

Doxorubicin (DOX) is a member of the anthracycline class of chemotherapeutic agents that are used for the treatment of many common human cancers, including aggressive non-Hodgkin’s lymphoma.[1,2] However, DOX is highly toxic in humans and can result in severe suppression of hematopoiesis, gastrointestinal toxicity,[3] and cardiac toxicity.[4] To date, several approaches, including delivery using liposomes (DOXIL),[5] have been developed to reduce the toxicity and enhance the clinical utility of this highly active antineoplastic agent.
Carbon nanotubes have unique mechanical, optical, and chemical properties, with broad potential biomedical applications, which include imaging and cancer therapeutics.[6–13] Carbon nanotubes have been used as novel in vitro delivery vehicles to effectively shuttle various biomolecules including drugs,[13,14] plasmid DNA,[15] and small interfering RNA (siRNA)[16,17] into cells by endocytosis.[18] The unique optical properties of single-walled carbon nanotubes (SWNTs) have been utilized for biological imaging[7–11] as well as cancer cell destruction.[19] Various research groups have studied the behavior of this material in animal models.[20–23] Suitably functionalized SWNTs have been found to be nontoxic in mice over several months, and can be gradually excreted by the biliary pathway from the reticuloendothelial system (RES) into feces, with the majority of SWNTs cleared within two months.[20,21] We explored the possibility of using supramolecular π–π stacking to load a cancer chemotherapy agent doxorubicin (DOX) onto branched polyethylene glycol (PEG) functionalized SWNTs for in vivo drug delivery applications. We found that our new drug formulation SWNT–DOX afforded a significantly enhanced therapeutic efficacy and a marked reduction in toxicity compared with free DOX and DOXIL.
It has been found that the surface of PEGylated SWNTs could be efficiently loaded with DOX by supramolecular π–π stacking (Figure 1a).[13] For the Hipco (high-pressure carbon monoxide) SWNTs used in this work, the binding energy of DOX on nanotubes is estimated to be approximately 48 kJmol−1 in water.[13] Branched PEG was used to achieve prolonged blood circulation time.[21] The SWNT–DOX complex had an average length of 100 nm and a diameter of 2–3 nm, as determined using AFM (Figure 1b). The loading ratio of DOX was determined by UV/Vis/near-infrared (NIR) absorption spectra (Figure 1c). Depending on the solution pH and DOX concentration, the DOX loading on nanotubes ranged from 1 to 4 grams DOX per gram of SWNTs.[13] We chose a loading of around 2.5 grams DOX per gram of SWNTs for the experiments in this work. We found that SWNT–DOX remained cytotoxic to cancer cells in vitro (Figure S1 in the Supporting Information), which is likely due to the release of DOX from the nanotubes inside cell endosomes and lysosomes under acidic conditions.[13]
Figure 1.
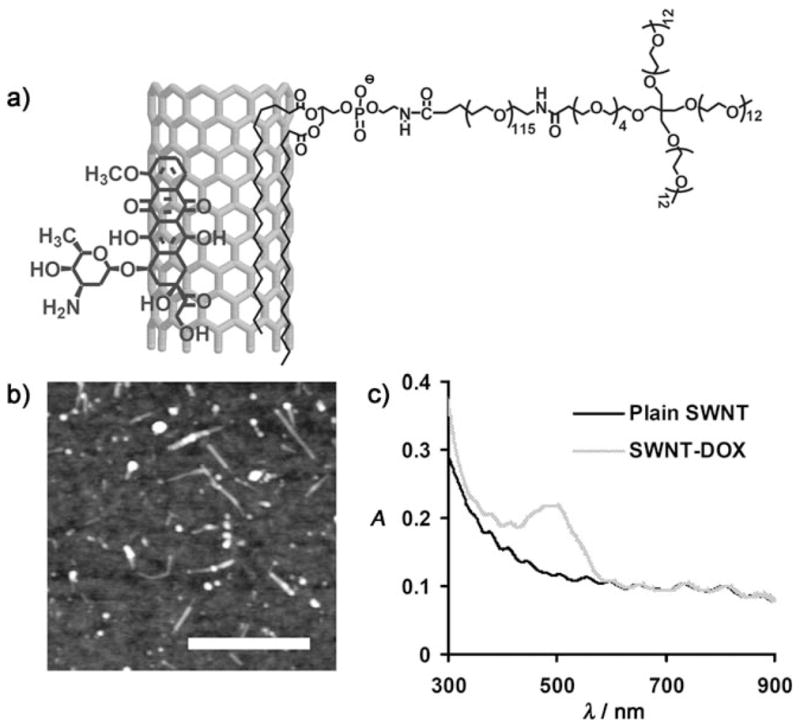
a) Representation of the SWNT–DOX complex. b) AFM image of SWNT–DOX complexes. The SWNT–DOX conjugates have an average length of 100 nm and a diameter of 2–3 nm. Scale bar: 250 nm. c) UV/Vis/NIR spectra of plain SWNTs and SWNT–DOX. The DOX loading was determined from the absorption peak at 490 nm.[13]
In order to investigate the in vivo pharmacokinetics and biodistribution, SWNT–DOX was injected via the tail vein into SCID mice bearing Raji lymphoma xenografts. Blood was drawn at different time points after injection, and the DOX concentrations were measured by fluorescence spectroscopy by following published protocols.[24] After being loaded onto the nanotubes, the DOX circulation half-life increased from 0.21 h for free DOX to 2.22 h for the SWNT–DOX formulation while the total area under the curve (AUC0–∞) also increased from 5.3 mghL−1 to 78.8 mghL−1 (Figure 2a). Mice were sacrificed 6 hours after injection, and the major organs were harvested and homogenized for DOX extraction.[24] The concentration of DOX in each organ was measured by fluorescence spectroscopy. The free DOX uptake by tumors was 0.68% of injected dose per gram of tissue (%IDg−1). The tumor uptake of DOX doubled to 1.51%IDg−1 for SWNT–DOX, which was likely due to the prolonged circulation of SWNT–DOX that facilitated the enhanced permeability and retention (EPR) effect observed with various nanomaterials (Figure 2b).[25] As expected, DOX accumulated in the RES including liver and spleen in the SWNT–DOX formulation. Despite the high RES uptake, SWNT–DOX did not show obvious hepatic toxicity from histology studies (Figure S4 in the Supporting Information). The combined biodistribution information of SWNTs determined by Raman scattering and DOX detected by fluorescence (Figure S2 in the Supporting Information) indicates that although most SWNT–DOX remains in the associated form in the first few hours after administration, the loaded DOX slowly dissociates from the nanotubes and is excreted via the kidneys. Most nanotubes are too large to be excreted via the kidneys and instead are slowly excreted via the biliary system into the feces.[21]
Figure 2.

Pharmacokinetics and biodistribution of two DOX formulations were studied by fluorescence spectroscopy. a) SWNT–DOX showed prolonged blood circulation compared with free DOX. Concentrations of DOX in blood from mice treated with free DOX and SWNT–DOX were measured by fluorescence spectroscopy at different time points after injection. b) SWNT–DOX had higher tumor-specific uptake and RES uptake than free DOX. Biodistribution of DOX in major organs of mice was measured 6 h after injection of free DOX and SWNT–DOX. Error bars were based on the standard error of the mean (SEM) of triplicate samples.
The in vivo therapeutic efficacy of the SWNT–DOX complex was also investigated. SCID mice bearing Raji lymphoma xenograft tumors were treated weekly with SWNT–DOX, free DOX, or DOXIL. The sizes of the subcutaneous tumors were measured over two weeks. The tumors in untreated controls rapidly increased by 7.53 ± 0.99 fold. The SWNT–DOX (5 mgkg−1) treated group showed a greater inhibition of tumor growth than free DOX at the equivalent dose (2.15 ± 0.16-fold tumor growth versus 2.90 ± 0.19, respectively, p =0.016; Figure 3a). Tumors in mice treated with plain SWNTs increased by 6.44 ± 0.42-fold, (versus untreated, p =0.34), thus indicating that SWNTs alone do not have therapeutic efficacy (Figure S3 in the Supporting Information). Mice treated with DOXIL (5 mgkg−1) developed severe treatment toxicity and mortality, as discussed below, but the DOXIL-treated mice that did remain alive after 14 days exhibited partial tumor regression to 0.88 ± 0.19 of the original size. Thus, loading of DOX onto SWNTs increased its therapeutic efficacy compared to free DOX, but not DOXIL.
Figure 3.

Supramolecular packing of DOX to SWNTs increased clinical efficacy in vivo. Raji tumor bearing SCID mice were treated with different DOX formulations once per week at day 0 and day 7. Tumor sizes and body weights were recorded three times per week. a) Tumor sizes of untreated (n =7), 5 mgkg−1 free DOX treated (n =10, 2 mice died in the second week), 5 mg kg−1 Doxil treated (n =5), 5 mgkg−1 SWNT-DOX treated (n =10) and 10 mgkg−1 SWNT-DOX treated (n =10) mice were measured during treatment. P values at 2 weeks: untreated versus DOX 5 mgkg−1, p =0.001; DOX 5 mgkg−1 versus SWNT–DOX 5 mgkg−1, p =0.016; DOX 5 mgkg−1 versus SWNT–DOX 10 mgkg−1, p <0.001; SWNT–DOX 5 mgkg−1 versus SWNT–DOX 10 mg kg−1, p =0.018. Mean tumor volume normalized to day 0 ± SEM is plotted. b) SWNT–DOX resulted in far less weight loss than DOX and DOXIL. Mean body weight normalized to day 0 ± SEM is plotted. c) Kaplan–Meier analysis. P values: DOX 5 mgkg−1 versus SWNT–DOX 5 mgkg−1 or 10 mgkg−1, p <0.001; DOXIL 5 mgkg−1 versus SWNT–DOX 5 mgkg−1, p =0.013; DOXIL 5 mgkg−1 versus SWNT–DOX 10 mgkg−1, p<0.001).
To evaluate the toxicity of the SWNT–DOX formulation, we measured the body weight of mice in each cohort. Mice treated with free DOX (5 mgkg−1) and DOXIL (5 mgkg−1) each exhibited a 19% and 17% decrease in weight within 2 weeks, respectively (Figure 3b), and appeared to be both thinner and weaker after treatment (Figure 4a, Figure S5 in the Supporting Information). Treatment with DOX and DOXIL resulted in 20% and 40% mortality, respectively. In marked contrast, mice treated with SWNT–DOX had stable weight and no mortalities. We note that SCID mice used in our study appeared to exhibit more DOX toxicity than as that reported for several other mouse strains including nude mice.[5,26] Thus, in our xenograft model of aggressive B cell lymphoma, loading of DOX onto SWNTs significantly attenuated the toxicity associated with free DOX and DOXIL.
Figure 4.

SWNT–DOX showed less in vivo toxicity than free DOX. a) Representative photos of mice from different groups were taken at the end of treatment. Mice became visibly thinner after treatment with free DOX. b) Gastrointestinal toxicity was observed in mice treated with free DOX but not in mice treated with SWNT–DOX. Histological sections of intestinal epithelium showed damage of the intestinal epithelium in the free DOX treated group. The arrows show the area of loss of columnar epithelial cells in tips of villi. Scale bars: 100 micrometers.
We considered that the reduced toxicity of SWNT–DOX might allow us to administer higher doses of DOX. Indeed, SWNT–DOX administered at a 10 mgkg−1 dose improved the treatment efficacy even further, as the tumor size at 2 weeks increased by only 1.64 ± 0.11-fold (Figure 3a). Furthermore, SWNT–DOX (10 mgkg−1) treatment resulted in neither a significant decrease in body weight nor an increase in mortality. In marked contrast, 10 mgkg−1 free DOX was a uniformly lethal dose within 2 weeks of treatment. Mouse tissues from SWNT–DOX (10 mgkg−1) did not exhibit gross microscopic gastrointestinal, cardiac, hepatic, or renal toxicity (Figure 4b and Figure S4 in the Supporting Information). However, when mice were treated with DOX alone (5 mgkg−1), disruption of the intestinal lining consistent with gastrointestinal mucositis was observed. Complete loss of columnar epithelial cells at the tips of villi occurred, thus exposing the underlying connective tissue core of lymphatic capillaries, blood capillaries and smooth muscle (Figure 4b). Therefore, by decreasing the gastrointestinal toxicity, SWNT–DOX also improved the dose intensity that could be achieved in mice, thus further enhancing the treatment efficacy.
Finally, to compare the overall clinical efficacy as defined by morbidity-free survival, a Kaplan–Meier analysis was performed. In this analysis, mice in each cohort were scored either when the tumor volume increased twofold, when body weight decreased by 10%, or when the mice died during treatment. In this model, we found that SWNT–DOX formulation was superior to DOX alone or DOXIL (Figure 3c). We conclude that supramolecular π–π stacking of a therapeutic agent onto SWNTs provides a novel method to efficiently deliver high-dose chemotherapy with an increase in clinical efficacy.
Our strategy has two critical advantages over a recently described approach in paclitaxel (PTX) delivery with SWNTs.[12] The first advantage is that DOX is directly loaded on the SWNT surface by simple noncovalent π–π stacking, thus achieving much higher drug loading capacity on nanotubes (up to 4 grams DOX per gram of SWNT), because of the ultrahigh surface area of SWNTs and the nature of noncovalent loading that is not limited by the number of available functional groups. It has been previously estimated that approximately 70–80% of the nanotube surface can be occupied by stacked DOX.[13] Our strategy can be easily extended to a wide range of lipophilic aromatic drugs, including daunarubicin, gefitinib, and camptothecin analogues.[27] The second advantage is that the DOX in the SWNT–DOX complex is stacked on the nanotube sidewalls and is thus protected by long branched PEG coating, which allows more stable drug loading and significantly prolonged blood circulation. These properties are not observed for the SWNT–PTX conjugate, in which the hydrophobic PTX is exposed to the external environment and reduces the circulation half-life of the conjugate.
There are several possible reasons why SWNT–DOX exhibits greater therapeutic efficacy and less toxicity to treated mice than equimolar amounts of free DOX. SWNT–DOX has a larger size that hampers its filtration through glomerulus, unlike free DOX, which is rapidly cleared out from blood circulation by renal excretion. The branched PEG coating attenuates the clearance of SWNT–DOX by macrophages in RES, thus causing SWNT–DOX to have a prolonged blood circulation half-life,[21] which allows repeated passing of drug conjugates through tumor vessels and increased tumor uptake by the EPR effect.
Our previous studies suggest that DOX-loaded SWNTs are stable at neutral pH but released in acidic environments.[13] The tumor microenvironment is slightly acidic,[28] which facilitates the dissociation of DOX from its SWNT carrier.[13] Normal organs and tissues, however, have a neutral pH, at which SWNT–DOX remains stable without releasing free DOX. SWNT–DOX that was detected in the intestinal tissues likely remained conjugated without release of free DOX because the epithelial cells that line the gastrointestinal tract maintain a neutral–alkaline environment.[29] Unlike DOX molecules, which can freely diffuse from blood vessels and reach intestinal tissues, including the epithelial intestinal lining, SWNT–DOX has a reduced ability to undergo such diffusion because of its larger size, which may limit access to intestinal tissues that are far from blood vessels in the intestine. This decrease in gastrointestinal epithelial toxicity is likely to contribute to the reduced toxicity of SWNT–DOX (Figure 4b). DOXIL has different pharmacokinetics, biodistribution, and toxicity profiles (e.g., skin toxicity)[30] than free DOX, However, our observation of different toxicity profiles between SWNT–DOX and DOXIL remains to be fully understood (Figure S5 in the Supporting Information). Further work is required to compare the clinical efficacy and toxic effects of SWNT-DOX and DOXIL at various doses.
In contrast to many other inorganic nanoparticles that contain toxic heavy metals, such as quantum dots, carbon nanotubes are composed purely of carbon atoms that are relatively nontoxic. Compared with other traditional drug carriers such as polymers and liposomes, SWNTs have valuable features that allow both the ability to image cancer cells as well as to deliver therapy. Near-infrared fluorescence, Raman scattering, or the photoacoustic contrast properties of SWNTs can be utilized for both in vitro and in vivo imaging.[7–11] The strong NIR optical absorption ability of SWNTs can be used for photothermal therapy,[19] which could be combined with chemotherapy delivered by nanotubes for enhanced treatment efficacy. In addition to passive tumor targeting, which relies on the EPR effect, active in vivo tumor targeting of SWNTs has been achieved by conjugation of targeting peptides[23] or antibodies[22] to nanotubes. Thus, carbon nanotubes have potential as an effective mechanism of drug delivery that could improve therapeutic efficacy and reduce drug-related toxicities while simultaneously serving as an imaging modality for cancer.
Experimental Section
DOX loading on functionalized SWNTs
Phospholipid-branched PEG (7 kDa) was synthesized as previously described.[21] Raw Hipco SWNTs (0.2 mgmL−1) were sonicated in a solution of phospholipid-branched PEG (0.2 mM) for 30 min with a cup-horn sonicator followed by centrifugation at 24000 g for 6 h to yield a suspension of SWNTs with noncovalent phospholipid-branched PEG coating in the supernatant.[13,16,21] Excess surfactant was removed by repeated filtration through a 100 kDa MWCO filter (Millipore) and extensive washing with water.
DOX loading onto PEGylated SWNTs was carried out by mixing DOX (0.5 mM) with the PEGylated SWNTs at a nanotube concentration of approximately 0.05 mgmL−1 (ca. 300 nM) at pH 8 overnight (ca. 18 hours). Unbound excess DOX was removed by filtration through a 100 kDa filter and washed thoroughly with water until the filtrate was no longer red (this color corresponds to free DOX). The formed SWNT–DOX complex was characterized as described previously[13] by UV/Vis/NIR absorbance spectra with a Cary 6000i spectrophotometer, and stored at 4°C.
In vivo circulation and biodistribution studies
Blood circulation was measured by drawing approximately 15 μL blood from the tail vein of Raji tumor bearing SCID mice after injection of free DOX or SWNT–DOX. The blood samples were dissolved in lysis buffer 1 (1% sodium dodecylsulfate (SDS), 1% Triton X-100, 40 mM tris(hydroxymethyl)aminomethane (tris) acetate, 10 mM ethylenediaminetetra-acetic acid (EDTA), 10 mM dithiothreitol (DTT) with brief sonication. The concentration of SWNTs in the blood was measured by using a Raman method.[21] DOX measurement was carried out following the protocol previously reported with minor modifications.[24] Briefly, DOX was extracted by incubating blood samples in HCl (0.75M, 1 mL) in isopropanol at −20°C overnight (ca. 18 hours). After centrifugation at 24000g for 15 min, the fluorescence of the supernatant was measured using a Fluorolog 3 fluorometer. Note that DOX loaded on SWNTs could be completely removed from the nanotubes by the extraction solution with around 100% recovery of fluorescence (the DOX fluorescence is quenched once loaded on nanotubes).
Biodistribution studies
Mice were sacrificed at 6 h after injection of free DOX or SWNT–DOX. The organs/tissues (0.1–0.2 g of each) were wet-weighed and homogenized in 0.5 mL of lysis buffer 2 (0.25M sucrose, 40 mM tris acetate, 10 mM EDTA) with a PowerGen homogenizer (Fisher Scientific). For DOX measurements, tissue lysate (200 μL) was mixed with Titron X-100 (10%, 100 μL). After strong vortexing, 1 mL of the extraction solution (0.75M HCl in IPA) was added and the samples were incubated at −20°C overnight (ca. 18 hours). After centrifugation at 24000g for 15 min, the fluorescence of the supernatant was measured.
Treatment of in vivo lymphoma xenograft model
SCID mice were subcutaneously injected with 10 million Raji cells. Treatment was initiated when the tumors reached an approximate size of 400 mm3 (2–3 weeks after tumor inoculation). Tumor-bearing mice were intravenously injected on a weekly basis with different formulations of DOX, including free DOX, SWNTs, SWNT–DOX, and DOXIL at 5 mgkg−1 of normalized DOX dose (or 10 mgkg−1 for SWNT–DOX) as well as related controls. The tumor sizes were measured by calipers three times a week and volume was calculated according to the formula ((tumor length) × (tumor width)2)/2. Relative tumor volumes were calculated as V/V0 (V0 is the tumor volume when the treatment was initiated). Mice were weighed with the relative body weights normalized to their initial weights. Tumors and organs were collected at the end of treatment, fixed in formalin, embedded in paraffin, and sectioned using a microtome. Standard hematoxylin and eosin (H&E) staining was carried for histological examinations.
Statistical analysis
Quantitative data were expressed as mean ± standard errors of the mean (SEM). Means were compared using Student’s t-test. P values less than 0.05 were considered statistically significant.
Supplementary Material
Footnotes
In memory of Evelyn Hagel
This work was supported in part by the National Institutes of Health (NIH)–National Cancer Institution (NCI) grants R01 CA135109-01 (H.D.), 1R01 CA89305-01A1 (D.W.F.), 3R01 CA89305-0351 (D.W.F.), 1R01 CA105102 Lymphoma Program Project (D.W.F.), NIH-NCI In Vivo Cellular and Molecular Imaging Center Grant P50 (D.W.F.), NIH–NCI Center for Cancer Nanotechnology Excellence Focused on Therapeutic Response at Stanford (H.D.), Burroughs Wellcome Fund (D.W.F.), the Damon Runyon Foundation (D.W.F.), the Leukemia and Lymphoma Society (D.W.F., A.C.F.), a Stanford Bio-X Initiative Grant (H.D., D.W.F.), an Enscyse grant (H.D.), and a Stanford Graduate Fellowship (Z.L.).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200902612.
Contributor Information
Dean W. Felsher, Email: dfelsher@stanford.edu.
Hongjie Dai, Email: hdai@stanford.edu.
References
- 1.Yustein JT, Dang CV. Curr Opin Hematol. 2007;14:375. doi: 10.1097/MOH.0b013e3281bccdee. [DOI] [PubMed] [Google Scholar]
- 2.DeVita VT, Jr, Young RC, Canellos GP. Cancer. 1975;35:98. doi: 10.1002/1097-0142(197501)35:1<98::aid-cncr2820350115>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 3.Morelli D, Menard S, Colnaghi MI, Balsari A. Cancer Res. 1996;56:2082. [PubMed] [Google Scholar]
- 4.Von Hoff DD, Layard MW, Basa P, Davis HL, Jr, Von Hoff AL, Rozencweig M, Muggia FM. Ann Intern Med. 1979;91:710. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 5.Gabizon A, Shmeeda H, Barenholz Y. Clin Pharmacokinet. 2003;42:419. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 6.Liu Z, Tabakman S, Welsher K, Dai H. Nano Res. 2009;2:85. doi: 10.1007/s12274-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De La Zerda A, Zavaleta C, Keren S, Vaithilingam S, Bodapati S, Liu Z, Levi J, Ma TJ, Oralkan O, Cheng Z, Chen X, Dai H, Khuri-Yakub BP, Gambhir SS. Nat Nanotechnol. 2008;3:557. doi: 10.1038/nnano.2008.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherukuri P, Bachilo SM, Litovsky SH, Weisman RB. J Am Chem Soc. 2004;126:15638. doi: 10.1021/ja0466311. [DOI] [PubMed] [Google Scholar]
- 9.Welsher K, Liu Z, Daranciang D, Dai H. Nano Lett. 2008;8:586. doi: 10.1021/nl072949q. [DOI] [PubMed] [Google Scholar]
- 10.Zavaleta C, Zerda AdL, Liu Z, Keren S, Cheng Z, Schipper M, Chen X, Dai H, Gambhir SS. Nano Lett. 2008;8:2800. doi: 10.1021/nl801362a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, Li X, Tabakman SM, Jiang K, Fan S, Dai H. J Am Chem Soc. 2008;130:13540–13541. doi: 10.1021/ja806242t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Cancer Res. 2008;68:6652. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Sun XM, Nakayama-Ratchford N, Dai HJ. Acs Nano. 2007;1:50. doi: 10.1021/nn700040t. [DOI] [PubMed] [Google Scholar]
- 14.Bianco A, Kostarelos K, Prato M. Curr Opin Chem Biol. 2005;9:674. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Wu DC, Zhang WD, Jiang X, He CB, Chung TS, Goh SH, Leong KW. Angew Chem. 2005;117:4860. doi: 10.1002/anie.200500042. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:4782. [Google Scholar]
- 16.Kam NW, Liu Z, Dai H. J Am Chem Soc. 2005;127:12492. doi: 10.1021/ja053962k. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Winters M, Holodniy M, Dai HJ. Angew Chem. 2007;119:2069. doi: 10.1002/anie.200604295. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:2023. [Google Scholar]
- 18.Kam NWS, Liu ZA, Dai HJ. Angew Chem. 2006;118:591. [Google Scholar]; Angew Chem Int Ed. 2006;45:577. [Google Scholar]
- 19.Shi Kam NW, O’Connell M, Wisdom JA, Dai HJ. Proc Natl Acad Sci USA. 2005;102:11600. doi: 10.1073/pnas.0502680102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schipper ML, Nakayama-Ratchford N, Davis CR, Kam NW, Chu P, Liu Z, Sun X, Dai H, Gambhir SS. Nat Nanotechnol. 2008;3:216. doi: 10.1038/nnano.2008.68. [DOI] [PubMed] [Google Scholar]
- 21.Liu Z, Davis C, Cai W, He L, Chen X, Dai H. Proc Natl Acad Sci USA. 2008;105:1410. doi: 10.1073/pnas.0707654105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDevitt MR, Chattopadhyay D, Kappel BJ, Jaggi JS, Schiffman SR, Antczak C, Njardarson JT, Brentjens R, Scheinberg DA. J Nucl Med. 2007;48:1180. doi: 10.2967/jnumed.106.039131. [DOI] [PubMed] [Google Scholar]
- 23.Liu Z, Cai W, He L, Nakayama N, Chen K, Sun X, Chen X, Dai H. Nat Nanotechnol. 2007;2:47. doi: 10.1038/nnano.2006.170. [DOI] [PubMed] [Google Scholar]
- 24.Laginha KM, Verwoert S, Charrois GJ, Allen TM. Clin Cancer Res. 2005;11:6944. doi: 10.1158/1078-0432.CCR-05-0343. [DOI] [PubMed] [Google Scholar]
- 25.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J Controlled Release. 2000;65:271. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 26.Huang SK, Mayhew E, Gilani S, Lasic DD, Martin FJ, Papahadjopoulos D. Cancer Res. 1992;52:6774. [PubMed] [Google Scholar]
- 27.Liu Z, Robinson JT, Sun XM, Dai H. J Am Chem Soc. 2008;130:10876. doi: 10.1021/ja803688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerweck LE, Seetharaman K. Cancer Res. 1996;56:1194. [PubMed] [Google Scholar]
- 29.Heitzmann D, Warth R. Physiol Rev. 2008;88:1119. doi: 10.1152/physrev.00020.2007. [DOI] [PubMed] [Google Scholar]
- 30.Uziely B, Jeffers S, Isacson R, Kutsch K, Wei-Tsao D, Yehoshua Z, Libson E, Muggia FM, Gabizon A. J Clin Oncol. 1995;13:1777. doi: 10.1200/JCO.1995.13.7.1777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.