

Abstract
The mucolipidoses are a heterogenous group of autosomal recessive neurodegenerative lysosomal storage disorders. Mucolipidosis type IV is rare, is seen predominantly in the Ashkenazi Jewish population, and usually presents with global neurodevelopmental delays in infancy, subtle corneal opacifications or clouding, and very slowly progressive neurodegeneration over many years. Elevation of serum gastrin is reported while x-rays of bone and joints and lysosomal studies are normal. We report two cases of mucolipidosis type IV in non-Jewish children who presented during infancy with non-specific global psychomotor delays, generalized hypotonia, and mild corneal abnormalities, but remained undiagnosed for years. A rare gene mutation in MCOLN1 was confirmed in one of our two patients in addition to the abnormal serum gastrin levels. More striking was the length of time these children eluded detection of their final diagnosis.
Keywords: mucolipidosis type IV, sialolipidosis, lysosomes, neurodegenerative disease, corneal clouding, gastrin, gene mutation, MCOLN1
INTRODUCTION
Mucolipidosis type IV (sialolipidosis) is a rare autosomal recessive neurodegenerative lysosomal storage disorder with an incidence of 1:10,000 [1]. The majority of reported cases have occurred in Ashkenazi Jews [2]. Phenotypic hallmarks of this condition include global psychomotor retardation in infancy, especially hypotonia, rarely spastic diplegia during the second decade [3], corneal clouding and/or opacification, substantial elevation of serum gastrin [4], distinctive brain abnormalities on brain magnetic resonance imaging (MRI) scan [5], iron deficiency anemia, coarsening of facial features as a late observation, and unexpectedly normal urine mucopolysaccharides, oligosaccharides, and plasma lysosomal hydrolase assays [1,3–7]. Organomegaly and skeletal findings are largely absent [1,4,8–12].
The cause of mucolipidosis type IV involves a mutation of the mucolipin-1 gene (MCOLN1) on the short arm of chromosome 19 [2]. Almost all tissues of the body are affected [10]. Positional cloning has determined that MCOLN1 falls within the Golgi microtubule associated protein locus located at 19p13.3 to 19p13.2 [13]. MCOLN1 is thought to be in the family of transient receptor potential calcium channels (homology to the transient protein receptor cation channel superfamily) [13]. Calcium efflux is associated with endocytosis, the hypothesized pathology in mucolipidosis type IV [13].
We report two unrelated Caucasian cases in non-Ashkenazi Jewish children who have typical yet subtle phenotypes described for this rare storage condition. One child was found to have a rare MCOLN1 gene mutation [14], but both children eluded detection of a specific diagnosis of mucolipidosis type IV over time.
CASE SUMMARIES
Case One
The patient is a 4–1/2-year-old non-Jewish Caucasian girl initially seen at one year of age for global neurodevelopmental delays. The mother’s prenatal course, term labor, and vaginal delivery were normal. Birth weight was 3.4 kilograms with normal Apgar scores. During her first year, she demonstrated delays in fine and gross motor development with generalized hypotonia as well as language delays. No seizures were reported. At one year, she could roll over, sit with support, use a pincer grasp, and babble. The maternal family history over three generations showed several individuals with physical or cognitive disabilities, including members with Smith-Magenis syndrome, Angelman syndrome, unexplained mental retardation, and a miscarriage with triploidy. There was no history of consanguinity.
Examination at one year included a weight of 11.2 kg (75th percentile), height of 76.2 cm (90th percentile), and head circumference of 45.5 cm (30th percentile) without specific dysmorphic features. Global neurodevelopmental delays including significant generalized hypotonia with a myopathic facies including a tented upper lip were present. Mild corneal haziness was initially evident only with magnification and illumination. The remainder of the physical exam was normal. Neurologic exam revealed mild delays in mental status, intact cranial nerves, severe gross motor delays with decreased muscle bulk, strength, and tone but no head lag. Normal coordination, deep tendon reflexes, and sensory responses were present.
A brain MRI at one year of age revealed nonspecific abnormal white matter hypomyelinization with no anatomic abnormalities. An electroencephalogram exhibited generalized slowing. An ophthalmologic evaluation including slit-lamp examination at 18 months of age confirmed very mild corneal crystallization/opacifications bilaterally with normal optic nerves and retinae.
At two years of age generalized hypotonia persisted with normal deep tendon reflexes but without any neurodevelopmental regression. Receptive language skills were her strength but she was unable to ambulate or speak effectively. A follow-up brain MRI revealed persistent hypomyelinization and mild hypoplasia of the corpus callosum. A serum gastrin was significantly elevated at 710 pg/ml (0–99pg/ml normal range). A skin biopsy was done and demonstrated vacuoles containing granular material and lipids compatible with a lysosomal storage disease. Prior extensive laboratory testing for neurometabolic-genetic disorders including plasma acylcarnitines, transferrin, lipid profile, and urine organic acids, sialic acid, mucopolysaccharides, and oligosaccharides was normal. Chromosome and subtelomere analysis, as well as myotonic dystrophy and Pelizaeus-Merzbacher gene analyses, were normal.
Following the finding of an elevated serum gastrin level, a diagnosis of mucolipidosis type IV was confirmed by finding heterozygous mutations in the MCOLN1 gene at 4 years of age. The mutation in her maternal allele was identified as p.T232P, a mutation previously reported in non-Ashkenazi families. Her paternal mutation revealed an insertion of a mitochondrial DNA fragment into the MCOLN1 gene, a type of mutation only reported previously in a Canadian patient [14]. This appears to be the initial report of this unique insertion of mitochondrial DNA fragment into the MCOLN1 gene being inherited from the father. We have been unable to link this family with the one previously reported family. She has continued to make slow neurodevelopmental progress without neuroregression. [Figure 1] Her vocabulary at age 6 years includes six words. She has persistent generalized hypotonia and poor fine and gross motor functions. She has no difficulty with eating, drinking from a cup, and using eating utensils with adaptive features. She stands with support wearing ankle-foot orthotics and is beginning to initiate steps.
Figure 1.

At 5 years of age Case 1 could sit while using an arm for minimal support, exhibited global neurodevelopment delays with no neuroregression, and had a normal phenotypic appearance with no obvious eye abnormalities.
Case Two
At presentation, the patient was an 11-year-old non-Jewish Caucasian girl with profound neurodevelopmental delays including no speech or ambulation. Her mother’s prenatal course, term labor and delivery were normal. Birth weight was 3 kilograms without neonatal problems. Global neurodevelopmental delays observed during infancy have persisted. She sat with support at nine months but has never walked. She can now say a few words, has normal hearing, is not toilet-trained, and has difficulty feeding herself. There is no history of neuroregression. Her past medical history revealed vesicoureteral reflux which has resolved, corneal clouding with optic atrophy and strabismus, and limb spasticity, more severe in her lower limbs, which required ankle-foot orthotics. Family history was noncontributory with no consanguinity.
Examination at 11 years included a weight of 23.6 kg (<3rd percentile), height of 116.2 cm (<3rd percentile), and head circumference of 48.5 cm (≤3rd percentile). She was non-verbal, non-ambulatory, profoundly globally neurologically impaired, and exhibited mild coarsening of her facial features and corneal opacifications. She was status postoperative scoliosis repair. She was alert with normal cranial nerve functions and central hypotonia with limb spasticity. She had generalized increased deep tendon reflexes, greater in her upper limbs, with grossly normal cerebellar and sensory function.
A brain MRI at 11 years of age revealed severe hypoplasia of the corpus callosum [Figure 2] with mild ventricular dilatation and significant widespread hypomyelinization. [Figure 3] A serum gastrin was elevated at 526 pg/ml (0–99pg/ml normal). Other extensive biochemical and genetic laboratory testing was normal. Molecular analysis of MCOLN1 for the common mutations seen in Ashkenazi Jews was normal, but this is not surprising considering her heritage.
Figure 2.
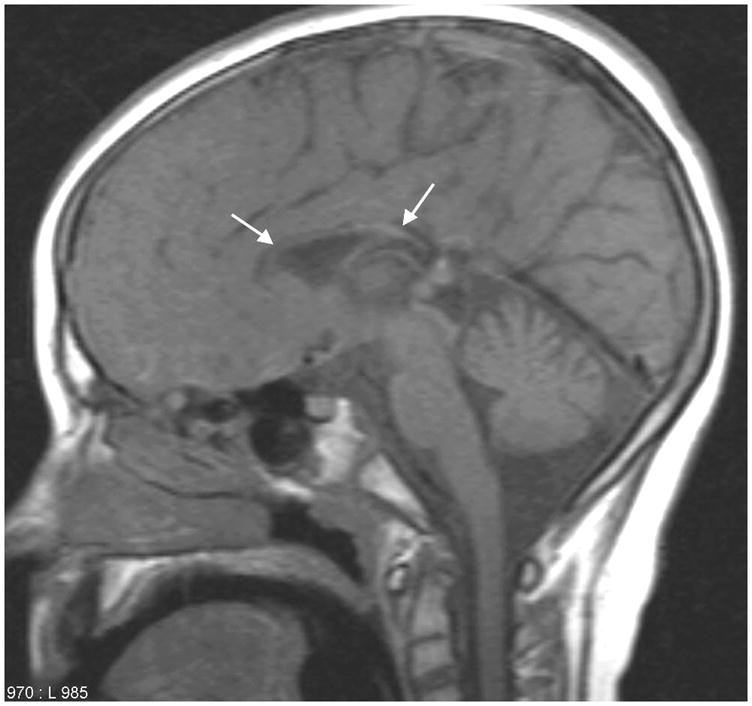
Sagittal T1-weighted magnetic resonance image (TE: 8, TR: 400, slice thickness 5mm) from Case 2 showing an extremely thin hypoplastic corpus callosum (white arrows) with mild enlargement of the cisterna magna.
Figure 3.
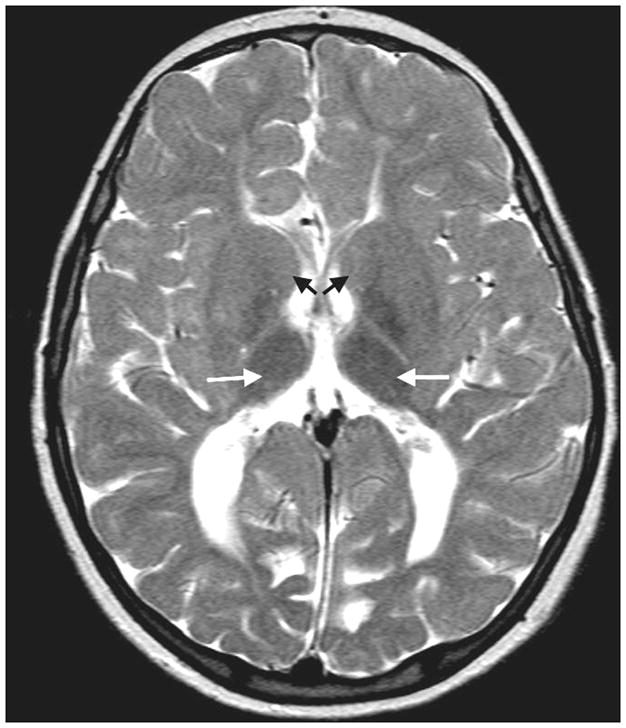
Axial T2 -weighted magnetic resonance image (TE: 90, TR: 3842, slice thickness 5mm) from Case 2 demonstrating the absence of myelinated fibers in the caudate heads (dark arrows) and adjoining putamen, and normal myelination in the thalami (white arrows). A diminished volume of white matter is present in the cerebral hemispheres and hypomyelination is present in the white matter extending out to include multiple subcortical U fibers. The ventricles are also mildly dilated.
DISCUSSION
Mucolipidosis type IV poses a significant diagnostic challenge. Of great significance diagnostically is the absence of gastric hydrochloric acid (achlorhydria) [4]. The mutated gene MCOLN1 involves cation efflux, especially calcium, through its close association with the protein receptor cation channel superfamily [13]. Calcium efflux is closely associated with normal endocytosis. The resultant endocytotic abnormality is believed to affect normal acid production in parietal cells of the gastric mucosa. This produces a constitutive achlorhydria that via a feedback pathway increases gastrin production, and in turn increases the serum gastrin [4,6]. Elevated serum gastrin may be one of the most specific diagnostic markers for this disorder, but is a test not usually recommended or done in the evaluation of children with global neurodevelopmental delays. Other medical etiologies of elevated serum gastrin levels occur at any age as a physiologic response to achlorhydria secondary to pernicious anemia, H. pylori infections, renal failure, atrophia gastritis, pyloric obstruction, antral G-cell hyperfunction, gastrinomas of the pancreas or gastrointestinal tract, Zollinger-Ellison syndrome, and the long-term use of proton pump inhibitor medications [15].
Mucolipidosis type IV is a unique lysosomal disorder in that it does not involve an abnormality of lysosomal hydrolases. The exact etiology is unknown, but it is felt to involve a disruption in sorting and/or transport involving the late endosome/lysosome endocytic process and/or compartment [13]. Examinations of tissues including skin, muscle, and liver show an accumulation of vacuoles containing granular material and lipids including phospholipids, phosphatidylcholine, sphingolipids, gangliosides, and mucopolysaccharides. These macromolecules are apparently transported into lysosomes at an increased rate rather than into the Golgi bodies for proper recycling into new cell membrane [13]. This explains why there is heterogeneity in the substances found within the lysosomes. Catabolism of these macromolecules is thought to be normal explaining the relative absence of significant coarse features, organomegaly, and presence of slow neurodevelopmental deterioration [8].
Ophthalmologically, patients usually have corneal clouding and/or opacities with gradual fibrous dysplasia of the cornea, retinal degeneration, and ultimately optic atrophy. Strabismus and photophobia are common findings [7]. Corneal opacifications may be absent or very subtle. Blindness is commonly due to optic degeneration [7]. A detailed examination by an ophthalmologist, even under anesthesia if necessary, is recommended as part of a complete evaluation.
The central nervous system commonly demonstrates cerebral and cerebellar atrophy with microcephaly [5]. Hypotonia, as well as hyperreflexia, spasticity, and dystonia can be seen during the slow progression of this disorder [8]. Independent normal ambulation and speech are rarely attained. Mental retardation is typical following infantile non-specific global neurodevelopmental delays. Diagnosis is commonly made in childhood but progression is slow during the first two to three decades. Our cases were not diagnosed until 4½ and 11 years of age. Motor and cognitive development rarely advance beyond the 13 to 15 month level. Motor function can stabilize with early intervention therapy and might even show some improvement. Genetic testing for the most frequent mutations can commonly be negative in non-Jewish persons. A useful web-based reference source for further detailed information on genotype-phenotype correlations and genetic laboratory testing is available through www.genetests.org [http://www.ncbi.nlm.nih.gov/sites/GeneTests/?db=GeneTests].
Although this is thought to be a rare disorder, its frequency may be greater than expected due to its milder storage disease phenotype, especially since urine oligosaccharides, mucopolysaccharides and plasma lysosomal hydrolases are uncharacteristically normal in this disorder. Psychomotor retardation and abnormal ophthalmologic findings are found in most patients with mucolipidosis type IV, but they can be subtle, especially during infancy and early childhood, and not as specific as in other lysosomal storage disorders [8,10]. A brain MRI showing a hypoplastic corpus callosum and/or white matter hypomyelination can be very supportive of the diagnosis and reason to pursue further work-up [5]. These prior findings support obtaining a serum gastrin level at this point in the evaluation as the most practical diagnostically significant test available to the clinician.
This report strongly reminds us that obtaining a serum gastrin is prudent as part of the cost-effective evaluation for mucolipidosis type IV in any infant or young child who has significant unexplained global neurodevelopmental delays (especially those who are hypotonic, nonambulatory, and nonverbal but are not necessarily exhibiting neuroregression), an abnormal MRI with corpus callosum thinning and diffuse white matter hypomyelination, and/or eye findings which may include subtle or gross corneal opacifications.
Acknowledgments
We acknowledge and thank Joel K. Curé, M.D., Raphael Schiffman, M.D., Maria G. Matheus, M.D., and Debra J. Hazen-Martin, Ph.D. for their contributions and insight, and Patti Broome for assistance with preparation of this manuscript.
Footnotes
This work was done in Greenville, Greenwood, and Charleston, South Carolina, as well as in Bethesda, Maryland. There are no conflicts of interest or financial disclosures to report with regard to this manuscript. No reprints are requested.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gordon RM, Marchese T. Mucolipidosis type IV: a rare genetic disorder: new addition to the Ashkenazi Jewish panel. J Midwifery Womens Health. 2004;49:359–60. doi: 10.1016/j.jmwh.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Bach G, Webb MBT, Bargal R, Zeigler M, Ekstein J. The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of three novel MCOLN1 mutations. Hum Mutat mutation in brief. 2005;856:1–5. doi: 10.1002/humu.9385. [DOI] [PubMed] [Google Scholar]
- 3.Bindu PS, Gayathri N, Yasha TC, Kovoor JME, Subasree R, Rao S, Panda S, Pal PK. A variant form of mucolipidosis IV: report on 4 patients from the Indian subcontinent. J Child Neurol. 2008;23:1443–6. doi: 10.1177/0883073808318537. [DOI] [PubMed] [Google Scholar]
- 4.Schiffmann R, Dwyer NK, Lubensky IA, Tsokos M, Sutliff VE, Latimer JS, Frei KP, Brady RO, Barton NW, Blanchette-Mackie EJ, Goldin E. Constitutive achlorhydria and mucolipidosis type IV. Proc Natl Acad Sci USA. 1998;95:1207–12. doi: 10.1073/pnas.95.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frei KP, Patronas NJ, Crutchfield KE, Altarescu G, Schiffmann R. Mucolipidosis type IV: characteristic MRI findings. Neurology. 1998;51:565–9. doi: 10.1212/wnl.51.2.565. [DOI] [PubMed] [Google Scholar]
- 6.Lubensky IA, Schiffmann R, Goldin E, Tsokos M. Lysosomal inclusions in gastric parietal cells in mucolipidosis type IV: a novel cause of achlorhydria and hypergastrinemia. Am J Surg Pathol. 1999;23:1527–31. doi: 10.1097/00000478-199912000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Riedel KG, Zwaan J, Kenyon KR, Kolodny EH, Hanninen L, Albert DM. Ocular abnormalities in mucolipidosis IV. Am J Opthalmol. 1985;99:125–36. doi: 10.1016/0002-9394(85)90220-x. [DOI] [PubMed] [Google Scholar]
- 8.Chitayat D, Meunier CM, Hodgkinson KA, Silver K, Flanders M, Anderson IJ, Little JM, Whiteman DA, Carpenter S. Mucolipidosis type IV: clinical manifestations and natural history. Am J Med Gen. 1991;41:313–8. doi: 10.1002/ajmg.1320410310. [DOI] [PubMed] [Google Scholar]
- 9.Weitz R, Kohn G. Clinical spectrum of mucolipidosis type IV. Pediatrics. 1988;81:602–3. [PubMed] [Google Scholar]
- 10.Amir N, Zlotogora J, Bach G. Mucolipidosis type IV: clinical spectrum and natural history. Pediatrics. 1987;79:953–9. [PubMed] [Google Scholar]
- 11.Bach G, Zeigler M, Schaap T, Kohn G. Mucolipidosis type IV: ganglioside sialidase deficiency. Biochem Biophys Res Comm. 1979;90:1341–7. doi: 10.1016/0006-291x(79)91183-5. [DOI] [PubMed] [Google Scholar]
- 12.Bach G. Minireview: Mucolipidosis type IV. Mol Genet Metab. 2001;73:197–203. doi: 10.1006/mgme.2001.3195. [DOI] [PubMed] [Google Scholar]
- 13.Bach G. Mucolipin1: endocytosis and cation channel-a review. Eur J Physiol. 2005;451:313–7. doi: 10.1007/s00424-004-1361-7. [DOI] [PubMed] [Google Scholar]
- 14.Goldin E, Stahl S, Cooney AM, Kaneski CR, Gupta S, Brady RO, Ellis JR, Schiffmann R. Transfer of a mitochondrial DNA fragment to MCOLN1 causes an inherited case of mucolipidosis IV. Hum Mutat. 2004;24:460–5. doi: 10.1002/humu.20094. [DOI] [PubMed] [Google Scholar]
- 15.Jensen RT. Pancreatic Endocrine Tumors. In: Goldman L, Ausiello D, editors. Cecil Medicine. 23. Philadelphia, PA: Saunders Elsevier; 2008. pp. 1482–4. [Google Scholar]