

Abstract
Background
Reactive oxygen species induce neuronal damage, and their role in reducing synaptic plasticity and function is beginning to be understood. Vitamin E is a potent reactive oxygen species scavenger, which has the potential to reduce oxidative damage encountered after traumatic brain injury (TBI). Brain-derived neurotrophic factor (BDNF) can facilitate synaptic function and support learning by modulating the CaMKII system, synapsin I, and cAMP-response element-binding protein (CREB). The elevation of superoxide dismutase (SOD) and Sir2 (silent information regulator 2) play an important role in resistance to oxidative stress.
Objective
We examined the possibility that vitamin E supplemented in the diet may help counteract the effects of TBI on the molecular substrates underlying synaptic plasticity and cognitive function in the hippocampus.
Methods
Rats were fed a regular diet with or without 500 IU/kg of vitamin E for 4 weeks (n = 6-8 per group) before a mild fluid percussion injury (FPI) was performed.
Results
FPI increased protein oxidation as evidenced by elevated levels of protein carbonyls and reduced levels of SOD and Sir2. In addition, FPI resulted in poor performance in the Morris water maze, which was accompanied by reduced levels of BDNF and its downstream effectors on synaptic plasticity, synapsin I, CREB, and CaMKII. Supplementation of vitamin E in the diet counteracted all the observed effects of FPI.
Conclusions
These results suggest that vitamin E dietary supplementation can protect the brain against the effects of mild TBI on synaptic plasticity and cognition, using molecular systems associated with the maintenance of long-term plasticity, such as BDNF and Sir2.
Keywords: traumatic brain injury, hippocampus, learning, BDNF, vitamin E
Introduction
Traumatic brain injury (TBI) is a major cause of disability, which emphasizes the necessity to develop means to decrease the short- and long-term effects of TBI. Dietary factors are emerging as potentially powerful and efficient agents to modulate the capacity of the brain to cope with synaptic dysfunction and cognitive impairment in response to challenging situations.1-4 Vitamin E has a powerful antioxidant action in many organs. It crosses the blood–brain barrier and accumulates with time at therapeutic levels in the CNS, which in turn may prevent spatial learning deficits, as seen in an animal model of aging,5 improve cognitive function, as seen in an animal model of Alzheimer's with repetitive concussive brain injury,6 and provide neuroprotection from many other types of insults.7-16
Our previous research showed that vitamin E reduced cognitive deficits resulting from consumption of a diet high in saturated fat.2 Oxidative stress may compromise cognitive function and neuroplasticity by disrupting brain-derived neurotrophic factor (BDNF) function.2 Given that TBI reduces levels of BDNF and behavioral plasticity,3,4 we hypothesize that vitamin E supplementation may protect the BDNF system from the deleterious effects of oxidative stress after TBI, thereby promoting resistance to cognitive impairment. The BDNF system enhances the function and viability of select neuronal populations, and its action appears to be crucial for maintaining molecular processes underlying cognitive function.17,18 BDNF promotes neuronal excitability19,20 and facilitates synaptic function.21-25 It is synthesized predominantly by neurons located in the hippocampus, a brain region intimately associated with learning and memory function.26-29 Synapsin I is a nerve terminal phosphoprotein involved in neurotransmitter release, axonal elongation, and maintenance of synaptic contacts,30,31 whose synthesis31 as well as phosphorylation32 are affected by BDNF. CREB, a transcription factor involved in learning and memory, is an important modulator of gene expression induced by BDNF.33 CaMKII is a signaling system required for learning and memory and is involved in the effects of exercise on learning and memory performance.17
The modulation of oxidative stress after TBI also seems to be related to the action of Sir2 (silent information regulator 2).34 Sir2 is a NAD+-dependent deacetylase involved in the metabolism of cellular energy and the control of gene expression. Sir2 may play important roles in stress resistance by mediating the elevation of antioxidant systems such as manganese superoxide dismutase (SOD).35 The current study aims to evaluate the action of vitamin E dietary supplementation after mild TBI. We examine the possibility that vitamin E may counteract behavioral dysfunction after TBI by involving elements associated with the control of cellular energy and synaptic plasticity.
Materials and Methods
Experimental Design and Tissue Preparation
Male Sprague-Dawley rats (Charles River Laboratories, Inc, Wilmington, MA) weighing between 200 and 240 g were housed in cages (2 rats per cage) and maintained in environmentally controlled rooms (22°-24°C) with a 12-hour light/dark cycle. The regular diet consisted of the standard chow (#5001, PMI Nutrition, USA) containing 42 IU/kg of vitamin E. The content of vitamin E in the experimental diet was 500 IU/kg. After acclimatization for 1 week on standard rat chow, one set of rats was randomly assigned to a regular diet (RD) with or without vitamin E (500 IU/kg) for 4 weeks. This concentration of vitamin E has been shown to provide beneficial effects on brain function in aged animals.14 The diets, fed ad libitum, were provided in powder form (TestDiet Inc, Richmond, IN) in a large bowl and contained a standard vitamin and mineral mix with all essential nutrients. After 4 weeks of the diet, some of the rats were exposed to a mild fluid percussion injury (FPI). After 1 week of the same diet postinjury, the rats (n = 6-8 within each group) were killed by decapitation, and their brains were rapidly dissected, frozen on dry ice, and stored at −70°C until use for biochemical analyses. All experiments were performed in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of California at Los Angeles Chancellor's Animal Research Committee.
Fluid Percussion Injury
The injury was performed as previously described.3,4 In brief, with the aid of a microscope (Wild, Heerburg, Switzerland), a 3.0-mm diameter craniotomy was made 3.0 mm posterior to the bregma and 6.0 mm lateral (left) to the midline with a high-speed drill (Dremel, Racine, WI). A plastic injury cap was placed over the craniotomy with silicone adhesive and dental cement. When the dental cement hardened, the cap was filled with 0.9% saline solution. Anesthesia was discontinued, and the injury cap was attached to the fluid percussion device. At the first sign of hind-limb withdrawal to a paw pinch, a mild fluid percussion pulse (1.5 atm) was administered. Sham animals underwent an identical preparation but with the exception of the lesion. Immediately after responding to a paw pinch, anesthesia was restored, and the skull was sutured. Neomycin was applied on the suture, and the rats were placed in a heated recovery chamber for approximately an hour before being returned to their cages.
Measurement of Oxidized Proteins
The amounts of oxidized proteins containing carbonyl groups were measured by using an OxyBlot kit (Intergen, Purchase, NY). Briefly, the protein sample (10 μg) was reacted with 1× dinitrophenylhydrazine (DNPH) for 15 minutes, followed by neutralization with a solution containing glycerol and β-mercaptoethanol. These samples were electrophoresed on an 8% polyacrylamide gel and electrotransferred to a nitrocellulose membrane. After blocking, the membranes were incubated overnight with a rabbit anti-DNPH antibody (1:150) at 4°C, followed by incubation in goat antirabbit IgG (1:300) for 1 hour at room temperature. After rinsing with buffer, the immunocomplexes were visualized by chemiluminescence using the ECL (enhanced luminal–based chemiluminescent) kit (Amersham Pharmacia Biotech Inc, Piscataway, NJ) according to the manufacturer's instructions.
Cognitive Testing
The cognitive testing was performed in a Morris water maze (MWM), as described previously.1,2,36 Briefly, the rats were trained in the water maze with 10 consecutive trials per day for 3 days, starting at day 5 after TBI. The rats were placed into the tank facing the wall from one of the equally spaced start locations that were randomly changed at every trial. Each trial lasted until the rat found the platform or for a maximum of 2 minutes. If the rat failed to find the platform in the allocated time, it was gently placed on the platform. At the end of each trial, the animals remained on the platform for 1 minute to make associations. The escape latencies to find the platform were recorded.
Enzyme-Linked Immunosorbent Assay (ELISA)
The ipsilateral hippocampal tissues were homogenized in a lysis buffer containing 137 mM NaCl, 20 mM Tris–HCl (pH 8.0), 1% NP40, 10% glycerol, 1 mM phenylmethyl-sulfonylfluoride, 10 g/mL aprotinin, 0.1 mM benzethonium chloride, and 0.5 mM sodium vanadate. The homogenates were then centrifuged, the supernatants were collected, and the total protein concentration was determined according to the Micro BCA procedure (Pierce, Rockford, IL), using bovine serum albumin as standard. BDNF protein was quantified using an ELISA kit (BDNF Emax ImmunoAssay System kit, Promega Inc, Madison, WI) according to the manufacturer's protocol.
Western Blot
The total proteins from ipsilateral hippocampal tissues were extracted as described above. Briefly, protein samples were separated by electrophoresis on an 8% polyacrylamide gel and electrotransferred to a nitrocellulose membrane. Nonspecific binding sites were blocked in Tris-Buffered Saline (TBS) overnight at 4°C, with 2% bovine serum albumin (BSA) and 0.1% Tween-20. Membranes were rinsed for 10 minutes in buffer (0.1% Tween-20 in TBS) and then incubated with anti-actin, anti-synapsin I, anti-CaMKII, anti-SOD (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA), followed by antigoat IgG horseradish-peroxidase conjugate (Santa Cruz Biotechnology), and anti-CREB, anti-Sir2 (1:1000; Cell Signaling Technology, Beverly, MA), followed by antirabbit IgG horseradish-peroxidase conjugate (Santa Cruz Biotechnology). After rinsing with buffer, the immunocomplexes were visualized by chemiluminescence using the ECL kit (Amersham Pharmacia Biotech Inc, Piscataway, NJ) according to the manufacturer's instructions. The film signals were digitally scanned and then quantified using NIH Image software. Actin was used as an internal standard for Western blot. The RD-fed rats with sham surgery were regarded as experimental controls for comparisons with other experimental groups.
Statistical Analysis
For MWM results, the latencies to reach the hidden platform were analyzed by a repeated measurement analysis of variance (ANOVA) followed by a Scheffe post hoc test, as described previously.36 For Western blot, the values were expressed as a ratio of actin value and then converted to percentage of sham group (presented as bar diagrams) and represented the mean ± standard error of the mean. The data were analyzed by ANOVA followed by Fisher's protected least significance post hoc test. Statistical differences were considered significant at P < .05.
Results
Effects of Vitamin E on Cognitive Function in TBI Animals
Our previous study revealed an impairment of cognitive function in TBI animals.3,4 We supplemented vitamin E in the diet (500 IU/kg) to determine whether vitamin E can provide protection from cognitive impairment following TBI. The learning performance assessed in the MWM showed the following latency values: 15.1 ± 1.7 s at day 1, 10.9 ± 1.2 s at day 2, and 8.1 ± 1.4 s at day 3 in the RD sham group; 24.2 ± 1.0 s at day 1, 16.9 ± 1.4 s at day 2, and 13.4 ± 1.3 s at day 3 in RD TBI rats; 18.4 ± 1.0 s at day 1, 13.1 ± 1.5 s at day 2, and 10.5 ± 1.3 s at day 3 in TBI rats supplemented with vitamin E. The results demonstrated that TBI rats performed worse, with longer escape latency to locate the platform in the MWM than the sham rats (P < .05; Figure 1). Furthermore, TBI rats supplemented with vitamin E showed significant improvement in cognition with shorter latency to find the platform compared with untreated TBI animals (Figure 1). In addition, there was no significant difference in swimming speed in all groups, consistent with our previous observations.3
Figure 1.
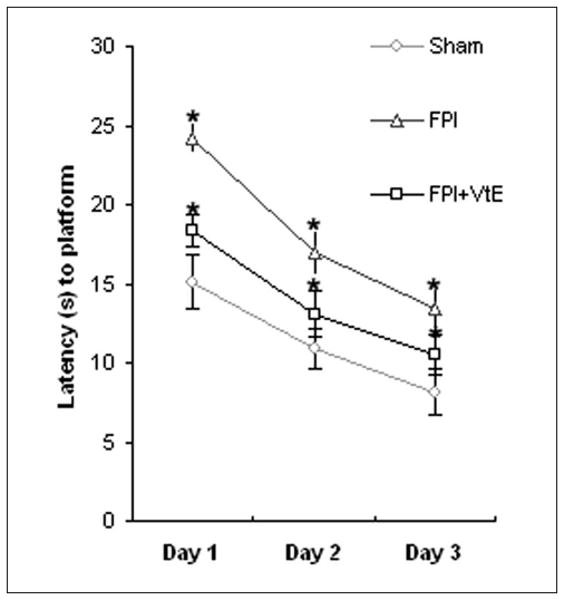
Vitamin E supplementation provides protection against cognitive disability in rats receiving FPI. Learning performance was scored as average of escape latencies to locate the platform in the Morris water maze. Supplementation with vitamin E resulted in significant improvement in cognition with shorter latency to find the platform in FPI rats compared with regular diet–treated FPI animals. *P < .05. VtE indicates vitamin E; FPI, fluid percussion injury.
Effects of Vitamin E on Oxidative Stress in TBI Animals
Oxidative damage was assessed by using Western blot analysis of DNPH-derivatized carbonyl groups on oxidized proteins, as shown in Figure 2A. The oxidized protein levels were significantly increased in TBI animals (139% of RD sham; P < .01; Figure 2B) relative to sham rats. However, TBI animals fed a diet containing vitamin E had a significantly lower level of oxidized proteins compared with untreated TBI and sham animals (44% vs 139% of RD sham; P < .01; Figure 2B).
Figure 2.
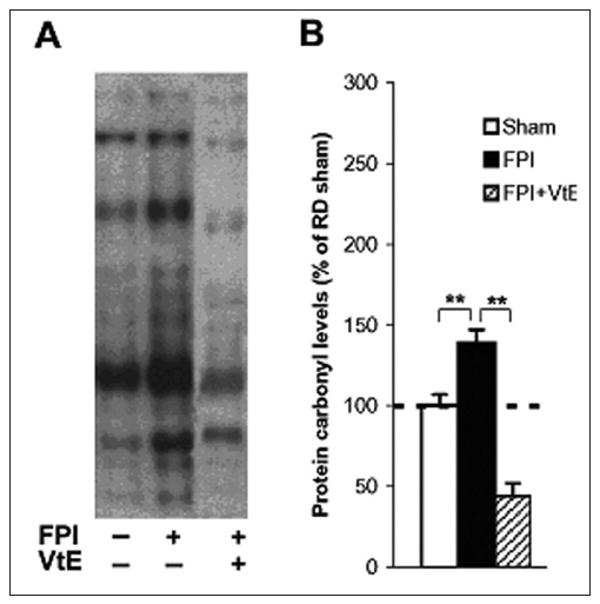
Measurements of oxidized protein levels in the hippocampus. (A) Representative example of OxyBlot bands from hippocampal tissue in each animal group. (B) FPI resulted in higher oxidized protein levels compared with sham animals, whereas supplementation of vitamin E in the diet dramatically reduced the FPI-induced elevation of protein carbonyl levels. The oxidized protein levels were determined using the OxyBlot kit. The values were converted to percentage of RD sham (mean standard error of the mean). **P < .01. VtE indicates vitamin E; FPI, fluid percussion injury; RD, regular diet.
Effects of Vitamin E on BDNF and CaMKII in TBI Animals
Accumulating evidence implicates BDNF in cognition and synaptic plasticity,19,21 and according to our previous study, BDNF levels are reduced in TBI animals.3,4 In the current study, dietary supplementation of vitamin E compensated for reduced BDNF levels in the hippocampus of TBI rats (Figure 3A).
Figure 3.
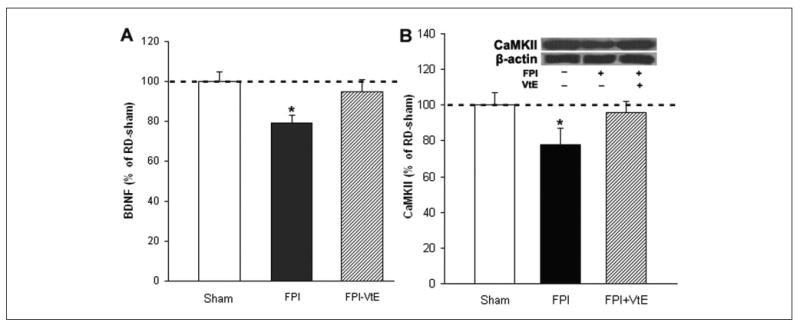
Vitamin E supplementation normalized levels of BDNF and CaMKII in the hippocampus of FPI rats. (A) FPI resulted in reduction of BDNF, whereas vitamin E supplementation reversed these effects. (B) FPI also resulted in reduction of CaMKII in RD rats, which was counteracted by vitamin E supplementation. BDNF protein levels were determined by ELISA. The values were converted to percentage of RD sham (mean standard error of the mean). *P < .05. VtE indicates vitamin E; FPI, fluid percussion injury; BDNF, brain-derived neurotrophic factor; ELISA, enzyme-linked immunosorbent assay; RD, regular diet.
We measured CaMKII levels based on its involvement in the effects of BDNF on hippocampal learning and memory.17 We found that TBI resulted in reduced CaMKII (78% of RD sham; P < .05; Figure 3B) in rats fed a RD. Vitamin E supplemented in the diet elevated the levels of CaMKII (96% of RD sham; Figure 3B) in TBI rats.
Effects of Vitamin E on BDNF's Downstream Effectors Synapsin I and CREB in TBI Animals
BDNF facilitates synaptic transmission and regulates gene expression through activation of synapsin I and CREB.31-33,37 We have previously shown that TBI may compromise cognitive abilities by affecting the action of BDNF on synaptic plasticity.3,4 To investigate whether vitamin E supplemented in the diet can protect against disrupted synaptic plasticity after TBI, we measured the protein expression of synapsin I in the hippocampus by Western blot analysis. The results showed that supplementation of vitamin E significantly increased levels of synapsin I in TBI rats as compared with that in untreated TBI animals (Figure 4A). Western blot analysis showed that vitamin E supplementation significantly increased levels of CREB in TBI rats as compared with the levels in untreated TBI animals (Figure 4B).
Figure 4.
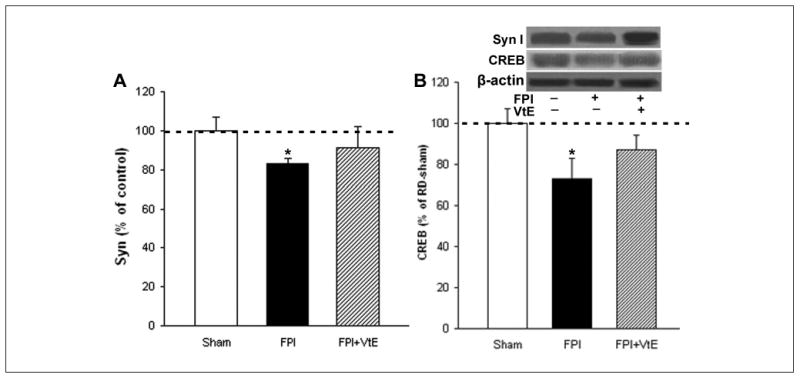
Effects of vitamin E supplementation on the levels of synapsin I and CREB in FPI rats. (A) Supplementation of vitamin E normalized levels of synapsin I in FPI rats compared with FPI rats fed a RD. (B) Supplementation of vitamin E also normalized levels of CREB in FPI rats. The levels of synapsin I were measured by Western blot analysis using actin as standard control. The values were converted to percentage of RD sham (mean standard error of the mean). *P < .05. VtE indicates vitamin E; FPI, fluid percussion injury; RD, regular diet.
Effects of Vitamin E on SOD and Sir2 in TBI Animals
We measured SOD levels based on its demonstrated ability to lower oxidative stress. We found that TBI reduced SOD (71% of RD sham; P < .05; Figure 5A), whereas vitamin E increased the levels of SOD (92% of RD sham; Figure 5A) in TBI rats, compared with untreated TBI rats. We also measured Sir2 levels because Sir2 has been shown to modulate SOD, thereby playing an important role in stress resistance and synaptic plasticity. We found that TBI resulted in reduced Sir2 (67% of RD sham; P < .05; Figure 5B) in rats fed a RD. Vitamin E elevated the levels of Sir2 (95% of RD sham; Figure 5B) in TBI rats as compared with the levels in untreated TBI rats. In addition, there was a significant correlation between Sir2 and SOD (r = .76; P < .01).
Figure 5.
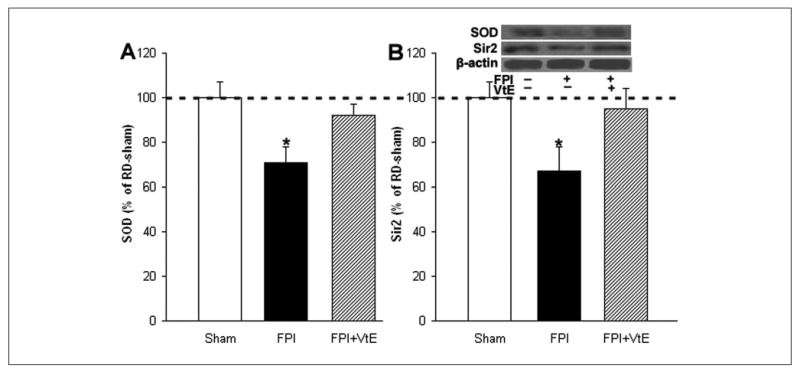
Effects of vitamin E supplementation on the levels of SOD and Sir2 in FPI rats. (A) Supplementation of vitamin E normalized levels of SOD in FPI rats compared with FPI rats fed a RD. (B) Supplementation of vitamin E also normalized levels of Sir2 in FPI rats. The levels of SOD and Sir2 were measured by Western blot analysis using actin as standard control. The values were converted to percentage of RD sham (mean standard error of the mean). *P < .05. VtE indicates vitamin E; FPI, fluid percussion injury; SOD, superoxide dismutase; Sir2, silent information regulator 2; RD, regular diet.
Discussion
TBI greatly elevates levels of oxidative stress, which seems to compromise BDNF production and function and reduce cognitive performance.3,4 Our current results show that dietary vitamin E supplementation significantly normalized levels of oxidative stress and counteracted cognitive decay in animals receiving TBI. In addition, the action of vitamin E was significant to stabilize levels of systems important for maintaining synaptic plasticity and cognition such as BDNF. In addition, the antioxidant capacity of vitamin E was corroborated by the normalization of SOD and Sir2, which are known for their antioxidant roles. These results suggest that oxidative stress buffering may promote resistance to the lowering of brain plasticity by TBI.
Vitamin E Reduces Oxidative Stress
We found that vitamin E supplemented in the diet 4 weeks before injury significantly reduced the oxidative damage seen in TBI animals. In the meantime, vitamin E increased molecules that may confer antioxidant capacity, such as SOD and Sir2, suggesting that this might mediate some of the effects evoked by vitamin E. Our results show that levels of SOD and Sir2 were reduced in conjunction with an increase in levels of protein oxidation after TBI. Sir2 has been shown to contribute to elevating antioxidant systems such as SOD, thereby playing an important role in stress resistance and synaptic plasticity.35 SOD is an important mitochondrial enzyme that acts as a scavenger of excessive reactive oxygen species in cells. In this study, we found that there is a significant correlation between Sir2 and SOD. Thus, the increase in Sir2 levels on vitamin E supplementation may be associated with elevated expression of SOD, which confers stress resistance after TBI. It is known that vitamin E has important neuroprotective actions against many types of insults.8-12 However, the roles played by SOD and Sir2 in the neuroprotection of vitamin E in TBI needs to be explored further. As discussed below, normalization of energy metabolism and oxidative stress can be important factors for maintaining synaptic function and cognition following injury.
Vitamin E–Reduced Oxidative Damage May Help Preserve Energy Homeostasis After TBI
An “energy crisis” after TBI may compromise synaptic plasticity and cognitive function. Sir2 can regulate transcription factors, including FOXO (Forkhead transcription factor) and PGC1α, that control energy homeostasis.35,38 However, Sir2 is reduced and associated with a decrease in a major cellular energy sensor AMPK (AMP-activated protein kinase) and its activated state (p-AMPK) in TBI animals.39 Furthermore, Sir2 downregulation is proportional to the increased oxidative stress and decreased SOD. In this study, we found that dietary supplementation of vitamin E can provide protection against reduction of Sir2 and SOD levels after TBI. Our previous study shows that omega-3 supplementation, which can provide antioxidant action, can reverse the decreased energy metabolic markers AMPK and uMtCK that are reduced by TBI.39 These findings implicate oxidative stress in the dysfunction of energy metabolism and subsequent synaptic and cognitive deficits after lesion. Vitamin E supplementation may preserve energy homeostasis after TBI by providing antioxidant action through upregulation of Sir2, SOD, and energy metabolic markers.
Vitamin E–Reduced Oxidative Damage May Mediate Improvement in Cognition and Plasticity in TBI
TBI results in excessive levels of oxidative stress and subsequent cognitive impairment, which may be related to a reduction in BDNF-mediated synaptic plasticity.3,4 The effects of vitamin E promoting a normalization of the cellular reactions that maintain cellular metabolism and oxidative stress may have contributed to the normalization of cognitive function in our study. It has been shown in animal models of aging that vitamin E supplementation can reduce cognitive deficits and symptoms of Alzheimer's disease.5,13-16
The mechanisms by which oxidative stress may affect cognitive function are not fully established, but recent evidence indicates that they may involve the action of BDNF. There is evidence indicating that excessive oxidative stress can cause a reduction in BDNF and subsequent decline in cognition and neuroplasticity.2 We have recently shown that the role of BDNF in maintaining synaptic plasticity is closely related to cellular energy metabolism, such that a disruption in energy homeostasis can affect synaptic plasticity and cognitive function.40,41 Given that oxidative stress is the by-product of aberrant energy metabolism, it may disrupt fundamental events that maintain BDNF-mediated synaptic plasticity and cognition. Recently, we have reported that other diets that have antioxidant capacity, such as those containing omega-3 fatty acids, can help improve neurological recovery after TBI involving BDNF.3 Accordingly, it is possible that BDNF modulation may be a common step in the action of antioxidants on synaptic plasticity, conferring resistance to cognitive dysfunction after TBI.
BDNF has been implicated in the regulation of synaptic plasticity19,21 and cognitive function.38 BDNF is synthesized predominantly by neurons located in the hippocampus, a brain region intimately associated with cognition.26,28,29,42,43 BDNF can facilitate synaptic function25,44,45 and regulate expression and/or activation of synapsin I31,32 and CREB.33,37 Our results demonstrated that supplementation of vitamin E in TBI rats increased the protein levels of BDNF, synapsin I, and CREB, which are involved in learning and memory events. The CaMKII is another signaling system critical for learning and memory.17 Our findings indicate that vitamin E modulates hippocampal CaMKII activation after TBI. CaMKII is activated by signaling at the TrkB receptors and can play an important role in BDNF-mediated cognitive enhancement.17 It is likely that the effects of vitamin E on BDNF-mediated synaptic plasticity play an important role in vitamin E counteracting the effects of TBI.
The Time Course of Vitamin E Treatment
Given the capacity of vitamin E to accumulate in lipid tissue components, there are concerns about what would be the minimal time of application before or after insults for a successful outcome. In our study, we maintained rats on a 500 IU/kg diet of vitamin E for 4 weeks before a mild TBI was inflicted. This treatment displayed significant beneficial effects on the cognition and plasticity after TBI. There is evidence indicating that dietary vitamin E treatment can significantly supplement existing vitamin E in the brain to promote neuroprotection. For example, it has been shown that vitamin E treatment for 4 weeks before repetitive concussive brain injury significantly increases brain vitamin E levels, decreases brain lipid peroxidation, and attenuates learning deficits in aging Tg2576 mice.6 The time course of this study was similar to ours. Another study shows that treatment with vitamin E at a dose of 500 IU/kg for 8 months retarded the onset of age-related cognitive behavior deficits.14 An even longer treatment with vitamin E for 12 months prevents spatial learning deficits in apo-E-deficient mice.5 Interestingly, a recent report shows that vitamin E treatment as early as 30 minutes before status epilepticus can reduce oxidative stress and partially inhibit chaperone-mediated autophagy in the brain at 24 hours after status epilepticus.46 Thus, the time period of weeks of treatment used in the current study seems within the range to expect a functional effect.
Conclusions
Our findings demonstrate that vitamin E supplementation can provide protection against cognitive deficits after TBI by involving BDNF-mediated synaptic plasticity, that is, synapsin I, CREB, and CaMKII. The action of vitamin E also influenced levels of SOD and Sir2, which play an important role in resistance to oxidative stress, suggesting that this free-radical scavenger can provide protection against oxidative damage after TBI. These results suggest that supplementation with vitamin E could help counteract some of the deleterious effects of TBI on neuroplasticity and cognition.
The powerful antioxidant capacity of vitamin E can be significant in providing protection against the cognitive deficits observed after TBI and may be other neurological disorders as well. The possibility that dietary habits may affect brain resistance to insults may shed light on the large variability observed in the results of clinical trials because individual participants can have great disparity in dietary habits.
Acknowledgments
Funding
This study was supported by NIH award NS 50465.
Footnotes
Declaration of Conflicting Interests
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
References
- 1.Molteni R, Wu A, Vaynman S, Ying Z, Barnard RJ, Gomez-Pinilla F. Exercise reverses the harmful effects of consumption of a high-fat diet on synaptic and behavioral plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience. 2004;123:429–440. doi: 10.1016/j.neuroscience.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 2.Wu A, Ying Z, Gomez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur J Neurosci. 2004;19:1699–1707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- 3.Wu A, Ying Z, Gomez-Pinilla F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J Neurotrauma. 2004;21:1457–1467. doi: 10.1089/neu.2004.21.1457. [DOI] [PubMed] [Google Scholar]
- 4.Wu A, Ying Z, Gomez-Pinilla F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp Neurol. 2006;197:309–317. doi: 10.1016/j.expneurol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Veinbergs I, Mallory M, Sagara Y, Masliah E. Vitamin E supplementation prevents spatial learning deficits and dendritic alterations in aged apolipoprotein E-deficient mice. Eur J Neurosci. 2000;12:4541–4546. [PubMed] [Google Scholar]
- 6.Conte V, Uryu K, Fujimoto S, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–764. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- 7.Behl C, Davis J, Cole GM, Schubert D. Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem Biophys Res Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 8.Erol FS, Topsakal C, Ozveren MF, et al. Protective effects of melatonin and vitamin E in brain damage due to gamma radiation: an experimental study. Neurosurg Rev. 2004;27:65–69. doi: 10.1007/s10143-003-0291-8. [DOI] [PubMed] [Google Scholar]
- 9.Ferri P, Cecchini T, Ciaroni S, et al. Vitamin E affects cell death in adult rat dentate gyrus. J Neurocytol. 2003;32:1155–1164. doi: 10.1023/B:NEUR.0000021909.84327.e8. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Polo RA, Soler G, Alvarez A, Fabregat I, Fuentes JM. Vitamin E blocks early events induced by 1-methyl-4-phenylpyridinium (MPP+) in cerebellar granule cells. J Neurochem. 2003;84:305–315. doi: 10.1046/j.1471-4159.2003.01520.x. [DOI] [PubMed] [Google Scholar]
- 11.Hara H, Kato H, Kogure K. Protective effect of alpha-tocopherol on ischemic neuronal damage in the gerbil hippocampus. Brain Res. 1990;510:335–338. doi: 10.1016/0006-8993(90)91386-u. [DOI] [PubMed] [Google Scholar]
- 12.Mishima K, Tanaka T, Pu F, et al. Vitamin E isoforms alpha-tocotrienol and gamma-tocopherol prevent cerebral infarction in mice. Neurosci Lett. 2003;337:56–60. doi: 10.1016/s0304-3940(02)01293-4. [DOI] [PubMed] [Google Scholar]
- 13.Fukui K, Omoi NO, Hayasaka T, et al. Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann N Y Acad Sci. 2002;959:275–284. doi: 10.1111/j.1749-6632.2002.tb02099.x. [DOI] [PubMed] [Google Scholar]
- 14.Joseph JA, Shukitt-Hale B, Denisova NA, et al. Long-term dietary strawberry, spinach, or vitamin E supplementation retards the onset of age-related neuronal signal-transduction and cognitive behavioral deficits. J Neurosci. 1998;18:8047–8055. doi: 10.1523/JNEUROSCI.18-19-08047.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray CA, Lynch MA. Dietary supplementation with vitamin E reverses the age-related deficit in long term potentiation in dentate gyrus. J Biol Chem. 1998;273:12161–12168. doi: 10.1074/jbc.273.20.12161. [DOI] [PubMed] [Google Scholar]
- 16.Sung S, Yao Y, Uryu K, et al. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer's disease. FASEB J. 2004;18:323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- 17.Vaynman S, Ying Z, Gomez-Pinilla F. The select action of hippocampal calcium calmodulin protein kinase II in mediating exercise-enhanced cognitive function. Neuroscience. 2007;144:825–833. doi: 10.1016/j.neuroscience.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Pinilla F, So V, Kesslak JP. Spatial learning induces neurotrophin receptor and synapsin I in the hippocampus. Brain Res. 2001;904:13–19. doi: 10.1016/s0006-8993(01)02394-0. [DOI] [PubMed] [Google Scholar]
- 19.Bolton MM, Lo DC, Sherwood NT. Long-term regulation of excitatory and inhibitory synaptic transmission in hippocampal cultures by brain-derived neurotrophic factor. Prog Brain Res. 2000;128:203–218. doi: 10.1016/s0079-6123(00)28018-7. [DOI] [PubMed] [Google Scholar]
- 20.Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- 21.Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 22.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherwood NT, Lo DC. Long-term enhancement of central synaptic transmission by chronic brain-derived neurotrophic factor treatment. J Neurosci. 1999;19:7025–7036. doi: 10.1523/JNEUROSCI.19-16-07025.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Natl Acad Sci U S A. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 26.Drapeau E, Mayo W, Aurousseau C, Le Moal M, Piazza PV, Abrous DN. Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2003;100:14385–14390. doi: 10.1073/pnas.2334169100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly A, Laroche S, Davis S. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J Neurosci. 2003;23:5354–5360. doi: 10.1523/JNEUROSCI.23-12-05354.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steffenach HA, Sloviter RS, Moser EI, Moser MB. Impaired retention of spatial memory after transection of longitudinally oriented axons of hippocampal CA3 pyramidal cells. Proc Natl Acad Sci U S A. 2002;99:3194–3198. doi: 10.1073/pnas.042700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugaya K, Chouinard M, Greene R, et al. Molecular indices of neuronal and glial plasticity in the hippocampal formation in a rodent model of age-induced spatial learning impairment. J Neurosci. 1996;16:3427–3443. doi: 10.1523/JNEUROSCI.16-10-03427.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brock TO, O'Callaghan JP. Quantitative changes in the synaptic vesicle proteins synapsin I and p38 and the astrocyte-specific protein glial fibrillary acidic protein are associated with chemical-induced injury to the rat central nervous system. J Neurosci. 1987;7:931–942. doi: 10.1523/JNEUROSCI.07-04-00931.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang T, Xie K, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jovanovic JN, Benfenati F, Siow YL, et al. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci U S A. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- 34.Wu A, Ying Z, Gomez-Pinilla F. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. J Neurotrauma. 2007;24:1587–1595. doi: 10.1089/neu.2007.0313. [DOI] [PubMed] [Google Scholar]
- 35.Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molteni R, Barnard RJ, Ying Z, Roberts CK, Gomez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–814. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- 37.Ying SW, Futter M, Rosenblum K, et al. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 39.Wu A, Ying Z, Gomez-Pinilla F. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. J Neurotrauma. 2007;24:1587–1595. doi: 10.1089/neu.2007.0313. [DOI] [PubMed] [Google Scholar]
- 40.Vaynman S, Ying Z, Wu A, Gomez-Pinilla F. Coupling energy metabolism with a mechanism to support brain-derived neurotrophic factor-mediated synaptic plasticity. Neuroscience. 2006;139:1221–1234. doi: 10.1016/j.neuroscience.2006.01.062. [DOI] [PubMed] [Google Scholar]
- 41.Gómez-Pinilla F. Brain foods: the effects of nutrients on brain function. Nat Rev Neurosci. 2008;9:568–578. doi: 10.1038/nrn2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mu JS, Li W, Yao ZB, Zhou XF. Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res. 1999;835:259–265. doi: 10.1016/s0006-8993(99)01592-9. [DOI] [PubMed] [Google Scholar]
- 43.Wilson IA, Ikonen S, Gureviciene I, et al. Cognitive aging and the hippocampus: how old rats represent new environments. J Neurosci. 2004;24:3870–3878. doi: 10.1523/JNEUROSCI.5205-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linnarsson S, Bjorklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 46.Cao L, Chen R, Xu J, Lin Y, Wang R, Chi Z. Vitamin E inhibits activated chaperone-mediated autophagy in rats with status epilepticus. Neuroscience. 2009;161:73–77. doi: 10.1016/j.neuroscience.2009.02.059. [DOI] [PubMed] [Google Scholar]