

Abstract
To fully describe the fold space and ultimately the biological function of membrane proteins, it is necessary to determine the specific interactions of the protein with the membrane. This property of membrane proteins that we refer to as structural topology cannot be resolved using X-ray crystallography or solution NMR alone. In this article, we incorporate into XPLOR-NIH a hybrid objective function for membrane protein structure determination that utilizes solution and solid-state NMR restraints, simultaneously defining structure, topology, and depth of insertion. Distance and angular restraints obtained from solution NMR of membrane proteins solubilized in detergent micelles are combined with backbone orientational restraints (chemical shift anisotropy and dipolar couplings) derived from solid-state NMR in aligned lipid bilayers. In addition, a supplementary knowledge-based potential, Ez (insertion depth potential), is used to ensure the correct positioning of secondary structural elements with respect to a virtual membrane. The hybrid objective function is minimized using a simulated annealing protocol implemented into XPLOR-NIH software for general use.
Keywords: Hybrid method, Membrane protein, Molecular modeling, Structural topology, PISEMA, Solid-state NMR
Introduction
Nearly all high-resolution membrane protein structures deposited in the protein data bank (PDB) have been determined by X-ray crystallography, solution NMR, and cryoEM. While these techniques offer unparalleled atomic resolution that guides the interpretation of membrane protein biological function, the coordinates generated by the refinement procedures do not include a topological dimension. There are now several examples in the literature, including mechanosensitive channels (Vasquez et al. 2008; Wang et al. 2008), nucleobase-cation-symport-1 transporters (Weyand et al. 2008), small multidrug resistant proteins (Bay et al. 2008), and potassium ion channels, that show protein function to be dictated by changes in the relative orientation of secondary structural elements with respect to the membrane (structural topology) rather than changes in the protein’s secondary structure. Furthermore, other classes of membrane proteins do not even adopt a compact tertiary structure; rather their fold space is determined by the intrinsic interactions with the lipid membrane. Since membrane protein topology plays a fundamental role in protein function, the failure to describe such interactions results in incomplete structural characterization (von Heijne 2006).
To mimic membrane protein environments, solution NMR spectroscopists utilize several different hydrophobic environments, including organic solvents, short chain lipid micelles, detergent micelles, or more recently, isotropic bicelles (Opella and Marassi 2004). The deleterious effects of organic solvents on membrane protein function are well-documented and researchers have now almost completely abandoned this avenue. On the other hand, detergent micelles present a viable alternative. There have been several examples of micellar reconstitutions that preserve membrane protein functional integrity, giving rise to high-quality NMR spectra for structural and interaction studies (Traaseth et al. 2008a, b; Zamoon et al. 2005). However, the intrinsic curvature of micelles, which may impact membrane protein structure and topology, is a significant concern (Chou et al. 2002). Small membrane proteins (30–200 residues), which account for most of the membrane proteins in several genomes, are especially vulnerable to changes induced by the membrane mimicking environment. While isotropic bicelles constitute an excellent membrane mimic and an alternative to micelles, the large size of the bicelle/protein complex results in substantially broader spectra than in detergent micelles (Poget and Girvin 2007; Poget et al. 2007; Sanders et al. 1994).
Solid-state NMR of membrane proteins in oriented lipid bilayers is one of the most accurate methodologies to directly investigate protein topology. Improvements in sample preparation and NMR methodology have provided higher resolution and sensitivity leading to topological and dynamic studies of multispan membrane proteins (De Angelis et al. 2006; Durr et al. 2007; Hallock et al. 2002; Opella and Marassi 2004; Park et al. 2006). However, these experiments rely on backbone measurements of chemical shift anisotropy (CSA) and dipolar coupling (DC) using separated local field (SLF) experiments [reviewed in (Ramamoorthy et al. 2004)] and heteronuclear correlation (HETCOR) spectroscopy (Ramamoorthy et al. 1999). The lack of side chain restraints has prevented complete structural characterization of membrane proteins using this technique.
A number of recent reports show that structure, topology, and oligomerization of membrane proteins are preserved both in lipid bilayers and in detergent micelles (Franzin et al. 2007; Mackenzie 2006; Moore et al. 2008; Stouffer et al. 2008). In some instances X-ray structures of membrane proteins are almost superimposable to NMR structures determined in micelles (Zhou et al. 2008). Therefore, a logical solution to the structure determination problem is to combine information from different experimental approaches into one unique structural refinement protocol that provides structure and topology simultaneously. Based on these considerations, we propose a method that combines the high-resolution information obtained from solution NMR on membrane protein samples reconstituted in detergent micelles with the structural data obtained by solid-state NMR on proteins reconstituted into lipid bilayers. In this work, we illustrate how to implement both distance and orientational restraints into a single target function. A depth of insertion potential from DeGrado and co-workers is also employed to embed helical segments into a virtual bilayer (Senes et al. 2007). The energy landscape is explored using the simulated annealing protocol implemented in XPLOR-NIH software (Schwieters et al. 2003) to determine the high-resolution structure and topology of membrane proteins simultaneously. We show the application of this method to monomeric phospholamban (PLN), a single pass membrane protein involved in cardiac muscle physiology (Traaseth et al. 2008a, b). To obtain monomeric PLN, we mutated the three cysteines in the transmembrane domain (C36A, C41F, and C46A). These mutations maintain the functional integrity of the protein (Zamoon et al. 2003). The solution NMR data was taken from the dodecylphosphocholine (DPC) studies from Zamoon et al. (2003), while the solid-state NMR restraints were derived from the work in mechanically aligned lipid bilayers by Traaseth et al. (2006, 2009) and Mascioni et al. (2002).
Methods and results
Energy terms in structural refinement
The hybrid solution and solid-state NMR target function (Etotal) is formulated as a linear combination of geometrical (Echem), solution NMR (Esol-NMR), and solid-state NMR (EssNMR) terms:
| (1) |
The geometrical and solution NMR potentials were available in XPLOR-NIH force field version 2.18 (Schwieters et al. 2003). Echem is the sum of bonding (Ebonds, Eangles, Eimproper) and non-bonding (Evdw) interactions, with adjustable weighting factors (w),
| (2) |
Esol-NMR is the sum of restraints from solution NMR experiments such as NOEs (ENOE), hydrogen bonds (EHBON), torsion angles (ECDIH), and an empirical torsion angle potential from a database (EDB; Kuszewski et al. 1996, 1997):
| (3) |
To include both CSA and DC data obtained from SLF solid-state NMR experiments such as PISEMA (polarization inversion spin exchange at the magic angle) (Wu et al. 1994), we used the penalty function EssNMR:
| (4) |
where wr is the relative weighting between CSA and DC and wPISEMA is the weighting of PISEMA potential both for CSA and DC.
To implement both CSA (ECSA) and DC (EDC) potentials, we used flat-well penalty functions as reported by Bertram et al. (2000):
| (5) |
| (6) |
σo,i (σError,i) and υo,i (υError,i) are the experimental data (error) for CSA and DC and σc,i, υc,i are calculated values associated with residue i. In our implementation, this potential energy function allows for structural refinement using different experimental errors and CSA tensor values for individual residues. Similar energy functions for CSA and DC have been used to refine the backbone structures of membrane proteins (Im and Brooks 2004; Ketchem et al. 1997, 1996; Lee et al. 2008; Nevzorov and Opella 2003).
Calculation protocol
Step 1: implementation of solution NMR restraints
In step 1, we used solution NMR data with geometrical restraints to define the secondary structural elements of monomeric PLN (Fig. 1).
Fig. 1.

Overview of the hybrid refinement protocol for the simultaneous determination of structure and topology of membrane proteins. Step 1: Starting from an extended structure, the simulated annealing protocol minimizes a target function containing only solution NMR data (NOEs, torsion angles, and hydrogen bonds). Step 2: The orientational constraints derived from solid-state NMR are included together with solution NMR restraints to obtain the correct orientation along the Z direction. Step 3: Depth of insertion is determined using rigid body minimization in the presence of the depth of insertion potential, keeping the helical orientation with respect to Z fixed. The resulting structural ensemble is refined using low temperature simulated annealing
The starting structure of monomeric PLN (the AFA-PLN mutant was used; C36A, C41F, C46A) was in an extended configuration, according to the simulated annealing protocol defined by Nilges et al. (1988).
Simulated annealing was carried out using the Echem and Esol-NMR potentials from Eq. 1. The system was cooled from 6,000 to 0 K in 5 K increments using 200 steps of torsion angle dynamics at each temperature. Both temperature and number of steps were optimized to achieve the lowest number of violations in the conformers generated. Note: the dihedral angle restraints for these calculations were generated from N, HN, Hα, C′, Cα, Cβ PLN chemical shifts using TALOS version 98.040.21.02 (Cornilescu et al. 1999).
200 steps of molecular dynamics were performed in torsion angle space followed by minimization in both torsion angle and Cartesian spaces.
At the conclusion of step 1, we generated 200 structures of AFA-PLN, where the 100 lowest energy structures were used for conformational analysis.
Similar to the conclusions by Zamoon et al. (2003), step 1 generates an ensemble of conformers with good convergence for the secondary structure elements, but that lacks precision in the structural overlay (see companion paper in Traaseth et al. 2009).
Step 2: implementation of solid-state NMR restraints
In step 2, we refined the structures obtained in step 1 with the solid-state NMR data (DC and CSA) using tensor 1 defined in Table 1.
Table 1.
Different tensor values (tensor 1 (Wu et al. 1995) and 2 (Page et al. 2008)) and the resulting topological angles
| Name | Tensor values |
Topological angles |
||
|---|---|---|---|---|
| (σ11, σ22, σ33) (ppm) | νNH (kHz) | θIb,II (°) | ρIb,II (°) | |
| Tensor 1 | (64, 77, 217) | 9.75 | 24 ± 2.6 | 203 ± 3.7 |
| Tensor 2 | (57.3, 81.2, 227.8) | 10.735 | 26 ± 2.9 | 204 ± 3.2 |
| Tensor 3 | (64, 77, 217) | 10.735 | 24 ± 2.6 | 202 ± 3.9 |
Note that tensor 3 is a combination of tensors 1 and 2
Starting from the ensemble of 100 lowest energy structures, simulated annealing calculations were carried out using all potential energy terms in Eq. 1 [Echem, Esol-NMR, EssNMR]. The initial annealing temperature was 3,000 K. Here, only torsion angle dynamics were applied to optimize the orientation of each individual peptide plane of AFA-PLN with respect to the Z-axis (corresponding to the lipid bilayer normal).
-
Cross-validation of CSA and DC restraints. To avoid overfitting the experimental data, we first optimized the solid-state NMR weighting factors (wPISEMA and wr) in Eq. (4) based on the cross-validation factor R described by Cross and co-workers (Kim et al. 2001).
(7) N is the number of data points used in the refinement protocol, while calculatedi, observedi and experimental errori are the calculated value, experimental value, and experimental error in CSA and DC for residue i. We fixed the weighting factors for Echem and Esol-NMR, and randomly partitioned the experimental data into two datasets: working (80% of data) and free (20% of data). The working dataset is included in the simulations, while the free dataset is not used and is back-calculated from the model. The work R (Rwork) indicates the fitting quality of the data included in the refinement, while the free R (Rfree) indicates the agreement between experimental and calculated data for residues with no CSA or DC restraints in the refinement protocol. In Fig. S1, we varied two factors: (1) wPISEMA from 0.1 to 1,000 for a total of ten independent calculations and (2) wr, the relative weight ratio of the DC/CSA potential, from 1/1 to 9/1. The Rwork and Rfree for CSA and DC were analyzed based on statistics for the 20 lowest energy structures from each of the calculations, as shown in Fig. S1. As wPISEMA becomes too large (>10), the quality of the structures decreases (Ebonds, Eangles, Eimproper, and Evdw become high) and the improvement in the R values is minimal. Similarly, when wr is >3, the geometrical and solution NMR penalties increase dramatically without improving the PISEMA R factor. When wr = 1, there is a large DC Rfree factor, indicating poor correlation with the experiments. Based on Fig. S1, the optimal weighting factor were found to be 5 for wPISEMA and 3 for wr, which is in agreement with the 10 (wPISEMA) determined by Cross and co-workers (Kim et al. 2001).
Using optimized weighting factor, a total of 400 structures were generated with the 100 lowest energy conformers selected for further analysis.
To evaluate the effects of the solid-state NMR restraints on refinement of the solution NMR structural ensemble, we defined three angles shown in Fig. 2: θ (tilt angle with respect to the Z axis or membrane normal), ρ (azimuthal rotation angle around helix axis), and χ [interhelical angle between domain Ia (residues 1–16) and the helix comprised of domains Ib and II (residues 23–52)]. The distributions of θ and ρ for the two helical domains in the 100 lowest energy conformers are shown in Fig. 3a. Noticeably, the orientation of the helix described by domains Ib and II is well-defined, with θIb,II = 23 ± 3° and ρIb,II = 205 ± 3°. The tilt angle of domain Ia is somewhat less defined with θIa = 95 ± 7°. While the solution NMR ensemble (Fig. 3b) fails to define the χ angle, the incorporation of CSA and DC restraints drastically reduces the conformational space allowed, confining χ between 70° and 125°. In contrast, the rotation angle (ρIa) for domain Ia is not well-defined using PISEMA data alone. This originates from several different factors: (a) helices with tilt angles ~90° have inherently clustered CSA (65–82 ppm) and DC (4–5 kHz) values, (b) helices with θ ~ 90° result in degenerate PISEMA spectra for ρ = 0° or 180°, and (c) the dynamic nature of domain Ia results in broad resonances (Metcalfe et al. 2005; Traaseth et al. 2006). While these spectroscopic difficulties were partially overcomed using several selectively labeled samples (see Traaseth et al. 2009), in order to determine the depth of insertion of the protein and fully resolve the orientation of the ρIa angle, we needed to incorporate a knowledge-based Ez potential (step 3).
Fig. 2.
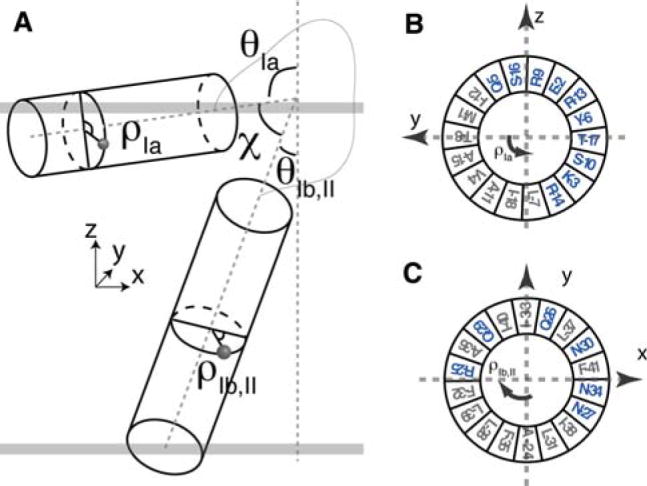
a Definition of (θ, ρ) describing the orientation of helix with respect to membrane normal Z. (θIb, II, ρIb,II) are the tilt and rotation angles for the domain Ib,II helix, while (θIa, ρIa) are the tilt and rotation angles for the domain Ia helix. The interhelical angle between the two domains is described by χ. b Helical wheel representation of the reference orientation of domain Ia where ρIa is defined to be zero. The N atom of T8 is aligned to +y axis. ρIa rotates counterclockwise viewing from the top of y–z plane. At ~90 degrees, the hydrophilic residues in blue point into bulk solution. c Reference orientation of transmembrane domain where ρIb,II is defined to be zero
Fig. 3.

a Distribution of θ and ρ angles derived from domain Ia and domains Ib and II. b Distribution of interhelical angle χ and ρIa and the comparison with the solution NMR ensemble
Step 3: incorporation of the knowledge-based Ez depth of insertion potential
The knowledge-based Ez potential by Degrado and coworkers (Senes et al. 2007) was used to embed the helical domains of AFA-PLN into a virtual bilayer. The Ez pseudo-energy was derived from the propensity of each amino acid in helical membrane proteins to insert into a lipid bilayer. This knowledge-based potential was derived from the statistical analysis of 24 crystal structures of helical membrane proteins. In our calculation, the helical domains were independently optimized under the knowledge-based Ez potential using rigid-body minimization, where the only degree of freedom is the translation of an entire domain along the Z-axis. Note that the loop connecting domain Ia with domain Ib was allowed to move and adjust the depth of insertion for all regions of AFA-PLN. The incorporation of the Ez term into the total energy function for simulated annealing calculations resulted in erroneous insertion of the protein in the virtual membrane irrespective of the weighting factor used (data not shown). This results from the step-function shape of the knowledge-based Ez potential which acts on each residue only along the Z axis. While this potential applied to single residues is not sufficient to embed the protein in the virtual membrane, the sum of the Ez potentials for all of the residues in a helix results in a unique minimum. Below we summarize the steps used to incorporate this knowledge-based potential into our calculations.
Definition of virtual bilayer. A virtual bilayer was built with the membrane normal parallel to the Z-axis and the origin in the center of the hydrocarbon core. The Z coordinate of the COM (center of mass) for each helical domain (using Cβ atoms since Ez potential depends only on Cβ coordinates) describes its insertion into the virtual bilayer.
Starting with the 100 lowest energy structures obtained after step 2, we minimized the domain insertion along the Z coordinate by rigid-body minimization using the Ez potential (i.e., the helical regions of domain Ia (residues 3–15) and domains Ib and II (residues 23–47) were held rigid). The use of the Ez potential helped to resolve the rotation angle ρIa ambiguity. Only structures with ρIa within the range of 60–150° gave reasonable minimized structures (Fig. S2). These angles are consistent with results from paramagnetic quenching experiments and NOEs connecting the hydrophobic face of domain Ia to the detergent micelle (Traaseth et al. 2008a, b; Zamoon et al. 2003). The introduction of the Ez potential confined the trans-membrane helix into a well-defined minimum, while the depth of insertion of the cytoplasmic domain was less defined with respect to the virtual membrane (Fig. 4). Regardless, the translational degeneracy was removed, resulting in one population of conformers with the hydrophilic residues pointing toward the bulk water, in agreement with the amphipathic nature of the cytoplasmic helix. Note that domain Ib in the N-terminal portion of the transmembrane domain protrudes outside the virtual membrane. This is in agreement with the hydrophilic nature of this domain and explains the hydrogen/deuterium exchange data (Zamoon et al. 2003). The flexible loop is also exposed to the solvent, which is in agreement with the dynamics probed by NMR spin relaxation measurements (Metcalfe et al. 2004).
In order to relax local geometries and improve the structure quality, we subjected the remaining structures to low temperature (300 K) simulated annealing using torsion angle and Cartesian molecular dynamics with full van der Waals interactions and all of the other restraints, but excluding the Ez potential. Although the knowledge-based Ez potential was not used during this annealing step, we verified that the structures did not deviate from the depth-of-insertion minima shown in Fig. 5b. From this ensemble, we selected 20 low energy conformers of PLN with no restraint violations, and deposited the structures into the protein data bank (PDB 2KB7, see Traaseth et al. 2009). The biological relevance of structures is discussed in our companion paper (Traaseth et al. 2009). Structural statistics are provided in Table S1.
Fig. 4.

Rigid-body minimization using the knowledge-based Ez potential. a Backbone cartoon representation of a selected PLN conformer before (upper panel) and after rigid-body minimization. b Comparison of the simulated CSA and DC before and after Ez minimization. The only discrepancies are due to residues located in the dynamic loop, which do not have CSA and DC restraints. c Representation of the Ez potential energy function for the domain Ib, II (blue) and domain Ia (red) helices. After minimization, both domains reside in the minima
Fig. 5.

a Conformational ensemble (structures and topologies) representing the 20 lowest energy structures in the virtual bilayer. Domain Ia is colored in red, and domains Ib and II are in blue. Hydrophobic side chains of domain Ia are shown in grey. Structure overlay is performed by rotating along Z and translating along X and Y, resulting in no changes in Ez energy and PISEMA data. b Position of the cytoplasmic and transmembrane domains with respect to the depth of insertion Ez potential. c Top view of a. d Distribution of θ and ρ angles in the final structural ensemble
Effects of different tensor components
One debate in the field surrounds the 15N CSA and 1H-15N DC tensor components used to model solid-state NMR data obtained from PISEMA-type experiments. To address this issue, we carried out a systematic conformational search for AFA-PLN domain Ib and II utilizing different tensor components shown in Table 1. We performed Steps 1–2 with low temperature annealing as discussed in the “Methods and results” section for each of the three tensors given in Table 1. Following energy minimization, the 20 lowest energy structures are selected with good agreement between the experimental and calculated DC and CSA using the three different tensors (Fig. 6). While small changes in local structure may occur, differences in the tensor values only marginally affect the overall protein topology. As shown in Table 1, changing the tensor components (tensors 2 and 3) from those used for the majority of this work (tensor 1), caused the tilt (θ) and azimuthal (ρ) angles to vary by 2.3° and 1.1°, respectively. Given the approximations necessary to calculate these angles for the transmembrane domain of PLN, these variations are negligible. In addition to the overall orientation, we also examined effects of different tensors on the local structures. Due to the nature of structural calculation, the bond angle, bond length and peptide plane planarity are all within acceptable errors of peptide geometry. We thus compared the Ramachandran angle (φ, ψ) for the transmembrane domain shown in Fig. S3. Although structures remained helical from all sets of simulations, there were considerable variations in these angles. These are likely due to the inclusion of different CSA and DC tensors.
Fig. 6.

a Comparison of the experimental and calculated CSA and DC for different values of the CSA tensor components shown in Table 1. The diagonal errors indicate ranges of ±5 ppm and ±0.5 kHz for CSA and DC, respectively. b Effects of the different CSA tensor components on the tilt and rotation angles for the helix defined by domains Ib and II of PLN. Tensors 1 (Wu et al. 1995), 2 (Page et al. 2008), and 3 are shown in red, black, and blue, respectively
Note that for glycine residues, a different set of tensor components needs to be used (Straus et al. 2003).
Sensitivity to PISEMA misassignments
Solid-state NMR methods to obtain orientational information from assigned PISEMA resonances are well-established. However, the assignment procedures for real membrane protein samples are still in their infancy. Although PISA wheels are a common way to assign resonances in well-dispersed spectra, most experimental PI-SEMA spectra contain inhomogenous broadening of resonances (due to mosaic spread and protein dynamics) making the assignments challenging. In spite of several computational attempts, these effects have not been fully rationalized (Quine et al. 2006; Straus et al. 2003). Therefore, selective labeling techniques constitute an important resource for resonance assignment. Unfortunately, if residues are on the same face of a helix, degenerate frequencies will be expected, further hampering the assignment procedure. To assess the sensitivity of our method to this problem, we have carried out two different calculations with two equiprobable assignments for the [15N-Ile] AFA-PLN PISEMA spectrum shown in Fig. 7b (see Traaseth et al. 2009). The assignment was carried out using a combinatorial search routine as described by Buffy et al. (2006). Using our hybrid method, we found that the two resonance assignments result in two conformational ensembles with similar tilt angles (24 ± 3° and 22 ± 2°), but different azimuthal rotation angles (203 ± 4° and 189 ± 3°). The conformational ensemble with the incorrect assignment (black assignment in Fig. 7) displayed covalent geometry violations at Ile45 for more than half of the structures, while also giving consistently higher conformational energy. This demonstrates that our hybrid method is sensitive to misassignment, and that it can be used to assist the assignment process.
Fig. 7.

Effects of the resonance misassignment on the calculation of the structural topology of PLN. a Plots of the distribution of the rotation angles for the two ensembles obtained from simulated annealing calculations. The correct assignments give rise to an average rotation angle of 203°, while the incorrect assignment gives rise to an average rotation angle of 189°. b Experimental PISEMA spectra with the two equiprobable assignments for the isoleucine residues of PLN derived from the combinatorial assignment procedures (see text). c Histogram representing the number of violations obtained for residues 44 and 45 in the conformational ensemble generated with the incorrect assignments
Static helix approach
PISEMA data has been used to calculate the tilt and rotation angles of helices based on a static ideal helix (Marassi and Opella 2002). In Fig. 8, we show the agreement between the tilt and rotational angles with the PISEMA data using a static approach. The potential energy landscape represented in Fig. 8 was obtained by calculating the least square fit between the experimental PISEMA data and an ideal poly-alanine α-helix. The lowest energy conformers generated with the hybrid method are located at the bottom of the minima defined by tilt and rotation angles for domains Ia and the helix comprised of domains Ib and II.
Fig. 8.

Projection of (θ, ρ) of the hybrid ensemble (20 lowest) for the two helical domains of PLN onto the PISEMA potential surfaces obtained using rigid helix fitting. Three different scoring functions are used to generate the potential surfaces: and These plots demonstrated that the topologies derived from the hybrid method correspond to the lowest energy minima identified in all of the PISEMA potential surfaces
This indicates that (1) the lowest energy conformers generated with the hybrid method are in remarkable agreement with the experimental data and (2) there is negligible discrepancy between the static fitting of the PISEMA data and a more refined conformational search procedure such as simulated annealing. A corollary to these considerations is the ideal nature of transmembrane helices embedded in lipid bilayers, demonstrated in several studies by Cross and co-workers using numerous membrane proteins (Page et al. 2008). Nevertheless, compared to the fitting with a rigid, ideal helix, the hybrid approach has the advantage of obtaining a high-resolution structure for both the backbone and side chains.
Discussion
The correlation between structure and function of complex biological systems requires the combination of several different techniques (biophysical and biochemical) to validate any mechanistic conclusions. The combination of methodologies, hybrid methods, is becoming quite common in structural biology (Cowieson et al. 2008). For instance, Clore and co-workers have used X-ray crystallographic data synergistically with solution NMR restraints to refine the structure of soluble proteins (Shaanan et al. 1992). More recently, solution NMR and small angle X-ray scattering (SAXS) data have been combined to refine the structures of large complexes and to resolve the intrinsic ambiguities of residual dipolar couplings (Gabel et al. 2006, 2008). Others have combined long-range distances from EPR with shorter range solution NMR distances for structure determination (Tamm et al. 2007). Finally, sparse solid-state NMR data have been used to complement X-ray structures to orient β-barrels within lipid bilayers (Mahalakshmi and Marassi 2008).
In this work, we combined structural restraints from solid-state and solution NMR into a global target function, which includes a depth of insertion potential (Senes et al. 2007). We applied this method to monomeric PLN, a small membrane protein that adopts an L-shaped conformation in lipids and whose tertiary fold is dictated by its strong interactions with the lipid membrane (see Traaseth et al. 2009). Solid-state and solution NMR have been previously used to study several membrane protein systems, demonstrating remarkable similarities of membrane protein structures in detergent micelles and lipid bilayers (Franzin et al. 2007; Gong et al. 2007; Hu et al. 2007; Opella and Marassi 2004; Page et al. 2007). Consistent with these findings, our calculations show that the structural restraints for both solution and solid-state NMR are compatible and can be used synergistically to define the structure and topology of PLN in lipid membranes. In addition, the depth of insertion potential gives a clear picture of the membrane location with respect to the protein that can be used as a starting point to embed it into an explicit lipid bilayer for further refinement. In fact, this final refinement step of the structural ensemble has already been adopted for soluble proteins (Calhoun et al. 2008; Kordel et al. 1997; Linge et al. 2003; Xia et al. 2002), and will be especially important for membrane proteins to (1) better define the fold space and (2) image protein–lipid interactions at the atomic level. Table S1 reports the statistics on the conformers generated with the hybrid method and a comparison with the conformational ensemble derived from the solution NMR data alone. It is clear that the introduction of the solid-state NMR data does not significantly affect the covalent geometry. The changes in the ideal geometry are within the allowed errors and all of the conformers have passed the conformational filters of the PDB deposition site. Also, the resolution of each single helical domain does not change. However, the overall resolution of the backbone changes dramatically. While in the conformational ensemble generated from the solution NMR data alone the rmsd for all of the backbone atoms was ~4.4 Å, the introduction of the solid-state NMR data drastically reduced the rmsd to ~2.3 Å. It is apparent that the interactions of PLN with the lipid membrane exemplified by the solid-state NMR data and the knowledge-base Ez potential limit the conformational space defining the structural topology of PLN. Nonetheless, the resolution achieved underscores the dynamic nature of PLN, a property that allows this small membrane protein to interact with several different binding partners.
Several other restraints obtained from biophysical and biochemical techniques can be easily incorporated in this protocol along the lines of the HADDOCK approach (Dominguez et al. 2003). In particular, it is important to measure depth of insertion experimentally using either paramagnetic quenching (Buffy et al. 2003) or protein/lipid REDOR measurements (Hong 2006; Mani et al. 2006; Toke et al. 2004) to complete the structural and topological characterization of membrane proteins within lipid bilayers. Although this approach is demonstrated for a small membrane protein, we are currently applying this method to the PLN pentamer (30 kDa), which will demonstrate the applicability to other multispan membrane proteins and complexes.
Conclusions
We developed a new computational protocol that is based on a total objective energy function (Etotal) that incorporates solution NMR data (distances, torsion angles, and hydrogen bonds), solid-state NMR data (orientational information: CSA and DC) and a depth of insertion knowledge-based potential (Ez) to determine the high-resolution structure and topology of membrane proteins. This hybrid energy function has been implemented into an XPLOR-NIH protocol and made available for general use. While this method is demonstrated for a small single pass membrane protein, the rapid progress made in sample preparation and spectroscopy of medium size membrane proteins will allow application to larger systems. The XPLOR-NIH scripts for the hybrid method are available for download at the authors’ website (www.chem.umn.edu/groups/veglia).
Acknowledgments
We would like to thank J. Hoch and B. Roux for helpful discussions, R. Bertram for sharing the DC and CSA python modules, C. Schwieters for help with the XPLOR-NIH code, and P. Gor’kov and others at the National High Magnetic Field Laboratory. This work was supported by grants to G.V. from the NIH (GM64742, HL80081, GM072701). This work was carried out in part using hardware and software provided by the University of Minnesota Supercomputing Institute.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10858-009-9328-9) contains supplementary material, which is available to authorized users.
Contributor Information
Lei Shi, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
Nathaniel J. Traaseth, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, 6-155 Jackson Hall, 321 Church St SE, Minneapolis, MN 55455, USA
Raffaello Verardi, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, 6-155 Jackson Hall, 321 Church St SE, Minneapolis, MN 55455, USA.
Alessandro Cembran, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
Jiali Gao, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA.
Gianluigi Veglia, Email: vegli001@umn.edu, Department of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA. Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, 6-155 Jackson Hall, 321 Church St SE, Minneapolis, MN 55455, USA.
References
- Bay DC, Rommens KL, Turner RJ. Small multidrug resistance proteins: a multidrug transporter family that continues to grow. Biochim Biophys Acta. 2008;1778:1814–1838. doi: 10.1016/j.bbamem.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Bertram R, Quine JR, Chapman MS, Cross TA. Atomic refinement using orientational restraints from solid-state NMR. J Magn Reson. 2000;147:9–16. doi: 10.1006/jmre.2000.2193. [DOI] [PubMed] [Google Scholar]
- Buffy JJ, Hong T, Yamaguchi S, Waring AJ, Lehrer RI, Hong M. Solid-state NMR investigation of the depth of insertion of protegrin-1 in lipid bilayers using paramagnetic Mn2+ Biophys J. 2003;85:2363–2373. doi: 10.1016/s0006-3495(03)74660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffy JJ, Traaseth NJ, Mascioni A, Gor’kov PL, Chekmenev EY, Brey WW, Veglia G. Two-dimensional solid-state NMR reveals two topologies of sarcolipin in oriented lipid bilayers. Biochemistry. 2006;45:10939–10946. doi: 10.1021/bi060728d. [DOI] [PubMed] [Google Scholar]
- Calhoun JR, Liu W, Spiegel K, Dal Peraro M, Klein ML, Valentine KG, Wand AJ, DeGrado WF. Solution NMR structure of a designed metalloprotein and complementary molecular dynamics refinement. Structure. 2008;16:210–215. doi: 10.1016/j.str.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou JJ, Kaufman JD, Stahl SJ, Wingfield PT, Bax A. Micelle-induced curvature in a water-insoluble HIV-1 env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J Am Chem Soc. 2002;124:2450–2451. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Cowieson NP, Kobe B, Martin JL. United we stand: combining structural methods. Curr Opin Struct Biol. 2008;18:617–622. doi: 10.1016/j.sbi.2008.07.004. [DOI] [PubMed] [Google Scholar]
- De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. Structure determination of a membrane protein with two transmembrane helices in aligned phospholipid bicelles by solid-state NMR spectroscopy. J Am Chem Soc. 2006;128:12256–12267. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Durr UH, Yamamoto K, Im SC, Waskell L, Ramamoorthy A. Solid-state NMR reveals structural and dynamical properties of a membrane-anchored electron-carrier protein, cytochrome b5. J Am Chem Soc. 2007;129:6670–6671. doi: 10.1021/ja069028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzin CM, Teriete P, Marassi FM. Structural similarity of a membrane protein in micelles and membranes. J Am Chem Soc. 2007;129:8078–8079. doi: 10.1021/ja0728371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel F, Simon B, Sattler M. A target function for quaternary structural refinement from small angle scattering and NMR orientational restraints. Eur Biophys J. 2006;35:313–327. doi: 10.1007/s00249-005-0037-3. [DOI] [PubMed] [Google Scholar]
- Gabel F, Simon B, Nilges M, Petoukhov M, Svergun D, Sattler M. A structure refinement protocol combining NMR residual dipolar couplings and small angle scattering restraints. J Biomol NMR. 2008;41:199–208. doi: 10.1007/s10858-008-9258-y. [DOI] [PubMed] [Google Scholar]
- Gong XM, Franzin CM, Thai K, Yu J, Marassi FM. Nuclear magnetic resonance structural studies of membrane proteins in micelles and bilayers. Methods Mol Biol. 2007;400:515–529. doi: 10.1007/978-1-59745-519-0_35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallock KJ, Henzler Wildman K, Lee DK, Ramamoorthy A. An innovative procedure using a sublimable solid to align lipid bilayers for solid-state NMR studies. Biophys J. 2002;82:2499–2503. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M. Oligomeric structure, dynamics, and orientation of membrane proteins from solid-state NMR. Structure. 2006;14:1731–1740. doi: 10.1016/j.str.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked transmembrane domain M2 proton channel from influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im W, Brooks CL., 3rd De novo folding of membrane proteins: an exploration of the structure and NMR properties of the fd coat protein. J Mol Biol. 2004;337:513–519. doi: 10.1016/j.jmb.2004.01.045. [DOI] [PubMed] [Google Scholar]
- Ketchem RR, Lee KC, Huo S, Cross TA. Macromolecular structural elucidation with solid-state NMR-derived orientational constraints. J Biomol NMR. 1996;8:1–14. doi: 10.1007/BF00198135. [DOI] [PubMed] [Google Scholar]
- Ketchem R, Roux B, Cross T. High-resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure. 1997;5:1655–1669. doi: 10.1016/s0969-2126(97)00312-2. [DOI] [PubMed] [Google Scholar]
- Kim S, Quine JR, Cross TA. Complete cross-validation and R-factor calculation of a solid-state NMR derived structure. J Am Chem Soc. 2001;123:7292–7298. doi: 10.1021/ja003380x. [DOI] [PubMed] [Google Scholar]
- Kordel J, Pearlman DA, Chazin WJ. Protein solution structure calculations in solution: solvated molecular dynamics refinement of calbindin D9 k. J Biomol NMR. 1997;10:231–243. doi: 10.1023/a:1018383102870. [DOI] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J Magn Reson. 1997;125:171–177. doi: 10.1006/jmre.1997.1116. [DOI] [PubMed] [Google Scholar]
- Lee J, Chen J, Brooks CL, III, Im W. Application of solid-state NMR restraint potentials in membrane protein modeling. J Magn Reson. 2008;193:68–76. doi: 10.1016/j.jmr.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- Mackenzie KR. Folding and stability of alpha-helical integral membrane proteins. Chem Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- Mahalakshmi R, Marassi FM. Orientation of the Escherichia coli outer membrane protein OmpX in phospholipid bilayer membranes determined by solid-state NMR. Biochemistry. 2008;47(25):6531–6538. doi: 10.1021/bi800362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani R, Tang M, Wu X, Buffy JJ, Waring AJ, Sherman MA, Hong M. Membrane-bound dimer structure of a beta-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry. 2006;45:8341–8349. doi: 10.1021/bi060305b. [DOI] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. Using pisa pies to resolve ambiguities in angular constraints from PISEMA spectra of aligned proteins. J Biomol NMR. 2002;23:239–242. doi: 10.1023/a:1019887612018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascioni A, Karim C, Zamoon J, Thomas DD, Veglia G. Solid-state NMR and rigid body molecular dynamics to determine domain orientations of monomeric phospholamban. J Am Chem Soc. 2002;124:9392–9393. doi: 10.1021/ja026507m. [DOI] [PubMed] [Google Scholar]
- Metcalfe EE, Zamoon J, Thomas DD, Veglia G. (1)H/(15)N heteronuclear NMR spectroscopy shows four dynamic domains for phospholamban reconstituted in dodecylphosphocholine micelles. Biophys J. 2004;87:1205–1214. doi: 10.1529/biophysj.103.038844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe EE, Traaseth NJ, Veglia G. Serine 16 phosphorylation induces an order-to-disorder transition in monomeric phospholamban. Biochemistry. 2005;44:4386–4396. doi: 10.1021/bi047571e. [DOI] [PubMed] [Google Scholar]
- Moore DT, Berger BW, DeGrado WF. Protein–protein interactions in the membrane: sequence, structural, and biological motifs. Structure. 2008;16:991–1001. doi: 10.1016/j.str.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevzorov AA, Opella SJ. Structural fitting of PISEMA spectra of aligned proteins. J Magn Reson. 2003;160:33–39. doi: 10.1016/s1090-7807(02)00138-6. [DOI] [PubMed] [Google Scholar]
- Nilges M, Gronenborn AM, Brunger AT, Clore GM. Determination of three-dimensional structures of proteins by simulated annealing with interproton distance restraints. Application to crambin, potato carboxypeptidase inhibitor and barley serine proteinase inhibitor 2. Protein Eng. 1988;2:27–38. doi: 10.1093/protein/2.1.27. [DOI] [PubMed] [Google Scholar]
- Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RC, Li C, Hu J, Gao FP, Cross TA. Lipid bilayers: an essential environment for the understanding of membrane proteins. Magn Reson Chem. 2007;45:S2–S11. doi: 10.1002/mrc.2077. [DOI] [PubMed] [Google Scholar]
- Page RC, Kim S, Cross TA. Transmembrane helix uniformity examined by spectral mapping of torsion angles. Structure. 2008;16:787–797. doi: 10.1016/j.str.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Prytulla S, De Angelis AA, Brown JM, Kiefer H, Opella SJ. High-resolution NMR spectroscopy of a GPCR in aligned bicelles. J Am Chem Soc. 2006;128:7402–7403. doi: 10.1021/ja0606632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poget SF, Girvin ME. Solution NMR of membrane proteins in bilayer mimics: small is beautiful, but sometimes bigger is better. Biochim Biophys Acta. 2007;1768:3098–3106. doi: 10.1016/j.bbamem.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poget SF, Cahill SM, Girvin ME. Isotropic bicelles stabilize the functional form of a small multidrug-resistance pump for NMR structural studies. J Am Chem Soc. 2007;129:2432–2433. doi: 10.1021/ja0679836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quine JR, Achuthan S, Asbury T, Bertram R, Chapman MS, Hu J, Cross TA. Intensity and mosaic spread analysis from PISEMA tensors in solid-state NMR. J Magn Reson. 2006;179:190–198. doi: 10.1016/j.jmr.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy A, Wu CH, Opella SJ. Experimental aspects of multidimensional solid-state NMR correlation spectroscopy. J Magn Reson. 1999;140:131–140. doi: 10.1006/jmre.1999.1827. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy A, Wei Y, Dong-Kuk L. PISEMA solid-state NMR spectroscopy. Ann Rep NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- Sanders CR, Hare BJ, Howard KP, Prestegard JH. Magnetically-oriented phospholipid micelles as a tool for the study of membrane-associated molecules. Prog Nucl Magn Reson Spectrosc. 1994;26:421–444. [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Senes A, Chadi DC, Law PB, Walters RF, Nanda V, Degrado WF. E(z), a depth-dependent potential for assessing the energies of insertion of amino acid side-chains into membranes: derivation and applications to determining the orientation of transmembrane and interfacial helices. J Mol Biol. 2007;366:436–448. doi: 10.1016/j.jmb.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Shaanan B, Gronenborn AM, Cohen GH, Gilliland GL, Veerapandian B, Davies DR, Clore GM. Combining experimental information from crystal and solution studies: joint X-ray and NMR refinement. Science. 1992;257:961–964. doi: 10.1126/science.1502561. [DOI] [PubMed] [Google Scholar]
- Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus SK, Scott WR, Watts A. Assessing the effects of time and spatial averaging in 15N chemical shift/15N–1H dipolar correlation solid state NMR experiments. J Biomol NMR. 2003;26:283–295. doi: 10.1023/a:1024098123386. [DOI] [PubMed] [Google Scholar]
- Tamm LK, Lai AL, Li Y. Combined NMR and EPR spectroscopy to determine structures of viral fusion domains in membranes. Biochim Biophys Acta. 2007;1768:3052–3060. doi: 10.1016/j.bbamem.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toke O, O’Connor RD, Weldeghiorghis TK, Maloy WL, Glaser RW, Ulrich AS, Schaefer J. Structure of (KIAGKIA)3 aggregates in phospholipid bilayers by solid-state NMR. Biophys J. 2004;87:675–687. doi: 10.1529/biophysj.103.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Buffy JJ, Zamoon J, Veglia G. Structural dynamics and topology of phospholamban in oriented lipid bilayers using multidimensional solid-state NMR. Biochemistry. 2006;45:13827–13834. doi: 10.1021/bi0607610. [DOI] [PubMed] [Google Scholar]
- Traaseth NJ, Ha KN, Verardi R, Shi L, Buffy JJ, Masterson LR, Veglia G. Structural and dynamic basis of phospholamban and sarcolipin inhibition of ca(2+)-ATPase. Biochemistry. 2008a;47:3–13. doi: 10.1021/bi701668v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Verardi R, Veglia G. Asymmetric methyl group labeling as a probe of membrane protein homo-oligomers by NMR spectroscopy. J Am Chem Soc. 2008b;130:2400–2401. doi: 10.1021/ja711499r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc Natl Acad Sci USA. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez V, Sotomayor M, Cordero-Morales J, Schulten K, Perozo E. A structural mechanism for MscS gating in lipid bilayers. Science. 2008;321:1210–1214. doi: 10.1126/science.1159674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7:909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- Wang W, Black SS, Edwards MD, Miller S, Morrison EL, Bartlett W, Dong C, Naismith JH, Booth IR. The structure of an open form of an E. coli mechanosensitive channel at 3.45 A resolution. Science. 2008;321:1179–1183. doi: 10.1126/science.1159262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand S, Shimamura T, Yajima S, Suzuki S, Mirza O, Krusong K, Carpenter EP, Rutherford NG, Hadden JM, O’Reilly J, Ma P, Saidijam M, Patching SG, Hope RJ, Norbertczak HT, Roach PC, Iwata S, Henderson PJ, Cameron AD. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science. 2008;322:709–713. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Ramamoorthy A, Opella SJ. High-resolution heteronuclear dipolar solid-state NMR spectroscopy. J Magn Reson Ser A. 1994;109:270–272. [Google Scholar]
- Wu CH, Ramamoorthy A, Gierasch LM, Opella SJ. Simultaneous characterization of the amide 1H chemical shift, 1H–15N dipolar, and 15N chemical shift interaction tensors in a peptide bond by three-dimensional solid-state NMR spectroscopy. J Am Chem Soc. 1995;117:6148–6149. [Google Scholar]
- Xia B, Tsui V, Case DA, Dyson HJ, Wright PE. Comparison of protein solution structures refined by molecular dynamics simulation in vacuum, with a generalized born model, and with explicit water. J Biomol NMR. 2002;22:317–331. doi: 10.1023/a:1014929925008. [DOI] [PubMed] [Google Scholar]
- Zamoon J, Mascioni A, Thomas DD, Veglia G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys J. 2003;85:2589–2598. doi: 10.1016/s0006-3495(03)74681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamoon J, Nitu F, Karim C, Thomas DD, Veglia G. Mapping the interaction surface of a membrane protein: unveiling the conformational switch of phospholamban in calcium pump regulation. Proc Natl Acad Sci USA. 2005;102:4747–4752. doi: 10.1073/pnas.0406039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cierpicki T, Jimenez RH, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. NMR solution structure of the integral membrane enzyme DsbB: functional insights into DsbB-catalyzed disulfide bond formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]