serines 274, 278, 281, and 285 markedly decreased RSK2 and NFAT3 binding (Fig. 5C). Furthermore, we found that pGFP-mtNFAT3FL failed to induce 3× NFAT-luciferase activity, which was significantly increased by pGFP-wtNFAT3FL in RSK2+/+ MEF cells (Fig. 5D). On the other hand, we found a 32-fold increase in 3× NFAT-luciferase activity induced by GFP-wtNFAT3FL in RSK2+/+ MEF cells compared with only a 7-fold increase in RSK2−/− MEF cells (Fig. 5D), which supports our previous findings. We then examined whether the activity of NFAT3 is mediated cooperatively by calcineurin activity and MEK-ERK-RSK2 signaling. The results indicated that NFAT3-mediated luciferase activity was suppressed by cyclosporin A (CsA) treatment in cells whether or not they were stimulated with A23187 (Fig. 5E). Moreover, the MEK1 inhibitor PD98059 partially inhibited 3× NFAT3-luciferase activity (Fig. 5F), suggesting that NFAT3 activation requires a cooperative effect of calcineurin and MEK-ERK-RSK2 signaling.
Figure 5. NLS1, the Ser/Pro repeat, and the polyproline regions spanning aa 261–365 play a key role in the nuclear localization and activation of NFAT3.
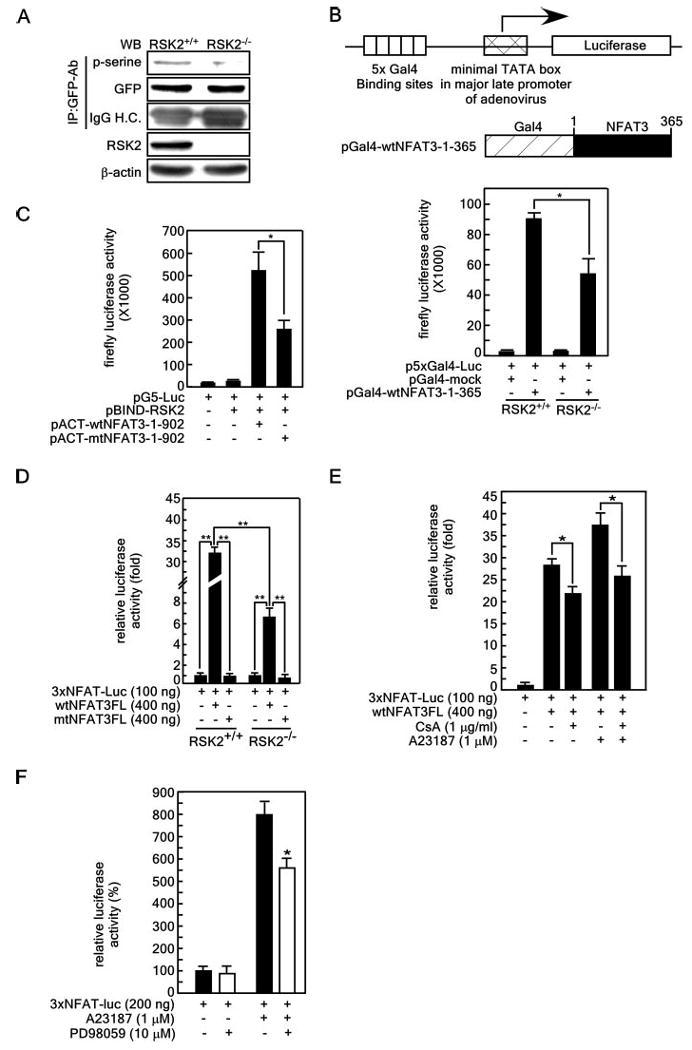
A, NFAT3-(261–365) is a physiological target of RSK2. The pGFP-NFAT3-(261–365) expression vector was transfected into RSK2+/+ and RSK2−/− MEF cells. Cells were cultured for 36 h in a 5% CO2 incubator, and then proteins were extracted with Nonidet P-40 cell lysis buffer. GFP-NFAT3-(261−365) was immunoprecipitated (IP) with anti-GFP antibody (GFP-Ab). The proteins were resolved by SDS-PAGE, transferred onto PVDF membranes, hybridized with an HRP-conjugated phosphoserine (p-serine)-specific antibody, and visualized using the ECL detection kit. WB, Western blot; IgG H.C., IgG heavy chain. B, RSK2 deficiency suppresses the N-terminal transactivation activity of NFAT3. pBIND-wtNFAT3-(1–365) was transfected with the luciferase reporter plasmid (containing five Gal4-binding sites located upstream of the minimal TATA box of the major late promoter in adenovirus) into RSK2+/+ and RSK2−/− MEF cells. Luciferase activity was analyzed at 36 h. A pBIND mock expression vector was transfected as a control. C, differential binding affinity of wild-type and mutant NFAT3 for RSK2.The binding affinity of wild-type and mutant NFAT3 for RSK2 was analyzed by mammalian two-hybrid assay. D, serine phosphorylation is required for NFAT3 activity. The 3× NFAT-luciferase (Luc) reporter plasmid was cotransfected with pGFP-wtNFAT3FL or pGFP-mtNFAT3FL into RSK2+/+ and RSK2−/− MEF cells, and 36 h after transfection, luciferase activity was measured. E, effect of CsA (a calcineurin inhibitor) on NFAT3 activity. The 3× NFAT-luciferase reporter plasmid was transfected with pcDNA3-FLAG-wtNFAT3FL Cells were treated with CsA and/or A23187for 24 h as indicated, and then firefly luciferase activity was analyzed. F, effect of the MEK1 inhibitor PD98059 on NFAT3activity.The3× NFAT-luciferase reporter plasmid was transfected; cells were treated for 24 h with A23187 (1 μm) and/or PD98059 (10 μm); and then firefly luciferase activity was analyzed. For B and F, firefly luciferase activity was normalized against Renilla luciferase activity (pRL-SV40). Each bar indicates the mean ± S.D. of values obtained from triplicate experiments, and significant differences were evaluated using Student's t test (*, p < 0.05;**, p < 0.001).
RSK2 Induces NFAT3-mediated Myotube Differentiation of C2C12 Myoblasts
To analyze the functional significance of RSK2-NFAT3 signaling, we searched the literature for information regarding skeletal muscle phenotypes associated with NFAT3 and/or NFAT4 deficiency. Calcineurin transgenic mice were reported to display cardiac hypertrophy, and N-terminally deleted NFAT3 transgenic mice show a loss of the cytosolic distribution of NFAT3 that is associated with cardiac hypertrophy (30). However, targeted disruption of NFAT4 (but not NFAT3) in calcineurin transgenic mice results in a significant reduction in transgenic calcineurin-induced cardiac hypertrophy (31). Moreover, NFAT4-null mice show decreases of ∼20 – 25% in extensor digitorum longus muscle, total number of myo-fibers, and muscle weight (32). In addition, ∼98% of double knock-out fetal mice die, suggesting a functional redundancy between NFAT3 and NFAT4 (33). However, no research group has reported on the specific function of NFAT3 in skeletal muscle, and therefore, we hypothesized that NFAT3 may also play an important role in skeletal muscle development. To investigate this idea, we first examined the protein level of NFAT3 in C2C12 cells (Fig. 6A). The results showed that total NFAT3 protein could be detected in C2C12 cells with anti-NFAT3 antibody (Santa Cruz Biotechnology catalog no. sc-13036). Notably, the phosphorylation of NFAT3 at serines 168 and 170 was very strong in C2C12 cells (determined using anti-phospho-NFATc4 (Ser168/170) antibody, Santa Cruz Biotechnology catalog no. sc-32630), indicating that C2C12 cells do indeed express NFAT3. In addition, we tested the efficiency of several commercially available anti-NFAT3 antibodies from different companies (data not shown) and found that the antibody used in these experiments provided the best results. Furthermore, a comparison of the expressed sequence tag profiles for NFAT3 and NFAT4 (NCBI UniGene Hs.77810 (NFATc4) and Hs.632209 (NFATc3)) revealed that NFAT3 and NFAT4 are expressed at the same level in skeletal muscle (data not shown). To continue to investigate of the interaction of NFAT3 and RSK2, we next introduced pGFP-wtNFAT3FLorpGFP-RSK2FL into C2C12 myoblasts. At 48 h after 2% HS stimulation, the nuclear localization of NFAT3 was increased, and then NFAT was relocalized to the cytoplasm at 72–96 h (Fig. 6B, left panels). Notably, the diffusible distribution of RSK2 was increased at 24 h after 2% HS stimulation, markedly increased at 48 h, and then redistributed as diffusible by 72 h (Fig. 6B, right panels). Western blotting results also indicated that the total NFAT3 protein level was decreased at 72 h after 2% HS induction (Fig. 6C). However, NFAT3 phosphorylation at serines 168 and 170 was substantially decreased within 48 h after 2% HS induction (Fig. 6C), indicating that the nuclear localization of NFAT3 might be increased. The total RSK2 protein level was increased at 24 h after 2% HS induction, sustained to 72 h, and decreased by 96 h (Fig. 6C). The phosphorylation of p90RSK was slightly increased at 24 h and sustained over the entire period of differentiation (Fig. 6C). Furthermore, results of co-immunoprecipitation experiments showed that endogenous RSK2 and NFAT3 binding was increased at 24 h after 2% HS induction, sustained until at least 48 h, and then decreased at 72–96 h (Fig. 6D). Notably, we found that the nuclear localization of endogenous RSK2 and NFAT3 was increased up to 48 h and then decreased at 72 h after 2% HS induction (Fig. 6E). The verification of muscle cell differentiation was confirmed by Western blotting with specific antibodies against differentiation marker proteins, including myogenin, MyoD, and myosin heavy chain (Fig. 6F). These results demonstrate that RSK2 and NFAT3 localization occurs at the same time during differentiation and thus might be involved in myoblast differentiation to myotubes.
To provide further evidence showing that RSK2 is involved in muscle cell differentiation, we examined C2C12 myoblast differentiation by introducing pcDNA3-FLAG-wtNFAT3FL or pcDNA4-RSK2FL individually or together into these cells. The differentiation of the transfectants was verified by immunocy-tochemistry to detect myosin heavy chain with monoclonal antibody MF20 and HRP-conjugated secondary antibody and by Hoechst nuclear staining. The results indicated that introduction of pcDNA3-FLAG-NFAT3FL induced differentiation slightly as illustrated by both the fusion index and myotube length compared with the mock-transfected control group (Fig. 6G, second left panel and right panels). Moreover, upon pcDNA4-RSK2FL transfection, the length of myotubes was increased compared with mock-transfected cells or cells transfected with NFAT3 alone (Fig. 6G, third left panel and right panels). Notably, the cotransfection of RSK2 and NFAT3 induced a significant increase in the fusion index and length of myotubes compared with mock-transfected cells or cells transfected with NFAT3 alone or RSK2 alone (Fig. 6G, fourth left panel and right panels). For these experiments, the same cell density was confirmed by Hoechst staining (Fig. 6G). The same transfection efficiency was confirmed by pGL3-luciferase control vector transfection into an identical experimental set of cells at the same time point of 2% HS induction (data not shown). These results provided strong evidence in support of our hypothesis that RSK2 plays a key role in skeletal muscle cell differentiation mediated by NFAT3.
The MEK1-ERK1-RSK2-NFAT3 Axis is a Key Signaling Pathway for Skeletal Muscle Cell Differentiation
The MEK-ERK1/2 pathway both promotes and inhibits muscle gene expression and regulates the activity of the MyoD/E protein dimer (34, 35), which strongly indicates that the ERK-RSK2 axis is involved in regulating muscle cell differentiation. We investigated the effect of PD98059, a MEK1 inhibitor, on muscle cell differentiation over a period of 96 h after induction of differentiation. The results indicated that, at 96 h, treatment with PD98059 suppressed differentiation to myotubes compared with untreated control cells (Fig. 7A). However, the effect of treatment with 10 μm PD98059 on C2C12 cell growth or viability was minimal (supplemental Fig. 2). We examined the direct effect of the ERK1/2-RSK2 signaling pathway on myotube differentiation by introducing small interfering RNA (siRNA) vectors against RSK2 (psi-RSK2), ERK1 (psi-ERK1), and ERK2 (psi-ERK2) into C2C12 cells. We observed that siRNA induced marked decreases in the respective endogenous protein levels (densitometry results: decreased RSK2 by 80%, ERK1 by 95%, and ERK2 by 60%) (Fig. 7B). The RSK2, ERK1, and ERK2 siRNA transfection markedly suppressed myotube differentiation compared with the mock siRNA transfection (Fig. 7C). Moreover, RSK2-induced C2C12 differentiation was also suppressed by ERK1 or ERK2 siRNA transfection. Furthermore, NFAT3 siRNA transfection suppressed ∼75% of the endogenous NFAT3 protein level (Fig. 7D) and markedly inhibited C2C12 differentiation to myotubes (fusion index and myotube length) (Fig. 7E). The same transfection efficiency was confirmed by pGL3-luciferase control vector transfection into an identical experimental set at the same time point for 2% HS induction (data not shown) as well as GFP mock transfection (supplemental Fig. 3). These results provide strong evidence showing that the ERK-RSK2 signaling pathway is required for myotube differentiation of C2C12 cells. On the basis of the results described in this study, we concluded that the MEK1-ERK-RSK2-NFAT3 signaling axis plays an important positive role in skeletal muscle cell differentiation.
Discussion
The NFAT family of transcription factors plays a critical role in skeletal muscle, cardiovascular, cartilage, neuronal, and immune system development (19, 32, 33, 36–40). Unlike calcineurin (41), NFAT proteins can be deactivated and localized in the cytoplasm through phosphorylation by several protein kinases, including glycogen synthase kinase-3, protein kinase A, p38, JNKs, and casein kinase (23,24,42–44). The finding that a new kinase, RSK2, is a positive regulator of NFAT3 activation has significant implications because of the fact that many of the same organ systems require calcineurin/NFAT signaling during embryonic development. This suggests a widespread utilization of calcineurin/NFAT signaling mechanisms in a broad spectrum of developmental and disease processes (45).
Originally, NFAT3 knock-out mice did not show a readily observable phenotype because of functional redundancy by NFAT4 (33). However, the NFAT3/NFAT4 double knock-out mice displayed 98% lethality at embryonic days 9.0∼11.5, but no major morphological defects were observed until about embryonic day 10.5. These animals were smaller and pale and displayed underdeveloped yolk sac vasculature, with many embryos having enlarged pericardial sacs. This suggests that NFAT3 most likely has an overlapping role with NFAT4 in normal development, thus supporting our hypothesis that NFAT3 is important in muscle cell differentiation. The consideration that only NFAT4 plays a key role in skeletal muscle development may not be accurate. The NFAT protein exists in a phosphorylated form in the cytoplasm, but translocates to the nucleus upon dephosphorylation in response to increased intracellular calcium levels and activation of the phosphatase calcineurin (46). Calcineurin activity is required for initiation of skeletal muscle differentiation (46), and CsA and FK506 (calcineurin inhibitors) prevent the nuclear localization of NFAT. Interestingly, CsA has been reported to have ameliorative effects on muscle weakness in Duchenne muscular dystrophy patients (47). Together, these results suggest that NFAT proteins may play functional roles in skeletal muscle development (48).
The nuclear localization of NFAT3 is known to be affected by phosphorylation by some kinases and by dephosphorylation by calcineurin activity. The p38 kinase phosphorylates NFAT3 at serines 168 and 170, and mutation of serines 168 and 170, which are located near the calcineurin-binding domain, increases nuclear localization (23). However, activated calcineurin has no effect on the subcellular distribution of either wtNFAT3 or NFAT3 mutated at the protein kinase A phosphorylation sites at serines 272, 273, 274, and 289, which are located adjacent to the NLS1 region (49), suggesting that the NLS1 region is not a target for calcineurin-mediated dephosphorylation.
Our results showed that wtNFAT3 activity was significantly inhibited in RSK2−/− MEF cells and that mutations of NFAT3 at RSK2 target amino acids almost blocked NFAT3 activity (Fig. 5D). Moreover, CsA and PD98059 (a MEK1 inhibitor) partially inhibited A23187-stimulated 3× NFAT-luciferase activity, indicating that NFAT3 localization and activation might be cooperative depending on RSK2 and calcineurin activities, which induce phosphorylation and dephosphorylation, respectively, at different site(s).
We have shown here that the phosphorylation of NFAT3 by RSK2 might have important functional implications because the activation of NFAT3 by RSK2 markedly enhanced multinucleated myotube differentiation of C2C12 myoblasts. Our results suggest that the RSK2-NFAT3 signaling pathway might be linked to skeletal muscle differentiation. Indeed, homozygous deletion and mutations of RSK2 are genetically linked to Coffin-Lowry syndrome, an X-linked disease characterized by cognitive disabilities, short stature, impaired heart function, skeletal anomalies, and abnormal characteristics of the face, trunk, and limbs (50, 51); skeletal muscle malfunction (52, 53); and possible defects of muscle differentiation. The involvement of RSK2 in the regulation of NFAT3 activation and nuclear localization provides a new understanding of a possible mechanism of pathological skeletal muscle malfunction. The identification of these targets may in turn contribute to the design of rational approaches, including drugs targeting the RSK2-NFAT signaling pathway, to treat these types of disorders.
Acknowledgments
We thank Dr. M. E. Greenberg (Division of Neuroscience, Children's Hospital, Harvard Medical School, Boston, MA) and Dr. J. A. Smith (Department of Pathology, Center for Cell Signaling, University of Virginia, Charlottesville, VA) for the RSK2 plasmid, Dr. R. J. Davis (Department of Biochemistry and Molecular Biology, University of Massachusetts Medical School, Worcester, MA) for pcDNA3-FLAG-JNK1, Dr. C W. Chow for the pcDNA3-FLAG-NFAT3 deletion mutants, and Dr. J. C Bruning (Institute for Genetics, Center for Molecular Medicine, Cologne, Germany) for the RSK2+/+ and RSK2−/− embryonic fibroblast cells. Anti-myosin heavy chain monoclonal antibody MF20 (developed by Donald A. Fischman) was obtained from the Developmental Studies Hybridoma Bank (developed under the auspices of NICHD, National Institutes of Health, and maintained by the Department of Biological Sciences of the University of Iowa (Iowa City, IA)). We also thank Andria Hansen for secretarial assistance.
Footnotes
This work was supported in part by the Hormel Foundation and National Institutes of Health Grants CA77646, CA88961, CA81064, and CA27502.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
The abbreviations used are: MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; RSKs, ribosomal S6 kinases; CREB, cAMP-responsive element-binding protein; NFAT, nuclear factor of activated T cell; aa, amino acid(s); JNK, c-Jun N-terminal kinase; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase kinase; MEF, mouse embryonic fibroblast; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; wt, wild-type; GST, glutathione S-transferase; GFP, green fluorescent protein; PVDF, polyvinylidene difluoride; HRP, horseradish peroxidase; HS, horse serum; NLS, nuclear localization signal; FL, full-length; CsA, cyclosporin A; siRNA, small interfering RNA.
References
- 1.Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearson G, Robinson F, Gibson TB, Xu BE, Karandikar M, Berman K, Cobb MH. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 3.Schaeffer HJ, Weber MJ. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho YY, Bode AM, Mizuno H, Choi BY, Choi HS, Dong Z. Cancer Res. 2004;64:3855–3864. doi: 10.1158/0008-5472.CAN-04-0201. [DOI] [PubMed] [Google Scholar]
- 5.Jones SW, Erikson E, Blenis J, Maller JL, Erikson RL. Proc Natl Acad Sci U S A. 1988;85:3377–3381. doi: 10.1073/pnas.85.10.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frodin M, Jensen CJ, Merienne K, Gammeltoft S. EMBO J. 2000;19:2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frodin M, Gammeltoft S. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 8.Nebreda AR, Gavin AC. Science. 1999;286:1309–1310. doi: 10.1126/science.286.5443.1309. [DOI] [PubMed] [Google Scholar]
- 9.Xing J, Ginty DD, Greenberg ME. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 10.Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S, Hanauer A, Allis CD. Science. 1999;285:886–891. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- 11.Blenis J. Proc Natl Acad Sci U S A. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis RJ. Mol Reprod Dev. 1995;42:459–467. doi: 10.1002/mrd.1080420414. [DOI] [PubMed] [Google Scholar]
- 13.Ward GE, Kirschner MW. Cell. 1990;61:561–577. doi: 10.1016/0092-8674(90)90469-u. [DOI] [PubMed] [Google Scholar]
- 14.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, Hanauer A, Karsenty G. Cell. 2004;117:387–398. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- 15.Cho YY, He Z, Zhang Y, Choi HS, Zhu F, Choi BY, Kang BS, Ma WY, Bode AM, Dong Z. Cancer Res. 2005;65:3596–3603. doi: 10.1158/0008-5472.CAN-04-3935. [DOI] [PubMed] [Google Scholar]
- 16.Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Science. 1988;241:202–205. doi: 10.1126/science.3260404. [DOI] [PubMed] [Google Scholar]
- 17.Baksh S, Widlund HR, Frazer-Abel AA, Du J, Fosmire S, Fisher DE, DeCaprio JA, Modiano JF, Burakoff SJ. Mol Cell. 2002;10:1071–1081. doi: 10.1016/s1097-2765(02)00701-3. [DOI] [PubMed] [Google Scholar]
- 18.Caetano MS, Vieira-de-Abreu A, Teixeira LK, Werneck MB, Barcinski MA, Viola JP. FASEB J. 2002;16:1940–1942. doi: 10.1096/fj.02-0282fje. [DOI] [PubMed] [Google Scholar]
- 19.Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Cell. 2003;113:657–670. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, Redondo JM. J Exp Med. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neal JW, Clipstone NA. J Biol Chem. 2003;278:17246–17254. doi: 10.1074/jbc.M300528200. [DOI] [PubMed] [Google Scholar]
- 22.Zaichuk TA, Shroff EH, Emmanuel R, Filleur S, Nelius T, Volpert OV. J Exp Med. 2004;199:1513–1522. doi: 10.1084/jem.20040474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang TT, Xiong Q, Enslen H, Davis RJ, Chow CW. Mol Cell Biol. 2002;22:3892–3904. doi: 10.1128/MCB.22.11.3892-3904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chow CW, Rincon M, Cavanagh J, Dickens M, Davis RJ. Science. 1997;278:1638–1641. doi: 10.1126/science.278.5343.1638. [DOI] [PubMed] [Google Scholar]
- 25.Yang T, Davis RJ, Chow CW. J Biol Chem. 2001;276:39569–39576. doi: 10.1074/jbc.M102961200. [DOI] [PubMed] [Google Scholar]
- 26.Ichida M, Finkel T. J Biol Chem. 2001;276:3524–3530. doi: 10.1074/jbc.M004275200. [DOI] [PubMed] [Google Scholar]
- 27.Yang TT, Xiong Q, Graef IA, Crabtree GR, Chow CW. Mol Cell Biol. 2005;25:907–920. doi: 10.1128/MCB.25.3.907-920.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Cho YY, Petersen BL, Bode AM, Zhu F, Dong Z. J Biol Chem. 2003;278:12650–12659. doi: 10.1074/jbc.M210368200. [DOI] [PubMed] [Google Scholar]
- 29.Rincon M, Flavell RA. Mol Cell Biol. 1997;17:1522–1534. doi: 10.1128/mcb.17.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Mol Cell Biol. 2002;22:7603–7613. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kegley KM, Gephart J, Warren GL, Pavlath GK. Dev Biol. 2001;232:115–126. doi: 10.1006/dbio.2001.0179. [DOI] [PubMed] [Google Scholar]
- 33.Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Cell. 2001;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- 34.Penn BH, Berkes CA, Bergstrom DA, Tapscott SJ. Mol Cell. 2001;8:245–246. doi: 10.1016/s1097-2765(01)00331-8. [DOI] [PubMed] [Google Scholar]
- 35.Perry RL, Parker MH, Rudnicki MA. Mol Cell. 2001;8:291–301. doi: 10.1016/s1097-2765(01)00302-1. [DOI] [PubMed] [Google Scholar]
- 36.Bushdid PB, Osinska H, Waclaw RR, Molkentin JD, Yutzey KE. Circ Res. 2003;92:1305–1313. doi: 10.1161/01.RES.0000077045.84609.9F. [DOI] [PubMed] [Google Scholar]
- 37.de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 38.Peng SL, Gerth AJ, Ranger AM, Glimcher LH. Immunity. 2001;14:13–20. doi: 10.1016/s1074-7613(01)00085-1. [DOI] [PubMed] [Google Scholar]
- 39.Ranger AM, Gerstenfeld LC, Wang J, Kon T, Bae H, Gravallese EM, Glimcher MJ, Glimcher LH. J Exp Med. 2000;191:9–22. doi: 10.1084/jem.191.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 41.Beals CR, Clipstone NA, Ho SN, Crabtree GR. Genes Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- 42.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Science. 1997;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 43.Sheridan CM, Heist EK, Beals CR, Crabtree GR, Gardner P. J Biol Chem. 2002;277:48664–48676. doi: 10.1074/jbc.M207029200. [DOI] [PubMed] [Google Scholar]
- 44.Zhu J, Shibasaki F, Price R, Guillemot JC, Yano T, Dotsch V, Wagner G, Ferrara P, McKeon F. Cell. 1998;93:851–861. doi: 10.1016/s0092-8674(00)81445-2. [DOI] [PubMed] [Google Scholar]
- 45.Schulz RA, Yutzey KE. Dev Biol. 2004;266:1–16. doi: 10.1016/j.ydbio.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 46.Friday BB, Horsley V, Pavlath GK. J Cell Biol. 2000;149:657–666. doi: 10.1083/jcb.149.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma KR, Mynhier MA, Miller RG. Neurology. 1993;43:527–532. doi: 10.1212/wnl.43.3_part_1.527. [DOI] [PubMed] [Google Scholar]
- 48.Abbott KL, Friday BB, Thaloor D, Murphy TJ, Pavlath GK. Mol Biol Cell. 1998;9:2905–2916. doi: 10.1091/mbc.9.10.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chow CW, Davis RJ. Mol Cell Biol. 2000;20:702–712. doi: 10.1128/mcb.20.2.702-712.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCandless SE, Schwartz S, Morrison S, Garlapati K, Robin NH. Am J Med Genet. 2000;95:93–98. doi: 10.1002/1096-8628(20001113)95:2<93::aid-ajmg1>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 51.Massin MM, Radermecker MA, Verloes A, Jacquot S, Grenade T. Acta Paediatr. 1999;88:468–470. doi: 10.1080/08035259950169909. [DOI] [PubMed] [Google Scholar]
- 52.Ishida Y, Oki T, Ono Y, Nogami H. Clin Orthop Relat Res. 1992;275:144–151. [PubMed] [Google Scholar]
- 53.Nelson GB, Hahn JS. Pediatrics. 2003;111:e197–e202. doi: 10.1542/peds.111.3.e197. [DOI] [PubMed] [Google Scholar]