

Abstract
Background
A replication-competent, attenuated, oncolytic herpes simplex virus-1, OncoVEXGALV/CD, has previously been engineered to express a fusogenic protein from the gibbon ape leukemia virus and cytosine deaminase/uracil phosphoribosyltransferase (CD/UPRT) which converts fluorocytosine (5-FC) to 5-fluorouracil (5-FU). OncoVEXGFP is an analogous vector that expresses enhanced green fluorescent protein.
Methods
We assessed the ability of OncoVEXGALV/CD and OncoVEXGFP to infect, replicate within, and lyse four head and neck squamous carcinoma (HNSCC) cell lines in vitro. The effects of adding 5-FC with OncoVEXGALV/CD were evaluated.
Results
HNSCC was permissive to GFP expression in100% of cells by OncoVEXGFP at a multiplicity of infection (MOI) of 1 after 48 hours, and supported logarithmic viral replication. Virus caused >60% cell death six days after exposure to virus at MOI 0.1 in three of the four cell lines. 5-FC failed to enhance cytotoxicity induced by OncoVEXGALV/CD at MOI 0.1. However, for the least sensitive SCC25 cell line, virus at MOI 0.01 was cytotoxic to only 4% of cells after six days, but was cytotoxic to 35% of cells with 5-FC.
Conclusions
OncoVEXGALV/CD efficiently infects, replicates within and lyses HNSCC at relatively low viral doses. Prodrug conversion by CD did not enhance therapy at viral doses which cause efficient cytotoxicity, but may have beneficial effects in less sensitive cell lines at low viral doses.
Introduction
The prognosis of patients with head and neck squamous cell carcinoma (HNSCC) remains essentially unchanged over the last 30 years, with approximately 60% of all patients dying within 5 years.1 Clinical research investigating combined radiation and chemotherapy regimens has led to improved organ preservation, but has not changed overall survival.2–5 Concurrent and adjuvant chemoradiation regimens used to achieve these goals are associated with functional morbidity, leading to xerostomia, loss of taste, and alterations in speech and swallowing.6, 7 Local and regional recurrence remains the primary source of failure for these patients.2–4, 8 There is a need for novel therapeutics aimed at improved local and regional control while minimizing toxicity.
Herpes simplex virus type 1 (HSV-1) is a double stranded DNA virus which can be genetically manipulated to have an attenuated ability to infect healthy cells, but retain the ability to infect and kill cancer cells. These viruses have been shown to be effective at infecting and lysing a variety of human cancer cell lines, including HNSCC. Because of a large, non-essential genome, HSV-1 can be extensively genetically altered. These alterations have included the insertion of reporter genes such as GFP or lacZ for fluorescent or histologic detection, or genes to improve the infectivity and cytotoxicity of oncolytic HSV-1. These have included insertion of genes encoding interleukins,9–14 GM-CSF,14–16 cytosine deaminase,17, 18 and fusogenic proteins.18, 19
Cytosine deaminase is a pyrimidine salvage enzyme derived from E. coli which, through hydrolytic deamination, converts cytosine to uracil. It also converts the antifungal 5-fluorocytosine (5-FC) to the chemotherapeutic agent 5-fluorouracil (5-FU). The cytotoxic effect of 5-FU is principally achieved by the conversion of 5-FU to 5-fluoro-dUMP (5-FdUMP) by uracil phosphoribosyltransferase (UPRT).20 5-FU is currently utilized in head and neck cancer therapy, and has activity as a radiosensitizer.4, 21 Side effects, particularly mucositis, can be increased with combined modality therapy2–4, 8 Such side effects might theoretically be diminished by localization of 5-FU. Tumor cells infected with a cytosine deaminase/UPRT producing virus can convert systemically administered 5-FC to 5-FU locally, reducing the systemic distribution of 5-FU.
The fusogenic membrane glycoprotein of the gibbon ape leukemia virus (GALV) may enhance cytotoxicity by forming large multinucleated syncytia without impairing viral replication.18, 19, 22 Oncolytic HSV-1 with genetic insertions of CD/UPRT or GALV demonstrate cytotoxicity in human malignant cell lines.17–20, 22, 23 Recently, a mutant HSV-1 was constructed that expresses both GALV and CD/UPRT (OncoVEXGALV/CD), and demonstrates cytotoxicity in colon, lung, and pancreatic cancer cell lines.18 In this study, we assess the utility of applying OncoVEXGALV/CD with and without 5-FC for treating HNSCC.
Methods and Materials
Cell Lines
Four human HNSCC cell lines, SCC15, SCC25, QLL1, and QLL2, were studied. Cell were grown in minimal essential medium (MEM) with 10% fetal calf serum (FCS) and penicillin and streptomycin. Cells were maintained at 37°C in 5% carbon dioxide.
Viruses
OncoVEXGFP and OncoVEXGALV/CD have been previously described.18 Briefly, both OncoVEXGALV/CD and OncoVEXGFP were constructed from the same wild-type HSV-1 strain, JS-1, with the genes encoding ICP34.5 and ICP47 completely deleted. The inserted genes replace both copies of ICP34.5 and are expressed from CMV or CMV and RSV promoters in OncoVEXGFP and OncoVEXGALV/CD respectively.
Viral Entry
SCC15, QLL1 and QLL2 cells were plated at 3×105 cells per well in 2.0 ml medium in 6 well plates overnight. OncoVEXGFP at MOI 1.0 was added in 100 ul MEM, and cells were digitally imaged along a time course using the an inverted microscope (Nikon TE300) with a green fluorescent filter and digital camera (SPOT RT Slider, Diagnostic Instruments Inc.) until 100% of cells expressed GFP.
Viral Cytotoxicity
Cell lines QLL1, QLL2, SCC15 and SCC25 were plated on 12 well plates at 2×104 cells per well in 1 ml media overnight. Cells were infected with OncoVEXGFP or OncoVEXGALV/CD at MOI 0, 0.1, 0.01, and 0.001 in 100 ul of medium. At daily intervals, supernatants were removed for viral titers, and cells were washed with PBS and lysed with Triton-X 1.35%. LDH was quantified using a Cytotox 96 kit (Promega, Madison, WI) and spectrophotometry (EL321e, Bio-Tek Instruments, Winooski, VT) at 490 nm. On the fourth day after infection, 1 ml of media was added. Results were expressed as percentage of cells surviving as compared to the untreated (MOI 0) control cells.
At day 6, additional wells of cells treated with virus at MOI 0.1 were fixed with 20% ethanol in PBS and stained with of 0.1% Crystal Violet for 10 minutes. Cells were washed with H2O, and plates were dried in room air. The plates were then digitally photographed using an inverted microscope (Nikon Eclipse TS100). All samples were assessed in triplicate.
Viral Replication
Supernatants from the cytotoxicity experiments were removed and frozen at −80°C for later plaque assays. Serial dilutions of supernatants were later added to confluent Vero cells on 6 well plates for 4 hours. Supernatants were removed and cells washed with 1 ml media. Two ml of 1% agarose was added to each well. After 48 hours, 2ml of 2% neutral red was added. After 24 hours plaques were counted. Experiments were performed in triplicate for each condition.
Viral Cytotoxicity + Prodrug Activation
QLL1 and QLL2 cells were plated at 2×104 cells per well in 2 ml MEM on 12-well plates overnight. Cells were infected with OncoVEXGALV/CD at MOI 0 or 0.1. 5-FC was added to a final well concentration of 600μmol/L or PBS added to control wells. LDH assays were performed on days 3–6 as described above.
To determine if the timing of 5-FC administration affects cytotoxicity, QLL1 cells were infected with OncoVEXGALV/CD at MOI 0.01. 5-FC was added to a final concentration of 2400 μmol/L at 0, 12, 24, and 48 hours after viral exposure, or PBS added to control wells. LDH assays were performed on days 3–6.
To assess prodrug activation at conditions adverse to viral oncolysis, the more resistant SCC25 was exposed to OncoVEXGALV/CD at an MOI of 0 or 0.05. PBS as control or 5-FC was added to a final concentration of 2400 μmol/L. LDH assays were performed on days 3–6. Each condition was assayed in triplicate.
Prodrug Activation Assay
The conversion of 5-FC to 5-FU was indirectly assayed by assessing the cytotoxic activity of conditioned media after viral inactivation. SCC25 cells were plated at 2.5×106 cells/well in 6-well plates in 2cc of medium and incubated overnight. OncoVEXGALV/CD was added at MOI 0.01 in 100 μl media for 24 hours. 5-FC was added to a concentration of 1200 μmol/L, or PBS added to control wells. Supernatants were removed after 24 hours and centrifuged at 800 rpm for 5 minutes. To inactivate virus, supernatant samples were heated to 90°C for 1 hour and treated with UV light at 10 pulses of 300 mJ/cm2 (Stratagene UV Stratalinker 1800).
Untreated SCC25 cells were plated at 2×104 cells/well in 12-well plates in 0.5 ml medium and incubated overnight. An additional 0.5 ml of each supernatant sample was then added. LDH assays were performed at the indicated days, using untreated cells in MEM+10% FCS as 100% viable controls. Samples were assayed in triplicate.
Statistical Analysis
All data are expressed as mean ± the standard error of the mean. Comparisons between two groups were made using the two-tailed student’s t-test.
Results
OncoVEXGFP efficiently infects and expresses GFP in HNSCC
Equal numbers of SCC15, QLL1, QLL2 cells exposed to OncoVEXGFP at MOI 1.0 were imaged with bright and darkfield photography. GFP was first visualized in SCC15 cells by 2 hours (data not shown). Progressively increasing percentages of GFP expression were observed under fluorescent microscopy (100X) over a time course. By 48 hours, 100% of remaining viable cells were fluorescent for all three cell lines (Figure 1), demonstrating efficient viral infection and gene expression by OncoVEXGFP. Viral cytotoxic effects also caused significant declines in viable cell number by 48 hours, most pronounced for SCC15, followed by QLL1.
Figure 1.
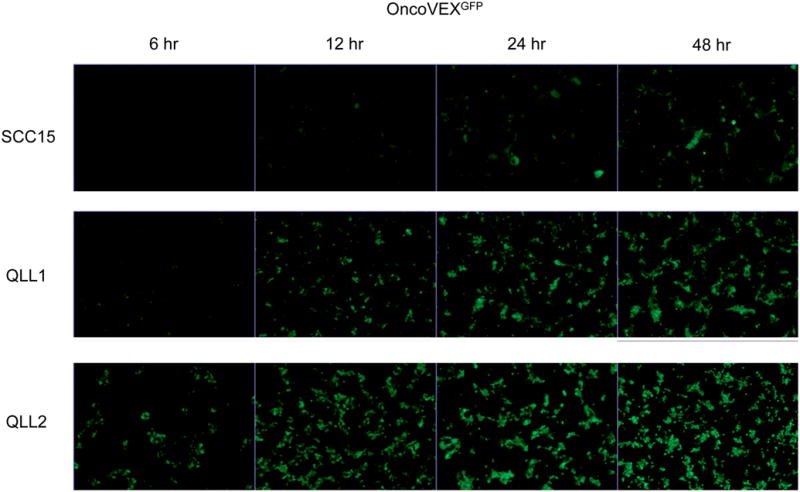
Equal numbers of cells from three HNSCC cell lines were infected with OncoVEXGFP at MOI 1. Progressively increasing percentages of GFP expression were observed under fluorescent microscopy (100X) over a time course. By 48 hours, 100% of remaining viable cells were fluorescent for all three cell lines, demonstrating efficient viral infection and gene expression by OncoVEXGFP. Viral cytotoxic effects also caused significant declines in viable cell number by 48 hours, most pronounced for SCC15, followed by QLL1.
OncoVEXGFP and OncoVEXGALV/CD replicate efficiently in HNSCC
SCC15, SCC25 QLL1 and QLL2 cell lines were treated with OncoVEXGFP or OncoVEXGALV/CD at MOI 0.01 and supernatants collected for plaque assays. All four HNSCC cell lines supported logarithmic replication by both viruses (Figure 2). SCC15 supported the most robust viral replication, with OncoVEXGFP titers rising dramatically from just 12.5 pfu to 2.88×106 pfu over a 3-day interval. Viral replication progressed slightly slower for QLL1, peaking at day 4, and was the latest for the least sensitive SCC25 cell line at day 5. Early viral replication was most pronounced for QLL2, reaching 3.43×104 pfu in 48 hours, but quickly leveled out in the subsequent days.
Figure 2.

Equal numbers of cells from four HNSCC cell lines were infected with OncoVEXGFP or OncoVEXGALV/CD at MOI 0.01. A time course of viral plaque assays demonstrates highly efficient, logarithmic viral replication by both viruses. A small, early replication advantage by OncoVEXGALV/CD over OncoVEXGFP was noted for SCC15 and SCC25 within the first viral replication cycle (days 1–3), although differences were lost once peak titers were reached. SCC15 and QLL2 supported the most rapid early viral replication, followed by QLL1 and SCC25.
Growth curves demonstrate an early advantage to the OncoVEXGALV/CD virus over OncoVEXGFP for SCC15 and SCC25 within the first viral replication cycle (days 1–3), although differences were no longer observed once peak titers were reached. Both viruses replicated at similar logarithmic rates in QLL1 and QLL2.
OncoVEXGFP and OncoVEXGALV/CD are cytotoxic to HNSCC in vitro
Head and neck squamous cell carcinoma lines QLL1, QLL2, SCC15, and SCC25 were treated with OncoVEXGFP or OncoVEXGALV/CD at varying MOI, and cell viability was assessed by LDH assay. Viral dose-dependent cytotoxicity was noted for all cell lines. SCC15 was most susceptible to viral cytotoxicity, followed by QLL1, QLL2, and SCC25 which was the least susceptible. With the exception of SCC25, cell lines showed >60% cytotoxicity by 6 days after exposure to either virus at MOI 0.1 (Figure 3). At a low viral dose of MOI 0.01, >70% of the highly sensitive SCC15 cells were dead by 6 days. OncoVEXGFP and OncoVEXGALV/CD exhibited similar cytotoxicity profiles.
Figure 3.

A. HNSCC cell lines were treated with OncoVEXGFP or OncoVEXGALV/CD at varying MOI, and cell viability was assessed over a time course by LDH assay. Viral dose-dependent cytotoxicity was noted for all cell lines. SCC15 was most susceptible to viral cytotoxicity, followed by QLL1, QLL2, and SCC25. All cell lines showed significant cytotoxicity by 5 days after exposure to either virus at MOI 0.1, although SCC25 was the least sensitive (Figure 3). At a low viral dose of MOI 0.01, >70% of the most sensitive SCC15 cells were dead by 6 days. OncoVEXGFP and OncoVEXGALV/CD exhibited similar cytotoxicity profiles. B. Crystal violet staining of cells treated with virus by day 6 qualitatively confirms the quantitative results from the LDH assays. The OncoVEXGALV/CD clearly induced a syncytial phenotype in QLL2 cells, creating multinucleated giant cells in contrast to the OncoVEXGFP virus. However, the identical cytotoxicity curves between these two viruses for all four HNSCC cell lines (A) demonstrate that GALV expression did not yield an additional cytotoxic benefit at these conditions.
Crystal violet staining of remaining viable cells at day 6 after exposure to virus at MOI 0.1 qualitatively confirms LDH assay results (Figure 3b). The OncoVEXGALV/CD at MOI 0.1, day 6, yielded a syncytial phenotype in QLL2 cells seen with crystal violet staining, in sharp contrast to the OncoVEXGFP virus. However, the identical cytotoxicity curves between the two viruses for these four HNSCC cell lines demonstrate that GALV expression did not induce any additional overall cytotoxic benefit.
5-FC enhances OncoVEXGALV/CD cytotoxicity of SCC25 at low MOI
The addition of the prodrug 5-FC at 600 μmol/L to QLL1 cells infected with OncoVEXGALV/CD at MOI 0.1 failed to enhance cytotoxicity as compared to cells that did not receive 5-FC (Figure 4a). QLL2 cells treated identically demonstrated a subtle trend towards increased cytotoxicity with the 5-FC at days 4 and 5, but this difference was not statistically significant. We considered the possibility that early 5-FU production interfered with viral replication and offset any additional benefit from the drug production. We therefore next delayed the timing of 5-FC addition to allow for viral replication, and increased its concentration to 2400 μmol/L. The addition of 5-FC at QLL1 cells at viral of MOI 0.01 showed no differences in cytotoxicity when the 5-FC is added at 12, 24, or 48 hours as compared to controls not receiving 5-FC.
Figure 4.

A. The addition of the prodrug 5-FC at 600 μmol/L to QLL1 cells infected with OncoVEXGALV/CD at MOI 0.1 failed to enhance cytotoxicity as compared to cells that did not receive 5-FC. B. QLL2 cells treated identically demonstrated a subtle trend towards increased cytotoxicity with the 5-FC at days 4 and 5, but this difference was not statistically significant. C. To account for the possibility that 5-FU production interfered with viral replication, the timing of 5-FC addition to QLL1 at MOI 0.01 was varied and the 5-FC concentration increased to 2400 μmol/L. There were still no differences in cytotoxicity when the 5-FC is added at 12, 24, or 48 hours as compared to controls not receiving 5-FC. D. As direct viral oncolysis might limit the ability of infected cells to adequately express CD/UPRT prior to cell death, we treated the least sensitive SCC25 cell line with OncoVEXGALV/CD at MOI 0.05, with or without 5-FC at 2400 μmol/L. Under these conditions, direct viral oncolytic effects were minimal, but we observed a significant enhancement of cytotoxicity by day 6, with an increase from 4% to 35% cytotoxicity appreciated with the addition of the 5-FC (p<0.01). E. To assess the production of active 5-FU by OncoVEXGALV/CD, we quantified the cytotoxic activity of conditioned media from SCC25 cells exposed to OncoVEXGALV/CD and 5-FC, after viral inactivation. LDH assays on SCC25 cells demonstrated 80% cytotoxicity by day 4 for the cells treated with conditioned media collected from cells treated with both OncoVEXGALV/CD and 5-FC. There were significant differences at all time points (p≤ 0.01) in comparison with the 5-FC or virus only control samples. The slightly reduced viability detected from the 5-FC or virus alone samples likely results from growth rate reduction effects from nutrient-depleted media.
We reasoned that the high sensitivity of SCC15, QLL1, and QLL2 to direct viral oncolysis might limit the ability of infected cells to adequately express CD/UPRT prior to cell death. To account for this possibility, we treated the least sensitive SCC25 cell line with OncoVEXGALV/CD at MOI 0.05, either with or without 5-FC at a concentration of 2400 μmol/L. Under these conditions, direct viral oncolytic effects were minimal. We observed a statistically significant enhancement of cytotoxicity by day 6, with an increase from 4% to 35% cytotoxicity appreciated with the addition of the 5-FC (Figure 4a).
OncoVEXGALV/CD infected cells convert 5-FC to 5-FU
To indirectly assess the production of active 5-FU by OncoVEXGALV/CD, we quantified the cytotoxic activity of conditioned media on SCC25 cells after viral inactivation. LDH assays demonstrated 80% less cell viability by day 4 for the cells treated with supernatant collected from SCC25 cells treated with both OncoVEXGALV/CD and 5-FC (Figure 4b). In contrast, supernatant from SCC25 cells treated with virus alone, or 5-FC alone, demonstrated approximately 20–25% less viability, with statistically significant differences at all time points (p<0.05) in comparison with the 5-FC samples. The reduced viability induced by the virus alone or 5-FC alone samples most likely relates to SCC25 growth rate reduction from the addition of the nutrient-depleted media, as compared to control SCC25 wells which received fresh media and were considered 100% viable for these comparative assays. Collectively, these studies suggest that there is active 5-FU produced by OncoVEXGALV/CD + 5-FC in SCC25 cells at these conditions.
Discussion
We demonstrate the ability of two attenuated, replication-competent, oncolytic herpes viruses (OncoVEXGFP and OncoVEXGALV/CD) to infect and lyse HNSCC cell lines in vitro. At an MOI of 1, OncoVEXGFP was able to completely infect all cells within 48 hours, and induced a significant amount of cell death for the SCC15 and QLL1 cell lines within this short period. Furthermore, both viruses exhibited efficient, logarithmic, replication after infecting cells at MOI 0.01. SCC15 supported the most rapid viral proliferation, followed by QLL1 and SCC25, although all supported similar peak titers. Even the least sensitive QLL2 permitted a >170-fold increase in viral titers over a 2 day period, demonstrating a highly favorable host cancer cell environment for viral production. From a clinical standpoint, this finding implies that high levels of progeny virus could be produced and released to infect and lyse adjacent cancer cells.
The relative sensitivity by these different cell lines to viral replication was similarly reflected in our viral cytotoxicity studies. Both viruses demonstrated significant cytotoxicity in three of the four cell lines at a relatively low concentration of MOI 0.1, and showed cytotoxic effects in a dose-response fashion. Furthermore, the highly sensitive SCC15 cell line was susceptible to nearly 80% cytotoxicity by day 6 at a very low MOI of just 0.01. The SCC25 cell line showed the least sensitivity to viral oncolysis. However, it is likely that higher viral MOI than those tested (0.1 and less) would yield more rapid and potent cytotoxicity, given the dose dependent effects observed and the high degree of infection achieved at MOI 1.
We did not note any difference between the oncolytic effects of OncoVEXGFP and OncoVEXGALV/CD. The GALV fusogenic membrane glycoprotein promotes syncytia formation, and we did identify multinucleated cells in samples infected by OncoVEXGALV/CD during our cytotoxicity studies with crystal violet staining. Although GALV glycoprotein expression has been shown to enhance viral cytotoxicity in some cell lines18,19,22, we did not observe any difference with GALV expression in these HNSCC cell lines.
The application of OncoVEXGALV/CD in the presence of 5-FC did not appear to offer a significant cytotoxic benefit over OncoVEXGFP at MOI’s that are able to induce significant cell death. There are several potential explanations for this finding. The most likely possibility is that the sensitive cells are infected and lysed before 5-FU, which requires cell division, has a chance to exert its cytotoxic effect. The second possibility is that 5-FU may be exerting an inhibitory effect on viral DNA replication. The CD/UPRT gene is under control of a constitutively-expressed early CMV promoter, so infected cells will express these proteins prior to the first replication cycle of the viral genome. Because 5-FU inhibits DNA synthesis and RNA production, viral protein production and genome replication could therefore be inhibited. Finally, 5-FU may not have particular activity for these four HNSCC cell lines. 5-FU is not typically used in isolation for HNSCC, and is most commonly employed as a radiosensitizer in combination with a platinum-based chemotherapeutic agent. 5-FU is effective as single agent therapy in only a small percentage of HNSCC.24, 25
To account for the first possibility, we tested OncoVEXGALV/CD with 5-FC in the least sensitive HNSCC cell line, SCC25, at a low MOI. At these conditions, where the virus by itself could only induce minimal cytotoxicity, we observed a modest enhancement of cytotoxicity with the addition of 5-FC. Prodrug assays of conditioned media suggested that active 5-FU had been produced. These findings imply that the addition of 5-FC to OncoVEXGALV/CD might be most beneficial at conditions where viral dosing must be limited. Such low dose viral application may be attractive from the standpoint of minimizing the potential for any viral-induced toxicity; the virus alone would be unable to induce cytotoxic effects, but in combination with 5-FC could elicit a therapeutic benefit. This strategy might also apply for clinical scenarios where enhancement of therapy is necessary less virally-sensitive tumor targets.
One obvious advantage of treating tumor sites directly with OncoVEXGALV/CD is combination with 5-FC is the localization of active 5-FU to these tumor sites, and the potential avoidance of toxicity associated with its systemic distribution. Similarly, the potential local administration of radiation therapy, which is has enhanced efficacy in combination with 5-FU, appears to be an attractive strategy to apply in combination with OncoVEXGALV/CD with 5-FC. Such applications of these viruses, using in vivo models, are potential future avenues of investigation.
This study demonstrates potent oncolytic efficacy by OncoVEXGFP and OncoVEXGALV/CD in HNSCC in vitro. We show the potential benefits and limitations of using a prodrug conversion strategy with OncoVEXGALV/CD in these cell lines. Although a benefit was observed only at conditions of minimal direct viral oncolysis, these findings may have potential application in clinical scenarios where minimal viral dosing is a goal or in treating tumor targets that are less sensitive to viral oncolysis. These findings support the further investigation of these novel viruses for HNSCC, and their potential application in vivo and in combination with radiation therapy.
Acknowledgments
DLP is supported by T32CA009685 from the National Cancer Institute. The project described was supported by Grant Number T32CA009685 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
RJW is supported by a Clinical Innovator Award from the Flight Attendant Medical Research Institute
Footnotes
Presented at the 7th International Conference on Head and Neck Cancer, July 23, 2008, San Francisco, CA.
References
- 1.Ries LAG MD, Krapcho M, Mariotto A, Miller BA, Feuer EJ, Clegg L, Horner MJ, Howlader N, Eisner MP, Reichman M, Edwards BK. SEER Cancer Statistics Review. 1975–2004 http://seer.cancer.gov/csr/1975_2004/
- 2.Bernier J, Domenge C, Ozsahin M, et al. Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N Engl J Med. 2004 May 6;350(19):1945–1952. doi: 10.1056/NEJMoa032641. [DOI] [PubMed] [Google Scholar]
- 3.Cooper JS, Pajak TF, Forastiere AA, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004 May 6;350(19):1937–1944. doi: 10.1056/NEJMoa032646. [DOI] [PubMed] [Google Scholar]
- 4.Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003 Nov 27;349(22):2091–2098. doi: 10.1056/NEJMoa031317. [DOI] [PubMed] [Google Scholar]
- 5.Numico G, Russi EG, Vitiello R, et al. Gemcitabine and cisplatin in a concomitant alternating chemoradiotherapy program for locally advanced head-and-neck cancer: a pharmacology-guided schedule. Int J Radiat Oncol Biol Phys. 2006 Nov 1;66(3):731–737. doi: 10.1016/j.ijrobp.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 6.Dirix P, Nuyts S, Vander Poorten V, Delaere P, Van den Bogaert W. The influence of xerostomia after radiotherapy on quality of life: Results of a questionnaire in head and neck cancer. Support Care Cancer. 2007 Jul 6; doi: 10.1007/s00520-007-0300-5. [DOI] [PubMed] [Google Scholar]
- 7.Eisbruch A, Lyden T, Bradford CR, et al. Objective assessment of swallowing dysfunction and aspiration after radiation concurrent with chemotherapy for head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2002 May 1;53(1):23–28. doi: 10.1016/s0360-3016(02)02712-8. [DOI] [PubMed] [Google Scholar]
- 8.Induction chemotherapy plus radiation compared with surgery plus radiation in patients with advanced laryngeal cancer. The Department of Veterans Affairs Laryngeal Cancer Study Group. N Engl J Med. 1991 Jun 13;324(24):1685–1690. doi: 10.1056/NEJM199106133242402. [DOI] [PubMed] [Google Scholar]
- 9.Bennett JJ, Malhotra S, Wong RJ, et al. Interleukin 12 secretion enhances antitumor efficacy of oncolytic herpes simplex viral therapy for colorectal cancer. Ann Surg Jun. 2001;233(6):819–826. doi: 10.1097/00000658-200106000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derubertis BG, Stiles BM, Bhargava A, et al. Cytokine-secreting herpes viral mutants effectively treat tumor in a murine metastatic colorectal liver model by oncolytic and T-cell-dependent mechanisms. Cancer Gene Ther Jun. 2007;14(6):590–597. doi: 10.1038/sj.cgt.7701053. [DOI] [PubMed] [Google Scholar]
- 11.Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A. 2000 Feb 29;97(5):2208–2213. doi: 10.1073/pnas.040557897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong RJ, Chan MK, Yu Z, et al. Angiogenesis inhibition by an oncolytic herpes virus expressing interleukin 12. Clin Cancer Res. 2004 Jul 1;10(13):4509–4516. doi: 10.1158/1078-0432.CCR-04-0081. [DOI] [PubMed] [Google Scholar]
- 13.Wong RJ, Chan MK, Yu Z, et al. Effective intravenous therapy of murine pulmonary metastases with an oncolytic herpes virus expressing interleukin 12. Clin Cancer Res. 2004 Jan 1;10(1 Pt 1):251–259. doi: 10.1158/1078-0432.CCR-0197-3. [DOI] [PubMed] [Google Scholar]
- 14.Wong RJ, Patel SG, Kim S, et al. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Hum Gene Ther. 2001 Feb 10;12(3):253–265. doi: 10.1089/10430340150218396. [DOI] [PubMed] [Google Scholar]
- 15.Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006 Nov 15;12(22):6737–6747. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 16.Malhotra S, Kim T, Zager J, et al. Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery Apr. 2007;141(4):520–529. doi: 10.1016/j.surg.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guffey MB, Parker JN, Luckett WS, Jr, et al. Engineered herpes simplex virus expressing bacterial cytosine deaminase for experimental therapy of brain tumors. Cancer Gene Ther Jan. 2007;14(1):45–56. doi: 10.1038/sj.cgt.7700978. [DOI] [PubMed] [Google Scholar]
- 18.Simpson GR, Han Z, Liu B, Wang Y, Campbell G, Coffin RS. Combination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor control. Cancer Res. 2006 May 1;66(9):4835–4842. doi: 10.1158/0008-5472.CAN-05-4352. [DOI] [PubMed] [Google Scholar]
- 19.Fu X, Tao L, Jin A, Vile R, Brenner MK, Zhang X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Mol Ther Jun. 2003;7(6):748–754. doi: 10.1016/s1525-0016(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 20.Tiraby M, Cazaux C, Baron M, Drocourt D, Reynes JP, Tiraby G. Concomitant expression of E. coli cytosine deaminase and uracil phosphoribosyltransferase improves the cytotoxicity of 5-fluorocytosine. FEMS Microbiol Lett. 1998 Oct 1;167(1):41–49. doi: 10.1111/j.1574-6968.1998.tb13205.x. [DOI] [PubMed] [Google Scholar]
- 21.Fu KK. Radiation Therapy With 5-fluorouracil in Head and Neck Cancer. Semin Radiat Oncol Oct. 1997;7(4):274–282. doi: 10.1053/SRAO00700274. [DOI] [PubMed] [Google Scholar]
- 22.Galanis E, Bateman A, Johnson K, et al. Use of viral fusogenic membrane glycoproteins as novel therapeutic transgenes in gliomas. Hum Gene Ther. 2001 May 1;12(7):811–821. doi: 10.1089/104303401750148766. [DOI] [PubMed] [Google Scholar]
- 23.Huber BE, Austin EA, Richards CA, Davis ST, Good SS. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc Natl Acad Sci U S A. 1994 Aug 16;91(17):8302–8306. doi: 10.1073/pnas.91.17.8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grem JL. Mechanisms of Action and Modulation of Fluorouracil. Semin Radiat Oncol Oct. 1997;7(4):249–259. doi: 10.1053/SRAO00700249. [DOI] [PubMed] [Google Scholar]
- 25.Wittes RE. Chemotherapy of head and neck cancer. Otolaryngol Clin North Am. 1980;13(3):515–520. [PubMed] [Google Scholar]