

Abstract
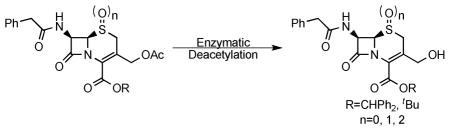
Cephalosporins remain one of the most important classes of antibiotics. A useful site for derivatization involves generation of and chemistry at the 3′-hydroxymethyl position. While 3′-acetoxymethyl substituted cephalosporins are readily available, deacetylation to access the free 3′-hydroxymethyl group is problematic when the carboxylic acid is protected as an ester. Herein we report that this important transformation has been efficiently accomplished using Candida antarctica lipase B. Although this transformation is difficult to carry out using chemical methods, the enzymatic deacetylation has been successful on gram scale, when the cephalosporin is protected as either the benzhydryl or t-butyl esters, and on the corresponding sulfoxide and sulfone of the t-butyl ester.
Cephalosporin antibiotics have been in use for more than 40 years and are still being employed to fight bacterial infections despite the rising incidence of resistance.1 Decades of intense research have given rise to an arsenal of synthetic methods that have allowed many analogs to be prepared, including four generations of cephalosporins that have reached clinical use.2 Although this research has led to many breakthroughs in cephalosporin-specific chemical methodology, accessing precursors for further elaboration is still not straightforward. Numerous derivatives have been made through modification at the 3′-hydroxymethyl group of cephalosporins. However, the deprotection of the common 3′-acetoxy precursor in the presence of the protected cephalosporanic acid, and without lactonization and/or double bond isomerization to give the Δ2 isomer, has yet to be accomplished in a high yielding and reproducible manner (Scheme 1). Herein we report a selective enzymatic approach to the deprotection of the cephalosporin acetoxy group, using commercially available Candida antartica lipase B (CAL B), on acrylic resin.
SCHEME 1.

Cephalosporin Acetate Deprotection
Commercially available 7-amino cephalosporanic acid (7-ACA) was converted, in reasonable yields, to 3 using a modified literature procedure to introduce the t-butyl ester4 and standard Schotten-Baumann conditions to install the phenylacetyl group as a simple representative side chain (Scheme 2). Despite considerable effort, the 3′-acetoxy substituent was unable to be removed in acceptable yields and in the absence of double bond isomerization using the following methods (see supporting information for details): saponification,4 KCN, HCl,5 TMSI/trifluoroacetate/pH 7.0,6 or bis(tributyltin) oxide.7 Based on these findings, enzymatic deacetylation methods were explored.
SCHEME 2.

Synthesis of Cephalosporin 3
Enzymatic deprotection of the cephalosporin acetoxy group has been demonstrated in the literature.8 Similar to chemical deacetylations,4,6,8c,d,f,9 the enzymatic deacetylation reactions have only been reported when the cephalosporin contains a free acid.8 When making modifications to the allylic alcohol it is usually more convenient and often necessary to have the acid protected as an ester. This is especially true for the synthesis of dual action cephalosporins, where the allylic alcohol is modified.8g, 10
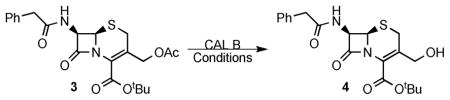
Initially a panel of nine lipases was screened for the enzymatic deprotection of the allylic alcohol of 3.11 This resulted in no product formation under various conditions. Conversely, extensive studies with CAL B provided an effective solution. Candida antarctica lipase B was chosen based on its broad utility and general experience in our group.12 After exploring solvents,13 temperatures, and adding n-butanol,14 cephalosporin 4 was obtained (Table 1). Interestingly, when water was used as the nucleophile, low yields or no reaction was observed. Alternatively, n-butanol was used to obtain the product in moderate yields, and the use of s-butanol was found to increase the conversion from 3 to 4 significantly.15 Further optimization showed that the addition of molecular sieves increased the efficiency and conversion of the reaction. In addition, different lots of CAL B gave different reaction rates based on the “loss on drying” reported by the manufacturer.16 These observations indicate that the reaction is sensitive to moisture. The optimized reaction can be performed on gram scale and a yield of 97%, after recrystallization, can be obtained.
TABLE 1.
Optimization of Cephalosporin 3 Deacetylation Using CAL Ba
 | ||||
|---|---|---|---|---|
| Entry | CAL Bb | Additivec | Timed | Conv.e |
| 1f | 50 | n-butanol | 4 | 0 |
| 2 | 50 | n-butanol | 4 | 48 |
| 3 | 50 | s-butanol | 4 | 85 |
| 4 | 50 | s-butanol, MS | 4 | 93 |
| 5 | 10 | s-butanol, MS | 4 | 74 |
| 6 | 10 | s-butanol, MS | 7 | 92 |
| 7 | 5 | s-butanol, MS | 4 | 21 |
| 8 | 5 | s-butanol, MS | 7 | 39 |
| 9g | 50 | s-butanol, MS | 4 | 97h |
All reactions were performed on 20 mg scale and at 50 °C, unless otherwise indicated, in 9:1 hexanes:THF, and carried out in an incubated shaker;
Mass %;
MS=4 Å molecular sieves;
Days;
% Conversions to product determined using 1H NMR spectroscopy and reported in %;
Reaction performed at 32 °C;
Reaction performed on 1g;
Isolated yield after recrystallization.
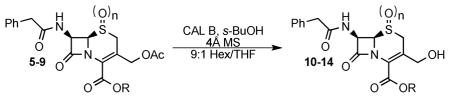
Several additional substrates were prepared to test the scope of the enzymatic deprotection. The substrates were prepared from 7-ACA using known and modified literature procedures. Acylation of 7-ACA using phenylacetyl chloride8e followed by protection of the acid using diphenyldiazomethane17 or p-methoxybenzyl bromide, prepared in situ,18 gave cephalosporins 6 and 7, respectively (Scheme 3). Cephalosporin 3 could be selectively oxidized to either sulfoxide 8 or sulfone 9 using varying reaction times and amounts of mCPBA (Scheme 4).4
SCHEME 3.

Synthesis of Cephalosporins 5–7
SCHEME 4.

Synthesis of Cephalosporins 8 and 9
These additional substrates were submitted to the previously optimized CAL B reaction conditions (Table 2). When the acid was not protected, such as in cephalosporin 5, no reaction was seen; this result was not disappointing as removal of the acetate from cephalosporin 10 and other free acid containing cephalosporins has been reported.8c,d,f,9 The most commonly used cephalosporin acid protecting group is the benzhydryl ester. One difficulty in working with cephalosporin 11 is that the alcohol can readily cyclize to form the corresponding lactone;20 this transformation can even occur when the acid is left unprotected.8a Surprisingly, cephalosporin 11 was obtained without cyclization to the lactone, and 100% conversion was observed. The low yield reported in Table 2 reflects the difficult isolation; recrystallization was inefficient and silica gel column chromatography caused lactone formation. Fortunately, the crude material was isolated very cleanly and was suitable for use. Cephalosporin 12, containing the PMB ester, gave complex mixtures and was not explored further. Both sulfoxide, 13, and sulfone, 14, also were substrates for CAL B; however, only moderate yields were obtained.
TABLE 2.
Scope of Enzymatic Deacetylation of Cephalosporins 5–9a
 | |||
|---|---|---|---|
| Compound | n | R | Yieldb(Conv.)c |
| 10 | 0 | H | N/A |
| 11 | 0 | Benzhydryl | 52 (100) |
| 12 | 0 | p-methoxybenzyl | N/A |
| 13 | 1 | t-butyl | 70 |
| 14 | 2 | t-butyl | 58 |
All reactions were performed in an incubated shaker with 50 mass % CAL B;
Isolated % yield;
% Conversions determined using 1H NMR spectroscopy.
In conclusion, we have developed an enzymatic transformation that allows the 3′-acetoxy group of cephalosporins, containing an ester protected carboxylic acid, to be removed in moderate to excellent yields. This methodology is especially useful in preparing cephalosporin analogues that require further modification at the allylic alcohol.
Experimental Section
(6R,7R)-TERT-Butyl 3-(hydroxymethyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate (4)
Compound 3 (1.007 g, 2.241 mmol) was added to a 1L Erlenmeyer flask. Tetrahydrofuran (12 mL) was added to dissolve the substrate. Then hexanes (108 mL) was added initially forming a gel which upon swirling formed a suspension. CAL B (502.0 mg), 4Å molecular sieves (1.0146 g), and s-butanol (4 mL, 43.604 mmol) were added and the flask was stoppered with a septum. The reaction was shaken in an incubated shaker at 50 °C for 4–6 days (solid that had dried on the sides of the flask was scraped back into the reaction each day). When the reaction was complete by TLC, the solid was dissolved in methylene chloride and the lipase and sieves were filtered off using vacuum filtration, and the solvent was removed in vacuo. The crude material was recrystallized from chloroform/cyclohexane in two crops to yield cephalosporin 4. The material was dried under vacuum with P2O5 to yield the product as a white solid (877.1 mg, 97%). mp 170–171.5 °C (lit. 174–176 °C);8e Rf 0.29 (5:1 DCM/EtOAc); IR (KBr) 3382, 1757, 1712, 1661 cm−1; 1H NMR (300 MHz, CDCl3): δ 1.51 (s, 9H), 2.76 (dd, J=6.5, 17, 1H), 3.51 (s, 1H), 3.57–3.70 (od, 2H), 3.84 (dd, J=10.5, 12.6, 1H), 4.44 (dd, J=4.2, 12.6, 1H), 4.89 (d, J=5.1, 1H), 5.84 (dd, J=4.8, 9.3, 1H), 6.15 (d, J=9, 1H), 7.23–7.28 (m, 2H), 7.29–7.39 (m, 3H); 13C NMR (125 MHz, CDCl3): δ 27.8, 28.0, 43.7, 57.2, 59.3, 62.4, 84.4, 127.1, 128.1, 129.5, 129.7, 130.1, 133.8, 161.8, 164.8, 171.4; HRMS (ESI-TOF) Calcd for C20H24N2NaO5S, 427.1298; Found, 427.1274.
(6R,7R)-Benzhydryl 3-(hydroxymethyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate (11)
Cephalosporin 6 (101.1 mg, 0.181 mmol) was dissolved in THF (1.5 mL) and hexanes (13.5 mL, HPLC grade) was added followed by 4Å molecular sieves (228.2 mg). The mixture became white and cloudy and some material adhered to the molecular sieves. The reaction was sonicated for a couple minutes to help solubilization. CAL B (52.3 mg), and s-butanol (0.5 mL, 6.746 mmol) were added. The reaction was shaken at 50 °C for 4 days in an incubated shaker. The reaction mixture was dissolved in methanol and the sieves and CAL B were removed using vacuum filtration. The crude NMR indicated that the reaction had gone to completion. The material was suspended in a minimal amount of EtOAc, cooled, and filtered to give a white solid (48.4 mg, 52%). mp: 175–176 °C (lit. 178–180 °C)20; IR (KBr) 3501, 1761, 1713, 1666 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 3.50–3.61 (od, J=14, 2H), 3.62 (s, 2H), 4.17–4.27 (m, 2H), 5.12 (d, J=4.5, 1H), 5.19 (t, J=5.5, 1H), 5.73 (dd, J=5, 8, 1H), 6.91 (s, 1H), 7.21–7.40 (m, 11H), 7.43 (d, J=7.5, 2H), 7.52 (d, J=7.5, 2H), 9.16 (d, J=8, 1H); 13C NMR (125 MHz, DMSO-d6): δ 25.6, 41.6, 57.7, 58.9, 59.8, 78.4, 122.0, 126.5, 126.6, 126.8, 127.8, 127.9, 128.3, 128.4, 128.6, 129.0, 134.4, 135.9, 140.0, 140.1, 160.9, 165.3, 171.0; HRMS (ESI-TOF) Calcd for C29H26N2NaO5S, 537.1460; Found, 537.1476.
(6R,7R)-TERT-Butyl 3-(hydroxymethyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate-5-oxide (13)
Cephalosporin 8 (261.4 mg, 0.565 mmol) was partially dissolved in anhydrous toluene (40 mL). CAL B (137.9 mg), s-butanol (1 mL, 10.901 mmol) and 4Å molecular sieves (520.3 mg) were added. The reaction was shaken at 50 °C in an incubated shaker. After shaking over 4 nights, the reaction mixture was dissolved in methanol, filtered to remove the lipase and the sieves, and the solvent was removed in vacuo. The material was purified using column chromatography. The material was preloaded on silca, and eluted using 2:1 CH2Cl2/EtOAc, until starting compound 8 was isolated (12.1 mg) and then EtOAc until product 13 was isolated as a white solid (286.8 mg, 70%). mp 192–195 °C (d) (lit. 198–199 °C)8e; Rf 0.15 (2:1 EtOAc/CH2Cl2); IR (thin film): 3506, 1778, 1695, 1661, 1029 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 1.49 (s, 9H), 3.54 (d, J=18.5, 1H), 3.55 (d, J=14, 1H), 3.69 (d, J=14, 1H), 3.86 (d, J=19, 1H), 4.08 (dd, J=5.5, 14, 1H), 4.43 (dd, J= 5.5, 14, 1H), 4.84 (d, J=4.5, 1H), 5.13–5.17 (m, 1H), 5.77 (dd, J=4.5, 8.5, 1H), 7.21–7.26 (m, 1H), 7.28–7.33 (m, 4H), 8.38 (d, J=8.5, 1H); 13C NMR (125 MHz, DMSO-d6): δ 27.5, 41.5, 45.3, 58.0, 60.2, 66.2, 82.5, 123.1, 125.6, 126.6, 128.3, 129.1, 135.9, 160.0, 164.1, 171.1; HRMS (ESI-TOF) Calcd for C20H24N2NaO6S, 443.1247; Found, 443.1243.
(6R,7R)-TERT-Butyl 3-(hydroxymethyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate-5,5-dioxide (14)
Cephalosporin 9 (99.9 mg, 0.209 mmol) was added to a 25 mL Erlenmeyer flask and was dissolved in THF (1.2 mL, from AcroSeal bottle) and hexanes (10.8 mL, HPLC grade) were added. Molecular sieves (4 Å, 199.9 mg), CAL B (50.0 mg), and s-butanol (0.4 mL) were added. The reaction was stoppered and shaken in an incubated shaker at 50 °C over 5 days. When complete by TLC, the reaction was dissolved in methylene chloride, the CAL B and molecular sieves were removed using vacuum filtration, and the solvent was removed in vacuo. The material was purified using column chromatography, loading in methylene chloride and eluting with 5:1 CH2Cl2/EtOAc. The product was isolated as a white solid (42.9 mg, 58%). mp 170.5–171.5 °C; Rf 0.16 (5:1 DCM/EtOAc); IR (KBr) 3512, 1782, 1718, 1660, 1332, 1158 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 1.47 (s, 9H), 3.56–3.64 (od, 2H), 4.01 (d, J=18.5, 1H), 4.17–4.27 (m, 2H), 4.26 (d, J=18.5, 1H), 5.26 (t, J=5 1H), 5.35 (d, J=4.5, 1H), 5.92 (dd, J=4.5, 8.5, 1H), 7.2–7.31 (m, 5H), 8.86 (d, J=8.5, 1H); 13C NMR (125 MHz, DMSO-d6): δ 27.4, 41.2, 50.8, 57.9, 59.0, 66.8, 83.0, 122.2, 126.5, 128.2, 129.2, 130.5, 135,6, 159.9, 164.1, 170.9; HRMS (ESI-TOF) Calcd for C20H25N2O7S, 437.1377; Found, 437.1377.
Supplementary Material
Acknowledgments
We acknowledge NIH (GM 025845 and AI 030988) for support of this research. We also acknowledge Dr. M. Joyce, Dr. W. Boggess and N. Sevova for assistance obtaining mass spectrometry data and both the Center for Environmental Science and Technology and the ND Mass Spectrometry and Proteomics Facility (CHE-0741793) at the University of Notre Dame. L.D.P acknowledges a Grace Fellowsip (2008–2009, UND).
Footnotes
Supporting Information Available: Experimental methods for compounds 1–3 and 5–9, and all 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Fischbach MA, Walsh C. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Walsh C. Antibiotics: Actions, Origins, Resistance. ASM Press; Washington, DC: 2003. [Google Scholar]; (c) Bryskier A. J Antibiot. 2000;53:1028–1037. doi: 10.7164/antibiotics.53.1028. [DOI] [PubMed] [Google Scholar]; (d) Singh GS. Mini Rev Med Chem. 2004;4:93–109. doi: 10.2174/1389557043487547. [DOI] [PubMed] [Google Scholar]
- 2.Morin RB, Gorman M, editors. Chemistry and Biology of β-Lactam Antibiotics. Vol. 1 Academic Press; New York: 1982. [Google Scholar]
- 3.Wang Y, Yuan H, Wright SC, Wang H, Larrick JW. BMC Chem Biol. 2001;1 doi: 10.1186/1472-6769-1-4. [Online] article 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Mangia A, Scandroglio A. Org Prep Proced Int. 1986;18:13–15. [Google Scholar]; (b) Stedman RJ. J Med Chem. 1966;9:444. doi: 10.1021/jm00321a056. [DOI] [PubMed] [Google Scholar]
- 5.Galeazzi R, Martelli G, Mabbili G, Orene M, Rinaldi S. Org Lett. 2004;6:2571–2574. doi: 10.1021/ol049146j. [DOI] [PubMed] [Google Scholar]
- 6.Mobashery S, Johnston M. Tetrahedron Lett. 1986;27:3333–3336. [Google Scholar]
- 7.Salomon CJ, Mata EG, Mascaretti OA. J Org Chem. 1994;59:7259–7266. [Google Scholar]
- 8.While there is extensive early patent literature related to similar transformations, the following references provide representative documentation of the efforts related to this type of cephalosporin chemistry. Jeffery JD’A, Abraham EP, Newton GGF. Biochem J. 1961;81:591. doi: 10.1042/bj0810591.Negi S, Yamanaka M, Sugiyama I, Komatsu Y, Sasho M, Tsuruoka A, Kamada A, Tsukada I, Hiruma R, Katsu K, Machida Y. J Antibiot. 1994;47:1507–1525. doi: 10.7164/antibiotics.47.1507.Carrea G, Corcelli A, Palmisano B, Riva S. Biotech Bioeng. 1996;52:648–652. doi: 10.1002/(SICI)1097-0290(19961220)52:6<648::AID-BIT2>3.0.CO;2-O.Saka Y, Abe T, Ohbayashi Y, Isaka K, Yamamoto K, Tani Y, Kato N. App Environ Microbiol. 1996;62:2669–2672. doi: 10.1128/aem.62.7.2669-2672.1996.Keltjens R, Vadivel SK, de Vroom E, Klunder JH, Zwanenburg B. Eur J Org Chem. 2001;13:2529–2534.Kidwap M, Dave B, Bhushan KR, Misra P, Saxena RK, Gupta R, Gulanti R, Singh M. Biocat Biotransform. 2002;20:377–379.Grant JW, Smyth TP. J Org Chem. 2004;69:7965–7970. doi: 10.1021/jo048970i.
- 9.(a) Okabe M, Sun RC. Synthesis. 1992;11:1160–1164. [Google Scholar]; b) Kukolja S. J Med Chem. 1968;11:1067–1069. doi: 10.1021/jm00311a035. [DOI] [PubMed] [Google Scholar]
- 10.Zhao G, Miller MJ, Franzblau S, Wan B, Möllmann U. Biorg Med Chem Lett. 2006;16:5534–5537. doi: 10.1016/j.bmcl.2006.08.045. [DOI] [PubMed] [Google Scholar]
- 11.Lipase kit from Aldrich containing: Lipase from Aspergillus, Lipase from Candida antarctica, Lipase from Candida cylindracea, Lipase from Mucor miehei, Lipase from Pseudomonas cepacia, Lipase from Pseudomonas fluorescens, Lipase from Rhizopus arrhizus, Lipase from Rhizopus niveus, Lipase from hog pancreas.
- 12.Mulvihill MJ, Gage JL, Miller MJ. J Org Chem. 1998;63:3357–3363. [Google Scholar]
- 13.Lanne C, Boeren S, Vos K, Veeger C. Biotechnol Bioeng. 1987;30:81–87. doi: 10.1002/bit.260300112. [DOI] [PubMed] [Google Scholar]
- 14.Anilkumar AT, Goto K, Takahashi T, Ishizake K, Kaga H. Tetrahedron: Asymmetry. 1999;10:2501–2503. [Google Scholar]
- 15.Miyazawa T, Hamada M, Morimoto R, Murashima T, Yamada T. Tetrahedron Lett. 2007;48:8334–8337. [Google Scholar]
- 16.The “loss on drying” reported for two different lots of CAL B was 0.31% and 1%. The lot with 0.31% loss on drying gave faster reaction times. See supporting information for a detailed study.
- 17.(a) Takaya T, Takasugi H, Murakawa T, Nakano H. J Antibiot. 1981;34:1300–1310. doi: 10.7164/antibiotics.34.1300. [DOI] [PubMed] [Google Scholar]; (b) Kumar S, Murray RW. J Am Chem Soc. 1984;106:1040–1045. [Google Scholar]
- 18.Mobashery S, Johnston M. J Org Chem. 1986;51:4723–4726. [Google Scholar]
- 19.Keltjens R, Vadivel SK, de Gelder R, Klunder AJH, Zwanenburg B. Eur J Org Chem. 2003;9:1749–1758. [Google Scholar]
- 20.Ganzález M, Rodríguez Z, Tolón B, Rodríguez JC, Velez H, Valdéz B, López MA, Fini A. IL Farmaco. 2003;58:409. doi: 10.1016/S0014-827X(03)00063-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.