

Abstract
Emerging evidence indicate novel modes of EGFR signaling, notably, one involves EGFR nuclear translocalization and subsequent gene activation. To date, however, the significance of the nuclear EGFR pathway in glioblastoma (GBM) is unknown. Here, we report that EGFR and its constitutively activated variant EGFRvIII undergo nuclear translocalization in GBM cells, in which the former event requires EGF stimulation and the latter is constitutive. To gain insight into the impact of nuclear EGFR on gene expression in GBM, we created isogenic GBM cell lines, namely, U87MG-vector, U87MG-EGFR and U87MG-EGFRdNLS that, respectively, express the control vector, EGFR and nuclear-entry defective EGFR with a deletion of the nuclear localization signal (NLS). Microarray analysis shows that 19 genes, including, cyclooxygenase-2 (COX-2), to be activated in U87MG-EGFR cells but not in U87MG-EGFRdNLS and U87MG-vector cells. Subsequent validation studies indicate that COX-2 gene is expressed at higher levels in cells with EGFR and EGFRvIII than those with EGFRdNLS and EGFRvIIIdNLS. Nuclear EGFR and its transcriptional co-factor STAT3 associate with the COX-2 promoter. Increased expression of EGFR/EGFRvIII and activated STAT3 leads to synergistic activation of the COX-2 promoter. Promoter mutational analysis identified a proximal STAT3-binding site that is required for EGFR/EGFRvIII-STAT3 mediated COX-2 gene activation. In GBM tumors, an association exists between levels of COX-2, EGFR/EGFRvIII and activated STAT3. Together, these findings indicate the existence of the nuclear EGFR/EGFRvIII signaling pathway in GBM and its functional interaction with STAT3 to activate COX-2 gene expression, thus linking EGFR-STAT3 and EGFRvIII-STAT3 signaling axes to pro-inflammatory COX-2 mediated pathway.
Keywords: EGFR, EGFRvIII, STAT3, COX-2, glioma, transcriptional regulation
Introduction
Over-expression of EGFR and/or its constitutively activated variant EGFRvIII is frequently found in glioblastoma (GBM), the most common and most malignant form of brain cancer, and is associated with tumorigenesis and more aggressive tumor phenotypes, such as, invasiveness and therapeutic resistance (1-3). EGFRvIII, a product of rearrangement of the EGFR gene with a deletion within the extracellular domain that is predominantly found in GBM, is more tumorigenic than wild-type EGFR (4, 5). Importantly, both EGFR- and EGFRvIII-mediated pathways are of a high biological complexity, including the existence of two major signaling modes, at the cell-surface and the other, in the nucleus (2, 6, 7).
In the cell-surface signaling mode, both EGFR and EGFRvIII function as receptor tyrosine kinases that activate signaling modules, such as, those mediated by PLC-γ, Ras, PI-3K and JAK2, that can contribute to tumorigenesis and more aggressive tumor behaviors (8, 9). On the other hand, signals such as EGFR ligands, oxidative stress and DNA damage stimulate EGFR nuclear transport (2). Nuclear EGFR is localized on the inner nuclear membrane (10, 11) and in the nucleoplasm (12-15). In the nuclear signaling mode, EGFR has three key functions: (1) gene transactivation, (2) tyrosine kinase and (3) protein-protein interactions (13, 15-19). Importantly, the level of nuclear EGFR predicts poor prognosis in patients with breast carcinomas (19), oropharyngeal and esophageal squamous cell carcinomas (19, 20) and ovarian cancer (21). Although nuclear detection of EGFR has been reported in highly proliferative normal tissues, such as, regenerating livers (22) and placenta (13), and in cancerous tissues (13, 19-21), its presence in GBM remains uninvestigated. A previous report showed that EGFRvIII is detected in the nucleus of normal glial cells and primary GBM specimens (23).
Nuclear EGFR, similar to HER2, activates gene expression via its transactivation domain (13, 24). Although EGFR lacks a DNA-binding domain, it has been shown to interact with DNA-binding transcription factors, such as, STAT3, E2F1 and STAT5, to induce expression of genes, including, inducible nitric oxide synthase (iNOS), B-Myb and aurora A, respectively, in breast cancer (15, 17, 25). A systemic approach has, however, yet been conducted to identify nuclear EGFR target genes. Knowledge of such genes will advance our understanding of the nature and impact of nuclear EGFR on cell physiology, provide novel insights into the role of nuclear EGFR in human cancers, and help clarify some of the molecular mechanisms underlying the observed association between nuclear EGFR and poor clinical outcome (19-21).
EGFR physically interacts and functionally cooperates with STAT3, at both cytoplasmic and nuclear levels. At the cytoplasmic level, via the two docking autophosphorylated tyrosines (Y1068 and Y1086), cell-surface EGFR interacts with STAT3 SH2 domain (26). This interaction leads to phosphorylation of STAT3 at Y705 and its activation. Cell-surface EGFRvIII also interacts with and phosphorylates STAT3. Importantly, we and others showed in cancers of breast, colon and skin that cell-surface EGFR cooperates with STAT3 to induce expression of TWIST (to facilitate epithelial-mesenchymal transition), VEGF (to promote angiogenesis) and Eme1 endonuclease (to reduce drug-induced DNA damage)(27-29). In primary breast carcinomas, co-expression of EGFR and activated STAT3 (Y705) is frequent, 39% (28). In primary GBMs, we found approximately one third of the tumors to concurrently express EGFR/EGFRvIII and activated STAT3-Y705 (30). At the nuclear level, EGFR interacts with STAT3 to activate expression of iNOS gene in carcinomas of breast and epidermoid (15).
The objectives of this current study, therefore, are to determine whether nuclear EGFR- and EGFRvIII- pathways are functional in human GBM and to identify the target genes of these pathways in GBM via an unbiased comprehensive approach. Our findings provide the evidence showing that GBM cell lines and primary specimens express nuclear EGFR/EGFRvIII. We also identified cyclooxygenase-2, COX-2, as a novel transcriptional target of the EGFR-STAT3 and EGFRvIII-STAT3 signaling axes and that nuclear EGFR/EGFRvIII is essential for COX-2 gene activation in GBM cells. COX-2 catalyzes the first step in the biosynthesis of the prostaglandins from arachidonic acid and plays a central role in the regulation of progression, invasiveness and angiogenesis of various cancers, including, gliomas (31). COX-2 inhibition has been shown to effectively target glioma xenografts (32) and consequently, COX-2 is an emerging target for anti-GBM therapy (33). Together, the findings shed light on the nature of the nuclear EGFR/EGFRvIII pathway in GBM and establish a novel link between the nuclear EGFR/EGFRvIII-STAT3 signaling pathways and COX-2-mediated inflammatory pathway in GBM.
Results
EGFR undergoes nuclear translocalization in human GBM cell lines and primary specimens
Using nuclear fractionation and western blotting, we showed that following EGF treatment, EGFR undergoes nuclear translocalization in human GBM T98G cells that express high levels of endogenous EGFR (Fig. 1A). Nuclear fractionation efficiency is indicated by the lack of cytoplasmic β-actin protein in the nuclear extracts (NE) and by the absence of the nuclear marker protein lamin B, in the non-nuclear extracts (NNE).
Figure 1. EGFR undergoes nuclear translocalization in human GBM cell lines and primary specimens.
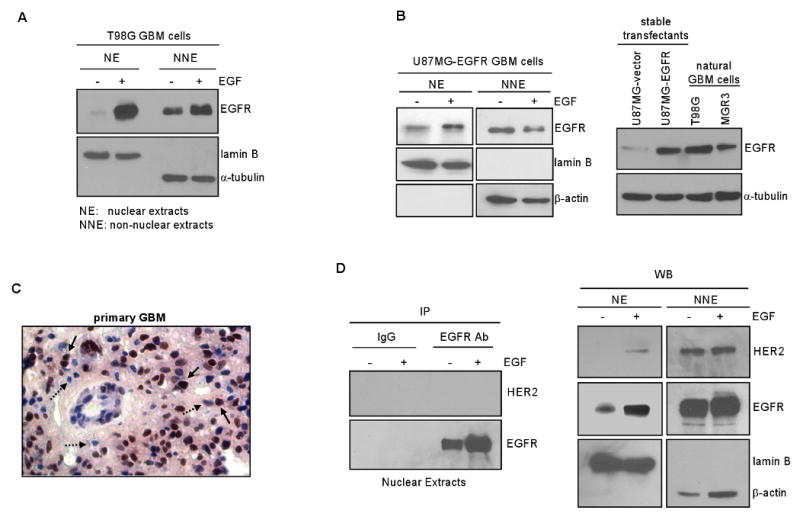
A. EGF induces EGFR nuclear transport in GBM cells with endogenous EGFR. Serum-starved human GBM T98G cells that express endogenous EGFR were stimulated with EGF (100 ng/ml) for 0 and 15 min, harvested and subjected to nuclear fractionation to obtain nuclear extracts (NE) and non-nuclear extracts (NNE). Both extracts were analyzed for EGFR, lamin B and α-tubulin expression via western blotting. Effectiveness of fractionation is indicated by the lack of cytoplasmic marker α-tubulin in NE and the absence of nuclear protein lamin B in NNE.
B. EGFR undergoes EGF-activated nuclear transport in GBM cells engineered to stably express EGFR. Left panel: U87MG-EGFR stable transfectant cells were similarly treated and analyzed, as described in panel A. Right panel: Western blotting shows U87MG-EGFR cells to express EGFR at the levels comparable to natural T98G and MGR3 GBM cells who express endogenous EGFR.
C. Nuclear EGFR is detected in a primary GBM specimen. We analyzed 12 primary GBM tumors for EGFR expression using IHC and found six tumors to express EGFR/EGFRvIII with one of them expressing significant levels of nuclear EGFR. EGFR-positive brown nuclei are pointed by solid arrows whereas EGFR-negative blue counterparts are marked by dashed arrows.
D. Nuclear EGFR is not in complex with nuclear HER2 in T98G cells. Using T98G GBM cells know to expression both EGFR and HER2, we treated the cells with EGF for 0 and 15 min, fractionated the cells into the nuclear and on-nuclear fractions, immunoprecipitated nuclear EGFR using an EGFR antibody and subjected the immunoprecipitates to western blotting to detect HER2. As shown in the left panel, HER2 was not co-immunoprecipitated with nuclear EGFR despite nuclear EGFR was effectively immunoprecipitated by the EGFR antibody. IgG did not pull down EGFR or HER2, indicating specificity. As shown by the western blotting in the right panel, HER2 underwent a modest level of nuclear translocalization after EGF stimulation with the majority of the HER2 protein in the non-nuclear fraction.
Similarly, in U87MG-EGFR stable transfectant cells, EGFR undergoes EGF-induced nuclear transport (Fig. 1B). U87MG-EGFR cells express equivalent levels of EGFR, compared to GBM cells with endogenous EGFR. EGF-induced EGFR nuclear translocalization in T98G cells was confirmed using immunofluorescence staining/confocal microscopy (Fig. S1 in Supplementary Data). The punctate staining pattern for nuclear EGFR, shown in Fig. S1, is consistent with what have been reported by many previous studies (18, 34). Nuclear HER2 aslo displays similar punctate staining rather than diffuse staining (35). To determine the presence of endogenous nuclear EGFR in GBM tumors, we analyzed 12 primary GBM specimens for EGFR expression using immunohistochemistry (IHC) with an antibody that is reactive to both EGFR and EGFRvIII. Six of the GBM tumors expressed EGFR/EGFRvIII with one of them showing significant levels of nuclear EGFR/EGFRvIII. In Fig. 1C, the solid arrows point to the brown-stained EGFR-positive nuclei and the dashed arrows to the blue EGFR-negative nuclei. Corroborating this observation, our earlier study found 6.9% (9/130) of primary breast carcinomas to express high levels of nuclear EGFR (19).
Given the facts that EGFR heterodimerizes with HER2 (8) and that HER2 undergoes nuclear translocalization (35), we examined whether nuclear EGFR and nuclear HER2 complex as heterodimers. Using T98G GBM cells know to express both EGFR and HER2, we immunoprecipitated nuclear EGFR using an EGFR antibody and subjected the immunoprecipitates to western blotting to detect HER2. As shown in Fig. 1D (left panel), we did not detect any HER2 signal despite nuclear EGFR was effectively immunoprecipitated by the EGFR antibody. As expected, IgG did not pull down EGFR or HER2, indicating specificity. Interestingly, HER2 underwent a modest level of nuclear translocalization after EGF stimulation, albeit the majority of the HER2 protein is in the non-nuclear fraction (Fig. 1D-right panel). Since we did not detect nuclear EGFR-HER2 heterodimers, EGF-induced HER2 nuclear transport may potentially be a result of nuclear transport of Her-3. This is supported by the facts that EGFR can transactivate HER3 (36), HER3 undergoes nuclear transport (37) and that HER2 is a preferred binding partner of HER3. Together, our results demonstrate for the first time that EGFR undergoes nuclear translocalization in human GBM cells and primary specimens.
Generation of GBM cells stably expressing mutant EGFR defective in nuclear entry
To gain insight into the nature of the nuclear EGFR pathway in GBM, we generated isogenic GBM cell lines that expressed a) wild-type EGFR and b) mutant EGFR, defective in its ability to translocate to the cell nucleus. For this, we engineered an EGFRdNLS mutant construct that carries the nuclear entry-defective mutant EGFR with a deletion of eight amino acids (aa 645-652) within the nuclear localization signal (NLS) region (Fig. 2A). Amino acid substitutions of the basic residues within this NLS region have been shown to abolish the ability of EGFR to enter the nucleus (15, 38). We subsequently established a U87MG stable transfectant line, U87MG-EGFRdNLS, to express EGFRdNLS. As indicated in Fig. 2B, U87MG-EGFRdNLS and U87MG-EGFR isogenic lines express equivalent levels of the respective EGF receptors. As expected, cells expressing EGFRdNLS did not contain nuclear EGFR, as shown by immunofluorescence staining/confocal microscopy (Fig. S2 in Supplementary Data) and nuclear fractionation/western blotting (Fig. 2C). In nuclear fractionation/western blotting analysis (Fig. 2C), nuclear fractionation efficiency is indicated by the absence of cytoplasmic β-actin protein in NE and by the lack of the nuclear protein lamin B in NNE.
Figure 2. Generation of GBM cells stably expressing mutant EGFR that is defective in nuclear entry.
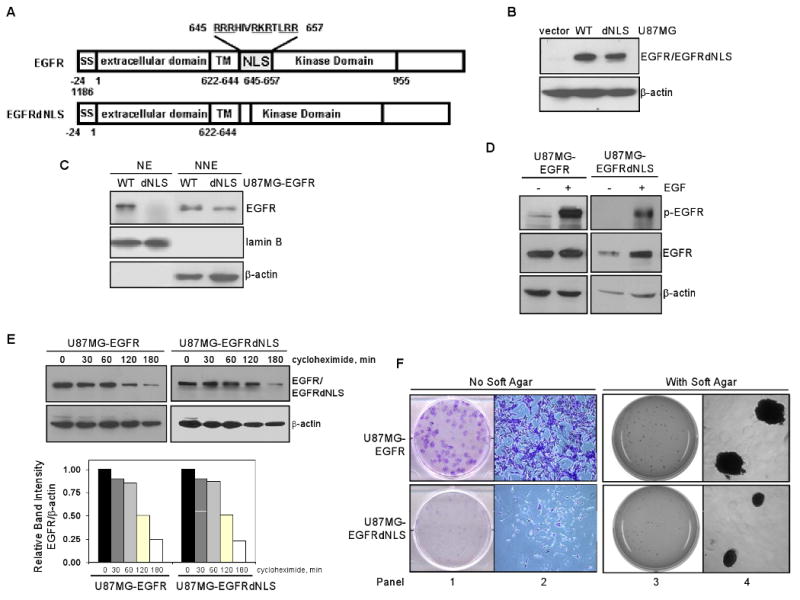
A. Structures of EGFR and EGFRdNLS, an EGFR mutant defective in nuclear entry. Top panel: EGFR contains a number of functional domains, namely, signal peptide (SS; aa -24 to -1), extracellular domain (1-621), transmembrane domain (TM; aa 622-644), NLS (aa 645-657) and kinase domain (658-955). Bottom panel: EGFRdNLS mutant contains a deletion of eight amino acids (aa 645-652) within the NLS region and a Ser insertion as aa residue 645. It has been shown that amino acid substitutions of the basic residues with this NLS region abolish the ability of EGFR to enter the nucleus (15, 38).
B. EGFR expression in isogenic U87MG-vector, U87MG-EGFR and U87MG-EGFRdNLS cells. Western blotting shows that U87MG-vector cells do not express Myc-tagged EGFR protein and in contrast, U87MG-EGFR and U87MG-EGFRdNLS express increased and comparable levels of EGFR/EGFRdNLS.
C. EGFRdNLS is absent in the cell nucleus. Efficiency of nuclear fractionation is indicated by the lack of cytoplasmic protein β-actin in NE and the absence of nuclear marker lamin B in NNE.
D. EGFRdNLS undergoes EGF-induced autophosphorylation similar to EGFR. Isogenic U87MG-EGFR and U87MG-EGFRdNLS cells were serum starved, stimulated with EGFR for 0 and 15 min and subjected to western blotting to determine levels of p-EGFR (Y1068; indicator of receptor activation), EGFR and β-actin.
E. EGFRdNLS has similar stability relative to EGFR. Isogenic U87MG-EGFR and U87MG-EGFRdNLS cells were treated with protein synthesis inhibitor cycloheximide (10 ug/ml) for 0-180 min and subjected to western blotting to determine levels of EGFR and β-actin. Intensity of the band signals were quantified using ImageJ software and protein half-life was subsequently computed.
F. Nuclear EGFR is important for the clonogenic growth of EGFR-expressing GBM cells. Colony formation assays were performed in the absence (left two panels) and presence (right two panels) of soft agar. U87MG-EGFRdNLS cells formed significantly less colonies than U87MG-EGFR cells. As shown by the 5X images in the second panel, the colony from U87MG-EGFR cells contained more cells and was with intensive staining whereas the one from U87MG-EGFRdNLS cells contained significantly fewer cells. As shown by the high-resolution images (fourth panel), U87MG-EGFR cells formed larger colonies compared to U87MG-EGFRdNLS cells in the presence of soft agar, suggesting that nuclear EGFR is important for the anchorage-independent growth of U87MG cells. U87MG-vector cells did not form colonies in either assay.
To demonstrate that EGFRdNLS retains its receptor tyrosine kinase function, we showed that EGFRdNLS undergoes EGF-induced autophosphorylation similar to EGFR (Fig. 2D). Using cells treated with the protein synthesis inhibitor, cycloheximide, we showed that the deletion within the NLS region did not alter the half-life/stability of EGFR (Fig. 2E). Importantly, results of colony formation assays (Fig. 2F) indicate that EGFRdNLS renders U87MG cells significantly less efficient in forming colonies in the absence (first and second panels) and presence of soft agar (third and fourth panels). U87MG-vector cells formed loose colonies in these assays. It is also noticeable that U87MG-EGFR cells formed larger colonies compared to U87MG-EGFRdNLS cells in the presence of soft agar (high-resolution images in Fig. 2F-fourth panel), suggesting that nuclear EGFR is important for the anchorage-independent growth of EGFR-expressing GBM cells. Taken together, these results demonstrate the utility of the isogenic U87MG-vector, U87MG-EGFR and U87MG-EGFRdNLS cell lines for subsequent gene expression profiling to identify nuclear EGFR target genes in human GBM cells.
The human COX-2 gene is a candidate nuclear EGFR target gene
We reason that nuclear EGFR target genes are regulated by EGFR but not by EGFRdNLS. Thus, microarray was performed to determine the expression levels of a number of human gene transcripts in the three isogenic U87MG-EGFR, U87MG-EGFRdNLS and U87MG-vector cell lines. In each of the three experiments, tumor cells were serum-starved for 24 hrs, treated without and with EGF for 4 hrs, total RNA extracted and submitted for microarray analysis. Data were analyzed by a data analyst in the Duke Microarray Facility. Here, we identified 19 genes that were significantly induced by EGF in U87MG-EGFR cells (Fig. 3A and Supplementary Table I; p<0.05), but not in U87MG-vector or U87MG-EGFRdNLS cells. Interestingly, COX-2 (arrow) is one of the potential nuclear EGFR target genes (Fig. 3A). Given the long-standing interest of our laboratory in pro-inflammatory pathways, we directed our validation effort at COX-2. The EGF-induced expression of COX-2 transcripts in U87MG-EGFR cells but not in U87MG-EGFRdNLS or U87MG-vector cells was confirmed by quantitative RT-PCR (Fig. 3B) and by regular RT-PCR and western blotting (Fig. 3C-left panel). Consistent with the microarray results, EGF significantly induced expression of COX-2 transcripts and protein in U87MG-EGFR cells but not in U87MG-vector or U87MG-EGFRdNLS cells (Fig. 3C-right panel). Consistent with the elevated transcript and protein levels, we showed the COX-2 promoter to be significantly activated by EGF in U87MG-EGFR cells but not in U87MG-vector or U87MG-EGFRdNLS cells (Fig. 3D). Conversely, EGFR kinase inhibitor Iressa suppressed the ability of EGF to induce COX-2 expression, as shown by RT-PCR (Fig. 3E-left panel) and to activate the COX-2 promoter (right panel) in U87MG-EGFR cells. These results indicate that the human COX-2 gene is a candidate nuclear EGFR target gene.
Figure 3. Gene expression analysis identified the human COX-2 gene as a candidate nuclear EGFR target gene.
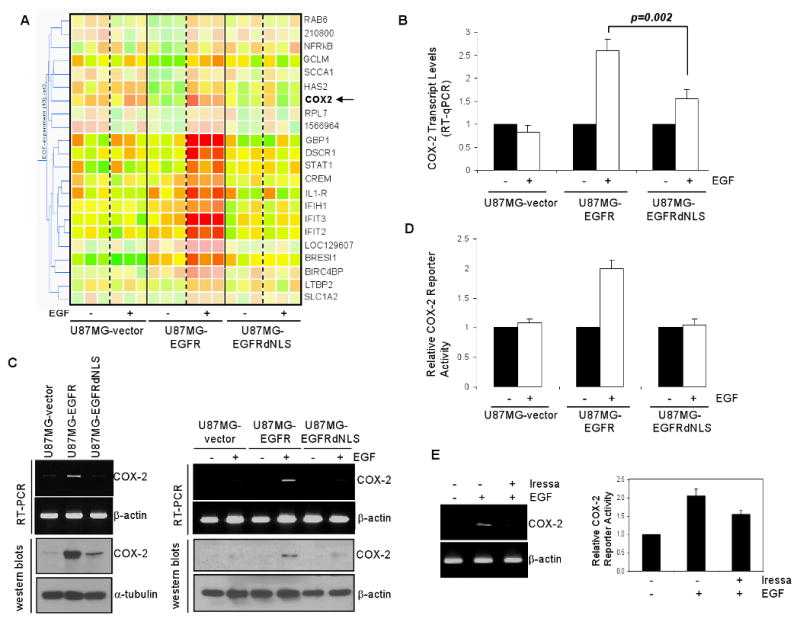
A. Microarray shows 19 genes to be significantly induced by EGF in U87MG-EGFR cells, but not in U87MG-vector or U87MG-EGFRdNLS cells. Cells were treated with and without EGF and the levels of expression of over 47,000 human gene transcripts were determined. Data represent results of three independent experiments. ANOVA was conducted to derive p-values. Clustering analysis included a total of 22 transcripts, with 19 known genes, that were significantly activated by EGF in U87MG-EGFR cells, but not in U87MG-vector or U87MG-EGFRdNLS cells (p<0.05). An arrow points to COX-2.
B. COX-2 transcripts are significantly up-regulated by EGF in U87MG-EGFR cells, but not in U87MG-EGFRdNLS or EGFR-vector cells. Microarray results was validated using RT-qPCR and the results show that the COX-2 transcription is significantly increased by EGF in U87MG-EGFR cells, but not in U87MG-EGFRdNLS or EGFR-vector cells.
C. U87MG-EGFR cells express higher levels of COX-2 transcripts and protein than U87MG-EGFRdNLS or EGFR-vector cells. Three U87MG stable transfectants were subjected to RT-PCR (top panels) and western blotting (bottom panels) to determine levels of COX-2 expression. In the left panels, cells were growth under regular growth conditions with 10% FCS. In the right panels, cells were serum-starved for 24 hrs and treated with EGF (100 ng/ml) for 0 and 4 hrs.
D. The human COX-2 promoter is significantly activated by EGF in U87MG-EGFR cells but not in U87MG-EGFRdNLS orU87MG-vector cells. A firefly luciferase reporter construct under the control of a 1-kb COX-2 promoter was transfected into the three U87MG isogenic cell lines. Twenty-four hrs later, the transfected cells were serum-starved for 24 hrs and stimulated with 100 ng/ml EGF for 0 and 4 hrs. A renilla luciferase vector, pRL-TK was co-transfected to control for transfection efficiency. Luciferase activity was determined as previously described (15). Relative reporter activity was determined by normalizing the firefly luciferase activity against that of the renilla luciferase.
E: EGFR kinase inhibitor Iressa reduced EGF-induced COX-2 expression and promoter activation. U87MG-EGFR cells were serum starved for 24 hrs in the presence of Iressa (25 uM) or 1% DMSO for 24 hrs, treated with EGF for 0 and 1 hr, and analyzed for COX-2 expression via RT-PCR (left panel). Furthermore, aliquots of cells were transfected with pCOX-2-Luc and pRL-TK. Twenty-four hrs after transfections, the cells were similarly treated as described for the RT-PCR experiments and subjected to luciferase assay.
EGFRvIII undergoes nuclear translocalization and activates the human COX-2 gene in GBM cells
Because endogenous EGFRvIII expression is not maintained in vitro, we used U87MG-EGFRvIII stable transfectants in these studies. Here, we found that EGFRvIII is constitutively detected in the nucleus of U87MG-EGFRvIII cells (Fig. 4A-left panel). Importantly, U87MG-EGFRvIII xenografts expressed similar levels of EGFRvIII compared to GBM xenografts with endogenous EGFRvIII (D-256 MG and D-270 MG; Fig. 4A-right panel), indicating that U87MG-EGFRvIII cells express physiological levels of EGFRvIII. To examine the role of nuclear EGFRvIII in COX-2 gene regulation, we created EGFRvIIIdNLS mutant that is defective in nuclear entry and established stable U87MG transfectants that express this mutant. As shown by Fig. 4B, U87MG-EGFRvIII and U87MG-EGFRvIIIdNLS cells expressed equivalent levels of EGFRvIII (left panel) and importantly, EGFRvIIIdNLS was absent from the nucleus (right panel). Using cycloheximide-treated cells, we showed that EGFRvIII and EGFRvIIIdNLS proteins have similar half-lives (Fig. 4C), indicating that the deletion of the NLS sequence did not affect receptor stability. In contrast to the observation in Fig. 2F, U87MG-EGFRvIIIdNLS cells formed colonies to the extent similar to U87MG-EGFRvIII cells (Fig. S3 in Supplementary Data). We further showed, using RT-PCR (Fig. 4D, top panel) and western blotting (Fig. 4D bottom panel), that COX-2 gene expression is significantly higher in U87MG-EGFRvIII cells than U87MG-EGFRvIIIdNLS and U87MG-vector cells.
Figure 4. EGFRvIII undergoes nuclear translocalization and activates the human COX-2 gene in GBM cells.
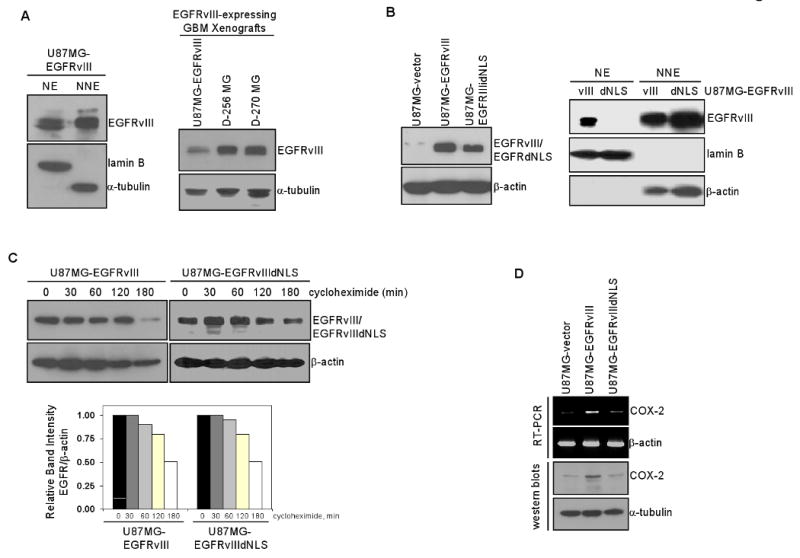
A. EGFRvIII is constitutively present in the nuclei of U87MG-EGFRvIII stable transfectants cells. Left panel: EGFRvIII is present in the nucleus of U87MG-EGFRvIII cells. Right panel: U87MG-EGFRvIII xenografts express EGFRvIII at the levels comparable to GBM xenografts with endogenous EGFRvIII (D-256 MG and D-270 MG).
B. Characterization of isogenic U87MG-EGFRvIII and U87MG-EGFRvIIIdNLS cells. Left panel: Western blotting shows U87MG-EGFRvIII and U87MG-EGFRvIIIdNLS cells to express equivalent levels of the receptors. Right panel: Nuclear fractionation and western blotting shows EGFRvIIIdNLS to fail to enter the cell nucleus.
C. EGFRvIII and EGFRvIIIdNLS proteins have similar half-life. Isogenic U87MG-EGFRvIII and U87MG-EGFRvIIIdNLS cells were treated with 10 ug/ml cycloheximide for 0-180 min and subjected to western blotting for EGFRvIII and β-actin. Band signals were quantified using ImageJ software and protein half-life was subsequently computed. The results indicate that NLS deletion does not affect EGFRvIII protein stability.
D. Levels of COX-2 transcripts and protein are significantly higher in U87MG-EGFRvIII cells than U87MG-EGFRvIIIdNLS and U87MG-vector cells. Top panel: RT-PCR. Bottom panel: Western blotting.
Nuclear EGFR-mediated activation of the human COX-2 gene is enhanced by STAT3
Although nuclear EGFR is known to have transactivational activity, it does not directly associate with DNA, and thus requires cooperation with DNA-binding transcription factors to regulate gene expression (13, 15). In previous studies, we showed that nuclear EGFR associates with STAT3 to induce iNOS gene expression (15). Similarly, nuclear EGFRvIII-STAT3 complex was shown to be present in glial cells and to be involved in their malignant transformation (23). Against this background, we examined whether nuclear EGFR and EGFRvIII cooperate with STAT3 to induce COX-2 gene expression in GBM cells. Using intracellular protein-DNA binding ChIP assay, we showed that nuclear EGFR and nuclear STAT3 associate with the COX-2 gene promoter and that the association was dependent on EGF stimulation (Fig. 5A). Fig. 5B shows that COX-2 promoter activity was modestly enhanced by EGFR alone and STAT3CA alone but significantly increased by the combination of EGFR and STAT3CA. STAT3CA is a constitutively activated STAT3 variant with two Cys substitutions that enable STAT3 molecules to dimerize spontaneously without phosphorylation at Y705 (39). Since STAT3CA is constitutively activated independent of EGF stimulation, we found EGF to modestly enhance (1.3-fold) the ability of EGFR and STAT3CA co-expression to activate the COX-2 promoter (data not shown). Similar to wild-type EGFR, the COX-2 promoter was also activated by EGFRvIII (Fig. 5C). Unlike the robust activation by the EGFR-STAT3CA combination, STAT3CA only modestly enhanced EGFRvIII-mediated activation of the COX-2 gene promoter. Consistent with these observations, EGFRdNLS-STAT3CA co-transfection had lower effects on the COX-2 promoter than the EGFR-STAT3CA combination (Fig. 5D), further providing evidence of an important role for nuclear EGFR in the activation of the COX-2 promoter by STAT3. EGFRvIIIdNLS-STAT3CA co-transfection activated COX-2 promoter similar to that observed for the EGFRvIII-STAT3CA combination. These findings are in line with several studies reporting that EGFRvIII preferentially activates the PI-3K/Akt signaling axis over the STAT3 and Ras/MAPK downstream signaling modules (40, 41).
Figure 5. STAT3 significantly enhances nuclear EGFR-mediated activation of the human COX-2 gene.
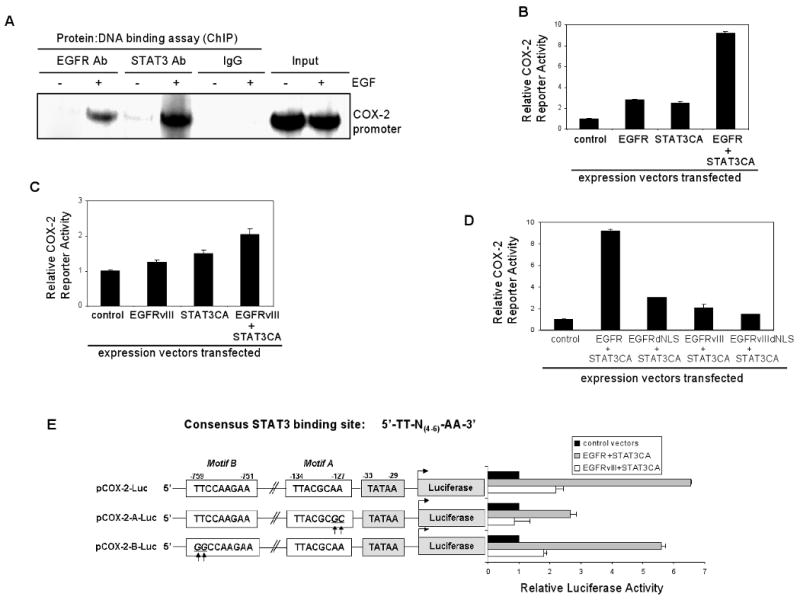
A. Nuclear EGFR and nuclear STAT3 associate with the COX-2 gene promoter in an EGF-dependent fashion. In intracellular protein-DNA binding ChIP assay, we used an anti-EGFR antibody (Ab) and an anti-STAT3 Ab in immunoprecipitation whereas normal IgGs were used as negative controls. Input chromatins were used as loading controls for PCR.
B. COX-2 promoter activity is enhanced by EGFR alone and STAT3CA alone but is significantly elevated by the combination of both. STAT3CA: constitutively activated STAT3 with two Cys substitutions that enable STAT3 molecules to dimerize spontaneously without phosphorylation. U87MG cells were transfected with pCOX-2-Luc and EGFR plasmid alone, STAT3CA plasmid alone or in combination for 48 hrs and then luciferase activity determined. pRL-TK vector was co-transfected as transfection efficiency control. Relative COX-2 reporter activity was determined by normalizing the firefly luciferase activity against that of the renilla luciferase. Three independent experiments were performed to derive means and standard deviations.
C. The COX-2 promoter is activated by EGFRvIII. The experiment was conducted as described in panel B. Although the COX-2 promoter is activated by EGFRvIII, unlike the potent activation by EGFR-STAT3CA combination, STAT3CA only modestly enhances EGFRvIII-mediated activation of COX-2 promoter.
D. Nuclear EGFR is important for EGFR-STAT3CA mediated activation of the COX-2 promoter. This was conducted as described in panel B. EGFRdNLS-STAT3CA co-transfection yields significantly lower activity than the EGFR-STAT3CA combination. In contrast, EGFRvIIIdNLS-STAT3CA co-transfection similarly activates the COX-2 promoter compared to EGFRvIII-STAT3CA combination.
E. The EGFR-STAT3 signaling axis activates the COX-2 promoter via the STAT3-binding site proximal to the TATAA Box. Consensus STAT3-binding site is listed on the top. Web-based search engine, TFSearch, identifies two putative STAT3-binding sites in the human COX-2 promoter, namely, the proximal motif (motif A; nt -134 to -127) and the distant motif (motif B; nt -759 to 751). pCOX-2-A-Luc construct carries the mutant COX-2 promoter with two nucleotide substitutions with the proximal motif. pCOX-2-B-Luc construct contains the mutant COX-2 promoter with two nucleotide substitutions with the distant motif. The results show that mutations within the proximal motif, but not motif B, significantly decreased the ability of EGFR-STAT3CA and EGFRvIII-STAT3CA to activate the COX-2 promoter.
To further investigate the structural requirement in the COX-2 promoter for EGFR/STAT3-mediated activation, we analyzed the human COX-2 promoter for consensus STAT3 binding motifs, 5′-TT-N(4-6)-AA-3′, using a web-based search engine, TFSearch (http://www.cbrc.jp/research/db/TFSEARCH.html), as we previously described (15, 42). The results of the search showed two putative STAT3 binding sites within the human COX-2 promoter (Fig. 5E-top panel). We mutated each motif by substituting two nucleotides and such that the TT-AA palindrome was disrupted. Using these mutant reporter constructs, we showed that mutations within the proximal STAT3-binding motif (motif A), but not in motif B, significantly decreased the ability of EGFR-STAT3CA and EGFRvIII-STAT3CA co-transfections to activate the COX-2 promoter (Fig. 5E). Collectively, the results summarized in Fig. 5 indicate that STAT3 cooperates with both EGFR and EGFRvIII to activate the COX-2 gene promoter, with the former more active than the latter.
Increased COX-2 expression is associated with EGFR/EGFRvIII and activated STAT3 in primary GBM specimens and xenografts
To determine the potential clinical relevance of the association between COX-2 and EGFR/EGFRvIII-STAT3, we examined 12 primary specimens of malignant gliomas and three GBM xenografts for the expression of COX-2, EGFR and p-STAT3 (Y705) via IHC. The results showed that 58.3% (7/12) of the primary malignant gliomas co-expressed EGFR and COX-2 and that levels of EGFR correlate with those of COX-2, as indicated by Chi-square analysis (p=0.03). In the three GBM xenografts we analyzed, we found COX-2 to be expressed at a higher level in the EGFR/EGFRvIII-positive, p-STAT3-positive U87MG-EGFR (data not shown) and D-256 MG GBM xenografts but not in U87MG xenografts, which have very low levels of EGFR and p-STAT3 (Fig. 6A). We further examined whether p-STAT3-expressing GBM cells express COX-2 using double fluorescence staining-coupled IHC. In these studies, p-STAT3 is indicated by red fluorescence and COX-2, by green fluorescence. Tumor nuclei were stained by DAPI (blue). As indicated by Fig. 6B, the majority of p-STAT3-positive nuclei (pink signals as merged products of red and blue fluorescence) in the D-256 MG xenograft expressed high levels of COX-2. Those cells with p-STAT3-positive nuclei but did not express COX-2 can potentially be due to the expression of a transcription repressor of COX-2. Noticeably, COX-2 is also expressed in cells without significant p-STAT3 expression, suggesting that factors other than STAT3 may account for COX-2 gene expression in these cells.
Figure 6. Increased COX-2 expression is detected in GBM xenografts with EGFR/EGFRvIII and activated STAT3.
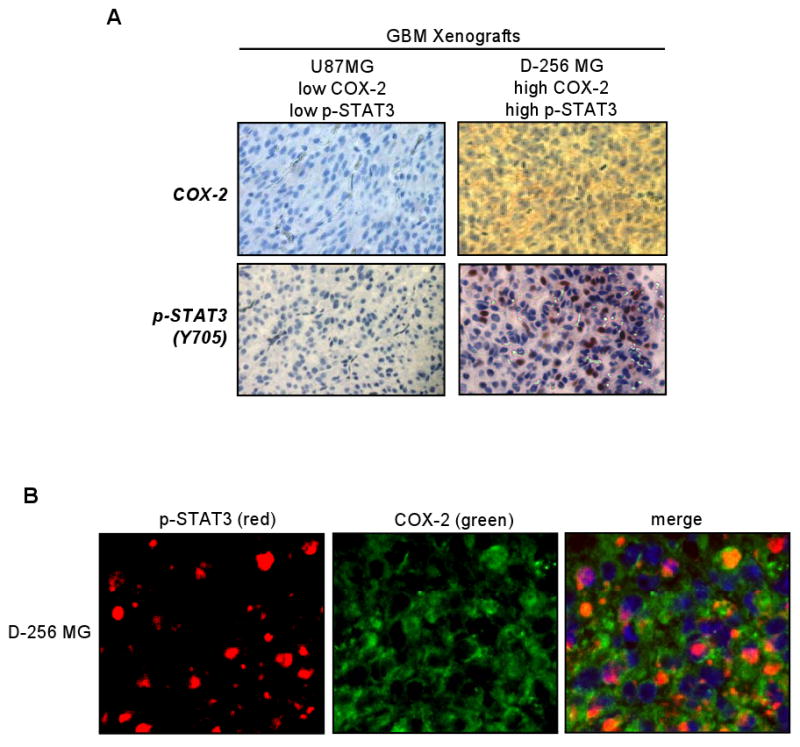
A. COX-2 is highly expressed in GBM xenografts with highly activated STAT3. Two GBM xenografts were analyzed for levels of COX-2 and p-STAT3 (Y705) via IHC. The D-256 MG GBM xenograft but not the U87MG xenograft expressed high levels of COX-2 and p-STAT3. The p-STAT3-positive nuclei are indicated by the brown signals whereas the negatively stained nuclei are in blue.
B. The majority of p-STAT3-expressing GBM cells in the D-256 MG xenograft express COX-2. The D-256 MG GBM xenograft was analyzed for co-expression of p-STAT3 and COX-2 using double fluorescence staining-coupled IHC. Red fluorescence: p-STAT3. Green fluorescence: COX-2. Nuclei: blue. p-STAT3-positive nuclei: pink signals as merged products of red and blue fluorescence. The majority of p-STAT3-positive nuclei expressed high levels of COX-2. COX-2 is also expressed in cells without significant p-STAT3 expression, suggesting that p-STAT3-independent mechanisms may account for the transcriptional activation of the COX-2 gene in these cells.
Discussion
Although over-expression of EGFR and EGFRvIII is a well-known characteristic of many human cancers, our understanding of EGFR- and EGFRvIII-associated tumor biology remains insufficient, which is likely due to the high degree of biological complexity that lies within these pathways (43). This is, in part, indicated by emerging evidence showing the ability of EGFR and EGFRvIII to undergo nuclear translocalization and in the cell nucleus, to regulate gene expression. To date, however, it is unknown whether the nuclear EGFR and EGFRvIII pathways are functional in GBM cells. In this study, we provide evidence that, for the first time, the nuclear EGFR and EGFRvIII pathways are functional in human GBM cells, the most common and deadliest brain malignancy in adults. Using an unbiased approach to identify genes that are targeted specifically by nuclear EGFR, but not by the cell-surface counterpart, we identified COX-2 as a nuclear EGFR target. Consistent with the known ability of nuclear EGFR to physically and functionally associate with STAT3 transcription factor, we found the nuclear EGFR-STAT3 signaling axis to transcriptionally activate the COX-2 gene in GBM cells. Collectively, these data provide a novel link between nuclear EGFR/EGFRvIII-mediated mitogenic pathway and the COX-2-associated inflammatory pathways in GBM.
COX-2 plays a central role in many important cellular processes, including, inflammatory response, tumorigenesis and tumor progression (31). Several studies have shown that EGFR and COX-2 are co-overexpressed in cutaneous squamous cell carcinoma (44) and lung adenocarcinoma (45). EGF has been shown to induce COX-2 expression through PI-3K, p38 and protein kinase C-δ (46, 47), albeit the exact transcriptional pathway that mediates the transcriptional activation of COX-2 was not determined under these conditions. Interestingly, a previous report showed that nuclear HER2 binds to and activates the human COX-2 gene promoter, leading to its transcriptional activation in breast cancer cells (35). The identified HER2 associated sequence (HAS) is a 20-bp element located at -1750 to -1731 bps of the human COX-2 promoter (35). Since the COX-2 reporter construct used in this study contains up to -1 kb of the COX-2 promoter, the observed EGF- and EGFR-induced COX-2 promoter activation (Figs. 3D-E, 5B) is independent of HER2 or HAS. Our co-immunoprecipitation results indicate that nuclear EGFR does not complex with nuclear HER2 in GBM cells analyzed, further supporting the notion of nuclear EGFR-dependent and nuclear HER2-independent activation of the COX-2 gene expression in GBM cells. Furthermore, our results indicate that nuclear EGFR and activated STAT3 cooperate synergistically to activate the COX-2 gene activity. This synergy may not apply to nuclear HER2-mediated COX-2 gene activation, given the fact that the HAS motif (5′-ATAAACTTCAAATTTCAGTA-3′) does not appear to contain a putative STAT3 binding site (TT-N(4-6)-AA-3′) and STAT3 is therefore unlikely to activate the COX-2 promoter through binding to HAS. The results of the present study combined with those of the study by Wang et al (35), together, unraveled an important link between the ErbB family of receptor tyrosine kinases, their nuclear translocalization and COX-2-mediated pro-inflammatory pathway.
To date, only four nuclear EGFR-targeted genes, namely, cyclin D1, iNOS, B-Myb and Aurora A, have been reported (13, 15, 17, 25). The identification and validation of the COX-2 gene as an additional transcriptional target of the nuclear EGFR and nuclear EGFRvIII pathways and the evidence that this activation of COX-2 by nuclear EGFR/EGFRvIII occurs in GBM cells growing in vitro and in vivo are significant. Unlike these previous studies, the current undertook an unbiased comprehensive approach to identify nuclear EGFR target genes using tumors expressing wild-type versus genetically modified EGFR molecules. Although we focused on COX-2 in this study, it is important to note that other genes have been identified in the microarray analysis. Ongoing studies are directed at these genes to gain further insight into their role in the malignant biology of tumors with active nuclear EGFR and EGFRvIII pathways.
STAT3 is highly activated in many human cancers, including, malignant gliomas (30, 48) and cancers of the breast and the head and neck (49, 50). Similar to increased COX-2 expression, STAT3 activation is associated with inflammation, more malignant tumor phenotype and poor prognosis (51, 52). Consequently, STAT3 and COX-2 both have emerged as targets for anti-GBM therapy (30, 53). The findings in this study linking STAT3 and COX-2 should contribute to a better understanding of their role in nuclear EGFR signaling-associated malignant biology.
Given that GBMs expressing EGFRvIII or activated EGFR are more refractory to therapy in comparison to those without EGFR activation, the findings could provide, potentially, the basis for novel combination therapies that simultaneously target EGFR/EGFRvIII and COX-2. Indeed, in pre-clinical and clinical studies, combination of inhibitors of EGFR and COX-2 have yielded promising results in advanced cutaneous squamous cell carcinoma, NSCLC, intestinal cancer and prostate cancer (44, 54-56). In summary, the results reported in this study define the COX-2 gene as a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes and provide new insight into the malignant biology of a subpopulation of human tumors with concurrently activated EGFR/EGFRvIII and STAT3 pathways. They also provide a potential rationale for combination therapy targeting EGFR/EGFRvIII, STAT3 and COX-2 pathways.
Materials and Methods
Cell Lines, Primary Gliomas, Xenografts and Reagents
Human GBM cell lines U87MG and T98G were from ATCC (Manassas, VA) whereas MGR3 cells were established in our laboratory from primary specimens. These cells were maintained in DMEM with 10% fetal calf serum. U87MG-vector, U87MG-EGFR and U87MG-EGFRvIII stable transfectant lines were previously established from the parental U87MG cells that express a very low level of EGFR (30). The stable transfectant cell lines were cultured in DMEM with 10% fetal calf serum and 1 mg/ml G418. GBM xenografts established in the flanks of nude mice were provided by the Preston Robert Tisch Brain Tumor Center at Duke University. Primary GBM specimens were obtained from Preston Robert Tisch Brain Tumor Center at Duke University and Imgenex (San Diego, CA). All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise stated. Rabbit polyclonal anti-EGFR antibody used in western blotting was purchased from Santa Cruz Biotech. (sc-03). The EGFR, EGFRvIII and STAT3CA expression vectors were generated in our laboratory (30) and expressed as Myc-tagged fusion proteins. Anti-Myc mouse monoclonal antibody used in immunofluorescence staining and confocal microscopy was purchased from Roche (Indianapolis, IN). Anti-lamin B mouse monoclonal antibody was from Calbiochem (San Diego, CA). β-actin and α-tubulin antibodies were obtained from Sigma. Rabbit polyclonal HER2 (29D8), p-EGFR (Y1068), p-STAT3 (Y705) and COX-2 antibodies were purchased from Cell Signaling (Danvers, MA).
Detection of Nuclear EGFR/EGFRvIII via Nuclear Fractionation and Western Blotting
This was performed as previously described (15). Cells treated per experimental procedures were collected, washed with PBS, and swelled in hypotonic buffer (25 mM Tris-HCl, pH 7.5, 5 mM KCl, 0.5 mM dithiothreitol, 1 mM PMSF and 0.15 m/ml aprotinin) for 20 min on ice. Following homogenization using a Dounce homogenizer, nuclei were pelleted and washed. To extract nuclear proteins from the isolated nuclei, we used an ultrasonic disruption step and sonication buffer containing 50 mM Tris-HCl and 0.15 m/ml aprotinin. To isolate non-nuclear extracts, the supernatant was exposed to 1% SDS and 0.1 % NP-40, centrifuged at 15,000g to remove cell debris and the resulting supernatant collected. Western blotting was conducted as previously described (30).
Immunohistochemical Staining of Primary Malignant Glioma Specimens for EGFR, Activated STAT3 and COX-2
The immunoperoxidase staining method used in these studies was a modification of the avidin-biotin complex technique, as described previously (30). Paraffin-embedded microsections (5 uM) of GBM primary specimens and xenografts were deparaffinized, dehydrated, and subjected to antigen retrieval in a microwave oven followed by incubation with 0.05% trypsin in PBS for 15 min at room temperature. Endogenous peroxidase activity was blocked by treatment of 0.3% hydrogen peroxide. The slides were incubated with 10% normal goat serum for 30 min and then with the EGFR mouse monoclonal antibody (1:50; Novocastra RTU-EGFR-384), COX-2 (1:25; Cell Signaling) and p-STAT3-Y705 (1:30; Cell Signaling) rabbit polyclonal antibodies at 4°C overnight. Following washes with PBS, the slides were incubated with biotinylated secondary antibodies and then with avidin-biotin-horseradish peroxidase complex. Detection was performed using 0.125% aminoethylcarbazole chromogen. After counterstaining with Mayer's hematoxylin (Sigma), the sides were mounted. Scoring was performed by a pathologist. H scores (histologic scores) for EGFR and COX-2 were computed from both % positivity (A%, A=1-100) and intensity (B=0-3) using the equation, H Score=A × B, according to a well-established IHC scoring system (30, 57).
Determination of Binding of Nuclear EGFR to Nuclear HER2 via Immunoprecipitation and Western Blotting
T98G cells treated without and with EGF (100 ng/ml) for 15 min were harvested and fractionated into nuclear and non-nuclear fractions. Nuclear extracts were subjected to immunoprecipitation and western blotting, in which the lysates were incubated with 1 μg anti-EGFR mouse monoclonal antibody (Ab-13, Neomarker) or control mouse IgG at 4°C overnight with gentle agitation (15). Following addition of protein G-agarose and incubation for 30 min at 4°C, protein G-agarose pellets were collected and washed for multiple cycles at 4°C. The washed immunoprecipitates were subjected to SDS-PAGE and western blotting, as described previously (58). Both nuclear and non-nuclear extracts were subjected to western blotting to detect levels of EGFR, HER2, lamin B (nuclear protein) and β-actin (cytoplasmic protein).
Plasmids, Transfection and Generation of NLS-deleted EGFR and NLS-deleted EGFRvIII Constructs and Stable Transfectants
The EGFR and EGFRvIII expression vectors, pCMV-Tag5A-EGFR and pCMV-Tag5A-EGFRvIII, were previously generated in our laboratory (30). To establish NLS-deleted EGFR and EGFRvIII, we first PCR amplified the N-terminal regions of EGFR (codons -24 to +644) and EGFRvIII (codons -24 to +377) by PCR, using a forward primer with the Hind III site: 5′-ATTAAGCTTCCACCATGCG ACCCTCCGGGA-3′ and a reverse primer with the Xba I site: 5′-TAATCTAGACATGAAGAG GCCGATCCC-3′. The PCR products were restricted with Hind III and Xba I and subcloned into compatible ends of pCMV-Tag5A plasmid (Stratagene). To create the C-terminal fragments of EGFR (codons +653 to +1186) and EGFRvIII (codons +378 to +919) fragments, we performed PCR with primers with sequences: 5′-TAATCTAGAACGCTGCGGAGGCTGCTG-3′ (forward primer with the Xba I site) and 5′-TAGGCTGGAGTGCT CCAATAAATTCACT-3′ (reverse primer with the Xho I site). To enable subcloning, a Ser codon was inserted at the 5′-end of the C-terminal fragments. The C-terminal fragments were restricted using Xba I and Xho I enzymes and ligated into the compatible ends of plasmids with the N-terminal EGFR/EGFRvIII. The resulting plasmids were designated as pCMV-Tag5A-EGFRdNLS and pCMV-Tag5A-EGFRvIIIdNLS, that encode EGFR and EGFRvIII receptors with a deletion of eight amino acids (aa 645-652 in EGFR) within the NLS region (see Fig. 2A). All transfections were performed with cells in exponential growth using FuGENE HD (Roche). Generation of stable transfectants was performed as previously described (30).
Determination of the Half-life of EGFR/EGFRvIII and Their Nuclear Entry-defective Mutants
Tumor cells exposed to 10 ug/ml cycloheximide for 0, 30, 60, 120 and 180 mins were harvested, total protein extracted and subjected to western blotting. A Myc-tagged mouse monoclonal antibody (Roche) was used to detect Myc-tagged EGFR, EGFRdNLS, EGFRvIII and EGFRvIIIdNLS fusion proteins.
Determination of Anchorage-independent Growth by the Clonogenic Growth Assays
Clonogenic growth assays were performed in 6-well cell culture plates with 1000 cells seeded per well, as previously described (15). For the experiments with soft agar, wells were pre-coated with 0.5% agarose as the bottom layer. The top layer contained cells in 0.3% agarose. Following 10-14 days, colonies were stained with crystal violet blue solution (Sigma) for 1 hr and counted. Triplicate wells were used for each cell line and three independent experiments were performed.
Identification of Nuclear EGFR Target Genes by GeneChip DNA Microarray
Total RNA extracted from the three U87MG stable transfectant lines, U87MG-vector, U87MG-EGFR and U87MG-EGFRdNLS, were used to examine their gene expression profile. This was conducted in the DNA Microarray Core Facility of Duke Institute of Genome Science & Policy using the human Genome U133 Plus 2.0 Array genechips (Affymetrix, Santa Clara, CA) containing over 47,000 gene transcripts. ANOVA was conducted to determine statistical significance.
Reverse Transcription-PCR (RT-PCR) and Quantitative PCR
Total RNA isolation and RT were conducted using the SV Total RNA Isolation System (Promega). RT-qPCR was performed in the Mx3005P qPCR System (Stratagene) using the SuperScript III platinum SYBR green one-step qRT-PCR system (Invitrogen), in which GAPDH gene was used as normalization controls. All experiments were in triplicate. Primer sequences were 5′-TTAATGAGTACCGCAAACGC-3′ and 5′-ACCAGAAGGGCAGGATACAG-3′ (COX-2) and 5′-ACTGCCAACGTGTCAGTGGT-3′ and 5′-GTGTCGCTGTTGAAGTCAGA-3′ (GAPDH). For standard RT-PCR, Superscript II First-Strand cDNA synthesis system (Invitrogen), was used and PCR performed with forward and reverse primers of 5′-TGTATGAGTGTGGGATTTGACC-3′ and 5′- GTGATCTGGATGTCAACACAT-3′ (COX-2) and 5′-GGCGGCACCACCATGTACCC-3′ and 5′-AGGGGCCGGACTCGTCATACT-3′ (β-actin).
Determination of COX-2 Promoter Activity by Mammalian Transfection of Reporter Vector and Luciferase Assay
The COX-2 reporter construct, pCOX-2-Luc, was purchased from Panomics (Fremont, CA) and contains a 1 kb human COX-2 promoter 5′ of the firefly luciferase reporter gene. A Renilla luciferase expression vector, pRL-CMV whose expression is controlled by the CMV promoter was used to control for transfection efficiency. Forty-eight hrs after transfection, the cells were lysed and luciferase activity measured using the Dual Luciferase Assay Kit in a TD 20/20 luminometer (Promega), as previously described (15). Relative luciferase activity was calculated by normalization of the firefly luciferase activity against that of the Renilla luciferase.
Chromatin Immunoprecipitation (ChIP) Assay to Measure Binding of Nuclear EGFR-STAT3 to the COX-2 Promoter
ChIP was performed using a ChIP Assay Kit (Upstate, Billerica, MA) as we described previously (15). An anti-EGFR mouse monoclonal antibody (Neomarkers) and an anti-STAT3 rabbit polyclonal antibody (Santa Cruz, C-20) were used in these experiments. Sequences of the primers for amplifying the COX-2 promoter are 5′-GGCTTACGCAATTTTTTTAAGGGGA -3′ (forward) and 5′-TGGTCGCTAACC GAGAGAACCTTCC-3′ (reverse). Control IgGs were used as negative controls for immunoprecipitation. Chromatin input was used as loading control for PCR.
Generation of Mutant COX-2 Promoter Reporters via Site-directed Mutagenesis
Site-directed mutagenesis was performed using QuikChange Mutagenesis Kit (Stratagene), according to the manufacturer's instructions. The pCOX-2-Luc luciferase reporter construct was used as the template. Sequences of the primers used to mutate the COX-2 promoter are 5′-CGGGCTTACGCGC TTTTTTTAAGGG-3′ and 5′-CCCTTAAAAAAAGCGCGTAAGCCCG-3′ (pCOX-2-A-Luc) and 5′-CCCGCCTCTCTGGCCAAGAAACAAG-3′ and 5′-CTTGTTTCTTGGCCAGAGAGGCGGG-3′ (pCOX-2-B-Luc).
Determination of p-STAT3 and COX-2 Co-expression via Double Immunofluorescence Staining-coupled IHC
Paraffin-embedded tumor sections were deparaffinized and subjected to epitope retrieval. The sections were blocked with 5% normal goat serum and 5% normal horse serum, and incubated with a mixture of the anti-p-STAT3 rabbit polyclonal antibody (1:50, Cell Signaling) and anti-COX-2 mouse monoclonal antibody (1:50, Santa Cruz) overnight at 4°C. After several washes, the sections were incubated with Texas red-conjugated goat anti-rabbit and fluoresin-conjugated horse anti-mouse secondary antibodies (1:200, Vector Lab). After mounted with VECTASHIELD Mounting Medium with DAPI (Vector Lab), the sections were examined under a Zeiss Axioskop2 plus fluorescence microscope.
Statistical Analysis
ANOVA, Chi-square analysis, Student t-test and regression analysis were performed using STATISTICA (StatSoft Inc., Tulsa, OK) and Microsoft Excel, as we previously described (30, 58)
Supplementary Material
Acknowledgments
This study was supported by NIH grant, K01-CA118423, and the Pediatric Brain Tumor Foundation (to H.-W. L).
References
- 1.Nakamura JL. The epidermal growth factor receptor in malignant gliomas: pathogenesis and therapeutic implications. Expert Opinion on Therapeutic Targets. 2007;11:463. doi: 10.1517/14728222.11.4.463. [DOI] [PubMed] [Google Scholar]
- 2.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–8. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Z, Bagheri-Yarmand R, Wang RA, et al. The epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 (Iressa) suppresses c-Src and Pak1 pathways and invasiveness of human cancer cells. Clin Cancer Res. 2004;10:658–67. doi: 10.1158/1078-0432.ccr-0382-03. [DOI] [PubMed] [Google Scholar]
- 4.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87:8602–6. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishikawa R, Ji XD, Harmon RC, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–31. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter G, Liao HJ. Trafficking of receptor tyrosine kinases to the nucleus. Experimental cell research. 2009;315:1556–66. doi: 10.1016/j.yexcr.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen S, Carpenter G, King L., Jr Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J Biol Chem. 1980;255:4834–42. [PubMed] [Google Scholar]
- 8.Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37 4:S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 9.Chien PS, Mak OT, Huang HJ. Induction of COX-2 protein expression by vanadate in A549 human lung carcinoma cell line through EGF receptor and p38 MAPK-mediated pathway. Biochem Biophys Res Commun. 2006;339:562–8. doi: 10.1016/j.bbrc.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 10.Cao H, Lei ZM, Bian L, Rao CV. Functional nuclear epidermal growth factor receptors in human choriocarcinoma JEG-3 cells and normal human placenta. Endocrinology. 1995;136:3163–72. doi: 10.1210/endo.136.7.7540549. [DOI] [PubMed] [Google Scholar]
- 11.Klein C, Gensburger C, Freyermuth S, Nair BC, Labourdette G, Malviya AN. A 120 kDa nuclear phospholipase Cgamma1 protein fragment is stimulated in vivo by EGF signal phosphorylating nuclear membrane EGFR. Biochemistry. 2004;43:15873–83. doi: 10.1021/bi048604t. [DOI] [PubMed] [Google Scholar]
- 12.Liao HJ, Carpenter G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol Biol Cell. 2007;18:1064–72. doi: 10.1091/mbc.E06-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin SY, Makino K, Xia W, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–8. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 14.Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem. 2006;98:1570–83. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- 15.Lo HW, Hsu SC, Ali-Seyed M, et al. Nuclear Interaction of EGFR and STAT3 in the Activation of iNOS/NO Pathway. Cancer Cell. 2005;7:575–89. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Dittmann K, Mayer C, Fehrenbacher B, et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280:31182–9. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 17.Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog. 2006;45:10–7. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- 18.Wang SC, Nakajima Y, Yu YL, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8:1359–68. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 19.Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005;65:338–48. [PubMed] [Google Scholar]
- 20.Psyrri A, Yu Z, Weinberger PM, et al. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res. 2005;11:5856–62. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- 21.Xia W, Wei Y, Du Y, et al. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog. 2009;48:610–7. doi: 10.1002/mc.20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marti U, Burwen SJ, Wells A, et al. Localization of epidermal growth factor receptor in hepatocyte nuclei. Hepatology. 1991;13:15–20. [PubMed] [Google Scholar]
- 23.de la Iglesia N, Konopka G, Puram SV, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–62. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie Y, Hung MC. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochem Biophys Res Commun. 1994;203:1589–98. doi: 10.1006/bbrc.1994.2368. [DOI] [PubMed] [Google Scholar]
- 25.Hung LY, Tseng JT, Lee YC, et al. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36:4337–51. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003;63:3923–30. [PubMed] [Google Scholar]
- 27.Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–8. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 28.Lo HW, Hsu SC, Xia W, et al. Epidermal Growth Factor Receptor Cooperates with Signal Transducer and Activator of Transcription 3 to Induce Epithelial-Mesenchymal Transition in Cancer Cells via Up-regulation of TWIST Gene Expression. Cancer Res. 2007;67:9066–76. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vigneron A, Gamelin E, Coqueret O. The EGFR-STAT3 oncogenic pathway up-regulates the Eme1 endonuclease to reduce DNA damage after topoisomerase I inhibition. Cancer Res. 2008;68:815–25. doi: 10.1158/0008-5472.CAN-07-5115. [DOI] [PubMed] [Google Scholar]
- 30.Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively Activated STAT3 Frequently Coexpresses with Epidermal Growth Factor Receptor in High-Grade Gliomas and Targeting STAT3 Sensitizes Them to Iressa and Alkylators. Clin Cancer Res. 2008;14:6042–54. doi: 10.1158/1078-0432.CCR-07-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenhough A, Smartt HJ, Moore AE, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–86. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 32.Nam DH, Park K, Park C, et al. Intracranial inhibition of glioma cell growth by cyclooxygenase-2 inhibitor celecoxib. Oncology reports. 2004;11:263–8. [PubMed] [Google Scholar]
- 33.Reardon DA, Quinn JA, Vredenburgh J, et al. Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer. 2005;103:329–38. doi: 10.1002/cncr.20776. [DOI] [PubMed] [Google Scholar]
- 34.Dittmann K, Mayer C, Kehlbach R, Rodemann HP. The radioprotector Bowman-Birk proteinase inhibitor stimulates DNA repair via epidermal growth factor receptor phosphorylation and nuclear transport. Radiotherapy and Oncology. 2008;86:375–82. doi: 10.1016/j.radonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 35.Wang SC, Lien HC, Xia W, et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–61. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Opresko L, Shankaran H, Chrisler W, Wiley HS, Resat H. HER/ErbB receptor interactions and signaling patterns in human mammary epithelial cells. BMC Cell Biology. 2009;10:78. doi: 10.1186/1471-2121-10-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW. c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002;157:929–39. doi: 10.1083/jcb.200109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem. 2007;282:10432–40. doi: 10.1074/jbc.M610014200. [DOI] [PubMed] [Google Scholar]
- 39.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 40.Huang PH, Mukasa A, Bonavia R, et al. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A. 2007;104:12867–72. doi: 10.1073/pnas.0705158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prigent SA, Nagane M, Lin H, et al. Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras-Shc-Grb2 pathway. J Biol Chem. 1996;271:25639–45. doi: 10.1074/jbc.271.41.25639. [DOI] [PubMed] [Google Scholar]
- 42.Lo HW, Stephenson L, Cao X, Milas M, Pollock R, Ali-Osman F. Identification and Functional Characterization of the Human Glutathione S-Transferase P1 Gene as a Novel Transcriptional Target of the p53 Tumor Suppressor Gene. Mol Cancer Res. 2008;6:843–50. doi: 10.1158/1541-7786.MCR-07-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang SC, Hung MC. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin Cancer Res. 2009;15:6484–9. doi: 10.1158/1078-0432.CCR-08-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jalili A, Pinc A, Pieczkowski F, Karlhofer FM, Stingl G, Wagner SN. Combination of an EGFR blocker and a COX-2 inhibitor for the treatment of advanced cutaneous squamous cell carcinoma. J Dtsch Dermatol Ges. 2008;6:1066–9. doi: 10.1111/j.1610-0387.2008.06861.x. [DOI] [PubMed] [Google Scholar]
- 45.Van Dyke AL, Cote ML, Prysak GM, et al. COX-2/EGFR expression and survival among women with adenocarcinoma of the lung. Carcinogenesis. 2008;29:1781–7. doi: 10.1093/carcin/bgn107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu K, Chang CM, Gao H, Shu HK. Epidermal growth factor-dependent cyclooxygenase-2 induction in gliomas requires protein kinase C-delta. Oncogene. 2009;28:1410–20. doi: 10.1038/onc.2008.500. [DOI] [PubMed] [Google Scholar]
- 47.Xu K, Shu HK. EGFR activation results in enhanced cyclooxygenase-2 expression through p38 mitogen-activated protein kinase-dependent activation of the Sp1/Sp3 transcription factors in human gliomas. Cancer Res. 2007;67:6121–9. doi: 10.1158/0008-5472.CAN-07-0141. [DOI] [PubMed] [Google Scholar]
- 48.Abou-Ghazal M, Yang DS, Qiao W, et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res. 2008;14:8228–35. doi: 10.1158/1078-0432.CCR-08-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia R, Bowman TL, Niu G, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 50.Masuda M, Suzui M, Yasumatu R, et al. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res. 2002;62:3351–5. [PubMed] [Google Scholar]
- 51.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–23. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res. 2007;13:1362–6. doi: 10.1158/1078-0432.CCR-06-2313. [DOI] [PubMed] [Google Scholar]
- 53.Reardon DA, Rich JN, Friedman HS, Bigner DD. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol. 2006;24:1253–65. doi: 10.1200/JCO.2005.04.5302. [DOI] [PubMed] [Google Scholar]
- 54.Chen L, He Y, Huang H, Liao H, Wei W. Selective COX-2 inhibitor celecoxib combined with EGFR-TKI ZD1839 on non-small cell lung cancer cell lines: in vitro toxicity and mechanism study. Medical oncology (Northwood, London, England) 2008;25:161–71. doi: 10.1007/s12032-007-9015-1. [DOI] [PubMed] [Google Scholar]
- 55.Buchanan FG, Holla V, Katkuri S, Matta P, DuBois RN. Targeting cyclooxygenase-2 and the epidermal growth factor receptor for the prevention and treatment of intestinal cancer. Cancer Res. 2007;67:9380–8. doi: 10.1158/0008-5472.CAN-07-0710. [DOI] [PubMed] [Google Scholar]
- 56.Jia RP, Xu LW, Su Q, et al. Cyclooxygenase-2 expression is dependent upon epidermal growth factor receptor expression or activation in androgen independent prostate cancer. Asian journal of andrology. 2008;10:758–64. doi: 10.1111/j.1745-7262.2008.00423.x. [DOI] [PubMed] [Google Scholar]
- 57.Camp RL, Rimm EB, Rimm DL. Met expression is associated with poor outcome in patients with axillary lymph node negative breast carcinoma. Cancer. 1999;86:2259–65. doi: 10.1002/(sici)1097-0142(19991201)86:11<2259::aid-cncr13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 58.Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69:6790–8. doi: 10.1158/0008-5472.CAN-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.