

Summary
Swe1 (Saccharomyces WEE1), the only “true” tyrosine kinase in budding yeast, is an Hsp90 client protein. Here we show that Swe1Wee1 phosphorylates a conserved tyrosine residue (Y24 in yeast Hsp90 and Y38 in human Hsp90α) in the N-domain of Hsp90. Phosphorylation is cell cycle-associated and modulates the ability of Hsp90 to chaperone a selected clientele, including v-Src and several other kinases. Non-phosphorylatable mutants have normal ATPase activity, support yeast viability, and productively chaperone the Hsp90 client glucocorticoid receptor. Deletion of SWE1 in yeast increases Hsp90 binding to its inhibitor geldanamycin, and pharmacologic inhibition/silencing of Wee1 sensitizes cancer cells to Hsp90 inhibitor-induced apoptosis. These findings demonstrate that Hsp90 chaperoning of distinct client proteins is differentially regulated by specific post-translational modification of a unique subcellular pool of the chaperone, and they provide a novel strategy to increase the cellular potency of Hsp90 inhibitors.
Introduction
Heat Shock Protein 90 (Hsp90) is an essential molecular chaperone in eukaryotic cells (Jackson et al., 2004; Neckers, 2007; Pearl and Prodromou, 2006). It creates and maintains the functional conformation of a subset of proteins that are referred to as “client proteins” - typically key components of multiple regulatory and signalling networks that mediate cancer cell proliferation, survival, and metastasis (Wandinger et al., 2008). Therefore Hsp90 has become an attractive target for new cancer therapeutics (Whitesell and Lindquist, 2005; Workman and de Billy, 2007), even though a full understanding of the chaperoning requirements of discrete clients remains poorly understood.
Hsp90 chaperone activity depends on ATP binding and hydrolysis (Obermann et al., 1998; Panaretou et al., 1998), which is coupled to a conformational cycle involving the opening and closing of a dimeric “molecular clamp” via transient association of Hsp90's N-terminal domains (Pearl and Prodromou, 2006). ATP binds to the N- domain of Hsp90 (Prodromou et al., 1997), which also binds the Hsp90 inhibitor geldanamycin (GA) (Grenert et al., 1997; Stebbins et al., 1997). Hsp90 ATPase activity is regulated by co-chaperones. For example, HopSti1 (Chang et al., 1997), p50Cdc37 (Lee et al., 2004; Vaughan et al., 2008), and p23Sba1 (McLaughlin et al., 2006; Picard, 2006) inhibit the Hsp90 ATPase cycle, while Aha1 (Panaretou et al., 2002), and Cpr6 (Johnson et al., 2007) stimulate it.
Hsp90 in solution does not have a single ‘relaxed’ conformation but exists as a continuum of conformations; ATP binding subtly shifts the equilibrium to favor transient formation/stabilization of a ‘tense’ state in which N-domains are dimerized (Graf et al., 2009). Recent studies have shown that the steady-state population density of these conformations is uniquely species dependent (Southworth and Agard, 2008). An earlier study reported that cancer cell Hsp90, in contrast to the bulk of the chaperone in non-transformed cells, preferentially adopts a tense (closed) conformation (Kamal et al., 2003). Together, these observations suggest that the dynamics of the Hsp90 chaperone cycle may be significantly influenced by epigenetic factors, including unique post-translational modifications (Scroggins and Neckers, 2007). Numerous literature reports identify Hsp90 as a phosphoprotein, and they show that Hsp90 phosphorylation impacts its function (Duval et al., 2007; Kurokawa et al., 2008; Lees-Miller and Anderson, 1989; Mimnaugh et al., 1995; Zhao et al., 2001).
Saccharomyces cerevisiae Wee1 (Swe1) is an Hsp90 client and also the only “true” tyrosine kinase in budding yeast (http://db.yeastgenome.org), (Aligue et al., 1994; Goes and Martin, 2001). Swe1Wee1 phosphorylates and inhibits the kinase activity of the main cell cycle cyclin-dependent kinase Cdc28p (human Cdc2), thereby regulating the G2/M transition (Booher et al., 1993; Harvey and Kellogg, 2003; Lew, 2003; McGowan and Russell, 1993).
Tyrosine phosphorylation of Hsp90 in yeast has not been reported previously. In the present study, we show that Swe1Wee1 directly phosphorylates a conserved tyrosine residue in the N-domain of Hsp90. Mutation of this residue to non-phosphorylatable phenylalanine did not affect Hsp90 ATPase activity, productive in vivo chaperoning of glucocorticoid receptor, or yeast viability, but did negatively impact chaperoning of certain Hsp90 clients including v-Src and heat shock factor (HSF). Identical effects on the chaperoning of these clients were observed in yeast expressing wild type (wt) Hsp90 but lacking Swe1. Further, purified Wee1 phosphorylated both yeast and human Hsp90 proteins on the same residue in vitro. Finally, GA bound preferentially to Hsp90 from swe1Δ yeast, and pharmacologic inhibition/silencing of Wee1 sensitized cancer cells to Hsp90 inhibitor-induced apoptosis.
Results
Swe1 phosphorylates Hsp90 in yeast
To evaluate Hsp90 tyrosine phosphorylation in Saccharomyces cerevisiae, we used the wt PP30 yeast strain in which both copies of endogenous Hsp90 (Hsp82 and Hsc82) are deleted and replaced with a single copy of Hsp82 that is His6 tagged at the N-terminus (yHsp90-His6) (Piper et al., 2003). Blotting with a phospho-tyrosine-specific antibody demonstrated constitutive tyrosine phosphorylation of yHsp90His6 after pulldown with Ni-NTA agarose (Figure 1A). Yeast Hsp90-His6 tyrosine phosphorylation was significantly reduced in cells treated with GA for 1 hr (Figure 1A).
Figure 1. Swe1 phosphorylates yHsp90.
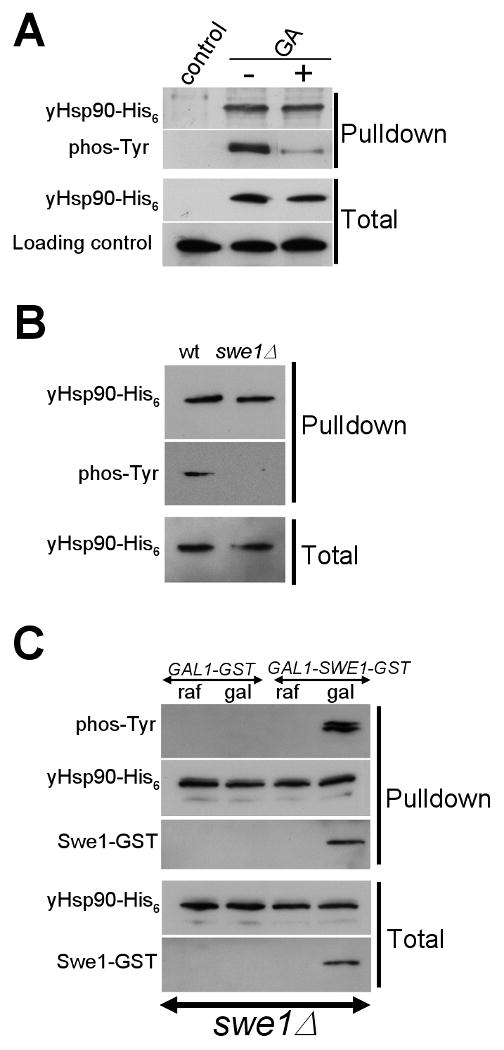
A) His6 tagged yHsp90 was precipitated from untreated or GA-treated (100 μM, 1 h) yeast cells and its tyrosine phosphorylation was detected with anti-phosphotyrosine antibody.
B) Deletion of SWE1 abolishes tyrosine phosphorylation of yHsp90.
C) Over-expression of GST tagged Swe1 in swe1Δ cells restores tyrosine phosphorylation of yHsp90. Swe1-GST was detected by anti-GST antibody.
Direct phosphorylation of Hsp90 by Swe1 has not been described previously. Swe1 expression is cell cycle-regulated, appearing most prominently in S-phase (Asano et al., 2005; Liu and Wang, 2006). We noticed that GA treatment for 1 hr, which significantly reduced yeast Hsp90 (yHsp90) tyrosine phosphorylation (Figure 1A), also decreased the S phase population (Figure S1).
To query further the possible involvement of Swe1 in tyrosine phosphorylation of Hsp90, we utilized swe1Δ cells. Although viable, these cells lack G2/M checkpoint controls and undergo premature entry into mitosis (Harvey and Kellogg, 2003; Lew, 2003). Hsp90-His6 tyrosine phosphorylation was undetectable in swe1Δ cells (Figure 1B).
In order to obtain further evidence for the Swe1 dependence of yHsp90 tyrosine phosphorylation, we expressed an N-terminally glutathione-S-transferase (GST) tagged Swe1 under the galactose inducible promoter GAL1 in swe1Δ cells. Swe1-GST expression was undetectable in cells grown in raffinose media (due to repression of the GAL1 promoter) (Figure 1C). Tyrosine phosphorylation of yHsp90-His6 was also not detected in these cells. Swe1-GST was induced by addition of galactose for 30 min, and this coincided with appearance of tyrosine phosphorylated yHsp90-His6 (Figure 1C). Additionally, Swe1-GST was co-precipitated with yHsp90-His6 (detected with anti-GST antibody). These data indicate that Swe1 mediates tyrosine phosphorylation of yHsp90.
Tyrosine phosphorylation of yHsp90 is cell cycle-associated and leads to Hsp90 ubiquitination and degradation by cytoplasmic proteasomes
As Swe1 synthesis and degradation is cell cycle regulated, we wished to determine whether cell cycle phase affected yHsp90 tyrosine phosphorylation. We examined the distribution of tyrosine-phosphorylated yHsp90 in cells arrested in G1-phase with α-factor, in S-phase with hydroxyurea (HU), or in M-phase with nocodazole (NOC). As shown in Figure 2A, yHsp90 tyrosine phosphorylation was more pronounced in S-phase than in an asynchronized log-phase population, and was absent or markedly reduced in G1 and M-phase-arrested cells (Figure 2A). These observations were confirmed by examining the temporal status of yHsp90 tyrosine phosphorylation in yeast cells released from G1 arrest by removal of α-factor. yHsp90 phosphorylation increased to a maximum after 40 min (Figure S2), which corresponds to S phase (Asano et al., 2005; Liu and Wang, 2006). These data indicate that yHsp90 tyrosine phosphorylation is cell cycle-associated and coincident with both Swe1 expression and Y19 phosphorylation of the known Swe1 substrate Cdc28 (Figure 2A).
Figure 2. yHsp90 tyrosine phosphorylation is cell cycle associated, occurs in the nucleus, and marks Hsp90 for ubiquitination and proteasome-mediated degradation.
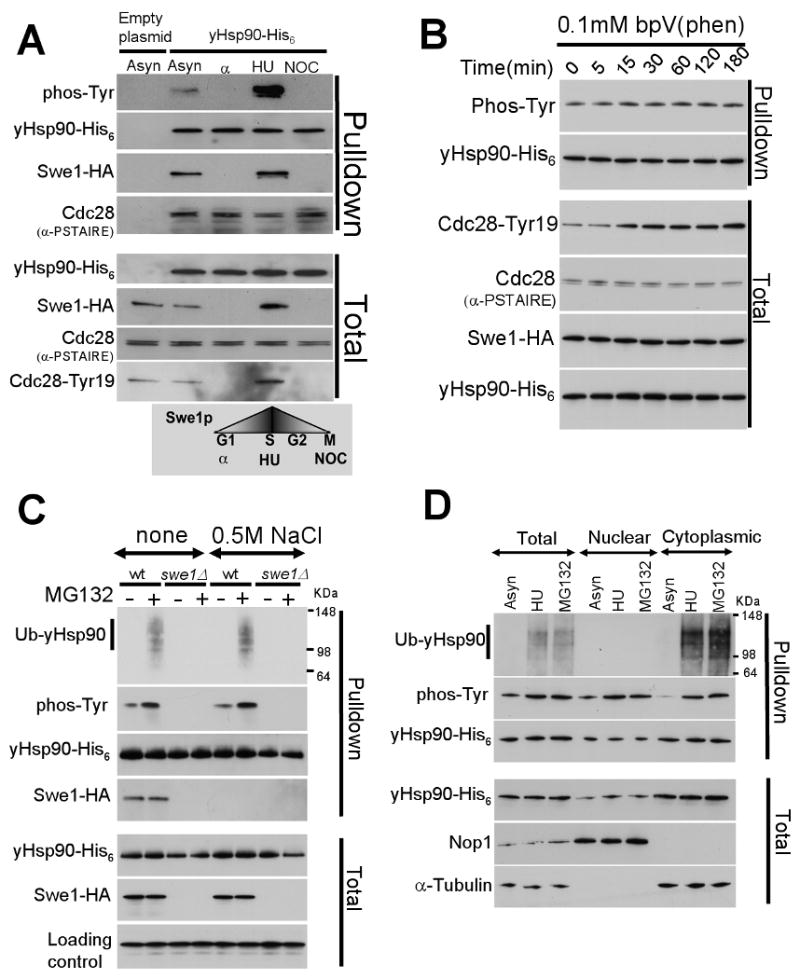
A) Yeast cells were arrested in G1 (with α-factor), in S-phase (with hydroxyurea, HU), and in M-phase (with nocodazole, NOC), and yHsp90 tyrosine phosphorylation was assessed. Cdc28 Tyr19 phosphorylation is shown as an indicator of Swe1 activity. Cdc28 interaction with yHsp90 was detected by α-PSTAIRE antibody.
B) Treatment of yeast with 0.1 mM bpV(Phen) did not increase tyrosine phosphorylation of yHsp90. Increase phosphorylation of Cdc28-Tyr19 was used as an indicator of phosphatase inhibition. Cdc28Cdc2 was detected by anti-PSTAIRE-antibody and Swe1-HA by anti-HA antibody.
C) Wild type or swe1Δ yeast cells expressing His6 tagged yHsp90 were treated with the proteasome inhibitor MG132 (50 μM for 1 h), and yHsp90 tyrosine phosphorylation and ubiquitination were assessed. Ub-yHsp90-His6 was detected using anti-ubiquitin antibody. The experiment was performed with and without salt stripping (0.5 M NaCl) of yHsp90 pulldowns.
D) Nuclear and cytoplasmic protein fractions were prepared from asynchronized (Asyn), S-phase arrested (HU), and MG132-treated cells. yHsp90-His6 pulldowns were blotted with anti-ubiquitin and anti-phosphotyrosine antibodies. Antibodies to Nop1 and α-tubulin were used to monitor purity of subcellular fractions.
The steady-state level of protein phosphorylation usually depends on the interplay between kinases and phosphatases. However, we were unable to identify a tyrosine phosphatase responsible for dephosphorylating yHsp90 when we screened the S. cerevisiae tyrosine phosphatase mutant collection from EUROSCARF (http://web.unifrankfurt.de/fb15/mikro/euroscarf/). To rule out the possibility of redundancy among yeast phosphatases, we treated yeast cells with the protein phosphotyrosine phosphatase inhibitor potassium bisperoxo(1,10-phenanthroline)oxovanadate (bpVphen) (Faure et al., 1995). Mid-log phase yeast cells were treated with 0.1 mM bpVphen for different times. While Y19 Cdc28 phosphorylation increased noticeably within 15 min, confirming the effectiveness of phosphatase inhibition, we observed no change in tyrosine phosphorylation of yHsp90 even after exposure of cells to bpVphen for 180 min (Figure 2B).
Given our inability to detect phosphatase-dependent dephosphorylation of yHsp90, even though no tyrosine phosphorylated species was detected in either M-phase or G1-phase-arrested cells, we next explored the possibility that tyrosine phosphorylation of yHsp90 may serve as a signal for its ubiquitination and subsequent proteasome-mediated degradation. Supporting this possibility, treatment of yeast cells with the proteasome inhibitor MG132, under conditions that caused neither a heat shock response nor cell cycle arrest (data not shown), resulted in an accumulation of tyrosine phosphorylated and ubiquitinated yHsp90 (top 2 panels, Figure 2C). To confirm that the signals obtained with the antibodies detecting phospho-tyrosine and ubiquitin were due to yHsp90 and not to adventitious proteins accompanying the Ni-NTA agarose pulldown of yHsp90-His6, we salt-stripped the pulldowns and repeated the blotting with identical results (Figure 2C). To demonstrate the efficacy of the salt-stripping procedure, we confirmed that it disrupted co-precipitation of Swe1-HA (Figure 2C). Lastly, yHsp90 ubiquitination was not observed in swe1Δ cells (Figure 2C), or in cells expressing only yHsp90-Y24F (see explanatory text below, Figure S2), supporting the hypothesis that tyrosine phosphorylation of yHsp90 precedes and is a prerequisite for its ubiquitination in yeast.
In order to determine the subcellular location of yHsp90 ubiquitination, we synchronized yeast cells in S-phase with HU, or we treated an asynchronous population with MG132. Yeast Hsp90-His6 was then precipitated from nuclear and cytoplasmic protein fractions and assessed for tyrosine phosphorylation and ubquitination status. Purity of nuclear and cytoplasmic protein fractions was assessed by blotting for the nuclear protein Nop1 and the cytoplasmic protein α-tubulin, respectively. Our data show that in untreated, asynchronized yeast cells most tyrosine phosphorylated yHsp90 is in the nucleus (Figure 2D). However, both S-phase arrest and proteasome inhibition caused an increase in tyrosine phosphorylated yHsp90 in both nuclear and cytoplasmic fractions, with the greatest increase appearing in the cytoplasm (Figure 2D). Importantly, only yHsp90 in the cytoplasmic fraction was ubiquitinated after either treatment (Figure 2D). These data are consistent with a model in which tyrosine phosphorylated yHsp90 is ubiquitinated and degraded in the cytoplasm. Our findings suggest that tyrosine phosphorylation of yHsp90 occurs primarily in the nucleus during S-phase, and that the tyrosine phosphorylated species exits the nucleus prior to its ubiquitination.
Hsp90 is phosphorylated by Swe1Wee1 on a single conserved tyrosine residue in the N-domain
In order to identify the tyrosine residue(s) that are phopshorylated by Swe1, we queried the crystal structures of yHsp90 individual domains as well as that of the full length dimer (Ali et al., 2006; Meyer et al., 2003). We identified Y24, Y47, Y508 and Y606 as tyrosine residues likely to be surface-exposed on yHsp90. After mutating each residue independently to the non-phosphorylatable amino acid phenylalanine (F), we expressed yHsp90-His6 harboring each mutation in yeast and we assessed its tyrosine phosphorylation status. Only Y24F mutation abrogated tyrosine phosphorylation of yHsp90 (Figure 3A).
Figure 3. Swe1Wee1phosphorylates a single conserved tyrosine residue in the Hsp90 N-domain in vivo and in vitro.
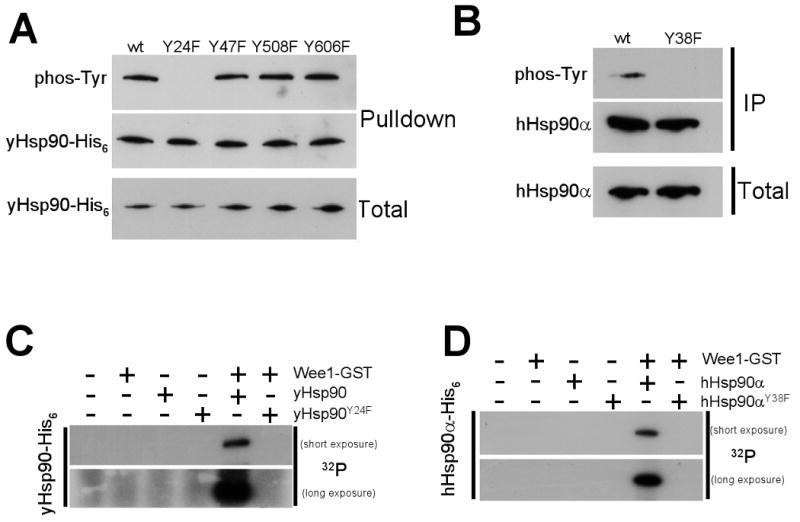
A) Mutation of tyrosine residues on the surface of yHsp90 to the non-phosphorylatable residue phenylalanine (F) revealed Y24 to be the sole site of Swe1 phosphorylation.
B) Mutation of the equivalent tyrosine residue (Y38F) in human Hsp90α abrogated its tyrosine phosphorylation in yeast.
C) & D) Insect-expressed and purified Wee1-GST phosphorylates bacterially expressed and purified yHsp90-His6 (C) and hHsp90α-His6 (D) in vitro. Wee1-GST was unable to phosphorylate either yHsp90-Y24F-His6 or hHsp90α–Y38FHis6 in this assay.
Tyrosine 24 in yeast Hsp90 is highly conserved in eukaryotic Hsp90 proteins, including human Hsp90α (hHsp90α, where the equivalent residue is Y38). However, it is not possible to specifically observe the phosphorylation status of Y38 in hHsp90α using a pan anti-phosphotyrosine antibody because hHsp90α is subject to tyrosine phosphorylation at several other residues (Duval et al., 2007; Scroggins and Neckers, 2007). Therefore, we used yeast strains that expressed either wt hHsp90α (Millson et al., 2007) or hHsp90α-Y38F as the sole Hsp90 protein. After immunoprecipitating hHsp90α, we probed Western blots with an anti-phosphotyrosine antibody. While wt hHsp90α was phosphorylated, hHsp90-Y38F was not, confirming Y38 as the sole phosphorylated tyrosine residue in hHsp90α expressed in yeast (Figure 3B).
Unlike budding yeast, the human genome contains 90 tyrosine kinase genes and five presumed tyrosine kinase pseudogenes. Therefore, in the absence of an antibody that uniquely recognizes tyrosine phosphorylated Y38, it is impossible to demonstrate Hsp90 tyrosine phosphorylation by Wee1 in human cells. Instead, we bacterially expressed and purified N-terminally His6-tagged yeast and human wt Hsp90, as well as yHsp90-Y24F and hHsp90α–Y38F (Figure S3). These proteins (bound to Ni-NTA agarose) were used as substrates in an in vitro kinase assay that included purified, active Wee1-GST. Under these conditions, Wee1-GST efficiently phosphorylated yeast and human wt Hsp90 proteins but not yHsp90-Y24F or hHsp90α–Y38F (Figure 3C, D). These results provide strong evidence that Wee1 can directly phosphorylate Y24 of yHsp90 and Y38 of hHsp90α in the absence of other proteins or co-chaperones. Further, Y24/Y38 is the only tyrosine residue of these Hsp90 proteins that is phosphorylated by Swe1Wee1.
Mutation of Y24 affects Hsp90 ATPase activity and N-domain dimerization
In order to gain further insight into the impact of Y24/Y38 phosphorylation on Hsp90 function, we mutated Y24 in yHsp90 and Y38 in hHsp90α to the phosphomimic residue glutamic acid (E). Unfortunately, these phosphomimic Hsp90 mutants were unable to support yeast viability (Figure 4A). We obtained similar results with alternative phosphomimic aspartic acid (D) mutants (Figure S4). The lethality of these mutants is likely due to a nearly complete lack of ATPase activity (Y24E < 3% of wt, Figure 4B) and failure to undergo N-domain dimerization in the presence of AMPPNP (a non-hydrolyzable ATP analog) (Figure 4C, D). In contrast, the non-phosphorylatable mutants Y24F (yHsp90) and Y38F (hHsp90) are viable (Figure 4A), display normal ATPase activity, and undergo AMPPNP-mediated N-domain dimerization (Figure 4 B - D). As Y24F and Y24E mutants bind AMPPNP with a Kd similar to that of wt yHsp90 (Figure 4B), it is likely that loss of ATPase activity of yHsp90-Y24E is due to lack of ATP-dependent N-domain dimerization and subsequent conformational events.
Figure 4. Mutation of Y24/Y38 impacts Hsp90 ATPase activity and N-domain dimerization.
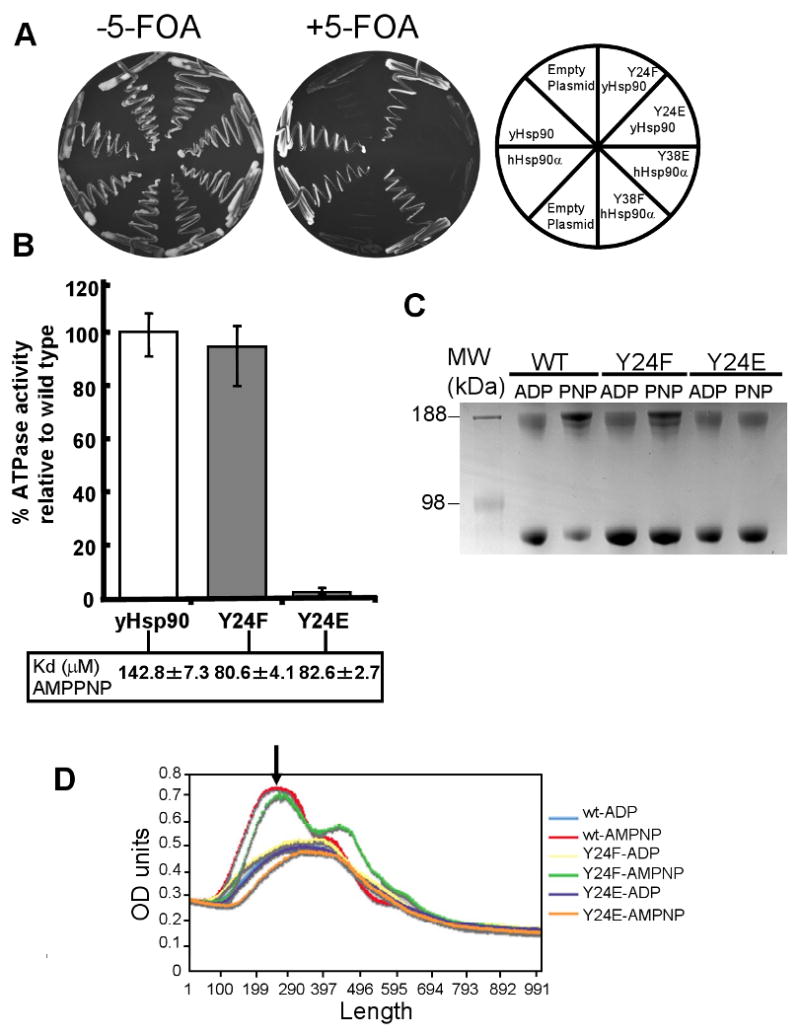
A) In vivo activity of phospho- and nonphosphomimic Hsp90 mutations. Phosphomimic mutants abolished viability, whereas the non-phosphorylatable mutants support yeast growth.
B) ATPase activity of bacterially expressed and purified wt yHsp90, yHsp90-Y24F, and yHsp90-Y24E proteins was determined as described in Methods. Kd of each protein for AMPPNP is shown in inset.
C) AMPPNP-dependent N-domain dimerization of wt yHsp90, yHsp90-Y24F, and yHsp90-Y24E proteins was determined as described in Methods. After crosslinking and polyacrylamide gel electrophoresis, N-domain dimerized Hsp90 runs with an apparent molecular weight of approximately 190 kD.
D) Averaged densitometry scans of results shown in Figure 4B and two additional experiments. Gels were scanned and where necessary normalized against total intensity. The peak representing the slowest migrating band (identified by the black arrow) indicates the degree of N-terminally dimerized wt or mutant Hsp90 protein.
Y24/Y38 mutation affects Hsp90 interaction with and chaperoning of distinct client proteins
Yeast Hsp90-Y24F supports viability and has normal ATPase activity but cannot be phosphorylated. Therefore, we utilized this protein to query the possible importance of Swe1-mediated Hsp90 phosphorylation for distinct aspects of chaperone function. Since Y24F mutation may engender structural alteration of Hsp90 unrelated to Y24 phosphorylation, we also used swe1Δ cells expressing wt yHsp90, in which we have shown that phosphorylation of Y24 is absent.
We first examined the interaction and stability of two endogenous yeast Hsp90 client proteins, the kinases Ste11 (a Raf-1 ortholog) (Louvion et al., 1998) and activated Mpk1/Slt2 (an Erk5 ortholog) (Millson et al., 2004). Ste11ΔN, a constitutively active allele that causes pheromone-independent activation of the mating pathway (under GAL1 promoter) was expressed in wt cells upon transfer from glucose to galactose media (Figure 5A). However we were unable to detect Ste11 in swe1Δ cells or in yeast expressing yHsp90-Y24F (Figure 5A). Mpk1/Slt2 MAP kinase activation strengthens its interaction with yHsp90 (Millson et al., 2004). We detected interaction of wt yHsp90 with Mpk1/Slt2 in cells stressed with 10 mM caffeine (activator of the cell wall integrity pathway in yeast). However, interaction of Mpk1/Slt2 with yHsp90-Y24F and yHsp90 from swe1Δ cells was significantly reduced (Figure 5B). These data strongly suggest that phosphorylation of yHsp90-Y24 is important for chaperoning of at least two endogenous kinase clients in yeast.
Figure 5. Swe1 expression and Y24/Y38 mutation affect Hsp90 chaperone function.
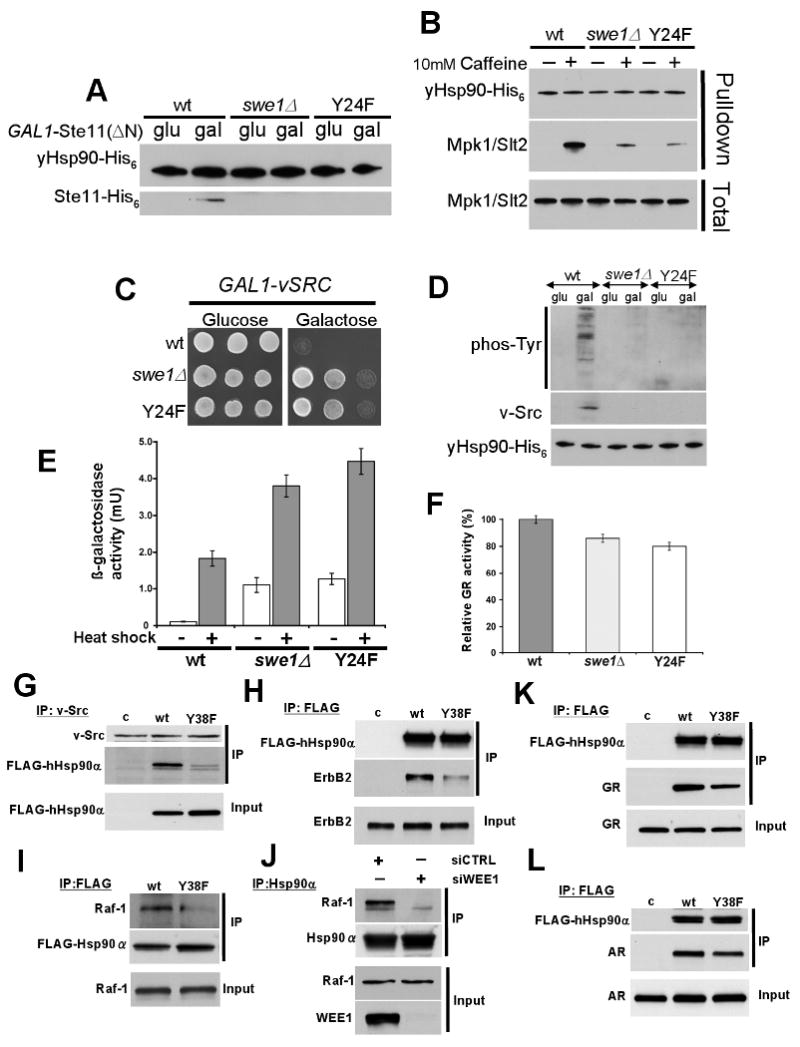
A) Wild type yeast, swe1Δ yeast, and yeast expressing yHsp90-Y24F containing Ste11ΔN-His6 (under control of GAL1 promoter) were grown on glucose- or galactose-containing media, and Ste11ΔN-His6 expression was examined.
B) yHsp90 from wt yeast, swe1Δ yeast, and yHsp90-Y24F were examined for interaction with Mpk1/Slt2 after cells were stressed with 10 mM caffeine (activator of Mpk1/Slt2 MAP kinase).
C) Growth of wt yeast, swe1Δ yeast, and yeast expressing Hsp90-Y24F containing v-SRC (under control of GAL1 promoter) on glucose- or galactose-containing media.
D) The same strains were analyzed for total phosphotyrosine and v-Src expression.
E) Heat Shock Element (HSE)-lacZ reporter was introduced into wt yeast, swe1Δ yeast, and yeast expressing yHsp90-Y24F; HSE activity was measured in unstressed (light bars) and heat shocked cells (40 min at 39°C, dark bars). The data are expressed as mean +/- standard deviation derived from three independent experiments.
F) GR activity was assessed in the same strains as above after transformation with a constitutive GR expression plasmid containing lacZ under the control of glucocorticoid response elements. Data are expressed as a percentage of the activity observed in wt (wt) cells, and are depicted as the mean +/- standard deviation derived from three independent experiments.
G) NIH 3T3 cells stably expressing v-Src protein were transfected with empty vector pcDNA3 (c), FLAG-hHsp90α (wt), or FLAG-hHsp90α-Y38F (Y38F) constructs. v-Src protein was immunoprecipitated and associated FLAG-Hsp90 was detected by immunoblotting.
H) SkBr3 cells were transfected with empty vector pcDNA3 (pc), FLAG-hHsp90α (wt), or FLAG-hHsp90α-Y38F (Y38F) constructs. FLAG-Hsp90 immunoprecipitates were probed for associated ErbB2 by immunoblotting.
I) PC-3 cells were transfected with Flag-tagged wt hHsp90α or hHsp90α-Y38F. After 24 h, endogenous Raf-1 associated with Flag IPs was monitored by Western blotting.
J) Wee1-specific siRNA was used to silence Wee1 in PC3 cells. At 72 h, endogenous Hsp90α was immunoprecipitated and associated Raf-1 was detected by Western blotting. Cells were treated identically with non-targeting siRNA as a control.
K) & L) COS7 cells were co-transfected with GR (I) or AR (J) and indicated FLAG-Hsp90 constructs. After lysis, proteins were immunoprecipitated with FLAG antibody-conjugated agarose; co-precipitating GR and AR were detected by immunoblotting.
The oncogenic tyrosine kinase v-Src is a well-known Hsp90 client protein (Nathan and Lindquist, 1995). Active v-Src expression in yeast requires Hsp90 and causes lethality due to de-regulated phosphorylation of yeast proteins. This has become a widely used reporter assay in yeast for analysis of Hsp90 chaperone function. Expression of v-Src under a galactose inducible GAL1 promoter in either swe1Δ cells or in yeast expressing yHsp90-Y24F as the sole Hsp90 failed to cause lethality, unlike v-Src expression in wt cells (Figure 5C). Further analysis revealed that v-Src protein and v-Src-mediated phosphorylation of total yeast proteins were clearly detectable in wt cells, but not in either swe1Δ cells or in yeast expressing yHsp90-Y24F as the sole Hsp90 (Figure 5D). Importantly, v-Src activation displayed the same cell cycle pattern as yHsp90 tyrosine phosphorylation (Figure S5). Thus, as with the endogenous kinases Ste11 and Mpk1/Slt2, Y24 phosphorylation status appears to be an important determinant for productive chaperoning of v-Src in yeast.
We next looked at the impact of yHsp90-Y24F or SWE1 deletion on the activity of two Hsp90-dependent transcription factors - heat shock factor (HSF) and glucocorticoid receptor (GR). It is generally held that Hsp90 binding serves to down-regulate HSF transcriptional activity (Zou et al., 1998). Mutation of Hsp90, or Hsp90 inhibitor administration, leads to induction of HSF activity in yeast, even in the absence of heat shock. (Hjorth-Sorensen et al., 2001; Piper et al., 2003). We measured HSF activity in swe1Δ yeast cells and in yeast expressing yHsp90-Y24F by transforming these cells with a plasmid-based lacZ HSF reporter, HSE-lacZ (see Supplemental Experimental Procedures). As shown in Figure 5E, both swe1Δ cells and yHsp90-Y24F mutants displayed elevated basal and heat-induced (39°C for 40 min) HSF activity compared to wt cells. These results indicate that Y24 phosphorylation contributes to the ability of yHsp90 to suppress HSF activity, consistent with the observation that, unlike the case in metazoans, HSF1 is constitutively present in the nucleus in yeast (Morimoto, 1998).
The mammalian steroid receptor GR is another well-characterized Hsp90-dependent transcription factor that provides a sensitive readout for Hsp90 function in yeast cells (Pratt et al., 2004). To assess the impact of Y24 phosphorylation on the chaperoning of GR, yHsp90-Y24F, swe1Δ, and wt cells were transformed with a plasmid constitutively expressing GR and carrying a glucocorticoid-regulated lacZ reporter gene. In this case, neither the swe1Δ cells nor the yHsp90-Y24F-expressing cells were significantly impaired for GR activity compared to wt cells (Figure 5F).
Since Swe1Wee1 phosphorylated human hHsp90α both in vitro and in vivo (Figure 3B, D), we assessed the impact of Y38F mutation on interaction of hHsp90α with several kinase and steroid receptor client proteins in mammalian cells. First, we transiently expressed either FLAG-tagged wt hHsp90α or FLAG-tagged hHsp90α-Y38F in v-Src-transformed NIH-3T3 fibroblasts. Following immunoprecipitation of v-Src, we detected robust interaction with FLAG-tagged wt hHsp90α, but its interaction with the Y38F mutant was markedly reduced (Figure 5G). We observed similar results with another Hsp90 kinase client, ErbB2. COS7 cells were co-transfected with either FLAG-tagged wt hHsp90α or Y38F mutant plasmids, and with plasmid expressing ErbB2. While we were able to co-immunoprecipitate ErbB2 with wt FLAG-hHsp90α, ErbB2 interaction with hHsp90α-Y38F was noticeably impaired (Figure 5H). Finally, we examined association of the endogenous client kinase Raf-1 with hHsp90α-Y38F in PC3 prostate cancer cells and we saw a similar reduced interaction compared to wt hHsp90α (Figure 5I). Importantly, these results were reproduced when we examined the impact of siRNA-mediated silencing of Wee1 on the endogenous Hsp90/Raf-1 interaction in PC3 cells (Figure 5J).
In contrast to these results, when we compared the interaction of wt hHsp90 and hHsp90-Y38F with the steroid receptors GR and androgen receptor (AR) in mammalian cells, we obtained results consistent with our data in yeast (Figure 5F). Y38F mutation minimally impacted hHsp90 interaction with either GR or AR (Figure 5K, L). Quantification of co-immunoprecipitated v-Src, ErbB2, GR, and AR with hHsp90-Y38F confirms the robustness of these observations (Figure S5).
Taken together, these data show that not all aspects of Hsp90 function or Hsp90-client interactions are uniformly affected by Y24/Y38 phosphorylation. While this modification significantly affects the interaction/activity of several kinases and suppression of HSF activity, it seems to have minimal significance for steroid receptor interaction/activity, for Hsp90 ATP binding and ATPase activity, and for support of yeast viability.
Hsp90 Y24/Y38 mutation impacts Hsp90 association with a specific subset of co-chaperones
Like post-translational modifications, co-chaperones also play an important role in regulating Hsp90 activity. Therefore, we examined the impact of Y24/Y38 phosphorylation status on co-chaperone interactions with Hsp90. Yeast cells expressing yHsp90His6 in the presence or absence of Swe1 and yeast cells expressing yHsp90His6-Y24F were subjected to affinity pulldown with Ni-NTA agarose, and associating co-chaperones were examined by Western blot analysis. yHsp90 from swe1Δ cells and yHsp90-Y24F interacted equivalently with Sti1Hop and Cdc37p50 compared to wt yHsp90. In contrast, Aha1 association with yHsp90-Y24F and with yHsp90 expressed in swe1Δ cells was completely abrogated, while the association of Sba1p23 was reduced compared to wt yHsp90 (Figure 6A, B). These results strongly suggest that Swe1-mediated phsophorylation of Y24 affects yHsp90 binding to a specific subset of co-chaperones, which may in turn affect the conformational dynamics of Hsp90.
Figure 6. Co-chaperone binding to non-phosphorylatable Hsp90 mutants.
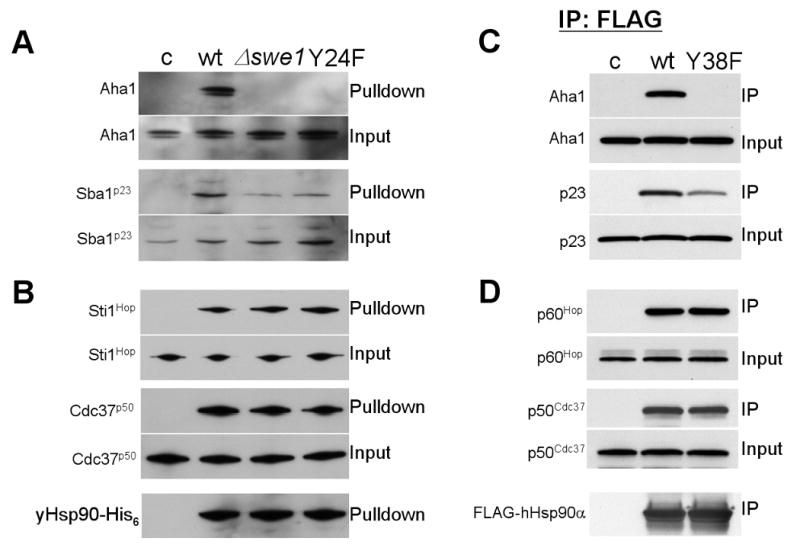
A) & B) yHsp90-His6 from wt or swe1Δ yeast and yHsp90His6-Y24F were precipitated and their interaction with co-chaperones was detected by immunoblotting.
C & D) COS7 cells were transfected with indicated FLAG-Hsp90 constructs (empty vector pcDNA3 [c], FLAG-hHsp90α [wt], or FLAG-hHsp90α-Y38F [Y38F]). FLAG-Hsp90 was immunoprecipitated and associated Aha1, p23, p60Hop and p50Cdc37 were detected by immunoblotting.
Comparable results were observed when we examined the interaction of hHsp90α-Y38F with mammalian co-chaperones. COS7 cells were transiently transfected with FLAG-hHsp90α or its Y38F mutant. Wild type hHsp90α interacted with Aha1, p23, p60Hop, and p50cdc37 (Figure 6C, D). However, hHsp90α-Y38F did not interact at all with Aha1 and its association with p23 was reduced, while interaction with p50cdc37 and p60Hop was not affected by this mutation (Figure 6C, D).
Pharmacologic inhibition/molecular knockdown of Wee1 sensitizes cancer cells to an Hsp90 inhibitor
Although the binding affinity of GA for purified wt and yHsp90-Y24F is comparable (Kdwt = 2.6 +/- 0.27 μM, KdY24F = 3.6 +/- 0.32 μM), we examined GA affinity bead binding to Hsp90 in lysates of yeast cells expressing yHsp90-Y24F, or in swe1Δ yeast lysates expressing wt yHsp90. To our surprise, when compared to wt yHsp90 in Swe1-expressing yeast cells, GA-bead binding to both yHsp90 in swe1Δ cells and to yHsp90-Y24F in Swe1-expressing cells was reproducibly increased, suggesting a contribution of epigenetic factors to GA binding in vivo that are not present in vitro (Figure 7A). We also observed increased GA-bead binding to hHsp90α-Y38F, compared to wt hHsp90α, from COS7 cell lysates (Figure 7B). Hsp90 binding to GA-beads is specific, as pretreating the protein lysates with 10 μM soluble GA completely prevented Hsp90 binding to GA-beads (Figure 7C). In contrast to GA binding, ATP binding to yHsp90 from each lysate was identical (Figure 7A). The lack of impact of the Y24F mutation on ATP binding was consistent with our in vitro data obtained with purified yHsp90 and yHsp90-Y24F proteins (Figure 4B).
Figure 7. Inhibition or silencing of Wee1 sensitizes cells to apoptosis induced by Hsp90 inhibitor.
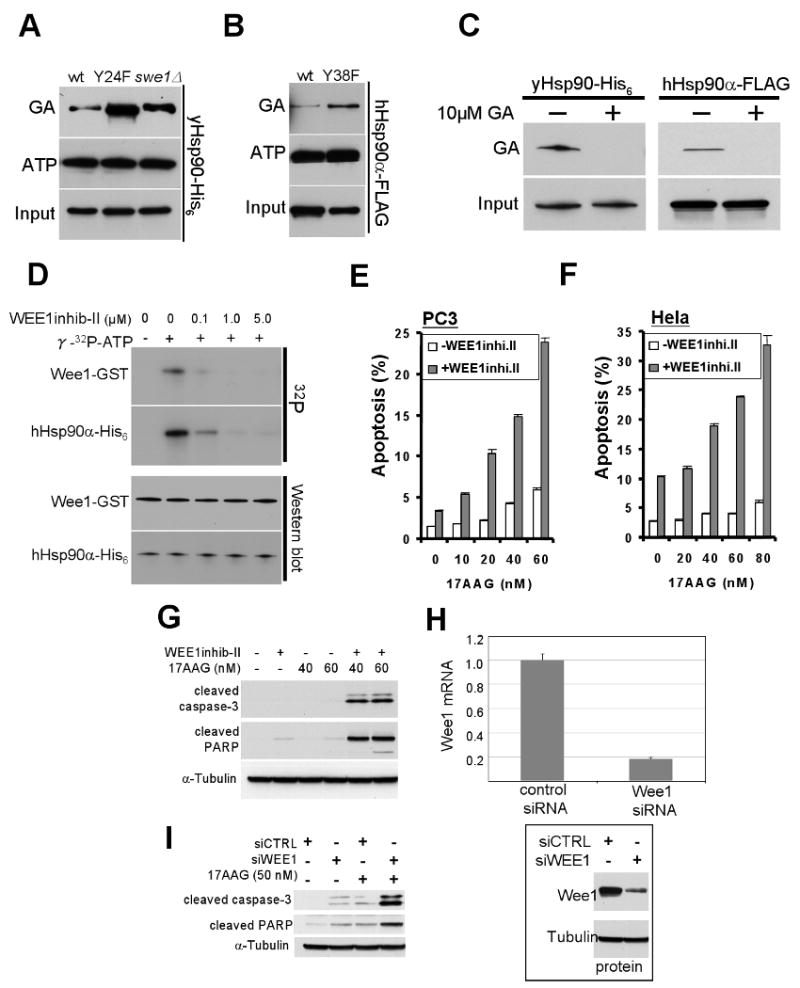
A) GA-bead pulldown of yHsp90 from yeast lysates of wt and swe1Δ cells, or of yHsp90-Y24F mutants; ATP-bead pulldown of yHsp90 from identical lysates is shown for comparison.
B) GA-bead pulldown of hHsp90-Y38F in lysates from mammalian cells. ATP binding is shown for comparison.
C) GA-bead pulldown of yHsp90 from yeast lysate or FLAG-hHsp90α from COS7 cell lysate. Both cell lysates were pre-incubated with 10 μM GA for 15 min prior to GA-bead pulldown to verify GA-bead specificity.
D) Wee1-GST, expressed and purified from insect cells, was used at 0.1 μM in the presence of γ32p-ATP. Wee1 inhibitor concentrations were as shown.
E) PC3 or (F) HeLa cells were treated with 2.5 uM or 5 uM Wee1 inhibitor, respectively, for 24 h. Cells were then treated with indicated concentrations of 17-AAG for an additional 48 h. Apoptosis was detected by FACS analysis as described in Methods.
G) PC3 cells were treated as above, and cleaved caspase-3 and cleaved PARP were detected by immunoblotting.
H) Effectiveness of Wee1 silencing was monitored by mRNA exxpression. Wee1 protein was also visualized by Western blotting with tubulin as loading control.
I) The experiment in (G) was repeated using 50 nM 17-AAG in Wee-1 silenced cells.
These observations suggested that inhibition/silencing of Wee1 might sensitize cancer cells to GA. To test this hypothesis, we first used a Wee1 inhibitor that is a potent competitor of ATP binding (Palmer et al., 2006). Using the in vitro assay described earlier, we titrated this compound to obtain a concentration that inhibited both Wee1 autophosphorylation and Wee1-mediated phosphorylation of hHsp90 (occurring at a concentration ≥1 μM, Figure 7D). Next, we treated two cancer cell lines, PC3 (prostate carcinoma) and HeLa (cervical carcinoma), with Wee1 inhibitor (2.5 μM for PC3 cells and 5 μM for HeLa cells) for 24 hrs, followed by 0 – 80 nM 17-AAG (a clinically well-tolerated Hsp90 inhibitor derived from GA) for an additional 48 h (in the continued presence of Wee1 inhibitor). At the end of the incubation period we quantified the percentage of apoptotic cells by flow cytometry (by measuring the increase in amount of sub-G1 cellular DNA content, shaded bars, Figure 7E, F). For comparison, cells were treated with 17-AAG alone (open bars, Figure 7E, F). Compared to either Wee1 inhibitor alone or 17-AAG alone, combined administration of both drugs had a greater than additive impact on apoptosis that was dose-dependently related to the concentration of 17-AAG.
We confirmed this observation in PC3 cells by assessing the abundance of cleaved caspase-3 and cleaved PARP, two cellular markers of apoptosis. Treatment as above with the combination of Wee1 inhibitor (2.5 μM) and 17-AAG (40 & 60 nM) noticeably increased the level of both apoptotic markers, while similar incubation with the individual drugs was ineffective (Figure 7G).
Finally, we repeated this experiment using Wee1-specific siRNA to silence Wee1 expression. Forty-eight hours after siRNA transfection, Wee1 mRNA and protein were reduced by 80 % (Figure 7H). We added 17-AAG (50 nM) for an additional 48 hours and monitored the abundance of cleaved caspase-3 and cleaved PARP. When contrasted with either Wee1 silencing alone or 17-AAG alone, Wee1 silencing combined with 17-AAG led to a marked increase of both apoptotic indicators (Figure 7I).
Discussion
We show here that Swe1Wee1 phosphorylates a conserved tyrosine residue in the N-domain of Hsp90 both in vivo and in vitro. This finding was unexpected as Swe1 is thought to singularly regulate Cdc28Cdc2. Our data suggest Swe1 phosphorylates yHsp90 in the nucleus, and that cytosolic relocation of phosphotyrosyl yHsp90 likely precedes its ubiquitination and degradation by the proteasome. We propose that proteasomal degradation of tyrosine phosphorylated Hsp90 is the “switching off” mechanism for this form of the chaperone, at least in yeast.
Our data identify Y24 as the target of Swe1 and the sole phosphotyrosine in yHsp90. This residue is conserved in higher eukaryotes and Swe1 is able to phosphorylate the equivalent tyrosine residue in human Hsp90α, Y38, when this protein is expressed in yeast. Further, purified Wee1 phosphorylated both yHsp90 and hHsp90α in vitro, but only on Y24/Y38. When expressed as the sole Hsp90 protein, yHsp90-Y24F supported yeast growth but caused Swe1 destabilization (data not shown, manuscript in preparation), with the cells behaving like swe1Δ mutants (e.g., having a short G2 and loss of DNA damage check point, unpublished data). This raises the possibility that Swe1 is among a subset of client proteins whose stability and function depend on association with tyrosine phosphorylatable Hsp90, even though this modification is dispensable for yeast viability.
yHsp90-Y24F failed to productively chaperone two endogenous yeast protein kinases, Ste11 and Mpk1/Slt2, as well as exongenous v-Src expressed in yeast. Likewise, hHsp90-Y38F interacted poorly with v-Src, ErbB2, and Raf-1 kinases in mammalian cells, and similar results were obtained for wt hHsp90 following silencing of Wee1. Additionally, non-phosphorylatable Hsp90 demonstrated elevated basal and heat-induced HSF transcriptional activity. This phenotype was reproduced in yeast expressing wt Hsp90 but lacking Swe1 expression. In contrast, hHsp90α-Y38F interaction with the transcription factors GR and AR in mammalian cells was unaffected, and yHsp90-Y24F productively chaperoned GR in vivo. Again, swe1Δ yeast displayed an identical phenotype. Taken together, these findings underscore the differential impact of Y24/Y38 phosphorylation/mutation on Hsp90 function, and they identify a unique mechanism by which the intracellular environment might specifically modulate Hsp90 activity toward individual clients.
We also found the interaction of non-phosphorylatable Y24F yHsp90 or Y38F hHsp90α with Aha1 to be lost and interaction with Sba1p23 to be significantly reduced both in yeast and in mammalian cells, suggesting that phosphorylation status of Y24/Y38 impacts the conformational dynamics of the Hsp90 chaperone cycle. This conclusion was strengthened by identical results obtained in swe1Δ yeast expressing wt yHsp90. As the in vitro binding constants of Aha1 to both wt and Y24F Hsp90 are similar (2.7 ± 0.34 vs 1.8 ± 0.33 μM, respectively), our data strongly suggest that Y24/Y38 phosphorylation status serves as a regulatable epigenetic modulator of Aha1 interaction with Hsp90.
Since yHsp90-Y24E and -Y24D mutants were not viable, we were not able to query the consequences of these phosphomimic mutations on Hsp90 function in vivo. While it is possible that tyrosine phosphorylation is restricted to a subpopulation of Hsp90 in vivo because this modification is incompatible with key aspects of chaperone function necessary for yeast viability, we cannot rule out the possibility that structural alterations accompanying these mutations are harmful for Hsp90 function. Y24 is among a group of residues that have been shown to form an interacting cluster in ATP-bound Hsp90 (Cunningham et al., 2008). The hydrophobic interactions established by this cluster are essential for N-domain dimerization and are necessary for active site formation and for ATPase activity (Cunningham et al., 2008; Morra et al., 2009). Indeed, while Swe1 is able to phosphorylate Hsp90-D79N, which exists in an open conformation at equilibrium (Y24 exposed to solvent), it is unable to phosphorylate Hsp90-E33A, which favors a closed (N-domain dimerized) conformation (Y24 buried in hydrophobic cluster, Figure S6). Thus, it is not surprising that yHsp90-Y24E protein has minimal ATPase activity and fails to undergo N-domain dimerization in the presence of AMPPNP, even though nucleotide binding to this mutant is comparable to that of wt yHsp90. In contrast, the ATPase activity of yHsp90-Y24F is unchanged from wt. These data emphasize the importance of the hydrophobic character of this residue, as a previous study found that the ATPase activity of yHsp90-Y24A was reduced nearly 5-fold compared to wt (Cunningham et al., 2008).
While it remains to be determined whether Y24/Y38 phosphorylation has a similar impact on Hsp90 ATPase activity as does phosphomimic mutation, the sum of our data predict that the hydrophobic interactions in which Y24/Y38 participate, and which are necessary for ATPase activity, are likely to be disrupted by tyrosine phosphorylation. The necessity of Y24/Y38 phosphorylation for chaperoning certain kinases and suppressing HSF activity is difficult to explain, but it may reflect a need of some client proteins for prolonged association with a conformationally restricted, ATPase-incompetent form of Hsp90.
Finally, we explored the impact of Y24/Y38 phosphorylation on the ability of Hsp90 to interact with the prototypical ATP binding pocket inhibitor GA. We found that, in yeast lysates, yHsp90-Y24F and hHsp90-Y38F bound more efficiently to GA-beads compared to wt Hsp90, and we verified these results using swe1Δ cells expressing wt Hsp90. In agreement with these data, the Hsp90 bound to GA-beads is not tyrosine phosphorylated (Figure S6). Neither Swe1 deletion nor Hsp90 mutation affected the binding of ATP. Next, we demonstrated that concomitant pharmacologic inhibition of Wee1 and Hsp90 induced significant apoptosis in two cancer cell lines, although neither drug alone was able to do so at the concentrations used. These results were confirmed by analysis of two apoptosis markers, cleaved caspase-3 and cleaved PARP. In each case, the drug combination was much more effective than either inhibitor alone. Silencing of Wee1, combined with Hsp90 inhibition, produced identical results. These data are consistent with the observation that both swe1Δ yeast and yeast expressing only yHsp90-Y24F are more sensitive to GA (data not shown), and they suggest that the inability of Hsp90 to chaperone specific clients, or to interact with specific co-chaperones, may enhance cellular sensitivity to Hsp90 inhibitors. Indeed, two previous studies have reported that knockdown of Aha1 and p23 enhance mammalian and yeast cell sensitivity, respectively, to Hsp90 inhibition (Forafonov et al., 2008; Holmes et al., 2008). Our data provide a novel approach to exploit these observations in the clinic, while at the same time identifying a unique post-translational modification of Hsp90 that differentially regulates chaperoning of specific client proteins.
Experimental Procedures
Yeast Strains and Growth Media
The yeast strains used in this study pp30-Swe1HA (SWE1HA6-HIS3MX6) and pp30-swe1Δ (SWE1HIS3MX6) were derived from pp30 (MAT a, trp1-289, leu2-3,112, his3-200, ura3-52, ade2-101, lys2-801, hsc82KANMX4, hsp82KANMX4), (Panaretou et al., 1998). Genetic manipulations and media conditions for both yeast and mammalian cells are detailed in Supplemental Data.
Protein Analysis and Immunoblotting
Total protein extracts were prepared and analyzed as previously described (Panaretou and Piper, 1996). Detailed protein precipitations and detection by Western blotting for both yeast and mammalian cells are presented in Supplemental Data.
Yeast Nuclear Extract
Nuclear and cytoplasmic protein extracts were prepared from yeast cells using a modified method on Dr. Steven Hahn's laboratory website (http://labs.fhcrc.org/hahn/Methods/biochem_meth/polii_nuc_ext.html). Additional details are found in Supplemental Data.
In Vitro Kinase Assay
Yeast Hsp90 and the Y24F mutant, as well as human Hsp90α and the Y38F mutant, were N-terminally His6 tagged using pRSETA plasmid. They were expressed in bacteria, and 2mg of protein extracts were incubated with 50 μl of Ni-NTA agarose (Qiagen). 200 ng of baculovirus expressed and purified Wee1-GST (Invitrogen) were used in the kinase assay. Wee1 kinase reactions were as previously described (Lees-Miller and Anderson, 1989)
Ste11ΔN, Mpk1/Slt2, v-Src, GR and HSE-LacZ Activation Assays
Ste11ΔN induction and Mpk1/Slt2 activation were analyzed as previously described (Louvion et al., 1998; Millson et al., 2004). Ste11ΔN plasmid is a gift of Jill Johnson. v-Src induction and activation were analyzed as previously described (Vaughan et al., 2008). Expressed v-Src protein was detected with EC10 mouse antibody (Millipore), and v-Src activity with 4G10 mouse anti-phosphotyrosine antibody (Millipore). GR assay was performed as previously described (Garabedian and Yamamoto, 1992), as was measurement of HSE-LacZ expression (Hjorth-Sorensen et al., 2001). Additional details are found in Supplemental Data.
Hsp90 ATPase Activity and Cross-Linking Assays
ATPase activity and Hsp90 cross-linking were measured as previously described (Panaretou et al., 1998). All activities are averages of three separate measurements.
Isothermal Titration Calorimetry (ITC) and Kd Determinations
ITC and Kd determinations were performed as previously described (Panaretou et al., 1998). Additional information can be found in Supplemental Data
Supplementary Material
Acknowledgments
We are grateful to Dr. Michael Snyder for Swe1-GST and Cdc14-GST constructs and to Dr. Daniel C. Masison for anti-Sti1 antibody. We are also grateful to Dr. Jill Johnson and Dr. Wanping Xu for discussion and helpful suggestions. This work was supported by the Intramural Research Program of the National National Cancer Institute (L.N.).
Footnotes
Highlights
• We have identified a novel regulatory tyrosine phosphorylation site on Hsp90
• Swe1Wee1 phosphorylates this tyrosine residue
• Phosphorylation of this site determines Hsp90 interaction with a subset of clients
• Swe1Wee1 silencing/inhibition sensitizes yeast & cancer cells to Hsp90 inhibitors
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aligue R, Akhavan-Niak H, Russell P. A role for Hsp90 in cell cycle control: Wee1 tyrosine kinase activity requires interaction with Hsp90. EMBO J. 1994;13:6099–6106. doi: 10.1002/j.1460-2075.1994.tb06956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Park JE, Sakchaisri K, Yu LR, Song S, Supavilai P, Veenstra TD, Lee KS. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. EMBO J. 2005;24:2194–2204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Nathan DF, Lindquist S. In vivo analysis of the Hsp90 cochaperone Sti1 (p60) Mol Cell Biol. 1997;17:318–325. doi: 10.1128/mcb.17.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Krukenberg KA, Agard DA. Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J Biol Chem. 2008;283:21170–21178. doi: 10.1074/jbc.M800046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval M, Le Boeuf F, Huot J, Gratton JP. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol Biol Cell. 2007;18:4659–4668. doi: 10.1091/mbc.E07-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure R, Vincent M, Dufour M, Shaver A, Posner BI. Arrest at the G2/M transition of the cell cycle by protein-tyrosine phosphatase inhibition: studies on a neuronal and a glial cell line. J Cell Biochem. 1995;59:389–401. doi: 10.1002/jcb.240590310. [DOI] [PubMed] [Google Scholar]
- Forafonov F, Toogun OA, Grad I, Suslova E, Freeman BC, Picard D. p23/Sba1p protects against Hsp90 inhibitors independently of its intrinsic chaperone activity. Mol Cell Biol. 2008;28:3446–3456. doi: 10.1128/MCB.02246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garabedian MJ, Yamamoto KR. Genetic dissection of the signaling domain of a mammalian steroid receptor in yeast. Mol Biol Cell. 1992;3:1245–1257. doi: 10.1091/mbc.3.11.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goes FS, Martin J. Hsp90 chaperone complexes are required for the activity and stability of yeast protein kinases Mik1, Wee1 and Swe1. Eur J Biochem. 2001;268:2281–2289. doi: 10.1046/j.1432-1327.2001.02105.x. [DOI] [PubMed] [Google Scholar]
- Graf C, Stankiewicz M, Kramer G, Mayer MP. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 2009;28:602–613. doi: 10.1038/emboj.2008.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272:23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- Harvey SL, Kellogg DR. Conservation of mechanisms controlling entry into mitosis: budding yeast wee1 delays entry into mitosis and is required for cell size control. Curr Biol. 2003;13:264–275. doi: 10.1016/s0960-9822(03)00049-6. [DOI] [PubMed] [Google Scholar]
- Hjorth-Sorensen B, Hoffmann ER, Lissin NM, Sewell AK, Jakobsen BK. Activation of heat shock transcription factor in yeast is not influenced by the levels of expression of heat shock proteins. Mol Microbiol. 2001;39:914–923. doi: 10.1046/j.1365-2958.2001.02279.x. [DOI] [PubMed] [Google Scholar]
- Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2008;68:1188–1197. doi: 10.1158/0008-5472.CAN-07-3268. [DOI] [PubMed] [Google Scholar]
- Jackson SE, Queitsch C, Toft D. Hsp90: from structure to phenotype. Nat Struct Mol Biol. 2004;11:1152–1155. doi: 10.1038/nsmb1204-1152. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Halas A, Flom G. Nucleotide-dependent interaction of Saccharomyces cerevisiae Hsp90 with the cochaperone proteins Sti1, Cpr6, and Sba1. Mol Cell Biol. 2007;27:768–776. doi: 10.1128/MCB.01034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kurokawa M, Zhao C, Reya T, Kornbluth S. Inhibition of apoptosome formation by suppression of Hsp90beta phosphorylation in tyrosine kinase-induced leukemias. Mol Cell Biol. 2008;28:5494–5506. doi: 10.1128/MCB.00265-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Shabbir A, Cardozo C, Caplan AJ. Sti1 and Cdc37 can stabilize Hsp90 in chaperone complexes with a protein kinase. Mol Biol Cell. 2004;15:1785–1792. doi: 10.1091/mbc.E03-07-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Anderson CW. Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J Biol Chem. 1989;264:2431–2437. [PubMed] [Google Scholar]
- Lew DJ. The morphogenesis checkpoint: how yeast cells watch their figures. Curr Opin Cell Biol. 2003;15:648–653. doi: 10.1016/j.ceb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang Y. The function and regulation of budding yeast Swe1 in response to interrupted DNA synthesis. Mol Biol Cell. 2006;17:2746–2756. doi: 10.1091/mbc.E05-11-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvion JF, Abbas-Terki T, Picard D. Hsp90 is required for pheromone signaling in yeast. Mol Biol Cell. 1998;9:3071–3083. doi: 10.1091/mbc.9.11.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan CH, Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993;12:75–85. doi: 10.1002/j.1460-2075.1993.tb05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin SH, Sobott F, Yao ZP, Zhang W, Nielsen PR, Grossmann JG, Laue ED, Robinson CV, Jackson SE. The co-chaperone p23 arrests the Hsp90 ATPase cycle to trap client proteins. J Mol Biol. 2006;356:746–758. doi: 10.1016/j.jmb.2005.11.085. [DOI] [PubMed] [Google Scholar]
- Meyer P, Prodromou C, Hu B, Vaughan C, Roe SM, Panaretou B, Piper PW, Pearl LH. Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol Cell. 2003;11:647–658. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- Millson SH, Truman AW, Racz A, Hu B, Panaretou B, Nuttall J, Mollapour M, Soti C, Piper PW. Expressed as the sole Hsp90 of yeast, the alpha and beta isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90beta generates sensitivity to the Hsp90 inhibitor radicicol. FEBS J. 2007;274:4453–4463. doi: 10.1111/j.1742-4658.2007.05974.x. [DOI] [PubMed] [Google Scholar]
- Millson SH, Truman AW, Wolfram F, King V, Panaretou B, Prodromou C, Pearl LH, Piper PW. Investigating the protein-protein interactions of the yeast Hsp90 chaperone system by two-hybrid analysis: potential uses and limitations of this approach. Cell Stress Chaperones. 2004;9:359–368. doi: 10.1379/CSC-29R1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimnaugh EG, Worland PJ, Whitesell L, Neckers LM. Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the hsp90 stress protein and the pp60v-src tyrosine kinase. J Biol Chem. 1995;270:28654–28659. doi: 10.1074/jbc.270.48.28654. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Morra G, Verkhivker G, Colombo G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLoS Comput Biol. 2009;5:e1000323. doi: 10.1371/journal.pcbi.1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–530. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J Cell Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer BD, Thompson AM, Booth RJ, Dobrusin EM, Kraker AJ, Lee HH, Lunney EA, Mitchell LH, Ortwine DF, Smaill JB, et al. 4-Phenylpyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione inhibitors of the checkpoint kinase Wee1. Structure-activity relationships for chromophore modification and phenyl ring substitution. J Med Chem. 2006;49:4896–4911. doi: 10.1021/jm0512591. [DOI] [PubMed] [Google Scholar]
- Panaretou B, Piper P. Isolation of yeast plasma membranes. Methods Mol Biol. 1996;53:117–121. doi: 10.1385/0-89603-319-8:117. [DOI] [PubMed] [Google Scholar]
- Panaretou B, Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretou B, Siligardi G, Meyer P, Maloney A, Sullivan JK, Singh S, Millson SH, Clarke PA, Naaby-Hansen S, Stein R, et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol Cell. 2002;10:1307–1318. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006;75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- Picard D. Intracellular dynamics of the Hsp90 co-chaperone p23 is dictated by Hsp90. Exp Cell Res. 2006;312:198–204. doi: 10.1016/j.yexcr.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Piper PW, Panaretou B, Millson SH, Trumana A, Mollapour M, Pearl LH, Prodromou C. Yeast is selectively hypersensitised to heat shock protein 90 (Hsp90)-targetting drugs with heterologous expression of the human Hsp90beta, a property that can be exploited in screens for new Hsp90 chaperone inhibitors. Gene. 2003;302:165–170. doi: 10.1016/s0378-1119(02)01102-2. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Galigniana MD, Morishima Y, Murphy PJ. Role of molecular chaperones in steroid receptor action. Essays Biochem. 2004;40:41–58. doi: 10.1042/bse0400041. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Scroggins BT, Neckers L. Post-translational modification of heat shock protein 90: impact on chaperone function. Expert Opin. Drug Discov. 2007;2:1403–1414. doi: 10.1517/17460441.2.10.1403. [DOI] [PubMed] [Google Scholar]
- Southworth DR, Agard DA. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Vaughan CK, Mollapour M, Smith JR, Truman A, Hu B, Good VM, Panaretou B, Neckers L, Clarke PA, Workman P, et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol Cell. 2008;31:886–895. doi: 10.1016/j.molcel.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem. 2008;283:18473–18477. doi: 10.1074/jbc.R800007200. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Workman P, de Billy E. Putting the heat on cancer. Nat Med. 2007;13:1415–1417. doi: 10.1038/nm1207-1415. [DOI] [PubMed] [Google Scholar]
- Zhao YG, Gilmore R, Leone G, Coffey MC, Weber B, Lee PW. Hsp90 phosphorylation is linked to its chaperoning function. Assembly of the reovirus cell attachment protein. J Biol Chem. 2001;276:32822–32827. doi: 10.1074/jbc.M105562200. [DOI] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.