

SUMMARY
Candida albicans is an opportunistic fungal pathogen causing life-threatening mucosal and systemic infections in immunocompromised humans. Using a murine model of mucosal Candida infection we investigated the role of the proinflammatory cytokine IL-1β in host-defense to Candida albicans. We find that the synthesis, processing and release of IL-1β in response to Candida are tightly controlled and first require transcriptional induction, followed by a second signal leading to caspase-1 mediated cleavage of the pro-IL1β cytokine. The known fungal pattern recognition receptorsTLR2 and Dectin-1 regulate IL-1β gene transcription, while the NLRP3 containing pro-inflammatory multiprotein complex, the NLRP3 inflammasome, controls caspase-1 mediated cleavage of pro-IL1β. Furthermore, we show that TLR2, Dectin-1 and NLRP3 are essential for defense against dissemination of mucosal infection and mortality in vivo. Therefore, in addition to sensing bacterial and viral pathogens, the NLRP3 inflammasome senses fungal pathogens and is critical in host defense against Candida.
Keywords: TLR, NLR, Innate Immunity, Pathogenesis, Mucosal, Fungal
INTRODUCTION
Opportunistic fungi cause life-threatening infections in immunocompromised hosts. In healthy individuals Candida ssp. have low pathogenicity; however, with depressed host immunity or when changes occur in oral microbial flora, overgrowth can occur, resulting in oropharyngeal candidiasis (OPC, also known as thrush) or denture stomatitis (Abu-Elteen and Abu-Alteen, 1998; Daniluk et al., 2006; Figueiral et al., 2007). Candida albicans is the leading cause of life-threatening fungal disease and ranks fourth among all bloodstream and nosocomial infections in the United States (Jarvis et al., 1995; Wisplinghoff et al., 2004).
Considerable progress has been made in recent years in our understanding of how pathogens are recognized by the innate immune system and how sensing translates into signaling and transcriptional regulation of immune response genes. Several classes of germline encoded pattern recognition receptors (PRRs) have now been implicated in innate defenses. These include the Toll-like Receptors (TLRs) (Takeda and Akira, 2005), the C-type lectin receptors (CLRs) (Huysamen and Brown, 2009), the RIG-like helicases (RLRs) (Yoneyama and Fujita, 2007), cytosolic DNA sensors (Burckstummer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Roberts et al., 2009; Takaoka et al., 2007) and members of the NOD-Like Receptor (NLR) family (Meylan et al., 2006). Recent evidence has implicated TLR2 and the CLR family member, Dectin-1 in fungal recognition, phagocytosis and the induction of inflammatory responses (Gantner et al., 2003; Herre et al., 2004; Kerrigan and Brown, 2009; Rogers et al., 2005).
IL-1β is a proinflammatory cytokine released by many cell types and is an important mediator of inflammation during infection and inflammation (Dinarello, 2002; Dinarello, 2005). The synthesis, processing and release of IL-1β are tightly controlled and require at least two distinct stimuli. An initial microbial stimulus through innate PRRs results in accumulation of intracellular stores of pro-IL-1β. A second stimulus activates a multiprotein complex containing one or more Nod-like receptors (NLRs), commonly referred to as the “inflammasome”, which controls the activation of caspase-1 and cleavage of pro-IL-1β, followed by release of the active mature 17-kDa IL-1β (Franchi et al., 2009; Meylan et al., 2006). The NLRs consist of a C-terminal LRR domain, a central nucleotide-binding oligomerization (NACHT) domain, and a variable N-terminal protein protein interaction domain which can be either a CARD, Pyrin or BIR domain (Franchi et al., 2009; Martinon and Tschopp, 2005). The best characterized inflammasome complex contains NLRP3, ASC (apoptosis-associated speck-like protein), CARDINAL and caspase-1 (Amer et al., 2006; Franchi et al., 2006; Miao et al., 2006; Miao et al., 2008; Sutterwala and Flavell, 2009; Sutterwala et al., 2007). The NLRP3 inflammasome is activated by many microbial stimuli including bacterial pore forming toxins, (Mariathasan et al., 2006; Martinon et al., 2004), by viruses such as Sendai virus (Kanneganti et al., 2006a), influenza (Allen et al., 2009; Ichinohe et al., 2009; Thomas et al., 2009) and adenovirus (Muruve et al., 2008) and by endogenous danger signals such as ATP and monosodium urate (Martinon et al., 2006; Martinon and Tschopp, 2004). Additional activators include indigestible particulates, like silica, asbestos, alum and β-amyloid (Dostert et al., 2008; Eisenbarth et al., 2008; Halle et al., 2008; Hornung et al., 2008). Although roles for IL-1α/β and also IL-18 have been shown in some animal models of fungal infection (Bellocchio et al., 2004; Mencacci et al., 2000; Netea et al., 2002; Steele and Fidel, 2002; Stuyt et al., 2004; Stuyt et al., 2002; Vonk et al., 2006), the role of NLRs and inflammasomes in host defense to fungal pathogens has not been addressed.
Here we investigate the role of IL-1β in host-defense to Candida albicans using a murine model of sustained mucosal infection. We show that TLR2 and dectin-1 control IL-1β gene transcription, while NLRP3, ASC and caspase-1 regulate processing of pro-IL-1β into the active mature 17kDa cytokine. Importantly, we also reveal a critical role for TLR2, as well as Dectin-1 and the NRLP3 inflammasome against disseminated infection and mortality in vivo. Our results therefore implicate the NLRP3 inflammasome and IL-1β production in the regulation of mucosal anti-fungal host defenses.
RESULTS
Candida albicans activates a caspase-1 dependent IL-1β response, which is dependent on the morphological stage of the organism
Candida ssp are dimorphic fungi that grow as yeast or hyphal forms depending on environmental conditions. We first examined IL-1β production by different morphological forms of C. albicans. Human PBMC were exposed to formalin fixed preparations of C. albicans grown to germ tube stage (exposed to hyphal permissive media for 2, 4 or 6 hours, respectively) or fully hyphal forms (>24 hours growth). As shown in Figure 1a, Candida induction of IL-1β from LPS primed PBMC was strongest in response to germ tube forms and comparable to ATP treated controls. Indeed, in cells that were incubated with Candida in the presence of the specific caspase-1 inhibitor Z-YVAD-fmk, a synthetic peptide that irreversibly and specifically inhibits caspase-1 activity, almost complete inhibition of IL-1β release was observed (Fig 1b). Similar data were obtained when THP-1 cells were examined (Supplemental Fig. 1). We also tested the presence of IL-1β in cellular supernatants by western blotting and found that only the cleaved IL-1β (i.e. the 17kD mature form) could be detected in the supernatants (Fig. 1c). Similar studies were performed using mouse bone marrow derived macrophages (BMDM) (Fig. 1d) which showed that C. albicans induced IL-1β release was also strongest for the germ tube forms. Cleaved IL-1β could be detected in the supernatants by western blotting (Fig. 1e).
Figure 1. In vitro IL-1β responses to Candida albicans are morphological stage dependent and blocked by caspase-1 inhibitor.

(a) IL-1β responses of normal PBMC primed for 3 h with LPS then stimulated with fixed preparations of C. albicans of different morphological stages at ~ 106/ml; (b) Inhibition of IL-1β responses after the addition of caspase-1 specific inhibitor, Z-YVAD-FMK; (c) Western blot of supernatants from PBMC stimulation probed with anti-IL-1β antibody; (d) IL-1β responses of bone-marrow derived macrophages from wild-type mice primed for 3 h with LPS then stimulated with C. albicans at ~ 106/ml; (e) Western blot of supernatants from BMDM stimulation probed with anti-IL-1β antibody, (f) and anti-caspase-1 antibody. (*** P < 0.001; ** P < 0.01; * P < 0.05)
The primary function of inflammasomes is to convert inactive procaspase-1 into the active, cleaved enzyme. Inflammasome complexes assemble upon activation by an appropriate stimulus leading to the multimerization of the adaptor molecule ASC. Subsequently, procaspase-1 is recruited to ASC via the interactions of the CARD domains of ASC and caspase-1. The induced proximity of caspase-1 leads to auto-cleavage of caspase-1. The two resulting subunits p10 and p20 assemble into the active caspase-1. The activation of caspase-1 was next examined by monitoring the cleavage status of caspase-1 in cell supernatants. To test whether Candida can directly activate the cleavage of caspase-1, we precipitated proteins from the supernatants of BMDM exposed to C. albicans. The caspase-1 p10 subunit could be detected in supernatants of C. albicans treated cells (Fig. 1f). Together, these data indicate that Candida can activate caspase-1 to release bioactive IL-1β, suggesting that a caspase-1 activating inflammasome complex is being activated. The mechanism by which C. albicans triggers these events is unknown.
IL-1β gene transcription is dependent on TLR2 and Dectin-1
The CLR, Dectin-1 is a receptor for fungal β-glucan and has been shown to cooperate with TLR2 for the transcriptional induction of inflammatory cytokine genes in response to C. albicans infection (Brown et al., 2003; Brown et al., 2002). First we defined the role of TLR2 and Dectin-1 in the upregulation and activation of IL-1β in response to C albicans. Peritoneal macrophages from wild-type, TLR2 or Dectin-1 deficient mice were stimulated with C. albicans. As shown in Figure 2, Candida induced IL-1β mRNA upregulation (Figure 2a) and protein release into the supernatants (Figure 2b) from wild-type macrophages. These responses were completely abrogated in Tlr2-/- as well as Dect1-/- deficient macrophages, demonstrating the importance of these receptors in fungal mediated IL-1β induction. Having established the role of Candida in the transcriptional induction of IL-1β, we next examined if Candida could lead to the processing of pro-IL-1β followed by its release into supernatants. Macrophages were first exposed to the fungal cell wall PAMPs, β-glucan and zymosan, or to LPS and then stimulated with C. albicans. Neither LPS, β-glucan nor zymosan alone were sufficient to drive IL-1β release as shown in Figure 2c. In contrast, when macrophages were additionally treated with C. albicans, IL-1β release was detected. Importantly, IL-1β release was detected in cells exposed to Candida alone and even higher levels of IL-1β were released in cells which had been exposed to β-glucan, zymosan or LPS. These data indicate that ligands which trigger TLR2 and Dectin-1 signaling cannot by themselves elicit IL-1β processing and release. The ability of Candida to trigger these events in both unprimed (media treated) and primed cells indicate that Candida can elicit IL-1β gene transcription (signal 1) and inflammasome activation (signal 2).
Figure 2. IL-1β responses to Candida albicans are dependent on TLR2 and dectin-1.

(a) Up-regulation of IL-1β mRNA by qPCR of macrophages from Tlr2-/-, Dect1-/- and wild-type mice stimulated for 8 h with C. albicans of different morphological stages at ~ 106/ml; (b) IL-1β protein levels in supernatants of macrophages from Tlr2-/-, Dect1-/- and wild-type mice stimulated overnight with C. albicans of different morphological stages at ~ 106/ml; (c) IL-1β responses of wild-type macrophages stimulated O/N with LPS at 500 ng/ml, β-glucan at 10 μg/ml or zymosan at 1 μg/ml alone, or primed for 6 h then removed and stimulated for an additional 8 h by C. albicans at ~ 106/ml. (*** P < 0.001; ** P < 0.01; * P < 0.05)
Candida albicans triggers IL-1β production via the NLRP3 inflammasome
To define the molecular mechanism regulating caspase-1 mediated processing of IL-1β, we monitored inflammasome activation by Candida in LPS primed macrophages. LPS-primed macrophages from wild-type mice or mice deficient in NLRP3 or ASC were stimulated with C. albicans and IL-1β release and cleavage examined by ELISA and western blotting, respectively. As shown in Figure 3a, IL-1β responses from macrophages deficient in NLRP3 and ASC were significantly reduced following treatment with all stages of C. albicans. LPS primed macrophages exposed to ATP induced high levels of IL-1β responses in wild-type macrophages as well as in macrophages lacking TLR2 or Dectin-1, but not in macrophages deficient in ASC, NLRP3 or Caspase-1 (Supplement Fig. 2a and b), as expected. In contrast, induction of TNF-α and KC in response to C. albicans, (which are dependent on TLR2/Dectin-1 signaling and not on the inflammasome) were comparable between wild-type and knockout cells (Supplemental Fig 2c and data not shown). We also examined IL-1β cleavage in cellular supernatants by Western blotting and found that the cleaved and active form of IL-1β could be detected in supernatants from wild-type but not Asc-/- cells (Figure 3b). We also confirmed that Candida induced IL-1β was dependent on Caspase-1 (Figure 3c). To determine if ATP was responsible for IL-1β production in response to Candida, macrophages from the ATP-gated ion channel receptor P2X7 deficient mice were stimulated with C. albicans. No difference in Candida induced IL-1β responses between the P2X7R deficient and wild-type cells were observed. As expected, a complete abrogation of LPS + ATP responses was seen (Figure 3d). Together these data show that C. albicans is able to induce IL-1β gene transcription in a TLR2 and Dectin-1 dependent manner, and that the processing of IL-1β is mediated by the NLRP3/ASC inflammasome.
Figure 3. IL-1β responses to Candida albicans are mediated by NLRP3, ASC and Caspase-1.

(a) IL-1β responses of macrophages from Asc-/-, Nlrp3-/- and wild-type mice primed for 4 h with 500 ng/ml LPS then stimulated with C. albicans at ~ 106/ml; (b) Western blot of supernatants from wild-type or Asc-/- macrophages probed with anti-IL-1β antibody; (c) IL-1β responses of macrophages from Casp1-/- and wild-type mice stimulated overnight with C. albicans at ~ 106/ml; (d) IL-1β responses of macrophages from P2X7R-/- and wild-type mice stimulated overnight with C. albicans at ~ 106/ml. (*** P < 0.001; ** P < 0.01; * P < 0.05)
IL-1β controls anti-fungal immunity in vivo
Having established the importance of TLR2, Dectin-1 and the NLRP3 inflammasome in controlling IL-1β production in macrophages, we assessed if this pathway was important in vivo. We first examined if C. albicans could induce IL-1β in vivo. To this effect, we established a murine model of mucosal infection to define the importance of IL-1β and IL-1 receptor signaling in vivo. To establish oral infection, mice were first treated with antibiotic containing drinking water to reduce the competing oral microflora. Next, a series of shallow scratches in the stratum corneum of the dorsal surface of the tongue were made in anesthetized mice using a sterile scalpel. Sterile PBS was applied for 3 h to allow the disruptions to partially heal then concentrated C. albicans, strain GDH2346 (NCYC 1467), originally obtained from a denture stomatitis patient at Glasgow Dental Hospital (McCourtie and Douglas, 1984) was applied to the oral cavity and allowed to remain in a saturated cotton ball for 4 to 6h with the animals under sedation, allowing for consistent and sustained colonization of the oral mucosa (Supplemental Fig. 3). There was no difference seen in the severity of infection, as determined by clinical scores, between mice infected with and without disruption of the stratum corneum, but we found that the additional disruption step led to more consistent infections with little variation in fungal colonization of the oral cavity between individual mice. Although colonization of the tongue was slightly lower in un-disrupted mice as measured by fungal culture, there was no difference in colonization of the gut (esophagus, stomach, duodenum, jejunum, ileum) or kidneys (a marker for systemic dissemination) as shown in Supplementary Fig. 5. Wild-type mice were infected and the epithelium harvested from the buccal surfaces for mRNA isolation and protein extraction. As shown in Figure 4, both IL-1β mRNA (Fig. 4a) and protein (Fig. 4b) levels were strongly induced in vivo in oral mucosa by C. albicans infection.
Figure 4. Protective role of IL-1β in a murine modal of oral infection with Candida albicans.

(a) Fold induction of IL-1β mRNA measured by quantitative real-time PCR by oral buccal epithelium after infection with C. albicans; (b) IL-1β (pg/ml) protein production in homogenized whole tongues of mice after infection with C. albicans; (c) Quantitative fungal burden of tongues, and (d) kidneys of Il-1r1-/- and wild-type (WT) mice after oral infection with C. albicans; (e) Fungal burden of tongues, and (f) kidneys of Casp1-/- and WT mice after oral infection with C. albicans; (g) Kaplan-Meier survival plots of WT, Il-1r1-/- and Casp1-/- mice after infection (P <0.0001); (h) Mean clinical severity score of Il-1r1-/-, Casp1-/- and wild-type after 3, 7, 14 or 21d of infection; (*** P < 0.001; ** P < 0.01; * P < 0.05)
To define the importance of IL-1β in mucosal fungal infection, wild-type or IL-1 receptor deficient mice (Il1r1-/-) were infected orally with C. albicans. After 3, 7, 14 and 21d of infection, mice were euthanized and clinically scored. Tongues and organs were removed asceptically and quantitative fungal burdens of tongues and kidneys determined. As shown in Figure 4c, fungal colonization of the oral cavity was significantly higher in the Il1r-/- mice at all time points. Neither wild-type nor Il1r-/- mice cleared the oral infection during the study period. In contrast to wild-type mice, the Il1r-/- mice showed dramatically higher levels of systemic dissemination, as measured by quantitative fungal burdens in the kidneys, at all time points measured (Fig. 4d). Survival was also impaired in the Il1r-/- mice with cumulative 63.7% survival after 21d compared to 97.0% in the wild-type mice (Fig. 4g). Clinical severity as assessed by gross scores at the experimental endpoint (Supplemental Fig. 3) was also significantly higher in Il1r-/- mice (Fig. 4h).
We also investigated the role of caspase-1 by infecting Casp1-/- or wild-type mice with C. albicans as above. As shown in Figure 4e, colonization of the oral cavity was similar between the Casp1-/- and wild-type mice at all time points, but slightly higher at 14d, and both strains failed to clear the oral infection during the study period. In contrast to oral colonization, the Casp1-/- mice showed greatly enhanced systemic dissemination, as measured by quantitative fungal burdens in the kidneys, with significance at 3d and 7d and a trend towards significance at 14 and 21d (Fig. 4f). Survival was also impaired in the Casp1-/- mice with cumulative 60.3% survival after 21d compared to 97.0% in the wild-type mice (Fig. 4g). Clinical severity as assessed by gross scores at the experimental endpoint was also significantly higher in Casp1-/- mice compared to wild-type mice at all time points (Fig. 4h).
The NLRP3 inflammasome controls anti-fungal immunity in vivo
Since ASC and NLRP3 control IL-1β production in response to C. albicans in macrophages, we next tested the importance of the NLRP3 inflammasome complex in anti-fungal defense in vivo. As shown in Figure 5a, mice deficient in ASC had higher oral colonization after infection with C. albicans, as measured by quantitative fungal burden of homogenized tongues. This correlated with higher gross clinical scores in the Asc-/- mice compared to wild-type (Fig 5d). Also, functional ASC is required for resistance to dissemination of infection as show in Figure 5b, with significantly higher fungal burdens in the kidneys compared with wild-type mice. Survival was also significantly impacted in the ASC deficient mice compared to wild-type (Fig. 5c). NLRP3 deficient mice infected with C. albicans showed similar oral colonization as compared to wild-type mice but enhanced dissemination, with significantly higher fungal burdens in the kidneys at 3 and 7d of infection. Clinical severity of infection was also worse in the NLRP3 deficient mice (Fig. 5g). We hypothesize that systemic dissemination of C. albicans occurs via translocation or invasion of the fungus through the mucosa of the small intestine which has been demonstrated in some models (Cole et al., 1988). To assess the impact of the NLRP3 inflammasome on intestinal colonization, organs were harvested and quantitatively cultured for fungal colonization. Both Asc-/- and Nlrp3-/- mice demonstrated higher fungal burdens of the stomach, duodenum, jejunum and ileum at 7d of infection compared to wild-type mice which appear to partially clear infection of the small bowel (Supplemental Fig. 4). Together, using a model of mucosal candidiasis, we show that activation of the NLRP3 inflammasome by C. albicans impacts colonization of the gut, dissemination of infection and survival in OPC.
Figure 5. NLRP3 inflammasome plays a critical role in host defense to mucosal infection with Candida albicans.

(a) Quantitative fungal burden of tongues, and (b) kidneys of Asc-/- and WT mice after oral infection with C. albicans; (c) Kaplan-Meier survival plots of WT and Asc-/- mice after infection (P = 0.0002); (d) Mean clinical severity score of Asc-/- and WT after 3, 7, 14 or 21d of infection; (e) Quantitative fungal burden of tongues, and (f) kidneys of Nlrp3-/- and WT mice after oral infection with C. albicans; (g) Mean clinical severity score of Nlrp3-/- and WT after 3 and 7d of infection. (*** P < 0.001; ** P < 0.01; * P < 0.05)
TLR2 and Dectin-1 control anti-fungal immunity in vivo
Since TLR2 and Dectin-1 regulated IL-1β production in response to C. albicans, we also determined the role of TLR2 and Dectin-1 in the murine model. As shown in Figure 6a, oral colonization was similar between wild-type and Dectin-1 deficient mice despite higher clinical scores (Fig. 6d). However, dissemination of infection occurred in the Dect1-/- mice (Fig. 6b) as well as significantly reduced survival (Fig. 6c). In TLR2 deficient mice, both enhanced oral colonization and dissemination of infection was also observed (Fig. 6e, f). TLR2 deficient mice also had significantly less survival and higher clinical scores than wild-type mice. Therefore, both TLR2 and Dectin-1 play an important role in host-defense to mucosal infection with C. albicans.
Figure 6. Innate PRRs enhance survival after oral infection with Candida albicans.

(a) Quantitative fungal burden of tongues, and (b) kidneys of Dect1-/- and wild-type (WT) mice after oral infection with C. albicans; (c) Kaplan-Meier survival plots of WT and Dect1-/- mice after infection (P = 0.0009); (d) Mean clinical severity score of Dect1-/- and WT after 3 or 7d of infection; (e) Quantitative fungal burden of tongues and (f) kidneys of Tlr2-/- and wild-type (WT) mice after oral infection with C. albicans; (g) Kaplan-Meier survival plots of WT and Tlr2-/- mice after infection (P = 0.0002); (h) Mean clinical severity score of Tlr2-/- and WT after 3, 7, 14 or 21d of infection. (*** P < 0.001; ** P < 0.01; * P < 0.05)
TLR2, Dectin-1 and the NLRP3 inflammasome control IL-1b production in vivo
Finally, circulating levels of IL-1β in serum from mice infected orally with C. albicans was quantified in all of these strains. Serum IL-1β responses to C. albicans infection was partially reduced in Tlr2-/- mice and completely abolished in Dect1-/- or double Dect1/Tlr2-/- mice, indicating a cooperative role for these receptors in regulating IL-1β production in vivo (Figure 7a, c). IL-1β production was undetectable in serum from Nlrp3-/- or Asc-/- mice (Figure 7b).
Figure 7. Serum IL-1β responses to in vivo infection with C. albicans is dependent on TLR2, Dectin-1 and the NLRP3 inflammasome.
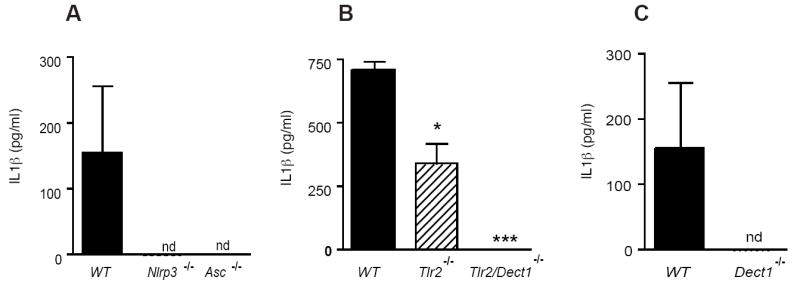
(a) IL-1β responses in serum collected from wild-type, Nlrp3-/- or Asc-/- mice orally infected with C. albicans; (b) IL-1β responses in serum collected from orally infected wild-type, Tlr2-/- or double Tlr2/dect1-/- mice; (c) IL-1β responses in serum collected from orally infected wild-type or Dect1-/- mice (*** P < 0.001; * P < 0.05)
DISCUSSION
Oral epithelial cells respond to fungal pathogens with the release of pro-inflammatory cytokines, including IL-1α/β, IL-8, TNF-α and antimicrobial peptides such as β-defensins (Dongari-Bagtzoglou and Kashleva, 2003; Dongari-Bagtzoglou et al., 2004; Lu et al., 2006; Mostefaoui et al., 2004). Although encoded by different genes, IL-1α and IL-1β share highly homologous structures and activate through the common receptor, IL-1 receptor 1 (IL-1R1) which functions as a heterodimer with the IL-1 receptor accessory protein (IL-1RAcP) and signals via the TIR domain containing adapter molecule MyD88 (Dinarello, 1998). Both IL-1α and IL-1β as well as IL-18 have been suggested to play an important role in host defense against C. albicans infections (Bellocchio et al., 2004; Mencacci et al., 2000; Stuyt et al., 2004; Stuyt et al., 2002; Vonk et al., 2006), although none has direct anti-fungal activity. IL-18, which is also processed by NLRs, has been shown to be upregulated in a caspase-1 dependent manner in response to Candida as well as mediating the development of protective Th1 immunity (Mencacci et al., 2000). It is likely that the NLRP3 inflammasome controls the processing of both IL-1β and IL-18 in response to fungal pathogens, as has been seen in the case of other activators of this inflammasome (Akita et al., 1997; Fantuzzi and Dinarello, 1999; Ghayur et al., 1997; Gu et al., 1997; Kuida et al., 1995).
Our studies reveal that C. albicans triggers pathways leading to the production of IL-1β. Further, we show that it acts both as an inducer of pro-IL-1β (signal 1) as determined by measuring IL-1β mRNA levels (Figure 2) and secretion of IL-1β in naive cells, a response that was dependent on both TLR2 and Dectin-1; and as an activator of the NLRP3 inflammasome to process pro-IL-1β (signal 2). The ability of fixed preparations to elicit this response suggests that fungal invasion and/or metabolic activity are not required. We utilized killed preparations for our in vitro studies in an effort to avoid IL-1β release due to Candida induced cell toxicity and death (which we did not observe in our in vitro studies). As shown previously, heat inactivation of Candida can unmask β-glucan structures which bind Dectin-1 (Gantner et al., 2005). To avoid this, we used formalin fixation to kill the organism without causing artificial exposure of β-glucans.
Our data show that both early germ tube and fully hyphal forms of Candida are capable of inducing IL-1β release in human PBMC and THP-1 cells. Release of cleaved functional IL-1β is confirmed by immunoblot analysis for the 17kDa active cytokine. Moreover, release of IL-1β, cleavage of IL-1β and cleavage of caspase-1 was also observed in both peritoneal and bone marrow derived macrophages from mice. The differential ability of the different forms of Candida to trigger signaling is consistent with published data on differential immune activation by different morphological stages of fungus (Bellocchio et al., 2004; Gantner et al., 2005). It is unclear why the studies recently published by another group did not reveal a role for the inflammasome in the production of IL-1β; however, there are substantial methodological differences between the two studies (van de Veerdonk et al., 2009). We show a profound suppression of IL-1β production in macrophages from NLRP3, ASC and Caspase-1 deficient mice. Most importantly, our studies using a mucosal model of Candida challenge show a significant phenotype in the inflammasome deficient mice providing in vivo support to our in vitro findings. Altogether, these data implicate first, that the inflammasome is activated and secondly that NLRP3, ASC and Caspase-1 mediate Candida induced IL-1β responses and anti-fungal defense both in vitro and in vivo. A recent publication using an intravenous challenge model and in vitro studies also supports a role for NLRP3 in IL-1β responses to Candida and is consistent with our findings (Gross et al., 2009).
Both TLR2 and dectin-1 are known to play an important role in fungal recognition and the induction of inflammatory responses. Dectin-1, which recognizes β-glucan in the fungal cell wall, has been shown to mediate phagocytosis of fungal pathogens in a partially Syk dependent manner, depending on the cell type studied (Herre et al., 2004; Kerrigan and Brown, 2009; Rogers et al., 2005). While TLR2 recognizes fungal PAMPs and mediates inflammatory responses, it is not required for phagocytosis; macrophages lacking TLR2, TLR4, or MyD88 internalize yeast particles normally (Gantner et al., 2003). We show that the fungal PAMPs β-glucan and zymosan, which bind Dectin-1 and TLR2 respectively, can prime cells for subsequent IL-1β release by C. albicans but by themselves are unable to elicit this response. C. albicans can therefore elicit pro-IL-1β induction and cleavage. This is achieved by its ability to trigger TLR2 and dectin-1 and activate the NLRP3 inflammasome to cleave pro-IL-1β into its biologically active form. IL-1β was induced by inactive forms of the fungi, indicating again that neither metabolic activity nor active invasion of cells was required. Early germ tube or hyphal forms were generally more stimulatory than the fully hyphal form in vitro. It is likely therefore that there is differential activation by C. albicans in the context of biofilms (i.e. fully hyphal forms) that colonize the outer layers of epithelium, as compared to yeast and early germ tube morphological forms more often associated with extravasating or early disseminated infection (Saville et al., 2008; Saville et al., 2003).
We developed a murine model of OPC where immunosuppression is not required and where animals develop the characteristic white patches and epithelial ulceration seen in humans with oral thrush. Our goal was to establish a model of sustained mucosal colonization that would closely mimic human oropharyngeal candidiasis. Histologically, the infections that we achieve are similar to human disease (Supplemental Figure 3). Our model is also unique as we observe low-level, chronic dissemination from a mucosal source in susceptible strains, which has not previously been modeled.
Using this model we have defined an essential role for IL-1β in host-defense to Candida infection. Increased fungal burdens were seen in the tongues of IL-1 receptor deficient mice. Moreover, dissemination or invasion of infection from the oral source was also dependent on IL-1β, with higher kidney fungal burdens (a marker for dissemination) and decreased survival in Il1r-/- mice. Candida can also be cultured from multiple organs in susceptible strains including the brain, lung, liver, bowel, stomach and spleen (data not shown). Using this physiologically relevant model of sustained oral infection, we also reveal the importance of TLR2, Dectin-1 as well as the NLRP3 inflammasome (caspase-1, ASC and NLRP3) in controlling IL-1β production, which determines resistance against dissemination and promotes survival of the infected host.
Key questions arising from our studies are how NLRP3 senses C. albicans and what component of the pathogen triggers the NLRP3 inflammasome. Numerous chemically and structurally diverse stimuli are now known to activate the NLRP3 inflammasome. These include pathogenic bacteria, viruses, as well as a broad range of microbial derivatives, bacterial pore forming toxins, small molecule immune activators, extracellular ATP, amyloid-β and various crystals (e.g. silica, asbestos) (Halle et al., 2008; Hornung et al., 2008; Kanneganti et al., 2006a; Kanneganti et al., 2006b; Martinon et al., 2004; Martinon et al., 2006). It remains unclear, however, how a single signaling molecule NLRP3 can detect such a broad range of stimuli. Because there is no evidence that all of these ligands bind directly to NLRP3, it has been suggested that activation is indirect. Our studies with P2X7R-deficient macrophages indicate that ATP release from Candida infected cells is not responsible for triggering NLRP3 activation. The generation of reactive oxygen species and cathepsin B release from damaged lysosomal compartments are additional proposed mechanisms for activation of the NLRP3 inflammasome. The contribution of cathepsin B has been found to be specific to NLRP3 activation by particulates such as silica and amyloid-β. Phagocytosis and lysosomal acidification are believed to be important for NLRP3 activation by these agents. Additional work is needed to elucidate the role of phagocytsosis, lysosomal disintegration and ROS generation in activation of the NLRP3 inflammasome by C. albicans.
EXPERIMENTAL PROCEDURES
Animals
Female C57BL/6 and Il1r-/- mice (ages 8-12 weeks) were purchased from Jackson Laboratories. Nlrp3-/- and Asc-/- mice were generated by Millenium Pharmaceuticals. Casp1-/- mice were generated by R. Flavell (Yale University). P2X7R-/- mice (C57BL/6 background) were originally provided by Pfizer Global Research and Development, Pfizer Inc. (Solle et al., 2001) and then backcrossed into a pure C57BL/6 background for >12 generations from a P2X7R-/- mouse strain. Animals were housed in filter-covered micro-isolator cages in ventilated racks. All animal studies have been approved by the Institutional Animal Care and Use Committee of Case Western Reserve University and University of Massachusetts.
Reagents
Cells were cultured in RPMI 1640 media (HyClone) containing 10% non-HI FCS (Atlanta Biologicals). β-glucan (isolated from Saccharomyces cerevisiae) and Z-YVAD-fmk were purchased from CalBiochem. Zymosan was obtained from Sigma. Macrophages were lysed using Radio-Immunoprecipitation Assay Buffer (Pierce) containing Halt Protease Inhibitor Cocktail (Thermo Scientific). Western Blots were probed using monoclonal 3ZD anti-IL-1β antibody (National Cancer Institute Biological Resources Branch) and polyclonal anti-capase-1 p10 antibody (Santa Cruz Biotechnology, Inc).
Fungal preparations
Candida albicans strain GDH2346, clinical strain originally isolated from a denture stomatitis patient (McCourtie and Douglas, 1984), was utilized for all in vivo and in vitro studies. Stocks were maintained on Sabouraud Dextrose (SD) agar at 4°C. Prior to challenge in the animal model, a suspension was made by transferring the fungus to SD broth (Difco) and incubating at 37°C with shaking for a period of 16 – 24 h. To obtain hyphal and intermediate germ tube forms, the Candida was grown in enriched media, consisting of RPMI + 10% Fetal Bovine Serum (FBS) for 2, 4 or 6 h (early, mid or late germ tube formations), or 24 h for fully hyphal forms (Supplemental Figure 6). For formaldehyde fixation, the preparations were washed in sterile PBS, fixed in 1% formaldehyde solution for 10 min and then washed 3 times in sterile PBS. After re-suspension in sterile PBS the preps were stored at -80°C until use.
Cell isolation and culture
Bone marrow derived macrophages were generated as described 32. Human PBMCs were isolated by from whole blood of healthy volunteers by density gradient centrifugation. Lysis of red blood cells was performed using red blood cell lysis buffer (Sigma). Experiments in PBMCs and macrophages were carried out at a cell density of 2×106 cells/ml. All primary cells and cell lines except THP-1 cells were cultured in DMEM supplemented with L-glutamine, ciprofloxacin (Cellgro) and 10% fetal calf serum (Hyclone). THP-1 cells were cultured in RPMI supplemented with 10% fetal calf serum (Hyclone), L-glutamine, sodium pyruvate (Cellgro), and ciprofloxacin. One day prior to stimulation, THP-1 cells were differentiated using 0.5 μM PMA for 3 h, washed three times and plated for stimulation. The caspase-1 inhibitor z-YVAD (10 mM) was added for 1 h prior to stimulation. Thioglycollate elicited peritoneal macrophages were isolated by adherence and stimulated at a density of 2×106 cells per well in a 6 well plate in RPMI 1640 (HyClone) with 10% FCS (Atlanta Biologicals). Cells were primed with 200 to 500 ng/mL low protein LPS for 3 to 4 h, followed by fixed Candida at ~106/ml for 8 h. Alternatively, naive cells were stimulated overnight with fixed Candida preparations. Supernatants were harvested and assayed for cytokine release. Cells were lysed in RIPA buffer and run on SDS-page gel and transferred to nitrocellulose, followed by probing for IL-1β and caspase-1. All experiments that were performed for Western blot analysis were carried out in serum free DMEM medium. ATP stimulations were carried out at 5 mM 1h prior to harvesting supernatants.
Murine model of OPC
Five days prior to challenge with C. albicans, the mice were placed on tetracycline (Fisher Scientific) containing drinking water (2.5 g/l). The mice were anesthetized i.p. with a cocktail of Ketamine (Vedco) and Acepromazine (Boehringer Ingelheim) and the dorsal surface of the tongue lightly scratched (limited to the superficial stratum corneum of the epithelial layer) using a sterile #10 blade scalpel to roughen the surface. A sterile, uniform-sized cotton packing was placed in the oral cavity and saturated with PBS to keep the mouth moist. After 3 h, the cotton was replaced and saturated with 100μl of 5.0 × 107/ml CFU suspension of Candida albicans yeast in PBS and left in place for approximately 4 – 6 h while the animals remain sedated. After a 3 – 28 d infection period, the mice were given a clinical score to assess the degree of oral infection (Supplemental Figure 3); and the tongues and other target organs were collected aseptically after euthanasia, weighed, and homogenized in sterile normal saline using a TissueLyser™ bead-beater homogenizer (Retsch) in individual sterile nuclease free tubes. Tenfold serial dilutions of the homogenate were made in sterile PBS and plated in triplicate onto SD agar, sealed and incubated for 48 h at 37°C and fungal colonies manually counted. Fungal burdens of the tissues are shown as log CFU per gram of tissue (absolute fungal data is included in Supplemental Table 1). To assess inflammatory responses in the oral mucosa, buccal epithelium was removed at necropsy by blunt dissection and placed in RNAlater™ (Qiagen) or PBS, homogenized and frozen at -80°C for mRNA isolation or protein assay respectively. For serum collection, whole blood was obtained from retro-orbital sinus into EDTA pre-coated tubes, centrifuged and serum removed and frozen.
Quantitative real-time PCR and enzyme-linked immunosorbent assay
Quantitative real-time PCR was done as described (see Supplementary Methods for additional details). Cytokines were measured in culture supernatants collected after 24 h of stimulation by ELISA (R&D).
Western blot analysis
Cell culture supernatants were precipitated by adding an equal volume of methanol and 0.25 volumes of chloroform, mixed and centrifuged at 20,000 × g for 10 min. The upper phase was discarded and 500 μl of methanol was added to the interphase. This mixture was centrifuged at 20,000 × g for 10 min and the protein pellet dried at 55 °C, resuspended in Laemmli buffer and boiled at 99°C for 5 min. Samples were separated by SDS-PAGE (15%) and transferred onto nitrocellulose membranes. As indicated, blots were incubated with rabbit polyclonal antibody to anti murine caspase-1 p10 (sc-514, Santa Cruz Biotechnology), rabbit polyclonal anti human caspase-1 p10 (sc-515, Santa Cruz Biotechnology), rabbit polyclonal anti human cleaved IL-1β (Asp116) (Cell Signaling) or rabbit polyclonal anti murine cathepsin B (R&D Systems).
Statistical Analysis
Data were analyzed using commercial software (GraphPad Software) and Student’s two-sample independent t-test used for statistical analysis. Comparison of two survival curves was done using the Logrank test. P values are presented where statistically significance was found (significance set at P < 0.05).
Supplementary Material
Acknowledgments
The authors are grateful to S. Akira (Osaka University, Osaka, Japan) for the gift of TLR2 deficient mice, to R. Flavell (Yale University) for providing caspase-1 deficient mice, to G. Dubyak for P2X7 receptor deficient mice, D. Hatala for tissue sectioning, H. Butler, K. Zongolowicz and A. Cerny for animal husbandry, M. Caprara for technical assistance and E. Kurt-Jones for advice and assistance. This work was supported by NIH R01DE018279 (to A.G.H.).
References
- Abu-Elteen KH, Abu-Alteen RM. The prevalence of Candida albicans populations in the mouths of complete denture wearers. New Microbiol. 1998;21:41–48. [PubMed] [Google Scholar]
- Akita K, Ohtsuki T, Nukada Y, Tanimoto T, Namba M, Okura T, Takakura-Yamamoto R, Torigoe K, Gu Y, Su MS, et al. Involvement of caspase-1 and caspase-3 in the production and processing of mature human interleukin 18 in monocytic THP.1 cells. J Biol Chem. 1997;272:26595–26603. doi: 10.1074/jbc.272.42.26595. [DOI] [PubMed] [Google Scholar]
- Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, Nunez G. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. 2006;281:35217–35223. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, Vecchi A, Mantovani A, Levitz SM, Romani L. The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol. 2004;172:3059–3069. doi: 10.4049/jimmunol.172.5.3059. [DOI] [PubMed] [Google Scholar]
- Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. Journal of Experimental Medicine. 2003;197:1119–1124. doi: 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SY, Gordon S. Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med. 2002;196:407–412. doi: 10.1084/jem.20020470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- Cole GT, Seshan KR, Pope LM, Yancey RJ. Morphological aspects of gastrointestinal tract invasion by Candida albicans in the infant mouse. J Med Vet Mycol. 1988;26:173–185. [PubMed] [Google Scholar]
- Daniluk T, Tokajuk G, Stokowska W, Fiedoruk K, Sciepuk M, Zaremba ML, Rozkiewicz D, Cylwik-Rokicka D, Kedra BA, Anielska I, et al. Occurrence rate of oral Candida albicans in denture wearer patients. Adv Med Sci. 2006;51(Suppl 1):77–80. [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16:457–499. doi: 10.3109/08830189809043005. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol. 2002;20:S1–13. [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1beta. Crit Care Med. 2005;33:S460–462. doi: 10.1097/01.ccm.0000185500.11080.91. [DOI] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Kashleva H. Candida albicans triggers interleukin-8 secretion by oral epithelial cells. Microb Pathog. 2003;34:169–177. doi: 10.1016/s0882-4010(03)00004-4. [DOI] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Kashleva H, Villar CC. Bioactive interleukin-1alpha is cytolytically released from Candida albicans-infected oral epithelial cells. Med Mycol. 2004;42:531–541. doi: 10.1080/1369378042000193194. [DOI] [PubMed] [Google Scholar]
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1 beta: two cytokine substrates for ICE (caspase-1) J Clin Immunol. 1999;19:1–11. doi: 10.1023/a:1020506300324. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiral MH, Azul A, Pinto E, Fonseca PA, Branco FM, Scully C. Denture-related stomatitis: identification of aetiological and predisposing factors - a large cohort. J Oral Rehabil. 2007;34:448–455. doi: 10.1111/j.1365-2842.2007.01709.x. [DOI] [PubMed] [Google Scholar]
- Franchi L, McDonald C, Kanneganti TD, Amer A, Nunez G. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol. 2006;177:3507–3513. doi: 10.4049/jimmunol.177.6.3507. [DOI] [PubMed] [Google Scholar]
- Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–128. doi: 10.1111/j.1600-065X.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. Embo J. 2005;24:1277–1286. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009 doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K, Okamura H, Nakanishi K, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science. 1997;275:206–209. doi: 10.1126/science.275.5297.206. [DOI] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herre J, Marshall AS, Caron E, Edwards AD, Williams DL, Schweighoffer E, Tybulewicz V, Reis e Sousa C, Gordon S, Brown GD. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood. 2004;104:4038–4045. doi: 10.1182/blood-2004-03-1140. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huysamen C, Brown GD. The fungal pattern recognition receptor, Dectin-1, and the associated cluster of C-type lectin-like receptors. FEMS Microbiol Lett. 2009;290:121–128. doi: 10.1111/j.1574-6968.2008.01418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis KG, Donnenberg MS, Kaper JB, Jarvis WR. Predominant pathogens in hospital infections. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. 2006a;281:36560–36568. doi: 10.1074/jbc.M607594200. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006b;440:233–236. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- Kerrigan AM, Brown GD. C-type lectins and phagocytosis. Immunobiology. 2009 doi: 10.1016/j.imbio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lu Q, Jayatilake JA, Samaranayake LP, Jin L. Hyphal invasion of Candida albicans inhibits the expression of human beta-defensins in experimental oral candidiasis. J Invest Dermatol. 2006;126:2049–2056. doi: 10.1038/sj.jid.5700346. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929–1934. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447–454. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- McCourtie J, Douglas LJ. Relationship between cell surface composition, adherence, and virulence of Candida albicans. Infect Immun. 1984;45:6–12. doi: 10.1128/iai.45.1.6-12.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencacci A, Bacci A, Cenci E, Montagnoli C, Fiorucci S, Casagrande A, Flavell RA, Bistoni F, Romani L. Interleukin 18 restores defective Th1 immunity to Candida albicans in caspase 1-deficient mice. Infect Immun. 2000;68:5126–5131. doi: 10.1128/iai.68.9.5126-5131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci U S A. 2008;105:2562–2567. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostefaoui Y, Bart C, Frenette M, Rouabhia M. Candida albicans and Streptococcus salivarius modulate IL-6, IL-8, and TNF-alpha expression and secretion by engineered human oral mucosa cells. Cell Microbiol. 2004;6:1085–1096. doi: 10.1111/j.1462-5822.2004.00420.x. [DOI] [PubMed] [Google Scholar]
- Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–107. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- Netea MG, Stuyt RJ, Kim SH, Van der Meer JW, Kullberg BJ, Dinarello CA. The role of endogenous interleukin (IL)-18, IL-12, IL-1beta, and tumor necrosis factor-alpha in the production of interferon-gamma induced by Candida albicans in human whole-blood cultures. J Infect Dis. 2002;185:963–970. doi: 10.1086/339410. [DOI] [PubMed] [Google Scholar]
- Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, Williams DL, Gordon S, Tybulewicz VL, Brown GD, Reis e Sousa C. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Saville SP, Lazzell AL, Chaturvedi AK, Monteagudo C, Lopez-Ribot JL. Use of a genetically engineered strain to evaluate the pathogenic potential of yeast cell and filamentous forms during Candida albicans systemic infection in immunodeficient mice. Infect Immun. 2008;76:97–102. doi: 10.1128/IAI.00982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saville SP, Lazzell AL, Monteagudo C, Lopez-Ribot JL. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot Cell. 2003;2:1053–1060. doi: 10.1128/EC.2.5.1053-1060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- Steele C, Fidel PL., Jr Cytokine and chemokine production by human oral and vaginal epithelial cells in response to Candida albicans. Infect Immun. 2002;70:577–583. doi: 10.1128/IAI.70.2.577-583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuyt RJ, Netea MG, van Krieken JH, van der Meer JW, Kullberg BJ. Recombinant interleukin-18 protects against disseminated Candida albicans infection in mice. J Infect Dis. 2004;189:1524–1527. doi: 10.1086/382955. [DOI] [PubMed] [Google Scholar]
- Stuyt RJ, Netea MG, Verschueren I, Fantuzzi G, Dinarello CA, Van Der Meer JW, Kullberg BJ. Role of interleukin-18 in host defense against disseminated Candida albicans infection. Infect Immun. 2002;70:3284–3286. doi: 10.1128/IAI.70.6.3284-3286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterwala FS, Flavell RA. NLRC4/IPAF: a CARD carrying member of the NLR family. Clin Immunol. 2009;130:2–6. doi: 10.1016/j.clim.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Flavell RA. The inflammasome in pathogen recognition and inflammation. J Leukoc Biol. 2007;82:259–264. doi: 10.1189/jlb.1206755. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Joosten LA, Devesa I, Mora-Montes HM, Kanneganti TD, Dinarello CA, van der Meer JW, Gow NA, Kullberg BJ, Netea MG. Bypassing pathogen-induced inflammasome activation for the regulation of interleukin-1beta production by the fungal pathogen Candida albicans. J Infect Dis. 2009;199:1087–1096. doi: 10.1086/597274. [DOI] [PubMed] [Google Scholar]
- Vonk AG, Netea MG, van Krieken JH, Iwakura Y, van der Meer JW, Kullberg BJ. Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis. 2006;193:1419–1426. doi: 10.1086/503363. [DOI] [PubMed] [Google Scholar]
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Fujita T. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007;18:545–551. doi: 10.1016/j.cytogfr.2007.06.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.