

Abstract
Primary mediastinal B-cell lymphoma (PMBL) is a separate entity of aggressive B-cell lymphoma, characterized by a constitutive activation of janus kinase - signal transducer and activator of transcription (JAK - STAT) signaling pathway, also observed in Hodgkin lymphoma. Although many cancers exhibit constitutive JAK - STAT pathway activation, mutations of STAT genes have not been reported in neoplasms. Here, we show that MedB-1 PMBL-derived and L1236 Hodgkin-derived cell lines and 20 of 55 (36%) PMBL cases harbor heterozygous missense mutations in STAT6 DNA binding domain, whereas no mutation was found in 25 diffuse large B-cell lymphoma samples. In 3 cases, somatic origin was indicated by the absence of the mutations in the non tumoral tissue. The pattern of STAT6 mutations was different from the classical features of somatic hypermutations. The mutant STAT6 proteins showed a decreased DNA binding ability in transfected HEK cells, but no decrease in expression of STAT6 canonical target genes was observed in PMBL cases with a mutated STAT6 gene. Although the oncogenic properties of STAT6 mutant proteins remain to be determined, their recurrent selection in PMBL strongly argues for their involvement in the pathogenesis of this aggressive B-cell lymphoma.
Keywords: Cell Line, Tumor; Female; Gene Expression Regulation, Neoplastic; genetics; Humans; Lymphoma, Large B-Cell, Diffuse; genetics; metabolism; Male; Mediastinal Neoplasms; genetics; metabolism; Mutation; genetics; Neoplasm Proteins; genetics; metabolism; Protein Structure, Tertiary; genetics; STAT6 Transcription Factor; genetics; metabolism; Signal Transduction; genetics
INTRODUCTION
Primary mediastinal B-cell lymphoma (PMBL) is recognized as a separate entity of aggressive B-cell lymphoma, with unique clinical, histological, and biologic features.1 This lymphoma presents as a mediastinal mass consisting of large B cells, that usually express little if any surface or cytoplasmic immunoglobulin and major histocompatibility complex class I and/or class II molecules and presumably derive from a subset of thymic B cells.2 PMBL displays a constitutive activation of the JAK/STAT signaling pathway as indicated by the presence of nuclear phosphorylated (P) STAT6,3 which is also observed in Hodgkin lymphoma.4,5 STAT6 expression was shown to regulate cell proliferation and survival in a PMBL derived cell line, MedB-1.6 These lymphomas harbor chromosomal gains of the JAK2 gene,7 and inactivating mutations of the Suppressor of Cytokine Signaling (SOCS)1 gene, 8 which are also commonly found in Hodgkin lymphoma.9,10 Although SOCS1 inactivation prevents STAT6 downregulation and thereby contributes to STAT6 activity,6 the primary mechanism responsible for STAT6 activation remains unclear. PMBL do not secrete the specific cytokines (i.e., Interleukin -4 or -13) classically involved in the activation of this factor, unlike classical Hodgkin lymphoma11,12 and JAK2 inhibition only partially prevented STAT6 activation in MedB-1.3
The analysis of STAT6 functional properties by directed mutagenesis has shown that a mutation in the SH2 domain (STAT6 V547A/T548A) leads to the constitutive activation of STAT6.13 This mutation changes the conformation of the protein and increases the stability of the monomeric and dimeric proteins, allowing STAT6VT mutant protein to undergo tyrosine phosphorylation, followed by DNA binding, in the absence of IL-4 stimulation. Transgenic mice expressing STAT6VT under control of the CD2 locus control region, which essentially directs expression to the T-cell compartment, show increased peripheral B cell and decreased T cell numbers,14 and develop, in a small proportion of cases, a spontaneous lymphoproliferative disease of T-cell or B-cell mixed phenotype.15 These data prompted us to analyze the sequence of STAT6 mRNA in MedB-1 and Karpas1106 PMBL derived cell lines and in a series of 55 primary tumor samples. We found heterozygous mutations targeting the DNA binding domain in one cell line and 36% of the samples, and therefore, analyzed the histological, clinical and molecular characteristics of the PMBL cases with a STAT6 mutation, as well as the functional properties of the mutant STAT6 proteins.
MATERIAL AND METHODS
Tissue samples selection and DNA or RNA extraction
This study was approved by the Ethic Committee of the University of Ulm, Germany and the institutional review board “comité de protection des personnes Ile de France IX”, Créteil, France. A series of 55 primary mediastinal B-cell lymphoma (PMBL) and 25 diffuse large B-cell lymphoma cases (DLBCL) were retrieved from the files of the Institute of Pathology, Ulm, Germany and of the Department of Pathology, Hospital Henri Mondor, Créteil, France. These lymphomas samples, initially diagnosed between 1984 and 2007, showed the histological features characteristic of these lymphoma types according to the recently updated WHO classification.1 The PMBL samples had been previously immuno-phenotyped extensively and were included in several publications that contributed to define PMBL characteristic features.3,8,16–19 Tumor infiltration of the frozen samples was checked on Hematoxylin Eosin Safran staining of tissue sections. Total RNA were extracted from frozen tissue samples with TRIZOL reagent (Invitrogen, Cergy Pontoise, France) or Qiagen RNeasy mini kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Genomic DNA was extracted with proteinase K digestion, phenol chloroform phases separation and ethanol precipitation.
Sequencing and quantitative Polymerase Chain Reaction (PCR)
Total RNA were reverse transcribed with Superscript II (Invitrogen, San Diego, CA) and poly-dT (500 ng) or random (300 ng) primers in a final volume of 20 μl, according to the manufacturer’s instructions, and diluted 1:5 or 1:10 for PCR. Genomic DNA and cDNA were amplified by PCR using primers and detailed protocol indicated in supplementary methods. Purified PCR products were sequenced using BigDye Terminator v3.1 Cycle sequencing Kits (Applied Biosystems, Courtaboeuf, France) and capillary electrophoresis (3130 Genetic analyser, Applied Biosystems). Sequences were analyzed with Seqscape software (Applied Biosystems) and standard electropherogram reading programs (Chromas Lite, http://www.technelysium.com). Nucleotide changes detected on one strand were confirmed by sequencing the reverse strand. STAT6 sequencing was performed on genomic DNA and cDNA in 25 cases (14 with and 11 without detected mutations), on cDNA only in 21 cases (4 with and 17 without detected mutations) or genomic DNA only in 9 cases (2 with and 7 without detected mutations), depending on the material available. The SOCS1 alterations (mutations or microdeletions) were assessed on genomic DNA and cDNA (5 cases), on cDNA only (5 cases), or genomic DNA only (43 cases).
Genomic tumoral DNAs from the PMBL cases were also used for the quantitative analysis of the JAK2 gene. Briefly, 50 ng DNA were amplified using the BioRad (Hercules, CA) Sybrgreen master mix and primers for JAK2 and for B2M (Table 1 in supplementary methods) in a total reaction volume of 20 μL in a BioRad I-cycler. Each PCR reaction was run in quadruplicate and the mean Ct values were used for further calculations. Relative JAK2 gene dosage was calculated according to the 2−ΔΔCT method.20 Genomic DNA isolated from lymphocytes of 4 different healthy donors and from 7 frozen reactive tonsils were used to calculate the mean normal JAK2 gene dosage. Tumor samples were considered to have a JAK2 gain or amplification if the relative JAK2 gene dosage was superior to mean normal + 3 Standard Derivation.
Transfection, generation of the HEK stable transfectants and reporter assays
Wild type (WT) and double mutant (DM) STAT6 expression vectors were obtained by PCR amplification of STAT6 coding sequence using cDNA from Ramos and MedB-1 cell line, respectively, and primers containing EcoR1 and XhoI restriction sides (underlined): STAT6 for:5′-CGACGTGGAATTCTGATGTCTCTGTGGGGTCTG-3′; STAT6 rev: 5′-CAGTGCTCGAGCTCACCAACTGGGGTTGGCCCT-3′). PCR products were inserted into the Multiple Cloning Site of pcDNA3.1 expression vector (Invitrogen, Cergy Pontoise, France) and verified by sequencing. Single mutants were obtained by directed mutagenesis of WT STAT6 expression plasmids with the GeneEditor in Vitro Site-Directed Mutagenesis System (Promega, Madison, USA). WT STAT6 and DM STAT6 expression plasmids or pcDNA-mock vector were transfected into HEK293 cell lines using Effectene Transfection Reagent (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. 24h post transfection, 800 μg/ml G418 (Invitrogen, Karlsruhe, Germany) was added to the medium and HEK293 cells were cultured in the selection medium for 2 weeks before isolation of single clones or pooling transfected cells. During analysis, the transfected cells were maintained under 600 μg/ml of G418.
Firefly luciferase reporter vector containing (N3-luc) or not (control-luc) 3 copies of the N3 Gamma Activated Site (GAS) from CSN2 (casein beta) promoter (TTCtagGAA) were obtained from Panomic (Panomic, Redwood City, CA). The N4-luc vector, comprising 3 STAT6 binding site from the SOCS1 promoter (TTCcgagGAA) upstream of the luciferase reporter gene was previously described.6 HEK293 cells were transiently co-transfected using Effectene Transfection Reagent with control-luc, N3-luc or N4-luc reporter constructs (1 μg) in combination with either mock or WT STAT6 or one of the mutated STAT6 expression vectors (1 μg). Forty ng of a Renilla luciferase reporter were added to each sample to normalize transfection efficiency. Luminescence intensity was measured using the luminometer Lumat FB12 (Berthold Technologies, Bad Wildbach, Germany) and luciferase dual assay kit (Promega, Madison, USA) according to the manufacturer’s protocol.
Western blotting and immunohistochemistry
Western blots were performed as previously described.6 Proteins were detected using antibodies against STAT6 (M-200; sc-1698) (Santa Cruz) 1:1000; P-STAT6 (Tyr641; 9361) (Cell Signaling, Beverly, MA) 1:1000; beta actin (clone AC-74), (Sigma-Aldrich, Saint Louis, MO) 1:5000; HDAC1 (H-51) (Santa Cruz) 1:1000.
Immunohistochemistry was performed on paraffin section of formalin fixed tissue or on frozen sections. Paraffin sections were pretreated for 20 min by steamer boiling in tris EDTA buffer (pH6). For detection of bound P-STAT6 antibody, the EnVision detection system (Dako, Hamburg, Germany) was used with diamonobenzidine as substrate.21 The detection of P-STAT6 protein was checked in control cells, i.e., interstitial fibroblastoid cells and a subset of endothelial cells, present in each tissue sample. Samples without positive control cells were disregarded. P-STAT6 staining of neoplastic cells was considered positive whenever more than 5 % of neoplastic cells had nuclear staining.
EMSA
N4 GAS probe from SOCS1 promoter (5′ AGGTCGACTTCCCAAGAACAGAG 3′ annealed with 5′ AGGCTCTGTTCTTGGGAAGTCGA 3′) and N3 GAS probe from the CSN2 (casein beta) promoter (5′ AGGAGATTTCTAGGAATTCAATCC 3′ annealed with 5′ AGGGGATTGAATTCCTAGAAATCT 3′) were prepared. One μg nuclear protein extracts from HEK293 stable pools (mock, WT STAT6 and DM STAT6) were incubated with 32P-labelled N4 or N3 GAS probes, with or without 1 μL anti-STAT6 antibody (S-20; sc-621) (Santa Cruz) or a 100 fold molar excess cold probe and were separated on a 4% native acrylamide gel.
RESULTS
Recurrent mutations of STAT6 DNA binding domain in MedB-1 cell line and PMBL primary tumors
We analyzed the entire coding sequence of STAT6 mRNA in MedB-1 and Karpas1106 PMBL derived cell lines. Direct sequencing of PCR products showed that Karpas1106 cells carried wild type (WT) STAT6 mRNA sequences, whereas MedB-1 cells exhibited two heterozygous missense mutations located in exon 12 (N417Y and N430T), which encodes part of the DNA binding domain. We amplified MedB-1 exon 12 genomic DNA and sequenced cloned PCR products. Seven clones showed both N417Y and N430T mutations and two were WT, indicating that both mutations were located on the same allele. These mutations were detected in the parental tumor from which this cell line was established,22 but were absent in fibroblasts isolated from the same patient.
To evaluate the relevance of these mutations in PMBL, we sequenced the region of the STAT6 gene encoding amino-acids L406 to R527 in 55 PMBL and 25 aggressive B-cell lymphomas of the diffuse large B cell type. The DNA binding domain of STAT6 was found to be mutated in 20 out of 55 (36 %) PMBL cases whereas no mutation was detected in the 25 diffuse large B cell lymphomas (Fisher’s exact test, p<0,0002). Interestingly, six PMBL cases had several missense mutations and one case had both a missense and a silent mutation (Figure 1a). The 28 mutations that we detected were heterozygous and targeted preferentially A:T base pairs (20/28, exact binomial test, p=0,0035) and AA, AG, AT or GA dinucleotides (Table 1). These mutations mostly affected exon 12, with four hotspots targeting amino acid 417, 419, 421 and 430, respectively (Figure 1b). Of note, D419 is the site of a known polymorphism (D419N), which was not observed in the 80 lymphoma samples analyzed. Sequencing of genomic DNA derived from non-tumoral tissue of two mutated PMBL cases revealed WT sequence, supporting a somatic origin of the mutations, as observed in MedB1/PMBL parental tumor. As Hodgkin lymphoma cell lines also exhibit STAT6 activation5 and share some features with PMBL in gene expression profiles,19,23 we sequenced the same region of STAT6 mRNA in 4 classical Hodgkin-derived cell lines. Strikingly, L1236 cell line, previously reported to have STAT6 gene amplification and overexpression24 showed only expression of a mutated STAT6 allele, encoding N417Y hotspot mutation and D419N polymorphic change (supplementary Figure), while no mutation was detected in L428, L540 and KMH2 mRNA sequences.
Figure 1. Heterozygous mutations in the DNA binding domain of STAT6 in PMBL.
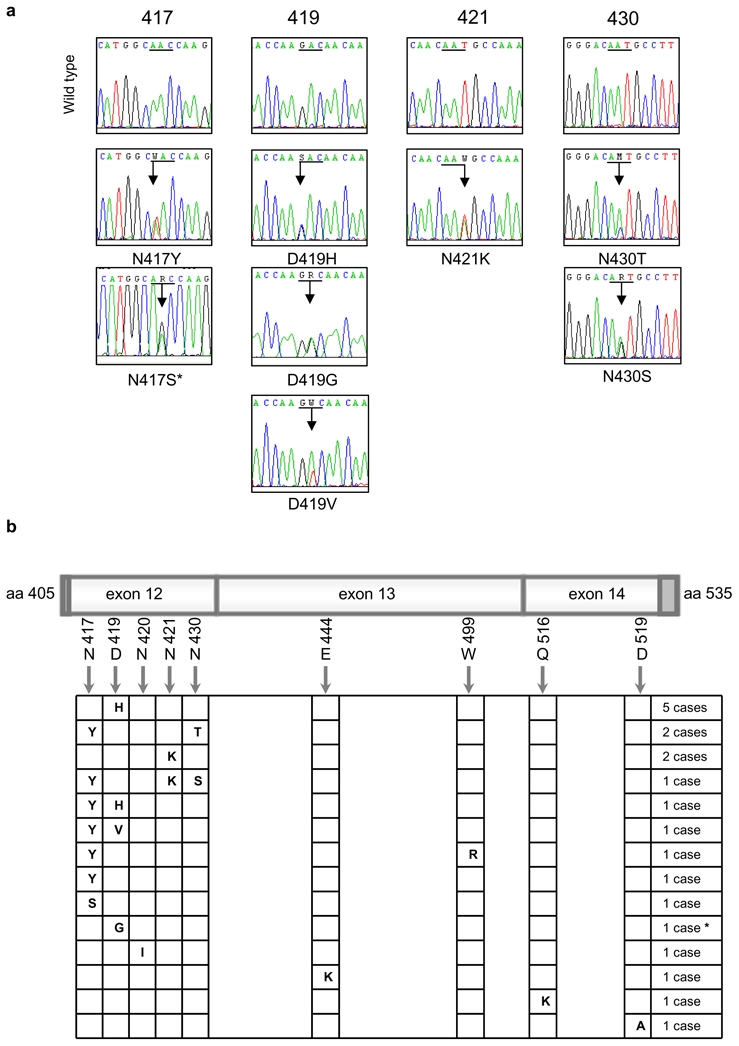
a) Electropherograms showing partial STAT6 cDNA sequences for the 4 most frequently mutated codons (underligned). Mixed nucleotides are lettered according to IUPAC international code. * This sequence was performed by PCR amplification of genomic DNA (no RNA available).
b) Schematic localization of mutations in the region correlating to the nucleotides 1216 to 1581, corresponds to exon 12 (minus 4 bases in the 5′ dark gray part), exon 13, and exon 14 (minus 26 bases in the 3′ dark gray part) of STAT6 in the 20 mutated PMBL cases. * This case presents both a missense mutation of codon 419 and a silent mutation of codon 423 (c.A1269G, according to NM_003153.3).
Table 1.
Mutation characteristics in STAT6 exons 12–14.
| Number of mutations observed | Number of motifs in sequence | Number of expected mutation * | Ratio observed/expected | P value ° | |
|---|---|---|---|---|---|
| Transversions/Transitions | 22/6 | - | 18,7/9,3 | 1,83 | 0,2301 |
| A:T base pairs | 20 | 158 | 12,1 | 1,65 | 0,003542 |
| G:C base pairs | 8 | 207 | 15,9 | 0,5 | |
| Dinucleotides | Number of mutations observed | Number of dinucleotide in sequence | Number of expected mutation | Ratio observed/expected | P value |
| AA | 13 | 20 | 3,077 | 4,225 | 0,0000007 |
| AC | 4 | 19 | 2,923 | 1,368 | 0,5198589 |
| AG | 7 | 20 | 3,077 | 2,275 | 0,0249483 |
| AT | 7 | 13 | 2,000 | 3,500 | 0,0014790 |
| CA | 8 | 27 | 4,154 | 1,926 | 0,0563318 |
| CC | 0 | 34 | 5,231 | 0,000 | 0,0068772 |
| CG | 0 | 9 | 1,385 | 0,000 | 0,3715297 |
| CT | 1 | 32 | 4,923 | 0,203 | 0,0509948 |
| GA | 10 | 23 | 3,538 | 2,826 | 0,0012213 |
| GC | 1 | 24 | 3,692 | 0,271 | 0,1614790 |
| GG | 0 | 33 | 5,077 | 0,000 | 0,0066356 |
| GT | 0 | 24 | 3,692 | 0,000 | 0,0410432 |
| TA | 0 | 2 | 0,308 | 0,000 | 1,0000000 |
| TC | 0 | 26 | 4,000 | 0,000 | 0,0254541 |
| TG | 5 | 42 | 6,462 | 0,774 | 0,6709954 |
| TT | 0 | 16 | 2,462 | 0,000 | 0,1554094 |
Expected mutation frequencies were calculated according to the sequence composition, assuming random targeting of the mutations at a 28/365 rate for single nucleotide changes and 56/364 rate for dinucleotide mutations.
Statistical comparisons were done with the exact binomial test, which compares the observed frequency to the expected one under null hypothesis. The dinucleotides which were significantly targeted by mutations are highlighted in bold.
Characteristics of the PMBL cases with a mutation of STAT6 gene
The 20 PMBL cases with a mutated STAT6 gene exhibited similar histological features (clear cells and fibrosis), and clinical characteristics as the 35 non-mutated ones (Table 2). Twelve PMBL cases previously shown to have a PMBL molecular signature, 19 had mutations in STAT6 gene (7 cases) or not (5 cases). Intriguingly, the seven cases with multiple STAT6 nucleotide changes presented a male predominance (M/F ratio = 5/2) and a younger age at diagnosis (median 32 years) compared to the 13 cases with single nucleotide changes (M/F ratio = 2/11, p=0.02, Fisher’s exact test; median age at diagnosis 41 years, p=0.07, Wilcoxon rank sum test), suggesting that a highly mutated phenotype might delineate a particular subset of PMBL patients.
Table 2.
Characteristics of the cases according to the presence or absence of a STAT6 mutation detected in exons 12–14.
| STAT6 Mutation (n=20) | no STAT6 mutation (n=35) | p value* | |
|---|---|---|---|
| median age at diagnosis (min-max) | 37.5 (22–59) | 32 (17–73) | 0,43 |
| females/males ratio | 13/7 | 15/20 | 0,16 |
| Ann Arbor stage at diagnosis | 0,71 | ||
| I-II | 11 | 16 | |
| III-IV | 3 | 7 | |
| unknown | 6 | 12 | |
| P-STAT6 staining | 0,13 | ||
| positive | 14 | 17 | |
| negative | 0 | 4 | |
| not evaluable | 6 | 14 | |
| SOCS1 gene alteration | 0,14 | ||
| present | 15 | 19 | |
| absent | 4 | 14 | |
| not determined | 1 | 2 | |
| JAK2 gene amplification | 0,76 | ||
| present | 7 | 15 | |
| absent | 10 | 15 | |
| not determined | 3 | 5 |
Age at diagnosis was compared with Wilcoxon rank test and the other characteristics were compared with Fisher’s exact test (using only informative cases).
P-STAT6 nuclear staining was detected in the nuclei of tumor cells in 31 of 35 (88%) cases that could be evaluated (Table 2). It was present in all STAT6 mutated cases, but also in a large fraction of non-mutated cases, indicating that activation of STAT6 can occur in the absence of mutation of the gene. SOCS1 gene deletions and/or mutations were observed in 34 of 52 (65%) PMBL cases. JAK2 gain or amplification was observed in 22 of 47 (47%) PMBL cases, at a similar frequency as that recently reported in a high resolution CGH array study.25 SOCS1 gene alterations and JAK2 amplifications were present both in STAT6 mutated cases and non-mutated cases (Table 2). Out of the 47 cases that could be analyzed for all three genes, five showed no genetic alteration, 20 showed one alteration, 17 showed two alterations and five cases had alterations of the 3 genes. Thus, it appears that the different genetic alterations of the JAK - STAT signaling pathway are not mutually exclusive in PMBL, suggesting additive effects.
Functional properties of STAT6 mutant proteins
In order to evaluate the functional properties of the mutated STAT6 proteins, we transfected the HEK293 STAT6 deficient cell line with expression vectors encoding either WT or N417/N430 double mutant (DM) STAT6, expressed in MedB-1/parental tumor and case 52. Independent stable pools and clones exhibited the same levels of basal and interleukin-4 (IL-4) induced STAT6 phosphorylation (Figure 2a). DM STAT6 transfectants showed a decreased DNA binding activity on both N4 and N3 GAS sites (TTC[N]xGAA), compared to WT STAT6 expressing cells, although similar amounts of STAT6 and P-STAT6 were detected in the nucleus of WT and DM STAT6 expressing cells (Figure 2b). We hypothesized that the diminished DNA binding activity of STAT6 mutants might affect their ability to activate the expression of N4- or N3- GAS driven reporters. As shown in Figure 2c, HEK293 cells transiently transfected with double mutant or with N417Y, D419H, N421K, N430T STAT6 single mutants displayed reduced N4- and N3- sites driven reporter expression, compared to WT STAT6 expressing cells, although achieving similar levels of P-STAT6 after IL-4 stimulation.
Figure 2. Functional properties of mutated STAT6 proteins in HEK transfectant cells.
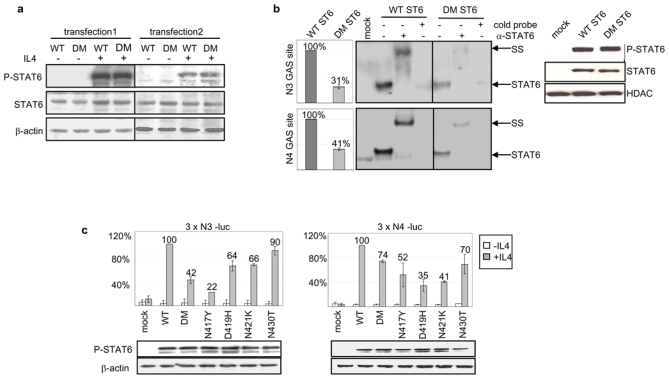
a) Transfected HEK293 cells expressing WT or N417/N430 double mutant (DM) STAT6, selected with G418 (transfection 1: pools, 2: single clones), were incubated (+) or not (−) with 10 ng/ml interleukin 4 for 24h. Western blots of total protein extracts (7μg) probed with antibodies against P-STAT6 (Tyr-641), total STAT6, and β-actin are shown.
b) Electrophoretic Mobility Shift Assay was prepared using 1μg nuclear extracts from mock transfected cells, WT or DM STAT6 HEK293 pools, stimulated with 10 ng/ml interleukin 4 for 6 h, and radiolabeled N3 GAS or N4 GAS probes. Incubation was performed with (+) or without (−) an antibody against STAT6 or an excess of cold probe. The protein/DNA complexes were electrophoresed on the same gel, and are assembled side by side in the figure for better comparison. The position of the STAT6 containing complexes and of the complexes supershifted (SS) by the antibody are indicated. Phosphorimager quantification of the radiolabelled complexes is shown in the left plots and Western blot analysis of P-STAT6 and HDAC, used as a loading control, in nuclear extracts is shown in the right plot.
c) HEK293 cells were co-transfected with expression vectors for WT STAT6, single or DM STAT6 mutants or a control mock-vector, together with 3 x N3-luc (left plot) or with 3 x N4- luc (right plot) luciferase reporter constructs. The diagrams show the mean percentage of luciferase activity, after normalization for transfection efficacy, in cells treated (grey bars) or not (white bars) with 10 ng/ml interleukin 4 for 20 h, compared to WT STAT6 expressing cells (set to 100%). Error bars shows the standard deviation (3 independent experiments). The amount of P-STAT6 in total protein extracts of interleukin 4 treated cells, detected by immunoblotting, is shown in the lower panel.
The decreased ability of STAT6 mutants to activate transcription was surprising considering the strong STAT6 signature previously reported in gene expression profiling studies of PMBL.19,23 We therefore compared the levels of expression of several genes which contain confirmed STAT6 binding motifs26 in the 5 non-mutated and the 7 mutated cases which had been analyzed with microarrays as part of a larger study.19 The levels of expression of these genes were similar in the two groups (Table 3), indicating that these mutations did not significantly impair the expression of these endogenous STAT6 targets in lymphoma cells.
Table 3.
Expression of loci containing one or more confirmed STAT6 binding motifs in PMBL cases.
| GENE | microarray ID | No STAT6 mutation (n=5)° | STAT6 Mutation (n=7)° | Fold Change # | Student t test p value* |
|---|---|---|---|---|---|
| FCER2 | 35220 | −1,26 | −0,24 | 2,02 | 0,26 |
| 17521 | −1,37 | −0,36 | 2,01 | 0,37 | |
| IL4 | 25233 | −0,37 | 0,03 | 1,32 | 0,32 |
| 34968 | −0,46 | −0,08 | 1,30 | 0,40 | |
| IL4R | 28597 | 1,05 | 1,41 | 1,28 | 0,15 |
| 19256 | 1,69 | 2,09 | 1,32 | 0,25 | |
| CCL17 | 29233 | −1,26 | −0,82 | 1,36 | 0,60 |
| CD40 | 27399 | 0,11 | 0,53 | 1,34 | 0,07 |
| 16913 | 0,10 | 0,45 | 1,28 | 0,12 | |
| BCL2L1 | 16454 | 0,02 | −0,15 | 0,89 | 0,30 |
| 24267 | 0,10 | −0,03 | 0,92 | 0,53 | |
| IL4I1 | 149016 | 2,25 | 2,40 | 1,11 | 0,75 |
| 149017 | 1,57 | 1,94 | 1,29 | 0,23 |
Mean expression levels (log2) in cases with (n=7) or without (n=5) STAT6 mutation (from Rosenwald, 2003).19
Fold change: ratio of mean expression in mutated cases relative to WT cases.
p values were not corrected for multiple comparisons.
DISCUSSION
PMBL is characterized by a constitutive activation of the JAK/STAT pathway, associated with SOCS1 gene defects and/or JAK2 gene amplification, leading to STAT6 phosphorylation and DNA binding activity. In this study, we identified mutations affecting the STAT6 DNA binding domains in 20 of 55 (36%) PMBL cases, but none of the 25 DLBCL tested. Although the constitutive activation of STAT proteins is a common finding in hematopoietic and non-hematopoietic cancers,27 this is the first observation of recurrent STAT gene mutations associated with human cancer. The seven PMBL cases with several nucleotide changes in STAT6 sequence were mostly young males, and 3 of them had a rapidly fatal disease, highlighting the need of larger studies to address the association of STAT6 mutation with clinical presentation and outcome. It is noteworthy that the L1236 classical Hodgkin lymphoma cell line, which harbors an amplification of the STAT6 gene24 and which depends on IL-13 signaling for cell proliferation and viability,28 had an identical hotspot mutation in STAT6 DNA binding domain. This finding extends the list of genetic alterations common to PMBL and classical Hodgkin lymphoma, such as REL and JAK2 genes gain/amplification,9 and SOCS1 gene deletions/mutations10 and supports the concept of a continuum between these two diseases, as acknowledged by the recognition of intermediate “grey zone” lymphomas in the 2008 updated WHO classification.1
Mutations of oncogenes have been shown to occur through aberrant somatic hypermutation (SHM) in DLBCL and PMBL.29 The mutations observed in STAT6 gene were strikingly different of the classical features of SHM.30 These mutations were mostly transversions, whereas SHM is characterized by transitions occurring twice as frequently as transversions; they mostly targeted A:T base pairs, whereas SHM affects mostly G:C base pairs; they were located 8,5 Kb from the transcriptional start site whereas SHM mutations are usually located 2 Kb from the start site. It is interesting to note that hypermutation of BCL6 regulatory region was previously reported to differ in PMBL and DLBCL.31 The pattern of somatic mutations reflects both their mechanism of origin and their selection during tumor growth. Since STAT6 mutations affect the coding sequence, the pattern of mutation may be biased by the selection process. Nevertheless, these results suggest that some atypical SHM or other mutational mechanisms may occur in PMBL.
In the last years, hypomorphic mutations of the STAT3 gene, most frequently located in the DNA binding domain, have been reported to be responsible for autosomic dominant and sporadic hyper-IgE syndrome, a primary immunodeficiency defect32 and mutations of the STAT1 gene have been associated with type I interferon immunodeficiency.33 These mutations were shown to be associated with impaired DNA-binding ability. In line with these observations, the analysis of the functional properties of STAT6 mutants identified in PMBL, also revealed an impaired function in HEK293 cells in EMSA and reporter assays. Three (N417, D419, and N421) of the 4 hotspot mutations are located in a region which was identified as an important DNA recognition element in the crystal structure of STAT134 and STAT3β35 dimers bound to DNA, and this may account for their diminished ability to activate STAT6 synthetic reporter constructs. The fourth hot spot (N430), which had a weak effect in these assays and was never observed as sole mutation, corresponds to a residue that is absolutely conserved in human and mouse STAT proteins.
The arguments for the involvement of STAT6 mutations in lymphomagenesis are i) their frequency in PMBL, indicating an in vivo selection during transformation and/or progression; ii) the presence of mutational hotspots; iii) the fact that they target an activated signaling pathway; iiii) their presence in a Hodgkin cell line with concordant gene amplification. However, the mechanisms through which these mutations affect tumor growth remain elusive. Although STAT6 mutants showed an impaired function on short synthetic promoters in a heterologous cell system, PMBL samples did not show a decreased expression of classical STAT6 targets. This apparent discrepancy may be due to the fact that the highly complex in vivo transcriptional regulation coordinated by STAT6 is not addressed in HEK cells transfections.
Altogether, our results show that the region encoding the DNA binding domain of STAT6 is the target of recurrent mutations in PMBL but not DLBCL. Further investigations are required to identify the mutational mechanisms involved and the oncogenic function associated with these mutations. This observation opens a wide field of investigation to determine the spectrum and functional consequences of STAT mutations in tumors, which are critical points in regard to the current development of anti-JAK targeted therapies.
Supplementary Material
Acknowledgments
This study was supported by a grant of the INCa and DAAD. OR was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG, RI 1915/1-1). We thank M. Dyer for providing the Karpas1106 cell line, M.C. Baglin (hospital Foch, Suresnes), B. Petit (CHU Limoges), M. Parrens (CHU Bordeaux) and J. Brière (hospital Saint-Louis, Paris) for providing PMBL samples. We are grateful to M. Baia for her technical help, to I. Dusanter and O. Bernard for helpful discussions and to P.H. Roméo for critically reading of the manuscript.
Footnotes
AUTHORSHIP
OR and CG contributed equally to this study; PM and KL are co-correspondong authors. OR, CG, and FC designed and performed experiments, interpreted the results and corrected the manuscript; KD, JM and GD performed experiments; JPJ performed statistical analysis, PM, PG selected and reviewed the lymphoma cases, contributed to the manuscript; KL designed and interpreted experiments, wrote the manuscript.
References
- 1.Swerdlow SH, Campo E, Harris NL. WHO classification of tumors of haematopoietic and lymphoid tissues. 4. Lyon, France: International Agency for Research on Cancer; 2008. [Google Scholar]
- 2.Barth TF, Leithauser F, Joos S, Bentz M, Moller P. Mediastinal (thymic) large B-cell lymphoma: where do we stand? Lancet Oncol. 2002;3:229–234. doi: 10.1016/s1470-2045(02)00714-3. [DOI] [PubMed] [Google Scholar]
- 3.Guiter C, Dusanter-Fourt I, Copie-Bergman C, et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood. 2004;104:543–549. doi: 10.1182/blood-2003-10-3545. [DOI] [PubMed] [Google Scholar]
- 4.Mottok A, Renne C, Willenbrock K, Hansmann ML, Brauninger A. Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood. 2007;110:3387–3390. doi: 10.1182/blood-2007-03-082511. [DOI] [PubMed] [Google Scholar]
- 5.Skinnider BF, Elia AJ, Gascoyne RD, et al. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2002;99:618–626. doi: 10.1182/blood.v99.2.618. [DOI] [PubMed] [Google Scholar]
- 6.Ritz O, Guiter C, Dorsch K, et al. STAT6 activity is regulated by SOCS-1 and modulates BCL-XL expression in primary mediastinal B-cell lymphoma. Leukemia. 2008;22:2106–2110. doi: 10.1038/leu.2008.85. [DOI] [PubMed] [Google Scholar]
- 7.Bentz M, Barth TF, Bruderlein S, et al. Gain of chromosome arm 9p is characteristic of primary mediastinal B-cell lymphoma (MBL): comprehensive molecular cytogenetic analysis and presentation of a novel MBL cell line. Genes Chromosomes Cancer. 2001;30:393–401. doi: 10.1002/1098-2264(2001)9999:9999<::aid-gcc1105>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 8.Melzner I, Bucur AJ, Bruderlein S, et al. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood. 2005;105:2535–2542. doi: 10.1182/blood-2004-09-3701. [DOI] [PubMed] [Google Scholar]
- 9.Joos S, Granzow M, Holtgreve-Grez H, et al. Hodgkin’s lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int J Cancer. 2003;103:489–495. doi: 10.1002/ijc.10845. [DOI] [PubMed] [Google Scholar]
- 10.Weniger MA, Melzner I, Menz CK, et al. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene. 2006;25:2679–2684. doi: 10.1038/sj.onc.1209151. [DOI] [PubMed] [Google Scholar]
- 11.Kapp U, Yeh WC, Patterson B, et al. Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. J Exp Med. 1999;189:1939–1946. doi: 10.1084/jem.189.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skinnider BF, Elia AJ, Gascoyne RD, et al. Interleukin 13 and interleukin 13 receptor are frequently expressed by Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 2001;97:250–255. doi: 10.1182/blood.v97.1.250. [DOI] [PubMed] [Google Scholar]
- 13.Daniel C, Salvekar A, Schindler U. A gain-of-function mutation in STAT6. J Biol Chem. 2000;275:14255–14259. doi: 10.1074/jbc.c000129200. [DOI] [PubMed] [Google Scholar]
- 14.Bruns HA, Schindler U, Kaplan MH. Expression of a constitutively active Stat6 in vivo alters lymphocyte homeostasis with distinct effects in T and B cells. J Immunol. 2003;170:3478–3487. doi: 10.4049/jimmunol.170.7.3478. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan MH, Sehra S, Chang HC, O’Malley JT, Mathur AN, Bruns HA. Constitutively active STAT6 predisposes toward a lymphoproliferative disorder. Blood. 2007;110:4367–4369. doi: 10.1182/blood-2007-06-098244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Copie-Bergman C, Boulland ML, Dehoulle C, et al. Interleukin 4-induced gene 1 is activated in primary mediastinal large B-cell lymphoma. Blood. 2003;101:2756–2761. doi: 10.1182/blood-2002-07-2215. [DOI] [PubMed] [Google Scholar]
- 17.Joos S, Otano-Joos MI, Ziegler S, et al. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood. 1996;87:1571–1578. [PubMed] [Google Scholar]
- 18.Paulli M, Strater J, Gianelli U, et al. Mediastinal B-cell lymphoma: a study of its histomorphologic spectrum based on 109 cases. Hum Pathol. 1999;30:178–187. doi: 10.1016/s0046-8177(99)90273-3. [DOI] [PubMed] [Google Scholar]
- 19.Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Moldenhauer G, Popov SW, Wotschke B, et al. AID expression identifies interfollicular large B cells as putative precursors of mature B-cell malignancies. Blood. 2006;107:2470–2473. doi: 10.1182/blood-2005-06-2502. [DOI] [PubMed] [Google Scholar]
- 22.Moller P, Bruderlein S, Strater J, et al. MedB-1, a human tumor cell line derived from a primary mediastinal large B-cell lymphoma. Int J Cancer. 2001;92:348–353. doi: 10.1002/ijc.1211. [DOI] [PubMed] [Google Scholar]
- 23.Savage KJ, Monti S, Kutok JL, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood. 2003;102:3871–3879. doi: 10.1182/blood-2003-06-1841. [DOI] [PubMed] [Google Scholar]
- 24.Feys T, Poppe B, De Preter K, et al. A detailed inventory of DNA copy number alterations in four commonly used Hodgkin’s lymphoma cell lines. Haematologica. 2007;92:913–920. doi: 10.3324/haematol.11073. [DOI] [PubMed] [Google Scholar]
- 25.Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105:13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 2006;17:173–188. doi: 10.1016/j.cytogfr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 27.Constantinescu SN, Girardot M, Pecquet C. Mining for JAK-STAT mutations in cancer. Trends Biochem Sci. 2008;33:122–131. doi: 10.1016/j.tibs.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Trieu Y, Wen XY, Skinnider BF, et al. Soluble interleukin-13Ralpha2 decoy receptor inhibits Hodgkin’s lymphoma growth in vitro and in vivo. Cancer Res. 2004;64:3271–3275. doi: 10.1158/0008-5472.can-03-3764. [DOI] [PubMed] [Google Scholar]
- 29.Rossi D, Cerri M, Capello D, et al. Aberrant somatic hypermutation in primary mediastinal large B-cell lymphoma. Leukemia. 2005;19:2363–2366. doi: 10.1038/sj.leu.2403982. [DOI] [PubMed] [Google Scholar]
- 30.Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol. 2006;6:573–583. doi: 10.1038/nri1896. [DOI] [PubMed] [Google Scholar]
- 31.Malpeli G, Barbi S, Moore PS, et al. Primary mediastinal B-cell lymphoma: hypermutation of the BCL6 gene targets motifs different from those in diffuse large B-cell and follicular lymphomas. Haematologica. 2004;89:1091–1099. [PubMed] [Google Scholar]
- 32.Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 33.Chapgier A, Boisson-Dupuis S, Jouanguy E, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006;2:e131. doi: 10.1371/journal.pgen.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 35.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.