

Abstract
Caffeine may very well be the most frequently ingested neuroactive drug in the world. Mechanistically, caffeine has been reported to affect cell cycle function, induce programmed cell death or apoptosis and perturb key cell cycle regulatory proteins. Although the effects of caffeine have been heavily investigated, much of the research data regarding caffeine's effects on cell cycle and proliferation seem ambiguous. One important factor may be that caffeine has been used experimentally in numerous cell types under a variety of conditions at concentrations ranging from micromolar to high millimolar. Physiologically, achieving experimental blood levels of caffeine would be extremely difficult without adverse side effects. Therefore, the relevance of experimental data obtained by using high concentrations of caffeine is not clear and may account for some of the discrepancies in the literature. This review attempts to reconcile data regarding the cellular effects of caffeine by examining reported effects on cell cycle, proliferation and apoptosis with careful attention to differences in experimental conditions and caffeine concentration utilized.
Keywords: Caffeine
1. Introduction
Caffeine (1,3,7-trimethylxanthine; Fig. 1) is a natural stimulatory compound that is ubiquitously present in many plants including cocoa beans, coffee beans, cola nuts, and tea leaves. Caffeine is also commonly used as a stimulant to prevent sleepiness and is found in several over-the-counter medications including some pain remedies. Because of its frequent and common consumption in tea, coffee and soft drinks, caffeine may very likely be the most frequently ingested neuroactive drug in the world [1]. For many years, caffeine has been generally believed to suppress cell proliferation [2], abolish chemical- or radiation-induced delays in cell cycle progression [3,4] and enhance the toxicity of radiation and anti-cancer agents [3,5,6]. Caffeine has also been shown to inhibit ultraviolet B (UVB)-induced skin cancer in mice [7] and recent work by Nomura et al. [8] showed that caffeine (0.5 mM) suppressed epidermal growth factor (EGF)-induced malignant cell transformation.
Fig. 1.
Chemical structure of caffeine.
Mechanistically, caffeine has been reported to affect cell cycle function, induce programmed cell death or apoptosis and perturb key regulatory proteins, including the tumor suppressor protein, p53 [9,10]. Following DNA damage, p53 has a major influence on whether a cell will live or die. Cells exposed to DNA damaging agents such as γ-radiation or many chemotherapeutic agents pause in their cell cycle progression to allow time for DNA repair. If the damage is too extensive, the affected cells will undergo apoptosis. Both cell cycle arrest and apoptosis are normally mediated by p53 activities. In general, cells expressing wildtype p53 arrest in G1, whereas cells that do not express p53 or express a mutant p53 arrest mainly in G2.
Although the effects of caffeine have been heavily investigated, much of the research data regarding caffeine's effects on cell cycle and proliferation seem ambiguous. One important factor may be that caffeine has been used experimentally in numerous cell types under a variety of conditions at concentrations ranging from micromolar to high mM. Physiologically, achieving a 2 mM blood level of caffeine would require the simultaneous consumption of over 100 cups of coffee [11]. Therefore, the relevance of experimental data obtained by using greater than 1 mM caffeine is not clear and may account for some of the discrepancies in the literature. This review will attempt to reconcile data regarding the cellular effects of caffeine by examining reported effects on cell cycle, proliferation and apoptosis with careful attention to differences in experimental conditions and caffeine concentration utilized.
2. Regulation of cell cycle: brief overview
A review of the cell cycle and its various components will facilitate an understanding of the reported effects of caffeine on cell cycle function, proliferation and apoptosis. The cell cycle consists of four distinct phases that include the S phase where DNA duplication occurs, the M phase or mitosis during which the DNA is separated and the cell divides, and two gap phases. Gap1 or G1 is the phase before DNA synthesis and G2 is the period before mitosis begins. Progression through the cell cycle depends on the presence of mitotic or growth factor stimulation. As long as stimulation occurs, cells will continue to proliferate, whereas in the absence of stimulation, cells stop dividing and enter the resting or quiescent state known as G0. At a certain point in G1, referred to as the restriction point or R, cell growth no longer requires mitotic stimulation and cells are irreversibly committed to enter the S phase [12].
Exposure of cells to DNA damaging agents such as γ-radiation and many chemotherapeutic agents induces a pause or arrest in their cell cycle progression at distinct points, called checkpoints, to allow time for DNA repair. The G1/S checkpoint delays entry into S phase and the G2/M checkpoint temporarily prevents entry into mitosis. Both checkpoints allow time for DNA repair and prevent replication of damaged DNA and propagation of genetic abnormalities. However, if the DNA damage is too extensive, the injured cells will undergo cell death or apoptosis. Both apoptosis and cell cycle arrest are normally mediated by the tumor suppressor protein p53.
Cyclin-dependent kinases (Cdks) control the individual checkpoints between successive phases of the cell cycle and are activated by their interaction with specific proteins called cyclins. The different cyclins and Cdks form complexes that are activated at distinct points in the cell cycle sequence. To assure a one-way cell cycle progression, the cyclin proteins are synthesized and degraded in a highly regulated and synchronized pattern throughout the cycle. When quiescent cells are stimulated to enter the cell cycle, cyclin D is the first cyclin protein produced and it associates with Cdk4 and/or Cdk6 to form an active complex during G1. Cyclin E is the second cyclin to be created and it associates with Cdk2. The expression of this complex peaks in late G1 and is required for entry into S phase [13].
During this same time period, two Cdk inhibitors, p21cip1 and p27kip1, are also synthesized. The p21cip1 and p27kip1 proteins appear early in G1 to promote the formation of the active cyclin D/Cdk4,6 complex but also delay the activation of the cyclin E/Cdk2 complex. Eventually, enough of the cyclin D/Cdk4,6 complex accumulates to suppress the p21cip1/p27kip1 inhibition of cyclin E/Cdk2, allowing the activation of this complex. DNA damage results in the elevation of p53, which causes an increased transcription of p21, which is a primary cause of G1/S delay (Fig. 2A).
Fig. 2.
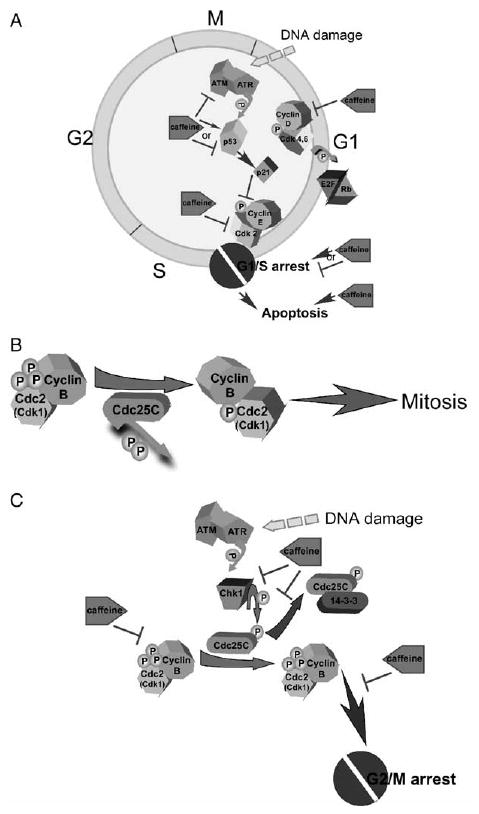
Effects of caffeine. (A) DNA-damage induces G1 arrest and caffeine has varying effects (see text for details). (B) G2 checkpoint— Under normal conditions, the cyclinB/Cdc2 complex is dephosphorylated and activated by Cdc25C and mitosis can proceed. (C) DNA-damage induces phosphorylation and activation of Chk1, which then phosphorylates and deactivates Cdc25C. This results in Cdc25C forming a complex with 14-3-3 and Cdc25C is not able to dephosphorylate and activate the cyclinB/Cdc2 complex. The net result is G2/M arrest to allow time for DNA repair. Caffeine acts on various components to reverse the G2/M arrest.
The cyclin E/Cdk2 and the cyclin D/Cdk4,6 complexes act together to phosphorylate and inactivate the retinoblastoma protein (Rb) and these phosphorylations of Rb are required for full progression into the S-phase. Active Rb is bound to the E2F transcription factor and phosphorylation of Rb results in the release of E2F, which allows E2F to facilitate the transcription of several important cell cycle proteins, including cyclin E and cyclin A.
Once cells enter S-phase, cyclin E is degraded and Cdk2 then associates with cyclin A, which is expressed at the G1/S boundary. Both cyclin E/Cdk2 and cyclin A/Cdk2 activities appear to be essential for the initiation and completion of DNA replication and activity of cyclin A/Cdk2 is required for progression through S phase [14]. Expression of cyclin B occurs after cyclin A synthesis and remains high throughout G2 and mitosis. Cyclin B associates with Cdc2 (Cdk1) but this complex is maintained in a phosphorylated, inactive but stabilized form until the G2/M boundary at which point the complex is dephosphorylated and activated by Cdc25 phosphatases. Both cyclin A and cyclin B now associate with Cdc2 (Cdk1) to promote entry into mitosis and cyclin A is then degraded. Entry into mitosis (G2–M) requires activated cyclin B/Cdc2 and exit from mitosis requires cyclin B degradation.
Phosphorylation of the cyclin/Cdk complexes results in their deactivation and most of the cyclin/Cdk complexes are activated by dephosphorylation mediated by the Cdc25 (A,B,C) phosphatases. For example, Cdc25A dephosphorylation of cyclin E/Cdk2 is required for S phase progression and Cdc25C dephosphorylation of cyclin B/Cdk1 is crucial for G2 to M progression (Fig. 2B). Cyclin D is expressed throughout the cell cycle as long as mitotic stimulation occurs. In the absence of stimulation, cyclin D is phosphorylated and inactivated by glycogen synthase kinase 3 (GSK3), which targets cyclin D for destruction by the proteosome. Cdc25 proteins are inactivated by phosphorylation by the checkpoint regulatory proteins, Chk1 and Chk2. Cdc25A phosphorylation leads to its degradation [15] and Cdc25C phosphorylation leads to its inactivation by the formation of a complex with 14-3-3 proteins in the cytoplasm [16] (Fig. 2C). Inactivation of Cdc25C results in an inability to activate the cyclin B1/Cdc2 complex and therefore prevents progression into the G2/M phase.
Caffeine has generally been reported to induce G1/S arrest and to reverse or abrogate the G1/S and G2/M checkpoint delay periods (Fig. 2B and C). Importantly, when caffeine is combined with DNA-damaging agents, the potency of the DNA-damaging agent is increased markedly [4,17–19]. In particular, studies have shown that reversal of DNA-damage-induced checkpoint function or arrest with caffeine [17,18] doubled the cytotoxicity of ionizing radiation (IR) in human cancer cell lines. Many of these studies suggested that the increased toxicity of IR or chemical agents is associated with caffeine's inhibition of the G2 checkpoint activity (i.e. a reversal of the agent-induced arrest), which would accelerate movement of the cell cycle through the G2/M checkpoint, thus leaving cells less time to repair DNA damage and eventually lead them to apoptosis [20]. The effect seems to be most potent in p53-deficient cells, which can only arrest in G2 [17,18]. Although caffeine's effects on G2 have been most widely documented, caffeine has also been reported to have variable effects at G1. Interestingly, caffeine appears to exert its effects on G1 through both p53-dependent and p53-independent mechanisms.
3. Effects of caffeine on cell cycle progression
3.1. G1/S
When DNA damage occurs, p53 is phosphorylated (Ser15) by the ataxia telangiectasia mutated (ATM) protein and the AT-related homolog, ATR (Fig. 2A) [21], which results in p53 stabilization and accumulation [22]. ATM and ATR also prevent p53 degradation through their phosphorylation of mouse double minute 2 (MDM2) [23], which disrupts MDM2's association with p53 thereby preventing p53 ubiquitination and degradation. In addition, ATM phosphorylates Chk2, which in turn phosphorylates p53 (Ser20), also disrupting the inhibitory binding of MDM2 [24]. An important p53 target gene is p21, the protein product of which suppresses activation of cyclin D/Cdk4/6 and cyclin E/Cdk2 and leads to G1 arrest (Fig. 2A) [25,26].
Many early studies indicated that caffeine directly induced G1 arrest in mammalian cells [3,27]. On the other hand, almost 15 years ago, Kastan et al. [22] reported that caffeine treatment (4 mM) blocked both the G1 arrest and the induction of p53 protein expression in myeloblastic leukemia (ML-1) cells after γ-irradiation. In contrast with these results [22], Qi et al. [28] recently reported that caffeine (5 mM) alone or in combination with IR induced p53-independent G1 phase arrest and enhanced apoptosis in human A549 lung adenocarcinoma cells. This differed from the effect of IR alone, which induced a p53-dependent G1 arrest. However, a lower concentration (0.5 mM) of caffeine alone or in combination with IR could not induce G1 arrest. On the other hand, caffeine at either 0.5 or 5 mM reversed IR-induced G2 arrest suggesting that caffeine may act by different mechanisms to impair IR-induced G1 and G2 checkpoints and that G2 may be more sensitive to caffeine's effects. This also strongly supports the idea that the experimental concentration of caffeine is extremely important. In this same study [28], IR-induced arrest at G1 was found to be strictly dependent on p53, however, caffeine could still arrest cells at G1 in p53-deficient cells, suggesting that p53 was not required for caffeine's effects. In addition, IR-induced arrest still occurred at G2 in p53-deficient cells. These investigators [28] also examined the effect of caffeine (5 mM) with or without IR on Cdk2 and Cdc2 activities. Caffeine, IR or a combination inhibited Cdk2 activity, which would explain the G1 arrest because inhibition of cyclin E/Cdk2 is known to lead to G1-phase arrest [25,26]. This work was supported by Gong et al. [29], who reported that during G1, inactive cyclin E accumulates and only those cells with cyclin E expression above a distinct threshold level can enter the S-phase of the cell cycle. In p53-deficient cells, only caffeine could inhibit Cdk2 activity suggesting that IR and caffeine acted by different mechanisms. In contrast to Qi et al. [28], we [30] have found that caffeine (8 mM) did not affect the activity of active cyclin E/Cdk2 in JB6 cells, suggesting cell type specificity. Further, Qin et al. [31] reported that MOLT-4 lymphocyte leukemia cells treated with 50 mM caffeine entered into S-phase with a level of cyclin E well below the threshold level for untreated cells. These investigators [31] suggested that caffeine activated Cdc25C, which in turn activated Cdk2 resulting in an increased rather than a decreased cell proliferation [31]. However, the relevance of these results may be debatable due to the extremely high concentration of caffeine utilized.
Lower levels of caffeine (0.25–1 mM) were shown to suppress cell cycle progression at the G0/G1 phase [30]. In this work, JB6 Cl41 mouse epidermal cells were synchronized at G0 by serum deprivation (36 h in 0.1% FBS) and then exposed to 5% fetal bovine serum (FBS) as a mitogenic stimulus. Treatment of cells with caffeine inhibited FBS-stimulated cell proliferation (IC50= 0.7 mM) by inducing G0/G1 arrest but without causing apoptosis. The inhibitory effect appeared to result from the indirect suppression of cyclin D1/Cdk4 activation and subsequent inhibition of Rb phosphorylation (Ser780, Ser807, Ser811). As indicated earlier, phosphorylation of Rb by Cdk4/6 and Cdk2 [32] occurs at the G0/G1 and G1/S transitions [33]. This phosphorylation leads to the disruption of the Rb/E2F transcription factor complex, resulting in the release of active E2F and its subsequent activation of many genes required for cell cycle progression through S phase [34]. Hashimoto et al. [30] also found that caffeine inhibited the time-dependent phosphorylation of protein kinase B (Akt Thr308) and its substrate, GSK-3β (Ser9). GSK-3β normally phosphorylates cyclin D1 (Thr286), which triggers cyclin D1 degradation, thus decreasing the activity of Cdk4 [35]. Because caffeine has also been reported to directly inhibit PI-3 kinase activity [36], Hashimoto et al. [30] suggested that the inhibitory effects of caffeine on cell growth signaling through Akt/GSK-3β may result from the direct inhibition of PI-3 kinase, which is upstream of Akt/GSK-3β.
Kaufmann et al. [37] showed that caffeine (2 mM) induced about a 75% inhibition of G1/S progression in diploid human fibroblast cells. They further found that ATM and p53 were not required because AT cells and cells with inactive p53 (HPV16E6) also arrested in G1 in the presence of caffeine. ATM-dependent phosphorylation of p53 and transactivation of p21Cip1/Waf1 were also not affected by caffeine [37]. Caffeine did not appear to inhibit cyclin B1/Cdk1, which promotes G2/M progression, nor cyclin E/Cdk2 or cyclin A/Cdk2, which are required for initiation of replication and progression through S. Instead, they [37] suggested that caffeine appeared to target the cyclin D1/Cdk4,6 complex agreeing with the work of Hasimoto et al. [30].
Additional studies have shown that treatment with caffeine resulted in a suppression of Chk1 activity. UVC, the carcinogen benzo(a)pyrene dihydrodiol epoxide, hydroxyurea, or psoralen plus UVA radiation have been reported to activate Chk1 [38–41], which phosphorylates and inactivates Cdc25A. Activation of Cdc25A is required for activation of cyclin E/Cdc2 and entry into S phase. Others have also shown that UV and IR can induce Cdc25A degradation and this degradation can be suppressed by caffeine [15]. Chk1 was activated in mouse fibroblasts treated with methyl methanesulfonate (MMS) and 4-amino-1,8-naphthal-imide (4-AN), an inhibitor of poly(ADP-ribose) polymerase (PARP) activity, but the activation could be suppressed by caffeine (1 mM) [42]. Overall, caffeine appears to have variable effects at the G1/S checkpoint. The specific effect is heavily dependent on experimental conditions, cell type and caffeine concentration. Generally, caffeine alone seems to induce cell cycle arrest and under some conditions apoptosis, by both p53-dependent and p53-independent mechanisms. At least two reports indicated that caffeine might act on the cyclinD1/Cdk4 complex resulting in G1/S arrest. On the other hand, several reports suggested that caffeine might also suppress DNA-damage agent-induced Chk1 activation or prevent Cdc25 inactivation thereby reversing G1/S checkpoint activation and driving cells into S phase.
3.2. G2/M
Thirty years ago, caffeine was first reported to directly inhibit DNA repair that resulted in DNA-damage induced cell death [43]. However, a few years later in 1982, Lau and Pardee [4] established that caffeine actually prevented G2-arrest that was induced after treatment with nitrogen mustard and allowed mitosis to occur before completing DNA repair, thereby causing apoptosis. As indicated earlier, p53 (Ser15) is phosphorylated by ATM and ATR when DNA damage occurs. However, even though G1 arrest is highly dependent on p53 activation, G2 arrest can still occur in p53-deficient cells. Besides p53, the checkpoint kinases, Chk1 and Chk2, are critical in the regulation of cell cycle checkpoint G2. Evidence indicates that Chk1 (Ser345, Ser317) is preferentially phosphorylated and activated by ATR [44] whereas Chk2 (Thr68) is activated and phosphorylated mainly by ATM [45,46]. Activated Chk1 and Chk2 phosphorylate and inactivate the Cdc25C (Ser216) phosphatase by facilitating its binding with the 14-3-3 protein (Fig. 2C) [45,47]. The 14-3-3 protein binds with and prevents the activation of the cyclin B/Cdc2 complex [48], thereby inhibiting mitosis. Thus, the net result of ATM/ATR activation and phosphorylation of Chk1/2 is G2/M arrest (Fig. 2C).
More than 35 years ago, caffeine was reported to sensitize cells to UV radiation toxicity [49]. The increased sensitivity was suggested to be associated with the ability of caffeine to reverse the G2-phase arrest that is normally observed in cells exposed to radiation or other DNA-damaging agents [50]. Presumably, disruption at the G2/M checkpoint by caffeine leaves cells less time to repair DNA damage by driving them through mitosis [4] resulting in apoptosis [20,51]. Vavrova et al. [52] found that caffeine (2 mM) eliminated IR-induced G2 phase arrest (up to 72 h). Apoptosis was induced by IR both in the presence and absence of caffeine at 2–4 days, but also at days 5–7 and 10 in the presence of caffeine [52]. High concentrations of caffeine have been shown to directly induce apoptosis [53] and the increased radiosensitivity of certain cells associated with caffeine has been reported to correspond to enhanced apoptosis [54,55]. Radiosensitivity is defined as the relative susceptibility of cells, tissues, organs or organisms to the harmful effect of IR. Caffeine has been reported to increase apoptosis and mitochondrial damage in human HL-60 and U937 cancer cells [56], human neuroblastoma cells (10 mM) [57], human pancreatic adenocarcinoma cells (4 mM) [58], human A549 lung adenocarcinoma cells (5 mM) [28], and mouse epidermal JB6 Cl41 cells (450 μM) [9]. Fernandez et al. [59] determined that caffeine at a concentration of 3 mM or less actually increased viability of Chinese hamster ovary cells. On the other hand, greater than 5 mM caffeine was cytotoxic. The higher concentrations corresponded with increased caspase-8 activity and DNA fragmentation, which generally agrees with the work of Jafari et al. [60] who reported that caffeine at less than 5 mM prevented apoptosis of alveolar macrophages from rat lung, whereas 5–20 mM caffeine induced apoptosis.
Arrest at G2 induced by DNA damaging agents has been reported to be associated with suppression of Cdc2 (Cdk1) activity [61]. Numerous studies indicated that caffeine inhibits the agent-induced G2 arrest by promoting an increased dephosphorylation and activation of Cdc2 thereby driving its activation in complex with cyclin B [62]. Winters et al. [63] also confirmed that caffeine increased the dephosphorylation of Cdc2, an event that would effectively override the inhibition of the cyclin/Cdk complexes and accelerate mitosis [64]. Qi et al. [28] found that Cdc2 activity was reduced in cells exposed to IR and the suppression was not only reversed by caffeine but Cdc2 activity seemed to be enhanced. Cdc2 is known to be inactivated by phosphorylation (Thr14/15) and activated by dephosphorylation by Cdc25C [65]. Qi et al. [28] reported that caffeine appears to act by inhibiting IR-induced Cdc2 phosphorylation (Thr15) and blocking the interaction of Cdc25C with 14-3-3, which would effectively prevent Cdc2 inactivation and push the cell cycle through the G2/M transition.
Some evidence suggests that caffeine might target cancer cells with little effect on normal cells. Jha et al. [66] examined caffeine's effect on G2-phase delays in three human normal cell lines (GM2149, GM4626, AG1522) and three human tumor cell lines (HeLa, MCF7, OVGI) after exposure to gamma rays. All six cell lines displayed similar G2 phase delays after radiation. However, the gamma-ray-induced G2-phase delays were eliminated by caffeine in the tumor cell lines but were unaffected in all of the normal cell lines [66]. Florensa et al. [67] found that caffeine did not eliminate hydroxyurea-induced arrest in non-transformed cell lines, including normal rat kidney (NRK) and NIH 3T3 fibroblasts. However, caffeine had variable effects on tumor cell lines, including a reversal of the hydroxyurea-induced G2-phase arrest in various colorectal cancer cells. Overall, most research data indicate that caffeine does indeed reverse DNA-damage-induced G2 arrest and drive cells through a fatal mitosis. Many reports suggested that the mechanism may be related to caffeine's ability to reactivate Cdc2 and Cdc25C activities. In addition, some data indicated that caffeine may preferentially target tumor cells while having little effect on normal cells.
4. Molecular targets for caffeine-induced cell cycle arrest
4.1. ATM and ATR
Initiating the appropriate checkpoint responses to double-strand DNA breaks requires the function of ATM and ATR [68]. This is supported by studies in ataxia telangiectasia (AT) patients and cells in which DNA damage cannot induce cell cycle arrest [69]. The cellular effects of caffeine resemble some defects observed in AT cells [70]. Although somewhat controversial, ATM (IC50 0.2 mM) and ATR (IC50 1.1 mM) have been identified as the primary molecular targets of caffeine in vitro [70]. ATM and ATR belong to the phosphatidylinositol-3 kinase (PI-3 kinase)-related kinase family [71]. PI-3 kinase has been implicated in signaling pathways important in tumor development [72] and caffeine has also been shown to inhibit various isoforms of PI-3 kinase (IC50 0.075– 1.0 mM) [36,70] and components of the PI-3 kinase signaling pathway, including Akt [73]. Besides ATM/ATR, other protein targets of caffeine may include p53 [9], mammalian target of rapamycin (mTOR; IC50 0.4 mM), DNA-dependent protein kinase (DNA-PK; IC50 <10 mM) and Chk1 (IC50 <5 mM) [70] but not Chk2 (Thr68), which was shown to be insensitive to caffeine (1–10 mM) [74].
Most of the evidence supporting ATM/ATR as primary targets of caffeine was obtained from in vitro assays or assays in which activation of ATM/ATR substrates was found to be suppressed by caffeine. Sarkaria et al. [70] reported that A549 cells exposed to γ-irradiation after treatment with caffeine (1 mM) displayed a reversal of the radiation-induced G2/M arrest and inhibition of γ-radiation and UV (50 J/m2)-induced phosphorylation of p53 (Ser15). Caffeine's effect on p53 was attributed to an inhibition of ATM/ATR phosphorylation of p53 (Ser15) in vitro. Blasina et al. [75] showed that treatment of HeLa cells with caffeine (2 mM) prior to irradiation indirectly decreased the radiation-induced activation of Chk2 presumably due to the direct inhibition of ATM, which has been reported to be required for radiation-induced activation of Chk2 [76]. Zhou et al. [77] found that G2/M arrest corresponded with an increased phosphorylation (Ser216) and inactivation of Cdc25C and that pretreatment with caffeine (2 mM) inhibited the phosphorylation. They also showed that ATM phosphorylation and activation of Chk2 (Thr68) was also suppressed by caffeine (2 mM) after IR exposure. Caffeine (10 mM) has also been shown to reverse resveratrol-induced activation of ATM/ATR-Chk1/2 in human ovarian carcinoma Ovcar-3 cells [78]. Caffeine actually appeared to act by directly suppressing the resveratrol-induced Chk1/Chk2-mediated phosphorylation and deactivation of Cdc25C and Cdc2.
In marked contrast, Cortez et al. [79] recently found that treatment of HCT116 cells with caffeine (1–16 mM) or hydroxyurea actually promoted an increased phosphorylation of ATM/ATR substrates, Chk1, replication protein A, Chk2 and p53. Caffeine and hydroxyurea also increased ATM autophosphorylation and Chk2 phosphorylation in hTERT-RPE1 cells suggesting an increased activation of ATM. With IR, ATM was maximally autophosphorylated and caffeine had no effect on that phosphorylation. In addition, caffeine consistently caused a slight increase in Chk2 (Thr68) and Chk1 phosphorylation induced by IR. Importantly, the G2/M arrest induced in response to ionizing radiation or hydroxyurea was abrogated by caffeine (1 mM) treatment without a corresponding decrease in ATM-ATR-dependent signaling. These data suggest that although caffeine has been reported to be an inhibitor of ATM-ATR kinase activity in vitro, it can block checkpoints without inhibiting ATM-ATR activation in vivo [79]. Of ATM, ATR and DNA-PK, DNA-PK has been suggested to be the most resistant to caffeine [70]. However, Block et al. [80] recently reported that caffeine inhibited immunoprecipitated and purified DNA-PK as well as cellular DNA-PK with an IC50 of 0.2–0.6 mM, suggesting that caffeine cannot accurately differentiate between ATM, ATR and DNA-PK-dependent phosphorylations.
4.2. p53
The tumor suppressor protein, p53, is believed to be the primary mediator of cell cycle arrest and induction of apoptosis in most cells lines in response to DNA damage. However, the research literature suggests that caffeine may exert its effects, including induction of apoptosis, not only through p53-dependent pathways [81], but also through p53-independent pathways that may involve activation and phosphorylation of Chk1 and Chk2 [64]. Alan Conney's group has been investigating the effects of topical and oral administration of caffeine in vivo for several years. They found that oral administration of caffeine alone (0.44 mg/ml) as the sole source of drinking fluid for 18–23 weeks inhibited the formation and size of non-malignant and malignant UVB-induced tumors in SKH-1 mice exposed to UVB (30 mJ/cm2) twice a week for 22–23 weeks [7]. They also found that prefeeding caffeine enhanced UV-induced increases in the number of p53-positive cells, p21-positive cells, and apoptotic sunburn cells in the epidermis [82]. Later work [83] confirmed that topical treatment with caffeine (6.2 μmol) following 20 weeks of UVB (twice/week) decreased the number of non-malignant and malignant skin tumors per mouse by 44%. Caffeine treatment increased caspase-3 positive cells in non-malignant skin tumors by 87% and in squamous cell carcinomas by 92% with no effect on normal epidermis [83]. In later work, topical application of caffeine (6.2 μmol) immediately after UVB (60 mJ/m2) in p53+/+ or p53-/- female mice enhanced the UVB-induced increase in apoptotic sunburn cells (6 h later) by 127 and 563%, respectively. The UVB-induced increase in apoptotic sunburn cells in p53−/− cells was 10–30% of that observed in p53+/+ mice. These results suggested that UVB-induced increases in apoptosis in the epidermis of wildtype mice occurred mostly through p53-dependent pathways and that topical application of caffeine enhanced UVB-induced increases in apoptosis by p53-independent pathways. This work supported earlier work from this group [84] in which they found that topical application of caffeine had only a small stimulatory effect on UVB-induced increases in the level of wild-type p53 protein and these changes were not related temporally to caffeine-induced increases in apoptotic cells. Most recently this group reported that UVB (30 mJ/cm2) exposure of female SKH-1 hairless mice two times a week resulted in the formation of cellular patches that expressed a mutated (but not wildtype) p53 protein [85]. Oral administration of caffeine (0.4 mg/ml) in drinking water inhibited UVB-induced formation of mutant p53 positive patches by about 40%. Topical applications of caffeine (6.2 μmol) starting immediately after discontinuation of UVB treatment enhanced the rate and extent of disappearance of the mutant p53-positive patches. Investigators concluded that the chemopreventive effect of caffeine or green tea might occur by a proapoptotic effect preferentially in early precancerous lesions [85]. Most recently, this group [86] irradiated female SKH-1 hairless mice with UVB (30 mJ/cm2) twice a week for 10–20 weeks and found that UVB resulted in the formation of cellular patches expressing a mutant but not wild-type p53 protein. The number and size of the patches increased progressively with continued UVB treatment but oral administration of caffeine (0.4 mg/ml) as the sole source of drinking fluid during irradiation with UVB, inhibited UVB-induced formation of mutant p53 positive patches. Topical applications of caffeine (6.2 μmol) once a day 5 days a week starting immediately after discontinuation of UVB treatment enhanced the rate and extent of disappearance of the mutant p53-positive patches[86]. This same group [87] also reported that oral treatment of mice with caffeine or green tea during chronic UVB-irradiation changed the mutation profile of the p53 gene in early mutant p53-positive epidermal patches, and topical applications of caffeine after discontinuation of chronic UVB-irradiation specifically eliminated patches harboring homozygous p53 mutations [87].
4.2.1. p-53-independent effects
A great deal of evidence suggests that caffeine exerts its most cytotoxic effects in cells that do not express p53 or express a non-functional p53 protein [88]. Powell et al. [18] have shown that p53-knockout fibroblasts exhibit greater caffeine-induced sensitivity to the deadly effects of IR compared with p53 wildtype cells. Yao et al. [55] found that irradiation induced apoptosis in wildtype thymocytes and bone marrow mononuclear cells but that p53-deficient cells were relatively resistant to IR, an effect that was attributed to a decreased Cdc2 activity. However, treatment with caffeine (4 mM) prevented the inactivation of Cdc2 and G2 arrest, and also induced apoptosis in p53-deficient cells with little additional effect on p53 wildtype cells. Higuchi et al. [54] found that caffeine (2 mM) reduced the radiation-induced G2 arrest in two rat yolk sac tumor cell lines each having a different p53 status. Apoptosis, which was not observed after either irradiation or caffeine treatment alone, was induced by irradiation in combination with caffeine in cells with a mutant-type p53 through a p53-independent pathway [54]. Janicke et al. [89] previously reported that caspase-3 deficient MCF-7 breast carcinoma cells [90] were especially resistant to IR-induced apoptosis although they harbor a functional p53 gene. Recently, this same group [91] reported that caffeine (1 mM) abrogated the IR-induced G2/M arrest in MCF-7 cell lines with or without caspase 3 expression but only induced apoptosis in MCF-7 cells with caspase 3 reintroduced. Caffeine sensitized MCF-7 caspase 3-expressing cells to IR-induced apoptosis independently of p53 activation and independently of caffeine's ability to reverse G2/M arrest [91]. Bache et al. [92] have previously shown that a strong irradiation-induced G2/M arrest was coupled with high clonogenic survival and a low rate of apoptosis after irradiation. More recently, they [93] reported that caffeine (4 mM) reduced the irradiation-induced G2/M block in human sarcoma cell lines (US8-93 and LMS6-93), which both have a mutated p53 but the abrogation of the block was not associated with a strong induction of apoptosis. Minemoto et al. [94] reported that treatment of FL-amnion cells (express wildtype p53) with adriamycin induced p53 activation. However, these cells arrested in G2 but not G1. Further investigation indicated that the p53 target protein, p21, was not expressed in these cells in spite of adriamycin-induced p53 accumulation. Besides its role in mediating G1 arrest [95], p21 is known to be involved in the G2/M transition [96]. Formation of the p21/cyclin B1/Cdc2 complex inhibits Cdc2 kinase activity [97], preventing cells from entering mitosis. The p21 protein can also interfere with the Cdc25C dephosphorylation and activation of Cdc2 to maintain G2/M arrest [98]. Adriamycin treatment of p21 deficient, p53 wildtype or p53 deficient cells resulted in G2 arrest suggesting that neither p53 nor p21 was required for G2 arrest induced by adriamycin [94]. Further, caffeine (2.5 mM) treatment activated Cdc2 and induced apoptosis in cells arrested at G2, thus reversing the adriamycin-induced block. Saito et al. [99] found that caffeine plus the adenovirus-mediated transfer of tumor suppressor phosphatase and tensin homologue deleted from chromosome 10 (PTEN) suppressed proliferation and induced apoptosis in numerous colorectal and prostate cancer cell lines (HCT116—p53 wildtype; HCT116— p53 deleted; SW480—p53 mutant; DLD-1—p53 mutant; CCD-18Co—p53 wildtype; Du145 prostate— p53 mutant; LnCAP—p53 wildtype/PTEN mutant) with variable p53 expression. PTEN normally suppresses PI-3 kinase/Akt signaling and its overexpression can induce apoptosis. Saito et al. [99] observed that all cell lines regardless of p53 status arrested in G2 except LnCAP (mutant PTEN), which was arrested in G1. Caffeine alone did not induce apoptosis but did eliminate the PTEN-induced G2/M arrest and down-regulated Akt in tumor cells and, notably, these effects were not observed in normal fibroblasts.
4.2.2. p-53-dependent effects
In contrast to the above-mentioned studies, some data suggested that radiosensitization or reversal of G2 arrest induced by caffeine is not selective for cells expressing non-functional p53. Takagi et al. [51] analyzed cell cycle and apoptosis after X-irradiation with and without caffeine in p53 wildtype (LCL) and mutant EBV-immortalized (LCL286A) lymphoblastoid cells, human leukemia p53 wildtype (KOPM46) and mutant p53 (KOPM28), and human leukemia HL60 cells, which do not express p53. All p53 mutant or p53-deleted cells displayed a G2 delay following X-IR and also resistance to apoptosis. Notably, treatment with caffeine (4 mM) and X-IR resulted in an abrogation of the G2 delay and enhanced radiation-induced apoptosis in p53 wildtype cells but not in p53 mutant cells. Caffeine also increased Cdc2 kinase activity but these changes were not associated with increased radiation-induced apoptosis [51]. Ribeiro et al. [100] showed that two bladder cancer lines (UCRU-BL-13 and UCRU-BL-28) expressing wildtype p53 failed to arrest at the G2 checkpoint after radiation, but caffeine still induced radiosensitization. In contrast, treatment of p53 mutant (UCRU-BL-17/2) cells reversed the radiation-induced G2 arrest but did not induce radiosensitization. No effects on radiosensitivity were seen in RT112 cells, which express a non-functional p53, at lower caffeine doses (2 mM) but at higher doses (4 and 10 mM) caffeine caused both abrogation of radiation-induced G2 arrest and increased radiosensitization. In none of the cell lines examined did caffeine treatment and/or irradiation result in apoptosis.
Low concentrations of caffeine (50–450 μM) induced apoptosis in JB6 Cl41 cell and the apoptosis was p53-dependent because it did not occur in p53 knockout cells [9]. Caffeine induced phosphorylation of p53 (Ser15) and increased p53 activation. Bax, a p53 target protein [101], and cleaved caspase 3, a key executioner of apoptosis expression also increased in a time and dose-dependent manner. Bax is a pro-apoptotic member of the Bcl-2 family of proteins and is known to form heterodimers with the anti-apoptotic Bcl-2 protein in vivo. The molar ratio of Bcl-2 to Bax has been shown to establish whether apoptosis is induced or inhibited in many tissues [102]. Caspase 3 is known to be one of the key executioners of apoptosis and its activation requires proteolytic processing of its inactive zymogen into activated p17 and p19 subunits [103]. Bax drives the release of cytochrome c from the mitochondria and cytochrome c release activates caspase 3 [104]. Dubrez et al. [105] reported that caffeine sensitizes the human non-small cell lung cancer H358 cell line to p53-mediated apoptosis by inducing mitochondrial translocation and a conformational change in the Bax protein. Caffeine (4 mM) was shown to decrease proliferation of NB4 promyelocytic leukemia cells, which express wildtype p53 [10]. The inhibition of proliferation was associated with an induction of G2/M phase cell cycle arrest followed by apoptosis mediated by a mitochondrial- and caspase 3-dependent pathway. Caffeine induced an increased expression and phosphorylation of p53 (Ser15), p21Cip1/Waf and the pro-apoptotic Bax protein, but had no effect on cyclin A, B, D, E, MDM2 or phosphorylation of p38. However, the inhibition was also associated with a reduced level of Cdc25C, which dephosphorylates and activates Cdc2 (Cdk1) [106]. Anti-sense inhibition of p53 reduced caffeine-induced G2/M phase cell cycle arrest and apoptosis [10]. Others have previously shown that caffeine significantly increased the cytocidal effects of DNA-damaging agents through the promotion of cell cycle progression by inhibiting delays in G2/M phase and particularly in a human osteosarcoma cell line expressing wild-type p53 [107]. Recently, Tsuchiya et al. [108] observed that Saos2 cells transfected with p53 had a slower growth rate than the Saos2 p53 deficient cell line. However, the Saos2/p53 line was two times more sensitive to cisplatin, an effect that was enhanced by 0.5 mM caffeine.
4.3. Other targets
Several studies indicated that caffeine has numerous effects other than those reported for cell cycle. For example, caffeine also inhibits the nucleotide exchange activity of RCC1 [109], alkaline phosphatase activity [110], and phosphodiesterase activity [111]. Caffeine has been shown to induce immediate early genes that are important regulators of cellular transcription [112]; to decrease cholesterol synthesis in glial cell cultures [113]; and to antagonize the action of adenosine in the metabolism of neurotransmitters [114]. Caffeine has been reported to inhibit cell cycle-dependent DNA repair induced by a variety of physical and chemical mutagens [115–117]. Caffeine inhibits the repair of gaps in daughter-strand DNA in UVC-damaged cells and the effects are especially notable in xeroderma pigmentosum (XP) variant cells [118].
5. Some controversies on the effects of caffeine on cell cycle
Earlier studies suggested that caffeine was a teratogen in rabbits, mice and rats, and placental blood flow and embryonic growth were also severely reduced by caffeine [114,119]. Others have also observed that in contrast to enhancing the killing of cells by IR and other DNA damaging agents, caffeine may have the opposite effect on the cytotoxic effect of certain chemotherapeutic agents such as paclitaxel [120]. Paclitaxel (Taxol) induces G2/M arrest and has been used in several clinical trials [121,122]. Although caffeine (20 mM) has been reported to enhance apoptosis induced by paclitaxel in human beast cancer cells (MCF-7) [123], Kitamoto et al. [120] found that combining caffeine (1 mM) with paclitaxel had no effect on the surviving fraction of A549 lung adenocarcinoma cells, but that higher concentrations (5 or 20 mM) clearly suppressed the cytotoxic effect of paclitaxel in a dose- and time-dependent manner. On the other hand, caffeine combined with paclitaxel suppressed cell proliferation. Caffeine alone caused early G1 accumulation and paclitaxel alone caused an early increase in G2/M and a decrease in G1, but the cell cycle changes induced by paclitaxel were suppressed by caffeine.
Deplanque et al. [124] reported that the sensitizing properties of caffeine probably did not result from a G2/M block override but rather from caffeine's inhibition of proliferation independent of p53 status. They contended that the presence of caffeine actually slowed down the cell cycle rather than reversing the G2/M arrest. Based on recent work, this group [125] concluded that in contrast to admitted concepts, and independently of p53 alteration, caffeine does not appear to be a chemo/radiosensitizer at reasonable therapeutic concentrations. This conclusion was based on their findings that caffeine (2 mM) suppressed cisplatin- or UVC-induced apoptosis in human thyroid papillary carcinoma K1 cells and also had no effect on clonogenic survival of these carcinoma cells. Others have shown that caffeine can protect against ionizing radiation or genotoxic treatments [126]. Caffeine was also reported to diminish the cytotoxic effects of adriamycin, ellipticine and mitoxantrone [127].
6. Concluding remarks
Caffeine may very well be the most frequently ingested neuroactive drug in the world. Mechanistically, caffeine has been reported to affect cell cycle function, induce programmed cell death or apoptosis and perturb key cell cycle regulatory proteins. Although the effects of caffeine have been heavily investigated, much of the research data regarding caffeine's effects on cell cycle and proliferation seem ambiguous. One important factor may be that caffeine has been used experimentally in numerous cell types under a variety of conditions at concentrations ranging from micromolar to high mM. At lower concentrations (<1 mM), caffeine has been reported to induce p53 phosphorylation and p53-dependent apoptosis associated with increased expression of pro-apoptotic Bax and caspase-3 [9]. On the other hand, low concentrations have also been reported to have disturbing effects on nervous system development [128]. At concentrations of 1–2 mM, caffeine may induce G1 arrest [28,30], whereas concentrations 2–4 mM appeared to block G1 arrest [22,37] and induce apoptosis [105]. Depending on concentration and status of p53 expression, caffeine has been reported to induce [9,10,28,51,52,57,91], not induce [93] or even protect against apoptosis [54,59,60,94,125]. ATM/ATR have been widely accepted as the primary protein targets of caffeine [70], but this contention has been disputed [79,80]. Although exceptions have been noted [124], most concentrations of caffeine almost always reverse the DNA-damage induced G2/M block [17,52,54,55,70,79,91]. However, higher concentrations of caffeine can directly induce G2 arrest [10]. Usually, but not always [51], the effect of caffeine is most potent in p53 deficient cells [18,54,94,99,100] making it an attractive agent for treating p53 deficient cancers. Caffeine at various concentrations generally seems to enhance the toxicity of IR and DNA-damaging chemical agents, but exceptions have been noted [120,126,127]. Unfortunately, the high levels of caffeine required may be too toxic for humans. This suggests that the development of caffeine analogs with biologic activity similar to the parent compound, but with less toxicity, could be very useful in combination with DNA-damaging agents for treating cancers.
Contributor Information
Ann M. Bode, Email: ambode@hi.umn.edu.
Zigang Dong, Email: zgdong@hi.umn.edu.
References
- 1.Coffee, tea, mate, methylxanthines and methylglyoxal, IARC working group on the evaluation of carcinogenic risks to humans. Lyon, 27 February to 6 March 1990. IARC Monogr Eval Carcinog Risks Hum. 1991;51:1–513. [PMC free article] [PubMed] [Google Scholar]
- 2.Levi-Schaffer F, Touitou E. Xanthines inhibit 3T3 fibroblast proliferation. Skin Pharmacol. 1991;4:286–290. doi: 10.1159/000210963. [DOI] [PubMed] [Google Scholar]
- 3.Tolmach LJ, Jones RW, Busse PM. The action of caffeine on x-irradiated Hela cells. I. Delayed inhibition of DNA synthesis. Radiat Res. 1977;71:653–665. [PubMed] [Google Scholar]
- 4.Lau CC, Pardee AB. Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc Natl Acad Sci USA. 1982;79:2942–2946. doi: 10.1073/pnas.79.9.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busse PM, Bose SK, Jones RW, Tolmach LJ. The action of caffeine on x-irradiated hela cells. Ii. Synergistic lethality. Radiat Res. 1977;71:666–677. [PubMed] [Google Scholar]
- 6.Busse PM, Bose SK, Jones RW, Tolmach LJ. The action of caffeine on x-irradiated hela cells. Iii. Enhancement of x-ray-induced killing during g2 arrest. Radiat Res. 1978;76:292–307. [PubMed] [Google Scholar]
- 7.Lou YR, Lu YP, Xie JG, Huang MT, Conney AH. Effects of oral administration of tea, decaffeinated tea, and caffeine on the formation and growth of tumors in high-risk skh-1 mice previously treated with ultraviolet b light. Nutr Cancer. 1999;33:146–153. doi: 10.1207/S15327914NC330205. [DOI] [PubMed] [Google Scholar]
- 8.Nomura M, Ichimatsu D, Moritani S, Koyama I, Dong Z, Yokogawa K, Miyamoto K. Inhibition of epidermal growth factor-induced cell transformation and akt activation by caffeine. Mol Carcinog. 2005;44:67–76. doi: 10.1002/mc.20120. [DOI] [PubMed] [Google Scholar]
- 9.He Z, Ma WY, Hashimoto T, Bode AM, Yang CS, Dong Z. Induction of apoptosis by caffeine is mediated by the p53, bax, and caspase 3 pathways. Cancer Res. 2003;63:4396–4401. [PubMed] [Google Scholar]
- 10.Ito K, Nakazato T, Miyakawa Y, Yamato K, Ikeda Y, Kizaki M. Caffeine induces g2/m arrest and apoptosis via a novel p53-dependent pathway in nb4 promyelocytic leukemia cells. J Cell Physiol. 2003;196:276–283. doi: 10.1002/jcp.10289. [DOI] [PubMed] [Google Scholar]
- 11.Lelo A, Miners JO, Robson R, Birkett DJ. Assessment of caffeine exposure: caffeine content of beverages, caffeine intake, and plasma concentrations of methylxanthines. Clin Pharmacol Ther. 1986;39:54–59. doi: 10.1038/clpt.1986.10. [DOI] [PubMed] [Google Scholar]
- 12.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci USA. 1974;71:1286–1290. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin e, a nuclear protein essential for the g1-to-s phase transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girard F, Strausfeld U, Fernandez A, Lamb NJ. Cyclin a is required for the onset of DNA replication in mammalian fibroblasts. Cell. 1991;67:1169–1179. doi: 10.1016/0092-8674(91)90293-8. [DOI] [PubMed] [Google Scholar]
- 15.Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J. Rapid destruction of human cdc25a in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 16.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and g2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of cdc25c on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 17.Russell KJ, Wiens LW, Demers GW, Galloway DA, Plon SE, Groudine M. Abrogation of the g2 checkpoint results in differential radiosensitization of g1 checkpoint-deficient and g1 checkpoint-competent cells. Cancer Res. 1995;55:1639–1642. [PubMed] [Google Scholar]
- 18.Powell SN, DeFrank JS, Connell P, Eogan M, Preffer F, Dombkowski D, et al. Differential sensitivity of p53(−) and p53(+) cells to caffeine-induced radiosensitization and override of g2 delay. Cancer Res. 1995;55:1643–1648. [PubMed] [Google Scholar]
- 19.Fingert HJ, Chang JD, Pardee AB. Cytotoxic, cell cycle, and chromosomal effects of methylxanthines in human tumor cells treated with alkylating agents. Cancer Res. 1986;46:2463–2467. [PubMed] [Google Scholar]
- 20.Murnane JP. Cell cycle regulation in response to DNA damage in mammalian cells: a historical perspective. Cancer Metastasis Rev. 1995;14:17–29. doi: 10.1007/BF00690208. [DOI] [PubMed] [Google Scholar]
- 21.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by atm in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 22.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 23.Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, et al. Atm-dependent phosphorylation of mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 25.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. P21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 26.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 cdk-interacting protein cip1 is a potent inhibitor of g1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 27.Pardee AB, James LJ. Selective killing of transformed baby hamster kidney (bhk) cells. Proc Natl Acad Sci USA. 1975;72:4994–4998. doi: 10.1073/pnas.72.12.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi W, Qiao D, Martinez JD. Caffeine induces tp53-independent g(1)-phase arrest and apoptosis in human lung tumor cells in a dose-dependent manner. Radiat Res. 2002;157:166–174. doi: 10.1667/0033-7587(2002)157[0166:citigp]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 29.Gong J, Traganos F, Darzynkiewicz Z. Threshold expression of cyclin e but not d type cyclins characterizes normal and tumour cells entering s phase. Cell Prolif. 1995;28:337–346. doi: 10.1111/j.1365-2184.1995.tb00075.x. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto T, He Z, Ma WY, Schmid PC, Bode AM, Yang CS, Dong Z. Caffeine inhibits cell proliferation by g0/g1 phase arrest in jb6 cells. Cancer Res. 2004;64:3344–3349. doi: 10.1158/0008-5472.can-03-3453. [DOI] [PubMed] [Google Scholar]
- 31.Qin J, Tao D, Chen X, Feng Y, Hu J, Reed E, et al. Down-regulation of cyclin e expression by caffeine promotes cancer cell entry into the s-phase of the cell cycle. Anticancer Res. 2004;24:2991–2995. [PubMed] [Google Scholar]
- 32.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, et al. The consensus motif for phosphorylation by cyclin d1-cdk4 is different from that for phosphorylation by cyclin a/e-cdk2. Eur Mol Biol Org J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- 33.Taya Y. Rb kinases and rb-binding proteins: new points of view. Trends Biochem Sci. 1997;22:14–17. doi: 10.1016/s0968-0004(96)10070-0. [DOI] [PubMed] [Google Scholar]
- 34.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 35.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin d1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foukas LC, Daniele N, Ktori C, Anderson KE, Jensen J, Shepherd PR. Direct effects of caffeine and theophylline on p110 delta and other phosphoinositide 3-kinases. Differential effects on lipid kinase and protein kinase activities. J Biol Chem. 2002;277:37124–37130. doi: 10.1074/jbc.M202101200. [DOI] [PubMed] [Google Scholar]
- 37.Kaufmann WK, Heffernan TP, Beaulieu LM, Doherty S, Frank AR, Zhou Y, et al. Caffeine and human DNA metabolism: the magic and the mystery. Mutat Res. 2003;532:85–102. doi: 10.1016/j.mrfmmm.2003.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joerges C, Kuntze I, Herzinge T. Induction of a caffeine-sensitive s-phase cell cycle checkpoint by psoralen plus ultraviolet a radiation. Oncogene. 2003;22:6119–6128. doi: 10.1038/sj.onc.1206613. [DOI] [PubMed] [Google Scholar]
- 39.Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, Kaufmann WK. An atr- and chk1-dependent s checkpoint inhibits replicon initiation following uvc-induced DNA damage. Mol Cell Biol. 2002;22:8552–8561. doi: 10.1128/MCB.22.24.8552-8561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian chk1 during DNA replication arrest: a role for chk1 in the intra-s phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo N, Faller DV, Vaziri C. Carcinogen-induced s-phase arrest is chk1 mediated and caffeine sensitive. Cell Growth Differ. 2002;13:77–86. [PubMed] [Google Scholar]
- 42.Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(adp-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J Biol Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 43.Arlett CF, Harcourt SA, Broughton BC. The influence of caffeine on cell survival in excision-proficient and excision-deficient xeroderma pigmentosum and normal human cell strains following ultraviolet-light irradiation. Mutat Res. 1975;33:341–346. doi: 10.1016/0027-5107(75)90209-2. [DOI] [PubMed] [Google Scholar]
- 44.Zhao H, Piwnica-Worms H. Atr-mediated checkpoint pathways regulate phosphorylation and activation of human chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaturvedi P, Eng WK, Zhu Y, Mattern MR, Mishra R, Hurle MR, et al. Mammalian chk2 is a downstream effector of the atm-dependent DNA damage checkpoint pathway. Oncogene. 1999;18:4047–4054. doi: 10.1038/sj.onc.1202925. [DOI] [PubMed] [Google Scholar]
- 46.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates chk2 in vivo and in vitro. Proc Natl Acad Sci USA. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the chk1 checkpoint pathway in mammals: linkage of DNA damage to cdk regulation through cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 48.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 49.Rauth AM. Evidence for dark-reactivation of ultraviolet light damage in mouse l cells. Radiat Res. 1967;31:121–138. [PubMed] [Google Scholar]
- 50.Labanowska J, Beetham KL, Tolmach LJ. Caffeine-induced modulation of the lethal action of X-rays on chinese hamster v79 cells. Radiat Res. 1988;115:176–186. [PubMed] [Google Scholar]
- 51.Takagi M, Shigeta T, Asada M, Iwata S, Nakazawa S, Kanke Y, et al. DNA damage-associated cell cycle and cell death control is differentially modulated by caffeine in clones with p53 mutations. Leukemia. 1999;13:70–77. doi: 10.1038/sj.leu.2401247. [DOI] [PubMed] [Google Scholar]
- 52.Vavrova J, Marekova-Rezacova M, Vokurkova D, Szkanderova S, Psutka J. Caffeine induces a second wave of apoptosis after low dose-rate gamma radiation of hl-60 cells. Radiat Environ Biophys. 2003;42:193–199. doi: 10.1007/s00411-003-0209-4. [DOI] [PubMed] [Google Scholar]
- 53.Efferth T, Fabry U, Glatte P, Osieka R. Expression of apoptosis-related oncoproteins and modulation of apoptosis by caffeine in human leukemic cells. J Cancer Res Clin Oncol. 1995;121:648–656. doi: 10.1007/BF01218522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Higuchi K, Mitsuhashi N, Saitoh J, Maebayashi K, Sakurai H, Akimoto T, Niibe H. Caffeine enhanced radio-sensitivity of rat tumor cells with a mutant-type p53 by inducing apoptosis in a p53-independent manner. Cancer Lett. 2000;152:157–162. doi: 10.1016/s0304-3835(99)00449-8. [DOI] [PubMed] [Google Scholar]
- 55.Yao SL, Akhtar AJ, McKenna KA, Bedi GC, Sidransky D, Mabry M, et al. Selective radiosensitization of p53-deficient cells by caffeine-mediated activation of p34cdc2 kinase. Nat Med. 1996;2:1140–1143. doi: 10.1038/nm1096-1140. [DOI] [PubMed] [Google Scholar]
- 56.Dai Y, Yu C, Singh V, Tang L, Wang Z, McInistry R, et al. Pharmacological inhibitors of the mitogen-activated protein kinase (mapk) kinase/mapk cascade interact synergistically with ucn-01 to induce mitochondrial dysfunction and apoptosis in human leukemia cells. Cancer Res. 2001;61:5106–5115. [PubMed] [Google Scholar]
- 57.Jang MH, Shin MC, Kang IS, Baik HH, Cho YH, Chu JP, et al. Caffeine induces apoptosis in human neuroblastoma cell line sk-n-mc. J Korean Med Sci. 2002;17:674–678. doi: 10.3346/jkms.2002.17.5.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gururajanna B, Al-Katib AA, Li YW, Aranha O, Vaitkevicius VK, Sarkar FH. Molecular effects of taxol and caffeine on pancreatic cancer cells. Int J Mol Med. 1999;4:501–507. doi: 10.3892/ijmm.4.5.501. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez MJ, Lopez A, Santa-Maria A. Apoptosis induced by different doses of caffeine on chinese hamster ovary cells. J Appl Toxicol. 2003;23:221–224. doi: 10.1002/jat.910. [DOI] [PubMed] [Google Scholar]
- 60.Jafari M, Rabbani A. Studies on the mechanism of caffeine action in alveolar macrophages: caffeine elevates cyclic adenosine monophosphate level and prostaglandin synthesis. Metabolism. 2004;53:687–692. doi: 10.1016/j.metabol.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 61.Blasina A, Paegle ES, McGowan CH. The role of inhibitory phosphorylation of cdc2 following DNA replication block and radiation-induced damage in human cells. Mol Biol Cell. 1997;8:1013–1023. doi: 10.1091/mbc.8.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poon RY, Chau MS, Yamashita K, Hunter T. The role of cdc2 feedback loop control in the DNA damage checkpoint in mammalian cells. Cancer Res. 1997;57:5168–5178. [PubMed] [Google Scholar]
- 63.Winters ZE, Ongkeko WM, Harris AL, Norbury CJ. P53 regulates cdc2 independently of inhibitory phosphorylation to reinforce radiation-induced g2 arrest in human cells. Oncogene. 1998;17:673–684. doi: 10.1038/sj.onc.1201991. [DOI] [PubMed] [Google Scholar]
- 64.Iliakis G, Wang Y, Guan J, Wang H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene. 2003;22:5834–5847. doi: 10.1038/sj.onc.1206682. [DOI] [PubMed] [Google Scholar]
- 65.Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6:877–882. doi: 10.1016/0955-0674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 66.Jha MN, Bamburg JR, Bernstein BW, Bedford JS. Caffeine eliminates gamma-ray-induced g2-phase delay in human tumor cells but not in normal cells. Radiat Res. 2002;157:26–31. doi: 10.1667/0033-7587(2002)157[0026:cegrig]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 67.Florensa R, Bachs O, Agell N. Atm/atr-independent inhibition of cyclin b accumulation in response to hydroxyurea in nontransformed cell lines is altered in tumour cell lines. Oncogene. 2003;22:8283–8292. doi: 10.1038/sj.onc.1207159. [DOI] [PubMed] [Google Scholar]
- 68.Abraham RT. Cell cycle checkpoint signaling through the atm and atr kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 69.Shiloh Y. Atm and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 70.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of atm and atr kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–4382. [PubMed] [Google Scholar]
- 71.Keith CT, Schreiber SL. Pik-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- 72.Krasilnikov MA. Phosphatidylinositol-3 kinase dependent pathways: the role in control of cell growth, survival, and malignant transformation. Biochemistry (Mosc) 2000;65:59–67. [PubMed] [Google Scholar]
- 73.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase b signalling: aktion on multiple fronts. Trends Biochem Sci. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 74.Li J, Stern DF. Regulation of chk2 by DNA-dependent protein kinase. J Biol Chem. 2005;280:12041–12050. doi: 10.1074/jbc.M412445200. [DOI] [PubMed] [Google Scholar]
- 75.Blasina A, Price BD, Turenne GA, McGowan CH. Caffeine inhibits the checkpoint kinase atm. Curr Biol. 1999;9:1135–1138. doi: 10.1016/s0960-9822(99)80486-2. [DOI] [PubMed] [Google Scholar]
- 76.Matsuoka S, Huang M, Elledge SJ. Linkage of atm to cell cycle regulation by the chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 77.Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, et al. Caffeine abolishes the mammalian g(2)/m DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
- 78.Tyagi A, Singh RP, Agarwal C, Siriwardana S, Sclafani RA, Agarwal R. Resveratrol causes cdc2-tyr15 phosphorylation via atm/atr-chk1/2-cdc25c pathway as a central mechanism for s phase arrest in human ovarian carcinoma ovcar-3 cells. Carcinogenesis. 2005 doi: 10.1093/carcin/bgi165. [DOI] [PubMed] [Google Scholar]
- 79.Cortez D. Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (atm) and atm- and rad3-related (atr) protein kinases. J Biol Chem. 2003;278:37139–37145. doi: 10.1074/jbc.M307088200. [DOI] [PubMed] [Google Scholar]
- 80.Block WD, Merkle D, Meek K, Lees-Miller SP. Selective inhibition of the DNA-dependent protein kinase (DNA-pk) by the radiosensitizing agent caffeine. Nucleic Acids Res. 2004;32:1967–1972. doi: 10.1093/nar/gkh508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- 82.Lu YP, Lou YR, Li XH, Xie JG, Brash D, Huang MT, Conney AH. Stimulatory effect of oral administration of green tea or caffeine on ultraviolet light-induced increases in epidermal wild-type p53, p21(waf1/cip1), and apoptotic sunburn cells in skh-1 mice. Cancer Res. 2000;60:4785–4791. [PubMed] [Google Scholar]
- 83.Lu YP, Lou YR, Xie JG, Peng QY, Liao J, Yang CS, et al. Topical applications of caffeine or (−)-epigallocatechin gallate (egcg) inhibit carcinogenesis and selectively increase apoptosis in uvb-induced skin tumors in mice. Proc Natl Acad Sci USA. 2002;99:12455–12460. doi: 10.1073/pnas.182429899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu YP, Lou YR, Li XH, Xie JG, Lin Y, Shih WJ, Conney AH. Stimulatory effect of topical application of caffeine on uvb-induced apoptosis in mouse skin. Oncol Res. 2002;13:61–70. [PubMed] [Google Scholar]
- 85.Lu YP, Lou YR, Peng QY, Xie JG, Conney AH. Stimulatory effect of topical application of caffeine on uvb-induced apoptosis in the epidermis of p53 and bax knockout mice. Cancer Res. 2004;64:5020–5027. doi: 10.1158/0008-5472.CAN-04-0760. [DOI] [PubMed] [Google Scholar]
- 86.Lu YP, Lou YR, Liao J, Xie JG, Peng QY, Yang CS, Conney AH. Administration of green tea or caffeine enhances the disappearance of uvb-induced patches of mutant p53 positive epidermal cells in skh-1 mice. Carcinogenesis. 2005;26:1465–1472. doi: 10.1093/carcin/bgi086. [DOI] [PubMed] [Google Scholar]
- 87.Kramata P, Lu YP, Lou YR, Cohen JL, Olcha M, Liu S, Conney AH. Effect of administration of caffeine or green tea on the mutation profile in the p53 gene in early mutant p53-positive patches of epidermal cells induced by chronic uvb-irradiation of hairless skh-1 mice. Carcinogenesis. 2005 doi: 10.1093/carcin/bgi162. [DOI] [PubMed] [Google Scholar]
- 88.Schafer J, Bachtler J, Engling A, Little JB, Weber KJ, Wenz F. Suppression of apoptosis and clonogenic survival in irradiated human lymphoblasts with different tp53 status. Radiat Res. 2002;158:699–706. doi: 10.1667/0033-7587(2002)158[0699:soaacs]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 89.Janicke RU, Engels IH, Dunkern T, Kaina B, Schulze-Osthoff K, Porter AG. Ionizing radiation but not anticancer drugs causes cell cycle arrest and failure to activate the mitochondrial death pathway in mcf-7 breast carcinoma cells. Oncogene. 2001;20:5043–5053. doi: 10.1038/sj.onc.1204659. [DOI] [PubMed] [Google Scholar]
- 90.Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 91.Essmann F, Engels IH, Totzke G, Schulze-Osthoff K, Janicke RU. Apoptosis resistance of mcf-7 breast carcinoma cells to ionizing radiation is independent of p53 and cell cycle control but caused by the lack of caspase-3 and a caffeine-inhibitable event. Cancer Res. 2004;64:7065–7072. doi: 10.1158/0008-5472.CAN-04-1082. [DOI] [PubMed] [Google Scholar]
- 92.Bache M, Dunst J, Wurl P, Frode D, Meye A, Schmidt H, et al. G2/m checkpoint is p53-dependent and independent after irradiation in five human sarcoma cell lines. Anticancer Res. 1999;19:1827–1832. [PubMed] [Google Scholar]
- 93.Bache M, Pigorsch S, Dunst J, Wurl P, Meye A, Bartel F, et al. Loss of g2/m arrest correlates with radiosensitization in two human sarcoma cell lines with mutant p53. Int J Cancer. 2001;96:110–117. doi: 10.1002/ijc.1002. [DOI] [PubMed] [Google Scholar]
- 94.Minemoto Y, Gannon J, Masutani M, Nakagama H, Sasagawa T, Inoue M, et al. Characterization of adriamycin-induced g2 arrest and its abrogation by caffeine in flamnion cells with or without p53. Exp Cell Res. 2001;262:37–48. doi: 10.1006/excr.2000.5072. [DOI] [PubMed] [Google Scholar]
- 95.Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, et al. P53-dependent and independent expression of p21 during cell growth, differentiation. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 96.Cayrol C, Knibiehler M, Ducommun B. P21 binding to pcna causes g1 and g2 cell cycle arrest in p53-deficient cells. Oncogene. 1998;16:311–320. doi: 10.1038/sj.onc.1201543. [DOI] [PubMed] [Google Scholar]
- 97.Dulic V, Stein GH, Far DF, Reed SI. Nuclear accumulation of p21cip1 at the onset of mitosis: a role at the g2/m-phase transition. Mol Cell Biol. 1998;18:546–557. doi: 10.1128/mcb.18.1.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ando T, Kawabe T, Ohara H, Ducommun B, Itoh M, Okamoto T. Involvement of the interaction between p21 and proliferating cell nuclear antigen for the maintenance of g2/m arrest after DNA damage. J Biol Chem. 2001;276:42971–42977. doi: 10.1074/jbc.M106460200. [DOI] [PubMed] [Google Scholar]
- 99.Saito Y, Gopalan B, Mhashilkar AM, Roth JA, Chada S, Zumstein L, Ramesh R. Adenovirus-mediated pten treatment combined with caffeine produces a synergistic therapeutic effect in colorectal cancer cells. Cancer Gene Ther. 2003;10:803–813. doi: 10.1038/sj.cgt.7700644. [DOI] [PubMed] [Google Scholar]
- 100.Ribeiro JC, Barnetson AR, Jackson P, Ow K, Links M, Russell PJ. Caffeine-increased radiosensitivity is not dependent on a loss of g2/m arrest or apoptosis in bladder cancer cell lines. Int J Radiat Biol. 1999;75:481–492. doi: 10.1080/095530099140410. [DOI] [PubMed] [Google Scholar]
- 101.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 102.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 hetero-dimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 103.Hoshi T, Sasano H, Kato K, Yabuki N, Ohara S, Konno R, et al. Immunohistochemistry of caspase3/cpp32 in human stomach and its correlation with cell proliferation and apoptosis. Anticancer Res. 1998;18:4347–4353. [PubMed] [Google Scholar]
- 104.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 105.Dubrez L, Coll JL, Hurbin A, Solary E, Favrot MC. Caffeine sensitizes human h358 cell line to p53-mediated apoptosis by inducing mitochondrial translocation and conformational change of bax protein. J Biol Chem. 2001;276:38980–38987. doi: 10.1074/jbc.M102683200. [DOI] [PubMed] [Google Scholar]
- 106.Lee MS, Ogg S, Xu M, Parker LL, Donoghue DJ, Maller JL, Piwnica-Worms H. Cdc25+ encodes a protein phosphatase that dephosphorylates p34cdc2. Mol Biol Cell. 1992;3:73–84. doi: 10.1091/mbc.3.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tomita K, Tsuchiya H. Caffeine enhancement of the effect of anticancer agents on human sarcoma cells. Jpn J Cancer Res. 1989;80:83–88. doi: 10.1111/j.1349-7006.1989.tb02249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tsuchiya H, Mori Y, Ueda Y, Okada G, Tomita K. Sensitization and caffeine potentiation of cisplatin cytotoxicity resulting from introduction of wild-type p53 gene in human osteosarcoma. Anticancer Res. 2000;20:235–242. [PubMed] [Google Scholar]
- 109.Nishijima H, Nishitani H, Saito N, Nishimoto T. Caffeine mimics adenine and 2′-deoxyadenosine, both of which inhibit the guanine-nucleotide exchange activity of rcc1 and the kinase activity of atr. Genes Cells. 2003;8:423–435. doi: 10.1046/j.1365-2443.2003.00644.x. [DOI] [PubMed] [Google Scholar]
- 110.Wharton W, Goz B. Induction of alkaline phosphatase activity in hela cells. Inhibition by xanthine derivatives and thermostability studies. Biochem Pharmacol. 1979;28:763–768. doi: 10.1016/0006-2952(79)90356-3. [DOI] [PubMed] [Google Scholar]
- 111.Wells JN, Miller JR. Methylxanthine inhibitors of phosphodiesterases. Methods Enzymol. 1988;159:489–496. doi: 10.1016/0076-6879(88)59048-1. [DOI] [PubMed] [Google Scholar]
- 112.Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- 113.Marret S, Delpech B, Girard N, Leroy A, Maingonnat C, Menard JF, Fessard C. Caffeine decreases glial cell number and increases hyaluronan secretion in newborn rat brain cultures. Pediatr Res. 1993;34:716–719. doi: 10.1203/00006450-199312000-00004. [DOI] [PubMed] [Google Scholar]
- 114.Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: Mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Brain Res Rev. 1992;17:139–170. doi: 10.1016/0165-0173(92)90012-b. [DOI] [PubMed] [Google Scholar]
- 115.D'Ambrosio SM. Evaluation of the genotoxicity data on caffeine. Regul Toxicol Pharmacol. 1994;19:243–281. doi: 10.1006/rtph.1994.1023. [DOI] [PubMed] [Google Scholar]
- 116.Harish SK, Guruprasad KP, Mahmood R, Vasudev V. Inducible protective processes in animal systems vi. Cross-adaptation and the influence of caffeine on the adaptive response in bone marrow cells of mouse. Mutagenesis. 2000;15:271–276. doi: 10.1093/mutage/15.3.271. [DOI] [PubMed] [Google Scholar]
- 117.Jiang X, Lim LY, Daly JW, Li AH, Jacobson KA, Roberge M. Structure-activity relationships for g2 checkpoint inhibition by caffeine analogs. Int J Oncol. 2000;16:971–978. doi: 10.3892/ijo.16.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lehmann AR, Kirk-Bell S, Arlett CF, Harcourt SA, de Weerd-Kastelein EA, Keijzer W, Hall-Smith P. Repair of ultraviolet light damage in a variety of human fibroblast cell strains. Cancer Res. 1977;37:904–910. [PubMed] [Google Scholar]
- 119.Ritter EJ, Scott WJ, Wilson JG, Mathinos PR, Randall JL. Potentiative interactions between caffeine and various teratogenic agents. Teratology. 1982;25:95–100. doi: 10.1002/tera.1420250113. [DOI] [PubMed] [Google Scholar]
- 120.Kitamoto Y, Sakurai H, Mitsuhashi N, Akimoto T, Nakano T. Caffeine diminishes cytotoxic effects of paclitaxel on a human lung adenocarcinoma cell line. Cancer Lett. 2003;191:101–107. doi: 10.1016/s0304-3835(02)00591-8. [DOI] [PubMed] [Google Scholar]
- 121.Kakolyris S, Mavroudis D, Tsavaris N, Souglakos J, Tsiafaki P, Kalbakis K, et al. Paclitaxel in combination with carboplatin as salvage treatment in refractory small-cell lung cancer (sclc): a multicenter phase ii study. Ann Oncol. 2001;12:193–197. doi: 10.1023/a:1008322932251. [DOI] [PubMed] [Google Scholar]
- 122.Kramer JA, Curran D, Piccart M, de Haes JC, Bruning PF, Klijn JG, et al. Randomised trial of paclitaxel versus doxorubicin as first-line chemotherapy for advanced breast cancer: quality of life evaluation using the eortc qlq-c30 and the rotterdam symptom checklist. Eur J Cancer. 2000;36:1488–1497. doi: 10.1016/s0959-8049(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 123.Saunders DE, Lawrence WD, Christensen C, Wappler NL, Ruan H, Deppe G. Paclitaxel-induced apoptosis in mcf-7 breast-cancer cells. Int J Cancer. 1997;70:214–220. doi: 10.1002/(sici)1097-0215(19970117)70:2<214::aid-ijc13>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 124.Deplanque G, Ceraline J, Mah-Becherel MC, Cazenave JP, Bergerat JP, Klein-Soyer C. Caffeine and the g2/m block override: a concept resulting from a misleading cell kinetic delay, independent of functional p53. Int J Cancer. 2001;94:363–369. doi: 10.1002/ijc.1478. [DOI] [PubMed] [Google Scholar]
- 125.Deplanque G, Ceraline J, Lapouge G, Dufour P, Bergerat JP, Klein-Soyer C. Conflicting effects of caffeine on apoptosis and clonogenic survival of human k1 thyroid carcinoma cell lines with different p53 status after exposure to cisplatin or uvc irradiation. Biochem Biophys Res Commun. 2004;314:1100–1106. doi: 10.1016/j.bbrc.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 126.Weiss JF, Landauer MR. Protection against ionizing radiation by antioxidant nutrients and phytochemicals. Toxicology. 2003;189:1–20. doi: 10.1016/s0300-483x(03)00149-5. [DOI] [PubMed] [Google Scholar]
- 127.Traganos F, Kapuscinski J, Darzynkiewicz Z. Caffeine modulates the effects of DNA-intercalating drugs in vitro: a flow cytometric and spectrophotometric analysis of caffeine interaction with novantrone, doxorubicin, ellipticine, and the doxorubicin analogue ad198. Cancer Res. 1991;51:3682–3689. [PubMed] [Google Scholar]
- 128.Marret S, Gressens P, Van-Maele-Fabry G, Picard J, Evrard P. Caffeine-induced disturbances of early neurogenesis in whole mouse embryo cultures. Brain Res. 1997;773:213–216. doi: 10.1016/s0006-8993(97)00938-4. [DOI] [PubMed] [Google Scholar]