

Summary
Influenza viruses exploit host cell machinery to replicate, resulting in epidemics of respiratory illness. In turn, the host expresses anti-viral restriction factors to defend against infection. To find host-cell modifiers of influenza A H1N1 viral infection, we used a functional genomic screen and identified over 120 influenza A virus-dependency factors with roles in endosomal acidification, vesicular trafficking, mitochondrial metabolism and RNA splicing. We discovered that the interferon-inducible trans-membrane proteins, IFITM1, 2 and 3, restrict an early step in influenza A viral replication. The IFITM proteins confer basal resistance to influenza A virus, but are also inducible by interferons type I and II, and are critical for interferon's virustatic actions. Further characterization revealed that the IFITM proteins inhibit the early replication of flaviviruses, including dengue virus and West Nile virus. Collectively this work identifies a new family of anti-viral restriction factors that mediate cellular resistance to at least three major human pathogens.
Introduction
Influenza epidemics exact a formidable toll on world health. Moreover, viral super-infections can produce antigenic shifting, resulting in more virulent pathogens (Monto, 2009). At present, the emergence of a novel influenza A H1N1 viral strain has created a pandemic, producing illness in over 200 countries and territories (World Health Organization Pandemic (H1N1) 2009 - update 75). Additionally, the related avian influenza A viral strain, H5N1, represents a potentially catastrophic global health risk (Maines et al., 2008).
The influenza A viral genome encodes for 11 proteins and consists of eight segments of negative single-stranded RNA (Lamb and Krug, 2001). Each sub-genomic segment is coated by viral nucleoprotein (NP) and bound to a single viral RNA-dependent RNA-polymerase holoenzyme (RdRp), composed of PA, PB1 and PB2 subunits. Infection begins with the binding of the viral hemagglutinin (HA) protein to sialyated host cell surface glycoproteins (Skehel and Wiley, 1995). Following endocytosis, viral particles are trafficked through both early and late endosomes, with the acidification of the latter compartment altering the conformation of HA, leading to host-viral membrane fusion, entry of the vRNPs into the cytosol (Sieczkarski and Whittaker, 2003) and nuclear import.
Once in the nucleus, the RdRp commandeers 5′ caps from host mRNAs to prime transcription of viral mRNA [vmRNA, (Bouloy et al., 1978)] a positive sense template for new viral genomes (vRNAs). The vRNAs are coated by NP and exported though the nuclear pore complex (NPC) by the viral factors M1 and NEP/NS2 (nuclear export protein) working in concert with the host nuclear export machinery. The viral envelope proteins HA, M2 and neuraminidase (NA) are translated on the rough endoplasmic reticulum (ER) and trafficked to the cell surface where they, along with the soluble factors M1, RdRp and eight distinct vRNPs, are packaged into budding virions.
To defend against infection, the host mobilizes factors to confront the virus. Interferons (IFN) orchestrate a large component of this anti-viral response (Takaoka and Yanai, 2006). Over 2000 gene products are induced after IFN stimulation, including the anti-viral effectors MxA, PKR, RIG-I, and 2′5′-OAS (Haller et al., 2009; Nakhaei et al., 2009; Takaoka and Yanai, 2006). However, many viruses deploy anti-IFN countermeasures, which for influenza A virus are primarily enacted by the viral protein, NS1 (Hale et al., 2008). To identify host factors that modify viral replication we undertook an siRNA screen.
Results
An siRNA Screen for Influenza A Virus Infection Modifying Host Factors
We used a single round infection screen of osteosarcoma cells (U2OS), to find host proteins that modify the lifecycle of influenza A virus A/Puerto Rico/8/34 H1N1 (PR8). After 12 h the cells were stained for surface expression of HA as an indirect surrogate marker for viral infection (Fig. 1A). This approach detects viral-host receptor binding, endocytosis and fusion of the virion, vRNP trafficking and nuclear import, the transcription, nuclear export and translation of the viral HA mRNA, and the trafficking of HA to the surface. The screen was optimized using siRNAs against NP and the host factor NXF1, an mRNA exporter required for virus replication (Ge et al., 2003; Hao et al., 2008). siRNAs against either NP or NXF1 resulted in inhibition of infection (NXF1 10 fold, NP 4-6 fold, Fig. 1A, B, S1A).
Fig. 1. The siRNA screen for influenza A virus infection modifying host factors.
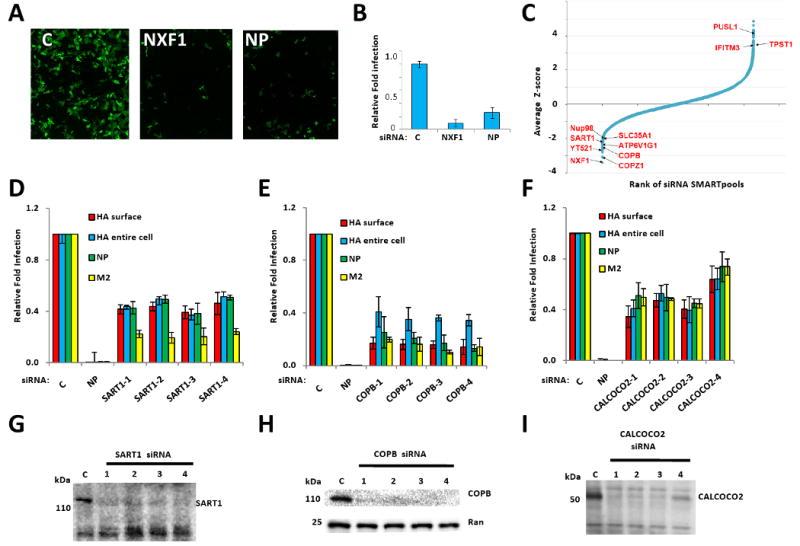
A) U2OS cells were transfected with the indicated siRNAs, then infected with influenza A virus (PR8) and immuno-stained 12 hours later for hemaggutinin (HA, green: HA). NP, siRNA targeting flu nucleoprotein, C, Nontargeting siRNA negative control. Magnification, 4×.
B) Quantification of samples in A. Relative fold infection is normalized to non-targeting (C) control. Values represent the mean ± SD, N=4.
C) The results of the screen are shown with the siRNA SMARTpools ranked in order of average Z-score, from lowest (decreased infection) to highest (increased infection). The position of known influenza A virus-host factors and several newly identified genes from the screen are indicated.
D, E and F) U2OS cells were transfected with the indicated siRNAs for 72 h, and then infected with PR8. Twelve hours after infection the cells were analyzed by IF for the following viral proteins, HA (surface or entire cell), NP and M2. Relative fold infection is normalized to non-targeting (C) control. Values represent the mean ± SD, N =4.
G, H and I) Western blots for cells in D, E and F. C, Non-targeting siRNA negative control. Ran levels are provided to demonstrate relative protein loading when cross-reacting bands were not present.
We screened the Dharmacon siRNA library in triplicate. siRNA pools were selected for further evaluation if the percentage of HA-positive cells was less than 55% of the plate mean, and cell numbers were not less than 40% of the plate mean. These criteria were fulfilled by 312 pools (1.7% of the total genes screened, Fig. 1C). Pools that increased HA expression > 200% of the plate mean were also selected for validation (22 pools, 0.1%). We next rescreened the four unique siRNAs from each pool separately. In this screen, 260 out of 334 total pools confirmed with at least one siRNA scoring and 133 confirmed with two or more siRNAs (40%), reducing the probability of off-target effects (Dataset S1A, B). We employed bioinformatics to identify networks and enriched gene sets with this gene set (Dataset S1C). Ninety two GO biological process terms, assigned to 109 genes, were significantly enriched (Dataset S2). Of these, 17 are non-redundant and assigned to less than 500 genes, suggesting they are informative and specific. The most significant terms include RNA splicing (22 genes, p=2e-12), proton transport (7 genes, p=2e-5) and mRNA transport (4 genes, p=9e-3, Fig. S1B). Analysis of GO molecular functions identified enrichment for 60 statistically significant terms assigned to 152 genes. Twelve terms were non-redundant and assigned to less than 500 human genes. The most significant terms include RNA binding (15 genes, p=0.014), ATPase activity (6 genes, p=0.008) and NADH dehydrogenase activity (4 genes, p=0.016).
Multiple biological pathways and complexes were also detected, concordant with known elements of the viral lifecycle (Fig. 2). Influenza A viral infection depends on sialic acid residues on the host cell surface, and we found that depletion of the sialic acid transporter, SLC35A1, decreased infection. Our screen confirmed the functional role of two small GTPases, RAB5A (surface internalization to early endosome trafficking) and RAB7L1 (late endosome trafficking), for viral infection (Sieczkarski and Whittaker, 2003). We also found that lowering RAB10 levels inhibited infection (Hao et al., 2008). RAB10 regulates the movement of endosomes generated from endocytosis downstream of RAB5 (Glodowski et al., 2007). Loss of each of 4 subunits of the multimeric vacuolar-ATPase proton pump (e.g. ATP6AP1, ATP6V0B, ATP6V1G1, ATP6V0E2) impeded infection, consistent with the low pH needed for fusion. Once released from the endosome, the vRNPs are transported into the nucleus though the NPC (Boulo et al., 2007), and multiple NPC factors were found.
Fig. 2. Integrated model of influenza A virus host factors.
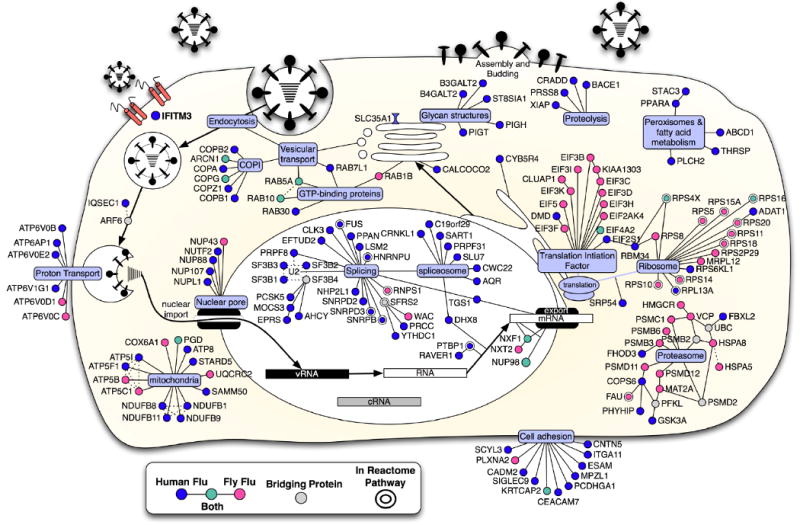
Using the influenza A virus lifecycle as a guide (Lamb and Krug, 2001), the candidate proteins from the human and fly screens were placed at the position most likely to be relevant to the virus using a database of annotations from Gene Ontology, KEGG, Reactome and OMIM (see methods). Computational mapping and supporting evidences were reviewed and refined manually (Dataset S1C-F). The known molecular functions of the host factors were determined with the use of bioinformatics and multiple data sets (gray ovals). Host factors identified in the human siRNA screen (blue); the human orthologues of proteins identified in the fly-based screen (pink), factors which were found in both human and fly screens (green), and bridging proteins that were not detected, but none-the-less generate potentially insightful interactions (gray). Double borders signify the candidate is present in the Reactome influenza A virus infection pathway (Vastrik et al., 2007). Solid lines between genes indicate a protein interaction in human or other speicies. Dotted lines indicate inferred interaction from literature or annotation. Viral RNA (vRNA), viral complementary RNA (cRNA).
Several splicing complexes were needed for viral HA protein surface-expression including three components of the U2 small nuclear RNP (snRNP), SF3B1, 2 and 3, and the U2 snRNP-interacting proteins, PRPF8, PTBP1, and FUS (Fig. 2, Dataset S1A, C). The U4/U6.U5 tri-snRNP, including SART1, was also required (Stevens et al., 2001). Four out of four siRNAs targeting SART1 resulted in lower levels of HA (surface-expressed and total protein), NP and M2 proteins (Fig. 1D, G). The decreased levels of all thee viral proteins, products of both spliced (M2) and unspliced (HA, NP) messages, suggests a general block in viral protein production with loss of SART1, perhaps secondary to effects on host protein expression.
The vesicular transport complex, coatomer 1 (COP1), scored with multiple components (p-value = 1e-7). COPI directs both retrograde intra-Golgi and Golgi to ER transport (Cai et al., 2007). Depletion of six of seven components of COPI (ARCN1, COPA, COPB1, COPB2, COPG, and COPZ1), inhibited HA surface expression, perhaps by interfering with secretion of the host cell receptor(s) and/or trafficking of HA protein to the cell surface. While COPB1 siRNAs decreased NP and M2 protein levels, they had a greater effect on surface expressed versus total HA levels, suggesting that less HA arriving at the cell surface was partly responsible for the phenotype (Fig. 1E, H). CALCOCO2 (NDP52) was also required for infection (Fig. 1F, I). CALCOCO2 localizes to the Golgi and interacts with the host proteins, TR6BP and Myosin VI, and may function in regulating secretion (Morriswood et al., 2007).
Identification of IFITM3 as an Influenza A Virus Restriction Factor
In the validation round, the depletion of four genes, interferon-inducible transmembrane protein 3 (IFITM3), PUSL1, TPST1, and WDR33, resulted in increased viral infection with two or more siRNAs (Dataset S1B). We focused on IFITM3 because of its link to interferon (Friedman et al., 1984). Eight out of eleven distinct siRNAs targeting IFITM3 increased infection, with the levels of knockdown correlating with the phenotype (Fig. 3A, B, S3A, Dataset S1B). Increased influenza A virus infection was also observed in primary lung fibroblasts after IFITM3 depletion (Fig. S3B) and Hela cells (Fig. S3C, E), with newly budded virus from Hela cells increased > 5 fold in titering assays (Fig. S3D). Lowering IFITM3 levels similarly increased infection by the influenza A H1N1 viral strain, WS/33 (Fig. 3I) but had no effect on HIV infection (Fig. S3F). Stable expression of a C-terminal HA-tagged protein, IFITM3-HA6R, lacking the 3′-untranslated region targeted by siRNA IFITM3-6, rescued resistance to the virus (Fig. 3D, E). Thus, IFITM3 is required for basal levels of cellular resistance to influenza A virus infection.
Fig. 3. IFITM3 silencing increases influenza A virus infection, and is required for the anti-viral actions of IFN.
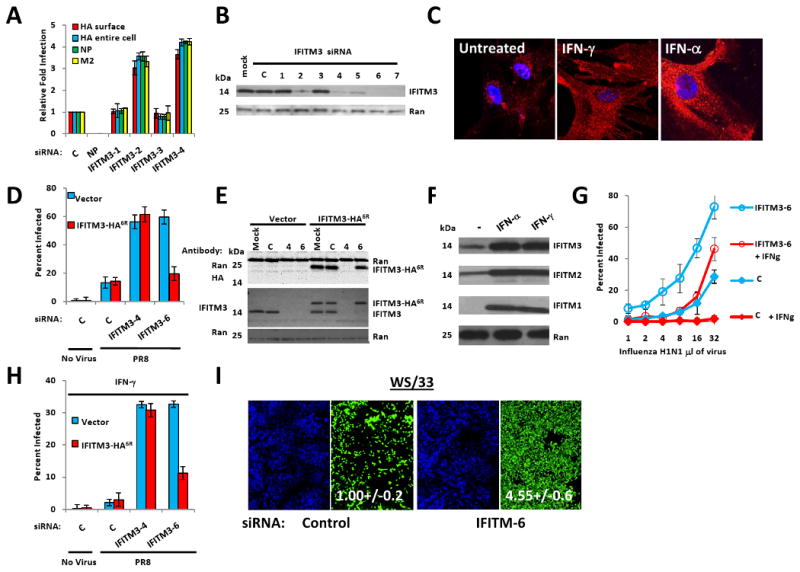
A) U2OS cells were transfected with the indicated siRNAs, then infected with PR8. Infection was assessed by IF for HA (surface or entire cell), NP or M2, 12 h after viral addition. Relative fold infection is normalized to non-targeting (C) control. Values represent the mean ± SD, N=3 throughout.
B) U2OS cells transfected with the indicated siRNAs were assessed for IFITM3 levels by Western blotting.
C) WI-38 human primary fibroblast cells stained for basal levels of IFITM3 expression (left panel), or after 24 h treatment with IFN-γ, or IFN-α (red: IFITM3, blue: nuclei), 63×.
D) U2OS cells stably expressing either IFITM3 with a C-terminal HA-epitope tag (IFTIM3-HA6R) lacking the target site for siRNA IFITM3-6, or the vector alone, were transfected with the indicated siRNAs (x-axis). After 72 h the cells were incubated without (no virus) or with influenza A (PR8) for 12 h, then stained for HA expression. The anti-HA antibody used to detect infection does not recognize the HA epitope tag on IFITM3-HA6R (no virus, uninfected control). C, non-targeting siRNA negative control.
E) U2OS cells stably expressing either IFTIM3-HA6R or vector alone were transfected with the indicated siRNAs and assessed 72 h after transfection by Western blot with the indicated antibodies.
F) U2OS cells were untreated (-), or incubated with either IFN-γ, or IFN-α. After 24 h the levels of IFITM1, 2 or 3 were checked by Western blot.
G) U2OS cells were transfected with the indicated siRNAs, then left untreated or incubated with IFN-γ 48 h later. After 24 h of IFN incubation, the cells were infected with increasing amounts of PR8. Twelve h after infection the cells were stained for HA expression.
H) IFITM3 is required for the anti-viral effect of IFN-γ. U2OS cells stably expressing either IFTIM36R or vector, were transfected with the indicated siRNAs (x-axis) and treated with IFN-γ 48 h later. After 24 h, cells were incubated without or with influenza A (PR8) and 12 h after infection cells were checked for HA surface expression.
I) IFITM3 loss enhances infection by the H1N1 strain, WS/33. U20S cells were transfected with the indicated siRNAs for 72 h, infected with WS/33 for 12 h, and stained for HA expression (green, blue: nuclei). Numbers indicate percent infection normalized to control /± SD, N=3.
The mRNAs for IFITM3, and the closely related and linked genes, IFITM1 and 2 (70%, and 91% amino acid identity, respectively, Fig. S6), are inducible by both IFN type I (α) and II [γ (Friedman et al., 1984; Lewin et al., 1991)] which we confirmed by immonofluorescence (IF) and Western blot (Fig. 3C, F). In unstimulated cells, the majority of IFITM3 resides in the ER (based on co-localization with sialic acid and N-acetylglucosamine-conjugated proteins stained by wheat germ agglutinin, WGA, Fig. S3G). IFN exposure, in contrast, triggers the distribution of IFITM3 in a vesicular pattern throughout the cell (Fig. 3C, Fig. S3H, I, 5D).
IFITM3 is Required for Interferon's Anti-viral Activity Against Influenza A Virus
In view of these dynamic changes, we examined IFITM3's functional role in the IFN response. Either IFN α or γ strongly decreased basal levels of influenza A virus infection in both U2OS or HeLa cells (Fig. 3G, H, S4A, B). The depletion of IFITM3 profoundly decreased the anti-viral actions of either IFN γ or α (Fig. G, H, S4A, B), but the inhibition was restored with the stable expression of IFITM3-HA6R (Fig. 3H). Thus we conclude that IFITM3 is required both for basal levels of resistance, as well as for the heightened defenses elicited by IFN γ and α.
IFITM1, IFITM2 and IFITM3 Inhibit the Early Replication of Influenza A Virus
We then tested if over-expression of IFITM3, or its paralogs, IFITM1 and 2, could alter viral infection. A549 lung epithelial cells were transduced with retroviruses expressing IFITM1, 2 or 3. Two days later, the transduced cells showed increased resistance to infection with influenza A viruses PR8 [H1(PR)], or H3N2 A/Udorn/72 [H3 (Udorn), Fig. 4A, B]. Profound restriction was also seen when IFITM3 was stably over-expressed in either A549, U2OS or primary Chicken fibroblast cells (ChEFs, Fig. 4C, D, E, F, S4C). In addition, the over-expression of IFITM3 in a canine cell line used for propagating influenza A viruses (Madin-Darby canine kidney (MDCK) cells), strongly inhibited the cytophathic effect of sequential rounds of viral infection (Fig. 4G, H), and also blocked infection by two current seasonal vaccine strains, A/Brisbane/59/07 H1N1 and A/Uruguay/716/07 H3N2, and by A/Aichi/2/68 H3N2, a viral isolate from the Hong Kong flu pandemic of 1968 (Fig S4D). This restriction was not universal, because IFITM proteins did not inhibit Moloney Leukemia virus (MLV, amphotropic envelope, Fig. 4A).
Fig. 4. The IFITM protein family restricts influenza A virus infection.
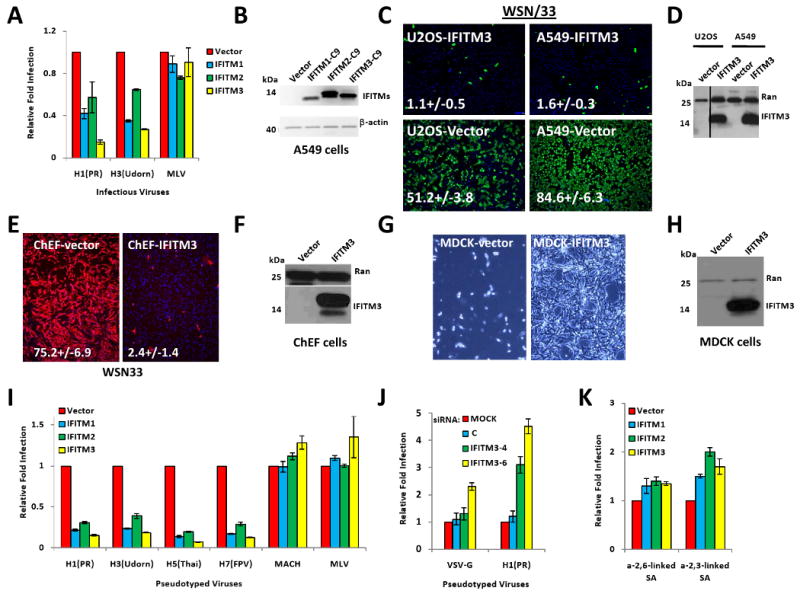
A) A549 cells were transduced with retroviruses containing C9-tagged cDNAs for the indicated IFITM proteins, or empty viral vector alone. After 2 d cells were infected with one of the following viruses: PR8 [H1(PR)], influenza A virus A/Udorn/72 (H3N2) [H3(Udorn)], or Moloney murine leukemia virus (MLV). 24 h after infection, cells were checked for HA surface expression by flow cytometry. Values represent the mean ± SD, N=3.
B) The expression of IFITM proteins in (A) was checked by Western using anti-C9 antibody. β-actin levels show protein loading.
C) A549 or U2OS cells stably over-expressing IFITM3 protein or vector alone, were infected with influenza A H1N1 WSN/33. 12 h. later, cells were fixed and stained for surface HA expression. Values represent the mean /± SD, N=3. (green: HA, blue: nuclei. 4×).
D) A549 and U2OS cells stably over-expressing IFITM3 were tested for expression by Western.
E) Primary Chicken fibroblast cells (ChEFs) stably over-expressing IFITM3 protein or vector alone, were infected with influenza A H1N1 WSN/33. 12 h. later, cells were fixed and stained for surface HA expression. Values represent the mean /± SD, N=3. (red: HA, blue: nuclei. 4×).
F) ChEF cells stably over-expressing IFITM3 were tested for expression by Western.
G) MDCK cells stably over-expressing IFITM3 protein or vector alone, were infected with influenza A H1N1 WSN/33 at a multiplicity of infection (moi) of 0.005. 72 h. later, cells were washed with fresh media, and then imaged live to assess cytopathic effect. Bright field images shown are representative of 4 independent experiments (10×).
H) MDCK cells stably over-expressing IFITM3 were tested for expression by Western.
I) A549 cells were transduced with retroviruses containing the indicated IFITM proteins, or empty vector. 48 h later, the cells were incubated with MLV-EGFP virus pseudotyped with the indicated envelope proteins. HA proteins from various influenza A virus strain including H1 (PR): A/PR/8/34 (H1N1), H3 (Udorn): A/Udorn/72 (H3N2), H5(Thai): A/Thailand2(SP-33)/2004 (H5N1), H7(FPV): A/FPV/Rostock/34 (H7N1), VSV-G: VSV G protein, MLV: MLV env protein, or MACH: Machupo virus glycoprotein. Viral entry is expressed as mean EGFP fluorescence relative to vector control cells, as measured by flow cytometry. Values represent the mean ± SD, N=3.
J) U2OS cells transfected with the indicated siRNAs for 72 h, were then incubated with MLV-GFP virus pseudotyped with the VSV-G or the HA protein of PR8, H1(PR). Entry, represented as percent green fluorescing cells relative to mock-transfected cells, was determined by IF microscopy two days post-infection. Values represent the mean ± SD, N=4. C, Non-targeting siRNA negative control.
K) A549 cells were transduced with the indicated retroviruses. 48 h later, the cells were tested for surface expression of sialic acid (SA). Values represent the mean ± SD, N=3.
To address where in the lifecycle the block was occurring, we used viral pseudoparticles. The pseudoparticles each contain an MLV genome encoding the enhanced green fluorescence protein (EGFP), however, each strain is uniquely coated with the envelope proteins from one of the following viruses: influenza A virus (strains H1, H3, H5, H7), Machupo virus (MACH), or MLV (Fig. 4I). Over-expression of each of the IFITM proteins blocked infection by all four influenza A enveloped pseudoviruses, with less restriction seen against VSV-G protein, and none against MLV (γ-retrovirus) or MACH (arenavirus) envelope proteins (Fig. 4I, S4E, F).
To complement these gain-of-function results, we depleted IFITM3 in U2OS cells, then infected with pseudoparticles expressing either influenza A virus envelope [H1(PR)] or or VSV-G (Fig. 4J). Decreased IFITM3 levels increased infection of the influenza A H1 pseudoviruses, with VSV-G entry elevated to a lesser extent only with the most potent siRNA, IFITM3-6 (Fig. 4J). Because the lifecycles of the pseudoviruses differ only in the means of entry mediated by their respective viral envelopes, these data are consistent with the IFITM proteins blocking influenza A virus infection early in the viral lifecycle, somewhere between and including viral-host receptor binding and entry of the vRNP into the cytosol.
Influenza A virus infection begins with the viral envelope proteins interacting with sialylated glycoproteins on the host cell's surface (Lamb and Krug, 2001). We found no reduction, and even a slight increase, in the levels of sialic acid (SA) with IFITM proteins over-expression, pointing away from a reduction in SA underlying the actions of the IFITM proteins (Fig. 4K). When the transduced cells were examined by flow cytometry, the N-terminal epitope tag was bound by antibody without membrane permeabilization, revealing that the N-terminus is extracellular (Fig. S5A). In addition, the C-terminus could also be detected in IF studies using non-permeabilized cells, demonstrating that it is also extracellular (Fig. S5B, lower right panel).
Deletion of the Murine Ifitm Locus Leads to Increased Influenza A Virus Infection in vitro
Human and murine IFITM proteins display a high degree of inter-species homology (Fig. S6). Thus, to examine the evolutionary conservation of IFITM protein function, we derived murine embryonic fibroblasts (MEFs) from a mouse strain, IfitmDel, deleted for all of the Ifitm genes (Ifitm1, 2, 3, 5 and 6, (Lange et al., 2008). In spite of the loss of these genes, the IfitmDel mice develop normally (Lange et al., 2008). Comparison of IfitmDel +/+, +/- and -/- MEFs revealed a marked increase in PR8 infection in the -/- cells (Fig. 5A). The differences in infection were more pronounced when the MEFs were cultured with IFN α or γ, prior to viral infection (Fig. 5A-C). Similar to human cells, we observed a vesicular staining pattern for Ifitm3 (Fig. 5D, 3C). Forced expression of Ifitm2 or 3 in the IfitmDel -/- cells restored resistance to influenza A H1N1 infection (Fig. 5E, F). We conclude that the Ifitm protein family accounts for a significant proportion of the anti-influenza actions of Type I and II IFNs in mice, and the majority of this function can be restored by the stable expression of Ifitm2 or 3.
Fig. 5. Ifitm knockout cells are more susceptible to influenza A H1N1 virus infection, and are protected by the reinstatement of Ifitm2 or 3 expression.
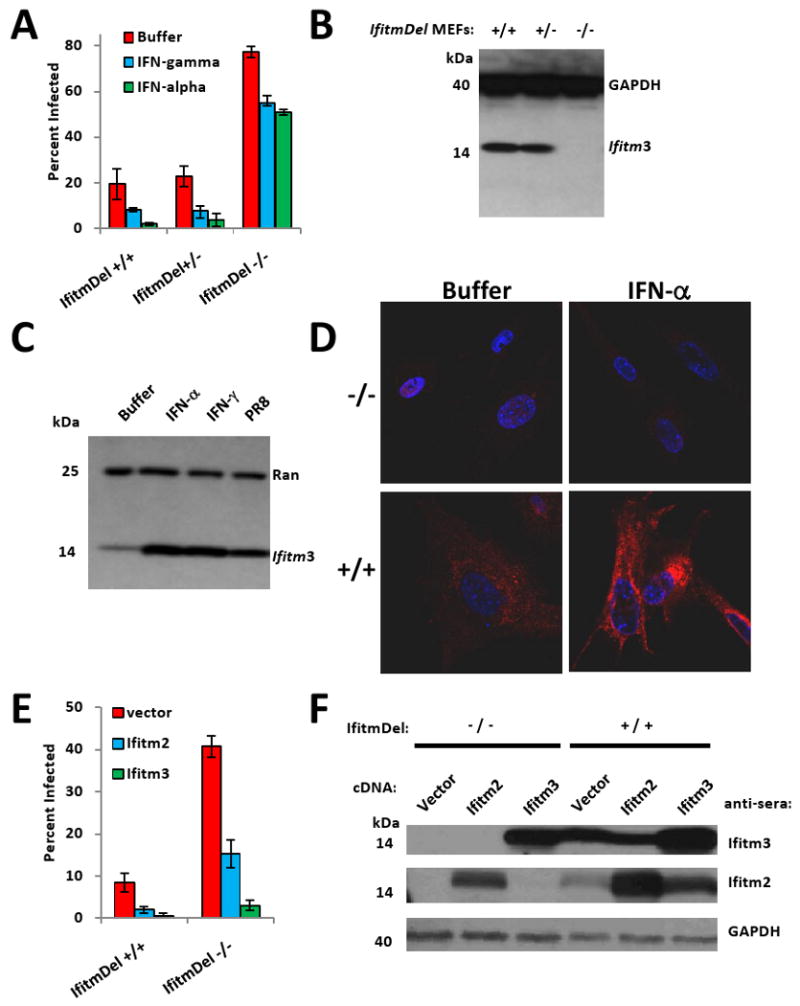
A) MEFs derived from the indicated IfitmDel mice, were left untreated (buffer), or treated with interferon-α or γ. After 24 h, the cells were incubated with influenza A virus H1N1 (PR8). Twelve h after infection, the cells were checked for HA surface expression. Values represent the mean ± SD, N=3.
B) MEFs from A) were assessed by Western blot for the presence of Ifitm3 protein. GAPDH levels are provided to show protein loading.
C) MEFs were left untreated (buffer), or incubated with either IFN-γ, IFN-α, or PR8 virus. After 24 h the levels of Ifitm3 were checked by Western blot.
D) IfitmDel +/+ MEFs, or IfitmDel -/- MEFs were incubated in the absence (buffer) or presence of IFN-α for 24 h, prior to staining with α-Ifitm3 (red: Ifitm3, nuclei: blue). 63×.
E) IfitmDel +/+ MEFs, or IfitmDel -/- MEFs stably expressing Ifitm2, 3 or the empty vector, were challenged with PR8 virus. 12 h later, the cells were fixed and imaged for HA expression. Values represent the mean ± SD, N=3.
F) The indicated MEFS were assessed for Ifitm2 and 3 expression by Western blot. GAPDH demonstrates protein loading.
IFITM3 Inhibits the Early Replication of West Nile Virus and Dengue Virus
We next explored the specificity of IFITM-mediated restriction by testing a panel of viral-like particles (VLPs) and pseudotyped viruses, each expressing a unique viral envelope protein. The VLPs expressed the envelope proteins of one of three flaviviruses, West Nile virus (WNV), yellow fever virus (YFV) or the Omsk hemorrhagic fever virus (OMSK). These VLPs can undergo a single round of infection, and are produced by transiently expressing the respective envelope proteins together with the WNV structural genes in cells stably expressing sub-genomic WNV replicons containing EGFP (Yoshii and Holbrook, 2009). As observed with influenza A pseudoparticles, all three VLPs were blocked by each of the IFITM proteins, demonstrating that these restriction factors impede first round infection (Fig 6A). Again, the IFITM proteins were found to inhibit VSV-G-mediated infection to a lesser extent (Fig. 6A). In contrast, pseudoparticles expressing the envelope proteins of three arenaviruses, lymphocytic choriomeningitis virus (LCMV), Lassa virus (LASV) and MACH, or the MLV retrovirus, were not affected by IFITM expression. We next tested the effects of IFITM protein levels on two pathogenic flaviviruses, WNV and DNV. The replication of the 2741 strain of WNV was dramatically decreased in either A549 or U2OS cells stably over-expressing IFITM3 (Fig. 6B, C). Furthermore, siRNA-depletion of IFITM3 protein also led to an increase in replication of both WNV (Fig. 6D, E) and DNV serotype 2 (New Guinea C strain, Fig. 6F, G). However, while IFITM3 did not inhibit hepatitis C virus (HCV), a more distantly related member of the Flaviviridae family, it did block influenza A virus infection in the same HCV-permissive liver cell line (Huh 7.5.1, Fig. S5D, E). We conclude that IFITM proteins restrict the replication of two additional human pathogens, DNV and WNV, and are likely to also limit YFV and OMSK infection based on the VLP data.
Fig. 6. The IFITM protein family restricts West Nile virus and dengue virus infections.
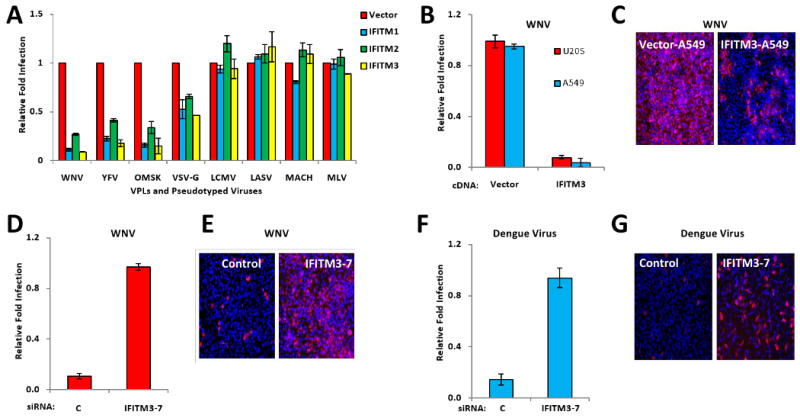
A) Vero E6 cells were transduced with retroviruses expressing the indicated IFITM proteins, or the empty viral vector. Two days later, the cells were incubated with flaviviral viral like particles (VLPs), expressing EGFP, and coated in envelope proteins from WNV, yellow fever virus (YFV) or Omsk virus (OMSK), or with EGFP-expressing MLV viruses pseudotyped with the indicated viral envelope proteins. Viral infection is expressed as mean EGFP fluorescence relative to vector control cells, as measured by flow cytometry 48 h post-infection. Values represent the mean ± SD, N=3.
B) A549 or U2OS cells stably expressing either IFITM3 protein or the vector alone (also shown in 4C, D), were infected with infectious WNV (strain 2741). 24 h. later, the cells were fixed and stained for viral E protein expression by IF. Values represent the mean ± SD, N=3
C) Images of A549 cells in B (red: WNV E protein, blue: nuclei), 4× magnification.
D) HeLa cells were transfected with the indicated siRNAs for 72 h, then infected with WNV. 24 h later, the cells were fixed and stained for viral E protein. Values represent the mean ± SD, N=3. C, Non-targeting siRNA negative control.
E) Images of HeLa cells in D (red: WNV E protein, blue: nuclei), 4× magnification.
F) HeLa cells were transfected with the indicated siRNAs for 72 h, then infected with dengue virus (New Guinea C strain). 30 h. post-infection, the cells were fixed and stained for viral E protein expression by IF. Values represent the mean ± SD, N=3. C, Non-targeting siRNA negative control.
G) Images of HeLa cells in F, 4×.
Discussion
Integrated Model of Influenza A Virus Host Factors
Although considerable knowledge exists regarding the function of viral proteins, the role of host factors in modifying infection is less understood. Therefore, we executed a genetic screen and identified over 120 human proteins needed by influenza A virus. The screen enriched for multiple host cell pathways including endosomal acidification, vesicular trafficking, mitochondrial metabolism, nucleocytoplasmic shuttling/mRNA export nuclear transport and RNA processing.
Our findings both support and extend those of a previous screen for influenza A virus-dependency factors using Drosophila cells (Hao et al., 2008). Among the factors identified in our primary screen are the human orthologs of 11 insect-cell host factors previously reported to be required for flu infection in fly cells (19.6% of the 56 identifiable human orthologues, Fig. 2, Dataset S1D). Within this common set, we recovered our positive control, NXF1, as well as NUP98, EIF4A2, ARCN1, COPG, PGD, RAB5A and RAB10. In addition to these exact candidate matches there was strong overlap between the screens within several biologic pathways and macromolecular complexes, and those of the Reactome's influenza A virus infection database (Fig. 2). This synthesis demonstrates the collective functional insights that unbiased large-scale mammalian and fly RNAi studies can provide in combination, and outlines many central functions and interactions required for the flu life cycle as well as new antiviral drug targets.
We have also constructed an enrichment analysis for host factors identified in the screen (Dataset S2), including a summary comparison of enriched GO categories for three whole genome screens for host factor modifiers of viral infection [HIV, (Brass et al., 2008), influenza A virus, this study, and WNV, (Krishnan et al., 2008)], wherein each screen's respective candidate gene list was analyzed separately for enriched GO categories. GO terms that were enriched for in one more of the screens are provided, with overlapping viral dependencies apparent for components involved in spliceosome activity, Golgi function, vesicular trafficking and proton transport (Dataset S2). These represent a core set of functions we now know to be shared among a very diverse set of viruses.
The IFITM Protein Family
Our screen identified the IFITM proteins as viral restriction factors. IFITM proteins were originally described 25 years ago based on their expression after IFN treatment (Friedman et al., 1984). The IFITM1, 2, 3 and 5 genes lie adjacently on chomosome 11. IFITM1, 2 and 3 are nearly ubiquitously expressed while IFITM5 is expressed in osteoblasts. The IFITM proteins have been ascribed roles in immune cell signaling, cell adhesion, oncogenesis, germ cell homing and maturation, and bone mineralization (Evans et al., 1993; Imai and Yoshie, 1993; Lange et al., 2003; Smith et al., 2006). However, with the exception of IFITM5's role in bone mineralization, we know of no other functional studies clearly demonstrating an additional function for an IFITM family member (Moffatt et al., 2008). Indeed, in our hands, transformed and primary cells either over-expressing or depleted for IFITM3 display no growth perturbations, and as noted, the IfitmDel mice develop and age normally (Lange et al., 2008).
The IFITMs belong to a protein domain super-family consisting of over 30 proteins, each possessing two trans-membrane domains and an intervening highly conserved intra-cellular loop (pfam04505, CD225, Interferon-induced transmembrane protein). Expression of members of the CD225 protein family have been reported in zebrafish, Xenopus, invertebrates, and bacteria, and homologs are found in frog, fish, fowl and mammals (mouse, rat, dog, swine, cow, primate, and human, Fig S6, S7). Now that an anti-viral role has been demonstrated, it will be interesting to determine if any of the related factors participate in host-pathogen interactions and, if so, how early in evolution this protein domain became associated with innate immunity.
The IFITM proteins are IFN-inducible Restriction Factors which Inhibit Influenza A and Flaviviral Infection at Entry
A vital component of the innate immune response to viral infection is mediated by restriction factors. The anti-viral action of the IFITM proteins was observed in primary human, chicken and mouse cells, in addition to multiple transformed cell lines, including dog cells permissive for influenza A virus replication. We also found that the IFITM proteins broadly inhibit the replication of all influenza strains tested, including two current vaccine strains. Furthermore, the IFITM proteins inhibit two highly pathogenic flaviviruses, WNV and DNV, and likely control YFV and OMSK, indicating a very general antiviral role. Previous work suggested that over-expression of human IFITM1 in murine fibroblasts partially blocked infection by VSV, but not by influenza A virus, although no loss of function experiments were reported (Alber and Staeheli, 1996). We also detected modest inhibition of VSV-G pseudoviruses but very strong inhibition of influenza A virus with all three IFITM proteins. However, the more distantly related hepacivirus, HCV, was not inhibited by IFITM3 levels (Fig. S5D, E), nor was HIV (Fig. S3F) or pseudoparticles bearing the envelope proteins of multiple arena viruses.
Although the basal levels of IFITM3 contribute to viral resistance, increased expression of IFITM proteins further enhances cellular defense. Consistent with this notion, we observed that depletion of IFITM3 resulted in loss of 40% to 70% of IFN's protective effect, with a similar diminution also detected in the IfitmDel mouse cells. Thus, IFITM proteins are critical for the innate immunity to influenza A virus afforded by IFNs. Thus these results contribute to our understanding of IFN action, in that they demonstrate that these anti-viral cytokines block viral entry by inducing the expression of the IFITM proteins.
Possible Mechanisms of IFITM Restriction of Influenza and Flavivirus Infection
As schematized in Fig. 7, the IFITM proteins could act at any stage of viral entry, for example by directly binding the virus and inhibiting interactions with host cell receptors (A), blocking receptor access (B), inhibiting endocytosis, preventing viral membrane fusion or rerouting vesicular traffic to a nonproductive end (C), acting as pattern recognition receptors on the cell surface or on endocytosed vesicles where they could signal to downstream anti-viral effectors (D). Logic would further predict that IFITM proteins could target a common step in the viral life cycle. While influenza A virus and flaviviruses bind distinct receptors, both are endocytosed through a clathrin-dependent pathway, although influenza A also has a clathrin independent pathway (Chen and Zhuang, 2008; Sieczkarski and Whittaker, 2002). After endocytosis, early endosomes containing influenza A virus fuse with late endosomes where a pH < 5.5 triggers the HA-directed fusion of the viral and endosomal membranes, permitting vRNP entry (Fig. 7, (Lamb and Krug, 2001). In contrast, flaviviruses undergo fusion in early endosomal compartments, and at a considerably higher pH of 6.5 (Krishnan et al., 2007; Sanchez-San Martin et al., 2009). Thus, a very general overlap involving clathrin-mediated endocytosis and trafficking into early endosomes, exists between influenza A virus and flaviviruses.
Fig. 7. IFITM proteins act as anti-viral restriction factors.
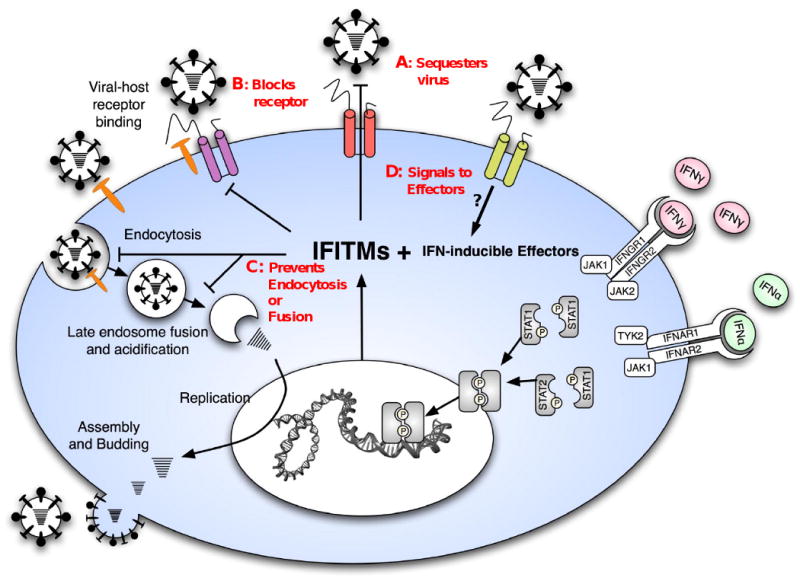
A schematic model of the influenza A virus life cycle, the induction of IFITM proteins and their role in blocking Influenza A virus infection. IFITM1, 2 and 3 are represented by the three multi-transmembrane proteins. Red legends indicate possible mechanisms of restriction, IFITM proteins may; A) sequester the incoming viruses at or near the surface, B) block viral receptors from interacting with host receptors (shown in orange), C) prevent endocytosis or viral membrane fusion D) act as receptors, and after binding the virus, signal to effectors.
However, these common entry steps are shared to varying degrees by several of the viruses not impacted by IFITM proteins. For instance, sparing of the arenaviruses (MACH, LCMV, LASV) argues against a block to general endocytosis or clathrin-mediated endocytosis. Moreover, like flaviviruses, the arenaviruses also traffic in early endosomes, where they undergo pH-dependent fusion, thereby leaving no unique entry step shared by the two viral families impacted by the actions of the IFITM proteins. However, we cannot rule out that these resistant viruses may have multiple entry or endosome escape pathways, or may have evolved to circumvent terminal rerouting by IFITMs. We would also note, however, that, co-culture of IFITM3 over-expressing cells with parent control cells did not confer protection, suggesting a cell-autonomous mode of action (data not shown). Therefore, higher resolution studies will now be required to determine the precise mode(s) of viral interference employed by the IFITM proteins.
We identified three additional candidate influenza A virus restriction factors, PUSL1, TPST1, and WDR33. It is possible that these might work together with IFITM proteins to execute viral resistance and it remains to be determined whether they block at the same point in the life cycle. WDR33 is an orphan WD40 repeat protein about which little is known. TPST1 is a golgi-localized transmembrane tyrosylprotein sulfotransferase which is known to sulfonate proteins destined for the cell surface or secretion (Hoffhines et al., 2006). PUSL1 shares homology with pseudouridylate synthase genes which modify RNA by which converting uridine into pseudouridine, a glycosylated form of uracil, in several cytosolic and mitochondrial tRNAs (Massenet et al., 1999). While further validation is warranted, it is tempting to speculate that PUSL1could potentially block influenza by directly modifying viral RNA.
The IFITM proteins and other innate cell-intrinsic defenders present opportunities not just for a greater understanding of fundamental questions, but also as tools to actively combat current and emerging pathogens. Variations in the basal and inducible levels of these factors as well as the dependency factors within a population might predict the severity of flu or flaviviral infections among individuals, or across species. The discovery of the roles of IFITM proteins in innate immunity has relevance to ongoing and future influenza pandemics. Not only could elucidation of the IFITM restriction mechanism prove important in designing new anti-viral therapies, but the proteins themselves could be used in multiple ways to fight influenza A, WNV and dengue virus. If IFITM proteins work on the plasma membrane, they could possibly be delivered to tissues susceptible to initial infection by liposomal transfer. Transgenic animals such as fowl or swine could be developed that over-express IFITM proteins to provide resistance to influenza A virus and other pathogens, thereby preventing the spread of these viruses, as well as limiting their ability to recombine with human influenza A strains to produce strains dangerous to human populations. Indeed, by creating animals with multiple transgenic restriction factors, we can confront the virus with a more intractable barrier. Finally, if IFITM proteins are also rate-limiting for influenza A virus infection in other organisms such as chickens, whose embryos are employed to passage attenuated viruses for vaccine production, the inhibition of IFITM protein expression could reduce the amount of time it takes to produce vaccine and thereby boost yields. This has been a critical issue confounding vaccine production in the current influenza pandemic. The discovery of the role of IFITM proteins as antiviral agents for multiple devastating pathogenic viruses has given us new insights into innate immunity and has provided us new tools with which to counter viral propagation in the future.
Experimental Procedures
siRNA screen: For the siRNA screen we employed an arrayed library targeting 17,877 genes (Dharmacon siARRAY siRNA Library (Human Genome, G-005000-05, Thermo Fisher Scientific, Lafayette, CO). siRNAs were transiently reverse transfected into the U2OS cells in triplicate at a 50 nM final concentration, using a final concentration of 0.32% Oligofectamine (Invitrogen) in a 384-well format. After 72 h, the medium was removed and the cells were infected with the Influenza A/Puerto Rico/8/34 (PR8, ATCC VR-1469), MOI of ∼ 0.2-0.3 in 40 uL complete media. After 12 h, media was removed, cells were fixed with 4% formalin and stained with anti-HA antibodies (Hybridoma HA36-4-5.2, Wistar Institute), followed by an Alexa Fluor 488 goat anti-mouse secondary at 1:1,000 (A11001, Invitrogen). Cells were imaged on an automated Image Express Micro (IXM) microscope (Molecular Devices) and analyzed using the Metamorph Cell Scoring software program (Molecular Devices Inc.). The validation round for single siRNAs was done as described previously (Brass et al., 2008) with 20 nM siRNA. All Dharmacon siRNAs and plasmids used for generating stable cell lines are shown in Dataset S1 and the Supplemental Experimental Procedures.
Cell lines and culture conditions: U2OS, A549, MDCK, 293T, Huh7.5.1, primary Chicken fibroblast cells (ChEFs, Charles River Labs), Vero E6 and HeLa cells were grown in DMEM (Invitrogen Cat#11965) with 10% FBS (Invitrogen). WI-38 cells were cultured in DMEM (Invitrogen Cat#10569), containing 1X MEM non-essential amino acids (Invitrogen Cat#11140, 10 mM stock/100×) and 15% FBS. Adult IfitmDel+/- mice (Lange et al., 2008) were intercrossed and fibroblasts (MEFs) derived from embryos at day 13.5 of gestation, as described previously (Nagy et al., 2003) and in the Supplemental Experimental Procedures.
Viral propagation and titration: influenza A (H1N1) A/PR/8/34 (ATCC VR-1469), influenza A (H1N1) A/WS/33 (ATCC VR-1520), influenza A (H1N1) A/WSN/33 and influenza (H3N2) A/Udorn/72 were propagated and viral infectivity was titrated as previously described (Huang et al., 2008). Hybrid Moloney/Amphotropic murine leukemia virus (MLV, ATCC VR-1450) was propagated in NIH3T3 cells. WNV (strain 2741) and DNV serotype 2 (New Guinea C strain) viruses were grown on Vero cells.
West Nile (WNV) and dengue (DNV) virus infections: West Nile (strain 2741) and dengue serotype 2 (New Guinea C strain) viruses were used to infect the IFITM3-silenced HeLa cells at an MOI of 0.1 for 24 or 30 h respectively, as reported previously (Krishnan et al., 2008). Infected cells were fixed in 4% PFA and immuno-stained with antibodies detecting viral E-proteins (Chemicon), and imaged by fluorescence microscopy (Zeiss). IFITM3 over-expressing or vector control-A549 or -U20S cells were infected with WNV at an MOI of one.
Influenza A virus and MLV infection: Influenza A virus A/PR/8/34 (H1N1) (MOI =5), A/Udorn/72 (H3N2) (MOI=1), and MLV were used to infect A549 cells expressing different IFITM proteins. 24 h later, infected cells were labeled with murine anti-influenza viral H1 IgG2a (C179), anti-influenza viral H3 IgG1 (F49) (Takara Bio. Cat#M145 and M146), or goat anti-MLV env polyclonal antibodies (ATCC), and stained with PE-conjugated anti -mouse or anti-goat secondary antibodies. Cells were then fixed with 1% formaldehyde and analyzed by flow cytometry.
VLP and Pseudotyped Virus: MLV-GFP pseudoviruses were made as described (Huang et al., 2006; Huang et al., 2008). Flavivirus VLP were as described (Hanna et al., 2005), except plasmids encoding structure proteins of WNV (strain NY99), yellow fever virus (strain D17), or Omsk hemorrhagic fever virus (Ref. Seq.: NP_878909.1) were used. VLP and pseudovirus entry in A549 or Vero E6 cells expressing IFITM proteins was assessed 2 d later by measuring GFP expression by flow cytometry. The infection level of siRNA-transfected U2OS cells after 2 d infection was determined by calculating the percent GPF positive cells by IF after fixation with 4% PFA and staining of nuclei with Hoechst 33342.
Intracellular HA-staining was performed as above with the exception that after PFA fixation, cells were incubated in 0.1-0.2% Tween 20 (Sigma), then blocked in 1% BSA with 0.3M glycine in D-PBS, prior to staining with the primary antibody. This identical protocol was used with staining for NP (Abcam, AA5H, ab20343, 1:1000), M2 (Abcam, 14C2, ab5416, 1:1000), Anti-HA7 from Sigma-Aldrich (Product code H 3663, which recognizes the HA nonapeptide tag on IFITM3R6, but not PR8's HA), and monoclonal antibody against Human Influenza A virus (H1N1, H2N2) (Takara, C179, Cat#M145, 1:1000), which recognizes the HA of WS/33, but not of PR8. Sialic acid staining has been described (Huang et al., 2006; Huang et al., 2008).
Enrichment Analysis
Statistical analysis of gene enrichment was performed using a hypergeometric distribution as decribed in the GOhyperGAll module of Bioconductor for gene ontology terms (Gentleman et al., 2004). A map of the viral lifecycle was created by connected keywords. Genes were mapped to these keywords using a database that integrates annotation information from UniProt (Bairoch et al., 2005), KEGG (Kanehisa et al., 2004), Reactome (Vastrik et al., 2007), Gene Ontology (Ashburner et al., 2000), NCBI GeneRIF (Mitchell et al., 2003) and OMIM Human orthologs were mapped to other species using NCBI HomoloGene (Wheeler et al., 2005)(see Supplemental Experimental Procedures for more details).
Supplementary Material
Acknowledgments
We thank the ICCB-Longwood; C. Shamu, S. Chang, S. Rudnicki, S. Johnston, K. Rudnicki, D. Wrobel, M. Ocana and Z. Cooper. Ragon Institute; L. Whiteman, K. Hartman, A. Piechocka-Trocha, J. Proudfoot, T. Diefenbach. We thank J. Philips, A. Mehle, M. Franti, F. Diaz-Griffero, J. Mabry (CDC) and J. Chou for helpful discussions. Funding support; A. L. B. (MGH GI Unit, Harvard Center for AIDS Research), M.N.K and E.F. (NIH grants AI 50031 and AI070343), Y.B and R.J.X. (CSIBD, Genetics and Genomics Core and NIH grant AI062773, DK060049, and DK043351), I.C., and M.R.F (NERCE U54 AI057159). D.J.A is supported by Cancer Research UK and the Wellcome Trust. L.vdW. is supported by a fellowship from the Kay Kendall Leukaemia Foundation. This study was supported by the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NIH grant U54 AI057159 to D Kasper). S.J.E. and E.F. are Investigators with the Howard Hughes Medical Institute.
Footnotes
Additional materials and methods are included in the supplement.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alber D, Staeheli P. Partial inhibition of vesicular stomatitis virus by the interferon-induced human 9-27 protein. J Interferon Cytokine Res. 1996;16:375–380. doi: 10.1089/jir.1996.16.375. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulo S, Akarsu H, Ruigrok RW, Baudin F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007;124:12–21. doi: 10.1016/j.virusres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Bouloy M, Plotch SJ, Krug RM. Globin mRNAs are primers for the transcription of influenza viral RNA in vitro. Proc Natl Acad Sci U S A. 1978;75:4886–4890. doi: 10.1073/pnas.75.10.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–682. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhuang X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc Natl Acad Sci U S A. 2008;105:11790–11795. doi: 10.1073/pnas.0803711105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SS, Collea RP, Leasure JA, Lee DB. IFN-alpha induces homotypic adhesion and Leu-13 expression in human B lymphoid cells. J Immunol. 1993;150:736–747. [PubMed] [Google Scholar]
- Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell. 1984;38:745–755. doi: 10.1016/0092-8674(84)90270-8. [DOI] [PubMed] [Google Scholar]
- Ge Q, McManus MT, Nguyen T, Shen CH, Sharp PA, Eisen HN, Chen J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci U S A. 2003;100:2718–2723. doi: 10.1073/pnas.0437841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glodowski DR, Chen CC, Schaefer H, Grant BD, Rongo C. RAB-10 regulates glutamate receptor recycling in a cholesterol-dependent endocytosis pathway. Mol Biol Cell. 2007;18:4387–4396. doi: 10.1091/mbc.E07-05-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- Haller O, Staeheli P, Kochs G. Protective role of interferon-induced Mx GTPases against influenza viruses. Rev Sci Tech. 2009;28:219–231. doi: 10.20506/rst.28.1.1867. [DOI] [PubMed] [Google Scholar]
- Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffhines AJ, Damoc E, Bridges KG, Leary JA, Moore KL. Detection and purification of tyrosine-sulfated proteins using a novel anti-sulfotyrosine monoclonal antibody. J Biol Chem. 2006;281:37877–37887. doi: 10.1074/jbc.M609398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang IC, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem. 2006;281:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang IC, Li W, Sui J, Marasco W, Choe H, Farzan M. Influenza A virus neuraminidase limits viral superinfection. J Virol. 2008;82:4834–4843. doi: 10.1128/JVI.00079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Yoshie O. C33 antigen and M38 antigen recognized by monoclonal antibodies inhibitory to syncytium formation by human T cell leukemia virus type 1 are both members of the transmembrane 4 superfamily and associate with each other and with CD4 or CD8 in T cells. J Immunol. 1993;151:6470–6481. [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R, Tsui M, Qian F, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan MN, Sukumaran B, Pal U, Agaisse H, Murray JL, Hodge TW, Fikrig E. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J Virol. 2007;81:4881–4885. doi: 10.1128/JVI.02210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RA, Krug RM. Orthomyxoviridae: The viruses and their replication. 4th. Philadelphia: Lippincott Williams and Wilkins; 2001. [Google Scholar]
- Lange UC, Adams DJ, Lee C, Barton S, Schneider R, Bradley A, Surani MA. Normal germ line establishment in mice carrying a deletion of the Ifitm/Fragilis gene family cluster. Mol Cell Biol. 2008;28:4688–4696. doi: 10.1128/MCB.00272-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange UC, Saitou M, Western PS, Barton SC, Surani MA. The fragilis interferon-inducible gene family of transmembrane proteins is associated with germ cell specification in mice. BMC Dev Biol. 2003;3:1. doi: 10.1186/1471-213X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin AR, Reid LE, McMahon M, Stark GR, Kerr IM. Molecular analysis of a human interferon-inducible gene family. Eur J Biochem. 1991;199:417–423. doi: 10.1111/j.1432-1033.1991.tb16139.x. [DOI] [PubMed] [Google Scholar]
- Maines TR, Szretter KJ, Perrone L, Belser JA, Bright RA, Zeng H, Tumpey TM, Katz JM. Pathogenesis of emerging avian influenza viruses in mammals and the host innate immune response. Immunol Rev. 2008;225:68–84. doi: 10.1111/j.1600-065X.2008.00690.x. [DOI] [PubMed] [Google Scholar]
- Massenet S, Motorin Y, Lafontaine DL, Hurt EC, Grosjean H, Branlant C. Pseudouridine mapping in the Saccharomyces cerevisiae spliceosomal U small nuclear RNAs (snRNAs) reveals that pseudouridine synthase pus1p exhibits a dual substrate specificity for U2 snRNA and tRNA. Mol Cell Biol. 1999;19:2142–2154. doi: 10.1128/mcb.19.3.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Aronson AR, Mork JG, Folk LC, Humphrey SM, Ward JM. Gene indexing: characterization and analysis of NLM's GeneRIFs. AMIA Annu Symp Proc. 2003:460–464. [PMC free article] [PubMed] [Google Scholar]
- Moffatt P, Gaumond MH, Salois P, Sellin K, Bessette MC, Godin E, de Oliveira PT, Atkins GJ, Nanci A, Thomas G. Bril: a novel bone-specific modulator of mineralization. J Bone Miner Res. 2008;23:1497–1508. doi: 10.1359/jbmr.080412. [DOI] [PubMed] [Google Scholar]
- Monto AS. The risk of seasonal and pandemic influenza: prospects for control. Clin Infect Dis. 2009;48 1:S20–25. doi: 10.1086/591853. [DOI] [PubMed] [Google Scholar]
- Morriswood B, Ryzhakov G, Puri C, Arden SD, Roberts R, Dendrou C, Kendrick-Jones J, Buss F. T6BP and NDP52 are myosin VI binding partners with potential roles in cytokine signalling and cell adhesion. J Cell Sci. 2007;120:2574–2585. doi: 10.1242/jcs.007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhaei P, Genin P, Civas A, Hiscott J. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol. 2009;21:215–222. doi: 10.1016/j.smim.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Sanchez-San Martin C, Liu CY, Kielian M. Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 2009;17:514–521. doi: 10.1016/j.tim.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002;76:10455–10464. doi: 10.1128/JVI.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieczkarski SB, Whittaker GR. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic. 2003;4:333–343. doi: 10.1034/j.1600-0854.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC. Influenza viruses and cell membranes. Am J Respir Crit Care Med. 1995;152:S13–15. doi: 10.1164/ajrccm/152.4_Pt_2.S13. [DOI] [PubMed] [Google Scholar]
- Smith RA, Young J, Weis JJ, Weis JH. Expression of the mouse fragilis gene products in immune cells and association with receptor signaling complexes. Genes Immun. 2006;7:113–121. doi: 10.1038/sj.gene.6364278. [DOI] [PubMed] [Google Scholar]
- Stevens SW, Barta I, Ge HY, Moore RE, Young MK, Lee TD, Abelson J. Biochemical and genetic analyses of the U5, U6, and U4/U6 × U5 small nuclear ribonucleoproteins from Saccharomyces cerevisiae. RNA. 2001;7:1543–1553. [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- Vastrik I, D'Eustachio P, Schmidt E, Gopinath G, Croft D, de Bono B, Gillespie M, Jassal B, Lewis S, Matthews L, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39. doi: 10.1186/gb-2007-8-3-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Church DM, DiCuccio M, Edgar R, Federhen S, Helmberg W, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005;33:D39–45. doi: 10.1093/nar/gki062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii K, Holbrook MR. Sub-genomic replicon and virus-like particles of Omsk hemorrhagic fever virus. Arch Virol. 2009;154:573–580. doi: 10.1007/s00705-009-0345-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.