

Abstract
Loss of up to four amino acids from the C terminus of the 1318 residue bacteriophage T7 gp16 allows plaque formation at normal efficiencies. Loss of five residues results in non-infective virions, and loss of twelve prevents assembly of stable particles. However, replacing the C-terminal seven with nineteen non-native residues allows assembly of non-infective virions. The latter adsorb and eject internal core proteins into the cell envelope but no phage DNA enters the cytoplasm. Extragenic suppressors of the defective gene 16 lie in gene 15; the mutant gp15 proteins not only re-establish infectivity, they fully restore the kinetics of genome internalization to those exhibited by wild-type phage. After ejection from the infecting particle, gp15 and gp16 thus function together in ratcheting the leading end of the T7 genome into the cytoplasm of the infected cell.
Keywords: bacteriophage, infection, ejected virion proteins, membranes, genome entry
INTRODUCTION
Bacteriophage T7 is the prototype of a large number of related members of the Podoviridae that infect enteric and other Gram-negative bacteria (Molineux, 2006a). The short, stubby tail of the T7-like phages is too short to span the cell envelope and, in order to eject the phage genome into the cell at the initiation of infection, virion proteins must first make a channel from the tip of the tail into the cell cytoplasm. T7 and all its close relatives code for three proteins that are known to form a cylindrical structure inside the phage head, and these proteins are ejected from the infecting virion into the cell envelope before the phage genome. The ejected proteins are thought to form a trans-envelope channel that connects the virion tail tip to the cell cytoplasm (Fig. 1A). T7 has therefore been described as having an extensible tail (Molineux, 2001), in contrast to the more familiar contractile tails of the Myoviridae. A quantification of T7 virion proteins estimated that the internal core contains about ten copies of gp14, eight copies of gp15, and four copies of gp16 (Kemp et al., 2005). About 18 copies of gp6.7 are also inside the capsid but it is not known if they form part of the internal core. In addition to their role in initiating infection, the internal core proteins are essential for morphogenesis of a mature infective virion (Serwer, 1976, Roeder and Sadowski, 1977). This duality of function makes it difficult to distinguish the effects of mutations in those genes on virus assembly from the subsequent infection process.
Fig. 1.

A. Schematic of a T7 virion before and after ejection of its internal core proteins. The location of the head protein gp6.7 is not known, and is not shown. B. Predicted secondary structure of the C terminus of gp16 by SOPM (Combet et al., 2000). Tubes represent predicted α-helices. Amino acids whose codons were mutated to amber in this study are underlined. The solid arrowhead indicates the 3′-proximal amber mutation that, when not suppressed, fails to produce stable capsids. The site of fusion of non-specific residues in gp16-CΔ7-F7, -F9, and –F19 (-Fm), which are predicted to be in a mainly extended conformation, is shown by the open arrowhead.
Reconstructions of the T7 particle revealed that the internal core is layered, with an overall cylindrical shape (Agirrezabala et al., 2005a,b). Gp6.7 was not considered in this reconstruction, and it is possible that some of the electron density associated with the internal core proteins gp14, gp15 and gp16 actually represents gp6.7. Gp14 lies at the base of the cylinder, closest to the portal protein, followed by gp15 and with gp16 at the top (Fig. 1A). No enzymatic activity has yet been described for gp14 or gp15 but the N-terminal domain of gp16 harbors a lytic transglycosylase activity (Moak and Molineux, 2004). T7 particles lacking muralytic activity develop normally in exponentially growing cells at 30°C but cannot undergo a successful infection at ≤ 20°C or when cells are entering stationary phase. Thus, even the Gram-negative cell wall can provide a barrier to efficient phage infection.
The central region of T7 gp16 is actively involved in transporting the phage genome into the infected cell. After phage adsorption and protein ejection, approximately 1 kb of the 40 kb genome is normally internalized by the cell (Zavriev and Shemyakin, 1982; Moffatt and Studier, 1988; García and Molineux, 1995). The mechanism of translocation of the leading part of the T7 genome is not yet understood in detail. Infecting virions harboring a mutant gp16 fail to terminate this initial stage of DNA ejection; more than 20 different amino acid substitutions affecting T7 gp16 have been shown to confer the same phenotype (García and Molineux, 1996; Struthers-Schlinke et al., 2000). Different mutants confer a similar rate of DNA internalization, constant across the entire 40 kb genome. It was concluded that the mutations were not directly affecting the DNA translocation mechanism but were simply inactivating the process normally terminating genome internalization after the leading ~1 kb has entered the cytoplasm. The rate of DNA translocation varies predictably with temperature, having the properties of an enzyme-catalyzed reaction that follows Arrhenius kinetics (Kemp et al., 2004). Thus, internalization of the T7 genome is not driven by any forces inside the virion that may result from the high packing density of DNA, or by a theoretical gain in entropy following genome translocation into the cell cytoplasm. Furthermore, as expected for an enzyme-catalyzed reaction, DNA translocation requires energy: collapsing the membrane potential while DNA is entering the cell cytoplasm immediately stops this mode of DNA translocation.
Subsequent to internalization of the leading ~1 kb, translocation of T7 DNA is normally coupled to transcription, first by E. coli and then by T7 RNAP (Zavriev and Shemyakin, 1982; Moffatt and Studier, 1988; García and Molineux, 1995). If transcription is blocked in the infected cell, genome internalization stops after the leading ~1 kb has entered. Three promoters for E. coli RNAP and one T7 promoter lie on this internalized segment, and transcription from these promoters normally pulls the genome into the cell. In the absence of T7 RNAP, E. coli RNAP internalizes the entire T7 genome at a constant 40 bp/sec at 30°C, approximately the same rate of rrn operon transcription in vivo (García and Molineux, 1995; Kemp et al., 2004; Vogel and Jensen, 1994). Similarly, if T7 RNAP is present in the cell prior to infection, it catalyzes genome internalization in a rifampicin-treated cell at about 250 bp/sec (García and Molineux, 1995), in good agreement with the optimal rate of transcription in vitro (Anand and Patel, 2006). There is thus no obvious energy requirement in pulling a phage genome into the infected cell beyond what is normally provided by NTP hydrolysis during transcription.
As part of our ongoing studies on the mechanism of T7 genome internalization, we have constructed variants of gp16 that lack its normal C terminus. We show that removal of the last four residues of the 1318 amino acid protein allows plaque formation but loss of five abolishes infectivity, even though virions are assembled and can be purified from cell lysates by normal procedures. Twelve or more C-terminal residues must be removed to prevent morphogenesis of stable virions. Using a mutant where the last seven residues of wild-type gp16 are replaced by non-native sequences, we show that the defective phage virions adsorb and eject their core proteins but no T7 DNA can be detected in the cell cytoplasm. Extragenic suppressors restoring normal infectivity lie in gene 15, suggesting that gp15 and gp16 cooperate in translocating the T7 genome into the cell.
RESULTS
The charged C terminus of gp16 is essential for infectivity
The secondary structure of gp16 is strongly predicted to be a series of α-helices (>50% of the total residues) separated by unstructured regions, and with only ~20% β-sheet. The very C terminus is expected to be largely unstructured; however, two mutations described below prevent the formation of the last predicted α-helix (Fig. 1B).
The C-terminal five residues of wild type gp16 are all charged, a feature conserved in the closest relatives of T7, and we had anticipated that they would prove critical either for phage viability or for virion assembly. Removal of two or four amino acids from the C terminus of gp16 (plasmids 16-CΔ2, 16-CΔ4, Table 1) allows complementation and thus plaque formation of Δ16 phage at close to normal efficiencies, relative to the full-length protein. However, plaques were distinctly smaller and more variable. The last four charged residues at the gp16 C terminus are therefore important but not essential for virion morphogenesis and/or infection. Removing five amino acids (gp16-CΔ5), thereby removing all charged residues, reduces the efficiency of plating (eop) by about three orders of magnitude. However, assembly of non-infective virions still occurs. A single round of Δ16 growth on cells supplying gp16-CΔ5 yields virions that can be purified through CsCl density gradients, but they are not able to infect cells productively. The positively charged C terminus of T7 gp16 therefore appears to be essential for infectivity.
Table 1.
Properties of mutant T7 gp16.
| Gene 16a | gp16 C-terminal Sequence (in Su− cells) | Particle formation | Δ16 EOPc |
|---|---|---|---|
| 16+ (1318 aa) | + | 1 (2) | |
| 16-CΔ2 | + | 1.1 (1) | |
| 16- CΔ4 | + | 0.75 (1) | |
| 16- CΔ5 | + | 1.9 × 10−3 (0.5) | |
| 16- CΔ6 | + | 2.0 × 10−6 (l) | |
| 16- CΔ9 | + | 4.0 × 10−6 (l) | |
| 16 CΔ11-F2b | + | 3.5 × 10−6 (l) | |
| 16- CΔ12 | − | <10−9 | |
| 16- CΔ19 | Q1299 | − | <10−9 |
| 16-CΔ7-F19c | + | 6.0 × 10−6 (2) | |
| 16-CΔ7-F9c | + | 2.2 × 10−6 (2) | |
| 16-CΔ7-F7c | + | 1.3 × 10−6(2) |
Gene 16 plasmids used to complement T7 Δ16. CΔn refers to the removal of n amino acids in Su− cells from the C-terminus of wild-type gp16. CΔn-Fm refers to the loss of n codons, relative to gene 16+, followed by m non-specific codons fused (F) in frame.
Isolated from the site-specific mutagenesis producing 16-CΔ9.
Relative to cells containing wild-type gene 16. Numbers in parentheses are the average plaque sizes in mm.
The loss of six or nine amino acids (gp16-CΔ6 and gp16-CΔ9) reduces the eop of Δ16 by a further three orders of magnitude to ~10−6 (Table 1). Most plaques were no larger than pinpricks, but phages can be propagated and are thus pseudorevertants that overcome the defect of the mutant gp16. However, no plaques (eop <10−9) were found using plasmids providing gp16-CΔ12 or gp16-CΔ19, where 12 or 19 amino acids were removed. These two proteins did not support the formation of T7 virions that could be purified by CsCl density gradients. The 16-CΔ12 or 16-CΔ19 mutations disrupt the last predicted α-helix in gp16 (Fig. 1B), which must therefore be important in the morphogenesis of stable T7 virions.
The complementing gene 16 had been cloned into the polylinker of the plasmid pWSK29, allowing straightforward constructions of deletions extending from the unique HpaI site seven codons upstream of the termination codon. Three constructs are shown in Table 1, where a tail of 19, 9, and 7 polylinker-derived amino acids are fused, in frame, after residue 1311 (gp16-CΔ7-F19, -F9 and -F7, respectively). All three plasmids allowed virion assembly following infection by T7 Δ16 but progeny phages were not infective (Table 1).
Subcellular location of T7 virion proteins after infection
The three T7 internal core proteins, gp14, gp15, and gp16 have been suggested to constitute the extensible tail of T7 (Fig. 1A), forming a channel across the cell envelope that can allow subsequent genome translocation into the infected cell (Kemp et al., 2005; Molineux, 2001). In order to demonstrate this directly, we infected 35S-labeled T7 into IJ1133 cells in the presence of rifampicin, where only ~1 kb of the genome enters the infected cell. Any re-localization of ejected proteins that may occur when genome internalization is complete should therefore be prevented. Infected cells were harvested and washed extensively to release T7 proteins from the cell surface, thereby separating them from ejected proteins that had penetrated the cell envelope. The aqueous eluate and a portion of the cell pellet were then subjected to SDS-PAGE. As previously demonstrated (Kemp et al., 2005), the three internal core proteins, gp14, gp15, and gp16 are found in the cell-associated fraction (Fig. 2A). The remainder of the cell pellet was lysed by passage through a French press and, following removal of unbroken cells, cellular membranes were isolated by centrifugation. The membrane pellet was extracted with Sarkosyl to separate the insoluble outer and soluble inner membrane proteins (Filip et al., 1973). Essentially all the gp14 remained insoluble, indicating that it had localized to the outer membrane. However, a portion of both gp15 and gp16 are solubilized, indicating that they may also be associated with the cytoplasmic membrane.
Fig. 2.

Elution and SDS-PAGE separation of 35S-labeled T7 proteins from infected IJ1133 cells. Virions contain gp16+. A. Lane 1: phage marker; lane 2: phage-infected cells; lane 3: phage proteins eluted from the surface of infected cells; lane 4: phage proteins retained by the infected cell; lanes 5 and 6: Sarkosyl-soluble (5) and -insoluble (6) membrane proteins from infected cells. B and C. Fractionation of 35S-labeled T7 proteins from infected HM130 cells, a protease-deficient strain. B. After elution of surface-bound material, infected cells were lysed, and the inner and outer membranes were fractionated on a sucrose gradient. NADH oxidase assays on gradient fractions define inner membrane fractions. Outer membrane fractions were localized after SDS-PAGE by immunoblotting using an anti-OmpA antibody. C. SDS-PAGE of infected cells before and after membrane separation.
An alternative procedure for distinguishing outer and inner membrane proteins is to subject a total membrane preparation to sucrose gradient centrifugation where the two membranes sediment differently (Osborn et al., 1972). In this experiment, we used a protease-deficient strain to prevent degradation of the ejected proteins gp6.7 and gp7.3. The profile of radioactive T7 proteins across the sucrose gradient is shown in Figure 2B, where assays for OmpA and NADH oxidase defined, respectively, the outer and inner membranes. Fractions from the gradient were pooled and analyzed by SDS-PAGE. The majority of the internal core protein gp14 co-sediments with the outer membrane, whereas both gp15 and gp16 spread across the entire gradient (Fig. 2C). This suggests that the two proteins form a complex that associates with both the outer and inner membrane, consistent with the idea that gp15 and gp16 span the periplasm. It is likely that gp15/16 remain associated with gp14 in the infected cell, as they obviously do in the internal core structure inside the mature phage particle. In support of this idea, electron microscopy of T7 particles in lysates has revealed partially emptied capsids with a 40–55 nm extension to the tail penetrating membrane vesicles (Serwer, 1978; Serwer et al., 2008). This needle-like tail extension was assumed to consist of the ejected core proteins.
In the protease-deficient host used in this experiment, both T7 gp6.7 and gp7.3 are found predominately in the outer membrane. This subcellular location might be expected for proteins that must be ejected from an infecting virion before the internal core proteins can leave the virion in order to breach the entire cell envelope.
Ejection of core proteins from virions containing gp16-CΔ7-F19
Virions containing gp16-CΔ7-F19 are non-infective. It was therefore of interest to determine whether the inability to make plaques was due to the failure of one or more of the internal core proteins to be ejected normally into the infected cell. Therefore, as described above for wild-type T7, we infected HM130 cells with phage containing labeled gp16-CΔ7-F19. Following the wash steps, the aqueous eluate and cell pellet were subjected to SDS-PAGE. All three internal core components, gp14, gp15 and the mutant gp16 are found in the cell-associated fraction (Fig. 3). Isolated membranes from the washed, mutant-infected cells were extracted with Sarkosyl to separate insoluble outer and soluble inner membrane proteins. As after infection by wild-type, essentially all the gp14 remained insoluble, indicating that it had localized to the outer membrane whereas a portion of both gp15 and gp16-CΔ7-F19, are solubilized. There is thus no difference between wild-type virions and those containing gp16-CΔ7-F19 in their ability to adsorb to cells and to eject their internal core proteins into the cell envelope.
Fig. 3.
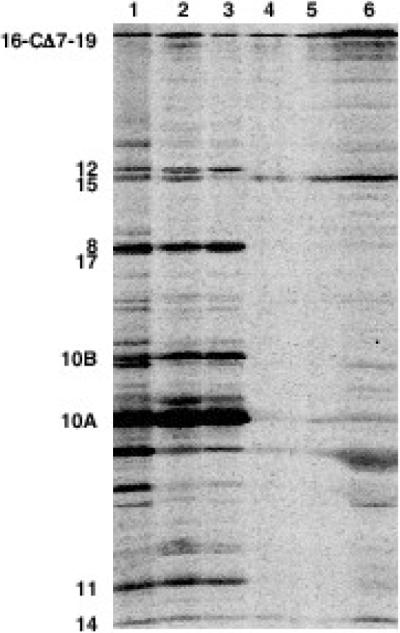
Elution and SDS-PAGE separation of 35S-labeled T7 proteins from infected HM130 cells. Virions contain gp16-CΔ7-F19. A. Lane 1: phage marker; lane 2: phage-infected cells; lane 3: phage proteins eluted from the surface of infected cells; lane 4: phage proteins retained by the infected cell; lanes 5 and 6: Sarkosyl-soluble (5) and -insoluble (6) membrane proteins from infected cells.
The failure of gp16-CΔ7-F19 to support infective phage production is therefore not a consequence of the mutant virion failing to eject the internal core proteins into the infected cell. Although they could be mislocalized in ways that this assay cannot detect, it is more likely that the internal core proteins fail either to make an open channel for DNA transport across the cytoplasmic membrane or to escort the leading genome end from the phage head into the cell.
T7 virions containing gp16-CΔ7-F19 are defective in DNA ejection
T7+ completely lyses infected cells by about 30 min of infection at 30°C (Fig. 4). In contrast, cells infected by mutant virions containing gp16-CΔ7-F19 continue to grow at the same rate as uninfected cells, and cell numbers increase in proportion to Klett value (data not shown). T7 particles containing gp16-CΔ7-F19 are therefore unable to kill cells, strongly suggesting that T7 genes are not being expressed. In turn this raises the possibility that translocation of the phage genome from the mutant virion into the infected cell is aberrant or may even fail to occur.
Fig. 4.

Lysis curves. Uninfected cells: dotted line, no plot symbol; Δ16 (gp16+): open squares; Δ16 (gp16-CΔ7-F19): open circles; Δ16,15-S560P,D614Y (gp16-CΔ7-F19): dotted line, closed triangles.
We therefore performed assays to measure the rates of genome translocation into the cell. In agreement with previous experiments (García and Molineux, 1995; Kemp et al., 2004), wild-type virions completely translocate their genome into chloramphenicol-treated cells in less than 20 min at 30°C (Fig. 5A). Also as expected, the gp16-I754T substitution, which permits DNA translocation in the absence of host transcription (Garcia and Molineux, 1996; Kemp et al., 2004; Struthers-Schlinke et al., 2000), led to complete genome internalization within 11 min at a constant 70 bp/sec at 30°C (data not shown).
Fig. 5.

A. Southern hybridization showing a time-course of T7 Δ16 [gp16+] genome internalization in chloramphenicol-treated IJ1133(pTP166) at 30°C. B. Southern hybridization showing that the genome of T7 Δ16 [gp16-I754T,CΔ7-F19] fails to enter chloramphenicol plus rifampicin-treated IJ1133(pTP166) cells. C. Southern hybridization showing that the genome of T7 Δ16 [gp16-CΔ7-F19] fails to enter chloramphenicol-treated IJ1133(pTP166). The ratio of infecting particles to cells was 0.4. Lanes labeled Sau were digested with Sau3A I as a control for the amount of T7 DNA in each experiment, all other samples were digested with DpnI. A schematic Sau3A I/Dpn I map of the phage genomes is shown at the bottom, A-4 denotes the 4 kb deletion of gene 16 that affects the Dpn I A fragment of wild-type T7 DNA.
However, virions containing gp16-I754T,CΔ7-F19 are completely defective in genome internalization. No genomic DNA is detected entering a rifampicin-treated cell even after 40 min of infection at 30°C (Fig. 5B). Similarly, no DNA enters a chloramphenicol-treated cell within 120 min when the infecting virion carries gp16-CΔ7-F19 (Fig. 5C). There is no significant loss of the full-length 36 kb Δ16 genome during these experiments. DNA is not, therefore, ejected from the mutant virion into the periplasm, where it would be degraded by endonuclease I, as is the DNA of superinfecting T4 (Anderson and Eigner, 1971). The failure of any DNA to enter the infected cell fully explains why cells infected by T7 virions containing gp16-CΔ7-F19 are not killed. The replacement of the seven C-terminal amino acids of gp16 with non-native residues therefore prevents DNA from entering the infected cell.
Suppressors of the defective gene 16 lie in gene 15
We selected mutants of T7 Δ16 that could be complemented by a plasmid providing gp16-CΔ7-F19. Four independent mutants, generically referred to as Δ16*, were obtained at an efficiency of ~10−6. By genome fragment swaps, all four contained at least one mutation to the right of the unique NcoI site in gene 15 and to the left of the EcoRI site, which in Δ16 replaces the entire gene 16. The NcoI-EcoRI fragment of each mutant Δ16* derivative was cloned on a plasmid; recombination between each plasmid and the parental Δ16 phage restored the ability to grow in the presence of gp16-CΔ7-F19 at a frequency at least four orders of magnitude higher than the appearance of spontaneous mutants. The cloned DNA therefore contains all necessary mutations for suppression of the infectivity defect, providing genetic evidence that genes 15 and 16 interact during the initiation of infection.
DNA sequencing performed directly on the mutant phage genomes showed that three of the Δ16* mutants contain the change gp15-D614Y, once as a single substitution, and in combination with gp15-S560P and gp15-M597I. The latter substitution was also found by itself. The double mutants grow better than the singles in the presence of gp16-CΔ7-F19 (no defect is apparent with any mutant in the presence of wild-type gp16), and it seems likely that spontaneous mutations conferring gp15-M597I or gp15-D614Y allow a small phage burst in the presence of gp16-CΔ7-F19. This first mutation then likely allowed additional growth cycles during which the second substitution arose. The specific infectivity (plaque-forming units/A260) of the Δ16* double mutants when virions contain gp16-CΔ7-F19 is comparable to that of wild-type phage. This result suggests that suppression of the defect in infection initiation due to the mutant gp16 is essentially complete. However, as phage titers in lysates are relatively low it is likely that a defect in virion assembly remains. We note that any detrimental effects of the negative charges on the C-terminal tail of gp16-CΔ7-F19 (which are opposite to the majority positive charges in wild-type gp16) must also be suppressed by the mutant gp15.
The gene 15 suppressor mutations also provide full infectivity to virions containing the shorter C-terminal tails of gp16-CΔ7-F7 and gp16-CΔ7-F9, even though they lack the negatively charged tail of gp16-CΔ7-F19. Interestingly, with the exception of the single suppressor mutation 15-D614Y, Δ16* phages grown in the presence of the two shorter gp16 fusion variants, which have the same or only two additional amino acids as the wild-type protein, yield plaques as large as phages grown in the presence of wild-type gp16 (Table 2). This observation suggests that virion assembly and infectivity are sensitive both to a longer gp16 and to the sequence of its C-terminal region. Support for this idea comes from growth of Δ16* in cells providing gp16-CΔ11-F2 and 16-CΔ9 where, again with the exception of 15-D614Y, large plaques are formed. In contrast, although complementation of Δ16* on cells providing longer gp16 variants was complete in that titers were essentially the same as with wild-type gp16, plaques were small.
Table 2.
Properties of gene 15 suppressor mutants.
| Gene 16 plasmid |
EOPa of Δ16 containing | |||
|---|---|---|---|---|
| 15-M597I,D614Y | 15-S560P,D614Y | 15-M597I | 15-D614Y | |
| Wild-type (1318 aa) | 1 (2) | 1 (2) | 1 (2) | 1 (1.5) |
| 16-CΔ2 | 0.88 (1) | 1.14 (1) | 1.05 (pp) | 0.56 (0.5) |
| 16-CΔ4 | 0.83 (0.5) | 0.91 (0.5) | 0.88 (0.5) | 0.43 (pp) |
| 16-CΔ5 | 0.79 (pp) | 0.64 (pp) | 0.92 (pp) | 1.12 (0.5) |
| 16-CΔ6 | 0.65 (pp) | 0.84 (pp) | 0.67 (pp) | 4×10−3 (pp) |
| 16-CΔ9 | 0.93 (3) | 0.94 (3) | 0.75 (2.5) | 2×10−3 (pp) |
| 16-CΔ11-F2 | 0.87 (3) | 0.73 (2.5) | 0.93 (2.5) | 0.04 (pp) |
| 16-CΔ12 | <10−9 | <10−9 | <10−9 | <10−9 |
| 16-CΔ19 | <10−9 | <10−9 | <10−9 | <10−9 |
| 16-CΔ7-F19 | 1.26 (2.5) | 0.93 (2.5) | 1.15 (2.5) | 0.58 (0.5) |
| 16-CΔ7-F9 | 1.00 (2) | 0.55 (3) | 0.92 (2.5) | 0.67 (1.5) |
| 16-CΔ7-F7 | 1.05 (2.5) | 0.74 (3) | 0.86 (2.5) | 0.81 (1.5) |
Relative to the same cells containing wild-type gene 16. Numbers in parentheses correspond to average plaque sizes in mm; pp = pinprick.
The gene 15 suppressor mutants were selected as allowing phage growth after replacement of the charged C terminus of gp16+ with non-native residues, and it appears that the mutant gp15 proteins have lower activity when part of the wild-type amino acid gp16 sequence, including some of those charges, is retained. However, they cannot suppress the virion assembly defect associated with the loss of 12 or more C-terminal residues.
The gene 15 suppressor mutations restore normal genome entry kinetics
The gene 15 mutations that suppress the plaque-forming defect associated with virions containing gp16-CΔ7-F19 must necessarily suppress the DNA ejection defect. Lysis curves, only one of which is presented in Figure 4, show that mutant phages have regained the ability to effect cell lysis with kinetics indistinguishable from wild-type. Nevertheless, it was still of considerable interest to determine whether rates of transcription-independent and transcription-mediated genome entry were the same as in a normal infection. Gp16 is known to function during internalization of the leading end of the phage genome, but a comparable role for gp15 has not yet been established.
In contrast to infections with virions containing gp16-CΔ7-F19 and wild-type gp15, the various mutant gp15 proteins all allow normal genome internalization. An experiment using virions containing gp15-M597I and gp16-CΔ7-F19, showing the kinetics of genome entry by E. coli RNA polymerase into chloramphenicol-treated cells, is presented in Figures 6A and 6C; the complete data set of all gp15 mutants with wild-type or gp16-CΔ7-F19 is summarized in Table 3. The time taken for the internal core proteins to form the trans-envelope channel and to internalize the leading 500 bp, which includes the major A1 promoter so that transcription-catalyzed genome entry can occur, is essentially the same whether the infecting virion contains a mutant gp15, and wild-type or gp16-CΔ7-F19. The rate of transcription-catalyzed genome entry is also unaffected; both parameters are therefore comparable to a wild-type infection. The only obvious defect is a low efficiency of genome entry by virions containing gp16-CΔ7-F19 and gp15-D614Y, an observation consistent with the low suppression activity of this gene 15 mutant.
Fig. 6.

Southern hybridizations showing time-courses of A: T7 Δ16 [gp15-M597I,gp16-CΔ7-F19] genome internalization at 30°C in chloramphenicol-treated IJ1133(pTP166) cells and B: T7 Δ16 [gp15-S560P,D614Y, gp16-I754T,CΔ7-F19] in chloramphenicol- plus rifampicin-treated cells. The ratio of infecting particles to cells was 0.4. Lanes labeled Sau were digested with Sau3A I as a control for the amount of T7 DNA in each experiment, all other samples were digested with DpnI. C: rates of genome entry from the experiments shown in A (open circles) and B (closed circles). A schematic Sau3A I/Dpn I map of the phage genomes is shown, A-4 denotes the 4 kb deletion of gene 16 that affects the Dpn I A fragment of wild-type T7 DNA.
Table 3.
Kinetics of E. coli RNA polymerase-catalyzed T7 genome internalization.
| Virion gp16 | Gene 15 | Genome entry by E. coli RNAP (bp/sec) | Time of bp #500 entry (sec) |
|---|---|---|---|
| Wild-type | Wild-type | 36/41 | 250 |
| CΔ7-F19 | Wild-type | 0 | N/A |
| Wild-type | M597I,D614Y | 39/40 | 200 |
| CΔ7-F19 | M597I,D614Y | 36 | 250 |
| Wild-type | S560P,D614Y | 37/40 | 200 |
| CΔ7-F19 | S560P,D614Y | 37 | 250 |
| Wild-type | M597I | 38/39 | 200 |
| CΔ7-F19 | M597I | 37 | 200 |
| Wild-type | D614Y | 40 | 200 |
| CΔ7-F19a | D614Y | ~40 | ~250 |
Data are approximate because <10% of genomes initiated infection.
In order to measure the kinetics of the transcription-independent mode of DNA translocation with the gp16 fusion protein, we used virions containing gp16-I754T,CΔ7-F19. The 16-I754T missense mutation allows the entire T7 genome to be internalized in the absence of transcription (Garcia and Molineux, 1996; Kemp et al., 2004; Struthers-Schlinke et al., 2000). The kinetics of transcription-independent genome internalization of one mutant virion (containing gp15-S560P,D614Y and gp16-I754T,CΔ7-F19) is shown in Figures 6B and 6C; the complete data set of all gp15 mutants with wild-type or gp16-I754T,CΔ7-F19 is summarized in Table 4. Extrapolation of the kinetic data to the point where DNA begins to enter the cell provides a measure of the time taken for the phage to adsorb to cells and to form an open channel across the cytoplasmic membrane. Virions carrying gp16-CΔ7-F19 and any of the gp15 mutant suppressor proteins restore the normal time of initiation of genome internalization. The rate of transcription-independent genome entry is also fully restored to the expected ~70 bp/sec across the entire genome. There is therefore a complete suppression of both the failure by gp16-CΔ7-F19-containing virions to open a channel for DNA transport across the cytoplasmic membrane and of the genome translocation mechanism itself. We conclude that gp15 and gp16 function together to internalize an infecting T7 genome into the cell.
Table 4.
Kinetics of transcription-independent T7 genome internalization.
| Virion gp16 | Gene 15 | Transcription-independent genome entry (bp/sec) | Time of bp #1 entry (sec) |
|---|---|---|---|
| I754T | Wild-type | 66/68 | 200 |
| I754T,CΔ7-F19 | Wild-type | 0 | N/A |
| I754T | M597I,D614Y | 66/67 | 150 |
| I754T,CΔ7-F19 | M597I,D614Y | 62 | 150 |
| I754T | S560P,D614Y | 69/65 | 150 |
| I754T,CΔ7-F19 | S560P,D614Y | 60 | 150 |
| I754T | M597I | 68/65 | 150 |
| I754T,CΔ7-F19 | M597I | 66 | 150 |
| I754T | D614Y | 71/69 | 200 |
| I754T,CΔ7-F19a | D614Y | ~60 | ~200 |
Data are approximate because <10% of genomes initiated infection.
DISCUSSION
Penetration of the outer membrane of a Gram-negative cell by a phage virion must precede its forming a channel for DNA transport across the cytoplasmic membrane. In principle, phages with long tail tips: e.g., T5, can perform both functions with the same structure, but Podoviridae must extend their tail – at least functionally - to breach the cell envelope. This extended tail must also prevent the periplasmic endonuclease I access to the entering genome, a feature that is automatically provided in phages whose tail tubes span the entire cell envelope. T7 extends its tail by ejecting the internal head proteins gp14, gp15, and gp16 into the cell prior to DNA transport into the cytoplasm. The channel created by the ejected phage proteins likely corresponds to the zones of adhesion between the cytoplasmic membrane and the outer envelope of phage-infected cells originally described by Bayer (1968).
In several phages, the leading end of the phage genome is known to be inserted partway down the tail tube, or at least into the neck region, during virion maturation (Chattoraj and Inman, 1974; Saigo, 1975; Saigo and Uchida, 1974; Thomas, 1974). Tail or neck insertion of the genome end may be a feature common to all members of the Myoviridae and Siphoviridae. If the genome end is not located in or close to the channel through the tail, it would be difficult for that end to find the channel and DNA ejection would be extremely inefficient. However, for T7-like phages, whose tails are extended when making a channel into the cell cytoplasm, inserting DNA into the stubby tail during assembly is unlikely, although association of the genome end with internal virion proteins that are ejected into the cell to extend the tail is not precluded. In mature T7, a central channel of ~30 Å diameter through the internal core lies coaxial with the channel through the portal (Agirrezabala et al., 2005a,b; Serwer, 1976). The channel is partially filled by what is likely the leading end of the phage genome. Similar structures have been observed in several phages that contain internal cores (Jiang et al., 2006; Lander et al., 2006; Leiman et al., 2007), and a comparable channel across the portal exists in SPP1 and ø29 (Lhuiller et al., 2009; Tang et al., 2008; Xiang et al., 2006).
Models of T7 tail extension by internal core proteins
The channel through the T7 portal decreases from 83 Å on the inner surface of the capsid shell down to 33 Å, and the channel through the length of the stubby tail has a comparable diameter (Agirrezabala et al., 2005a,b). Assuming that the portal does not grossly change its structure during the initiation of infection, a 33 Å channel is too small to allow the internal core to exit the head intact. Several copies of each internal core protein are ejected from the head to make the trans-envelope channel and it is likely that they enter the infected cell in a controlled, sequential manner. It is not obvious how the orderly ejection of multiple proteins from the head, through the portal and tail, is achieved.
A 33 Å channel is also likely too small to allow the internal proteins to pass through in a fully-folded conformation. The 1318 aa gp16 is predicted to consist of α-helices separated by unstructured regions, and we previously suggested that the protein might preserve some secondary structure during ejection from the virion (Molineux, 2001). Gp16 must refold rapidly in the periplasm to provide lytic transglycosylase activity, which is essential to locally degrade the cell wall under adverse conditions of growth (Moak and Molineux, 2000), but there are no appropriate chaperone proteins in the periplasm to aid in the refolding process. Gp14 and gp15 are also predicted to consist largely of α-helices separated by unstructured regions and they may likewise be ejected in a partially unfolded state. The backbone of an amino acid α-helix has a diameter of 5 Å, but a realistic effective diameter that still requires some flexibility of side chains is ~8 Å. If the phage genome is not ejected from the head at the same time, twelve extended protein molecules that maintained only α-helices, connected by flexible regions, could pass simultaneously through the 33 Å constriction of the portal and then through the tail (Fig. 7). This is just sufficient for the 10–12 copies of gp14 in the internal core to exit and, separately, for the four gp16 and eight gp15 molecules. The channel through the T7 portal must increase in diameter to ~40 Å to allow 12 internal core protein molecules and a 20 Å diameter DNA duplex to exit the head simultaneously. Such an increase would not be unprecedented; significant structural changes have been described in both the portal and the tail tube during SPP1 DNA ejection (Cuervo et al., 2007; Lhuiller et al., 2009; Plisson et al., 2007).
Fig. 7.

Schematic diagrams of A: a 33 Å channel through the T7 portal accommodating 12 partially unfolded protein molecules; B: a 40 Å portal channel with a DNA molecule and 12 partially unfolded protein molecules. A possible arrangement of the four gp16 and eight gp15 molecules is indicated.
One possibility is that the extended tail is formed stepwise. Gp14 lies at the base of the internal core in nature virions, and is ejected first (perhaps after gp6.7 has exited), establishing a channel across the outer membrane. Gp15 and gp16 would then pass through this channel and enter the periplasm. T7 gp16 would likely be the second protein ejected from the virion because lytic transglycosylase activity may be important before the cytoplasmic membrane is breached. However, muralytic activity is carried on the gp15 homolog in some T7-like relatives (Lavigne et al., 2004; Moak and Molineux, 2004), and thus the order of ejection in different phages may vary, or perhaps both proteins are ejected simultaneously (e.g., Fig.7). Regardless, all core proteins probably maintain some interactions in order to coordinate the overall ejection process.
An alternative idea is that the order of ejection from the capsid is gp16, either alone or together with gp15, followed by gp14. In this model, the internal core inverts its orientation as its constituent proteins are ejected from the virion. T7 gp16 and/or gp15 would dissociate from the upper part of the mature internal core, pass by gp14 and travel through the portal and tail, and then breach the outer membrane, leading the way into the periplasm and across the cytoplasmic membrane. Gp14, the protein closest to the portal in the mature virion, would trail gp15 and gp16 out of the head, anchoring itself just beyond the tail in the outer membrane (Fig. 1A).
We imagine that gp15 and/or gp16 surround the leading genome end inside the virion and escort the DNA across the cell envelope. The two proteins may form a tube, preventing attack by endonuclease I. This again would make an infection by T7 comparable to that of long-tailed phages where the tail tube (albeit pre-formed) surrounds the DNA. Interestingly, T7 structures with a 40–55 nm tube-like tail extension, long enough to span the entire cell envelope, have been observed in lysates (Serwer et al., 2008). The internal diameter of the tube was estimated to be 30 – 40 Å, more than sufficient to accommodate a duplex DNA.
Does gp16 escort DNA from the virion into the cell?
The C terminus of gp16 is highly charged, suggesting that it could bind the genome end and escort it into the cell. However, removing four of the five charges has little effect on infectivity, and although removal of all five yields defective particles, removal of the adjacent non-polar residue from the C terminus (gp16-CΔ6) causes a much greater defect (Table 1). It is then to be expected that fusion proteins replacing the last seven amino acids of gp16 with non-native sequences would yield non-viable virions. Such virions adsorb to but do not kill cells and do not even affect their growth rate. It was therefore surprising that defective particles would eject internal core proteins apparently normally into the cell. This raises the question of what happens to the genome, which is no longer prevented from exiting the virion by internal head proteins. Despite the high forces commonly assumed to be associated with DNA condensed to a density of ~500 mgs/ml, the genome appears to remain inside the capsid shell. DNA cannot be detected in the infected cell cytoplasm, nor is it degraded in the periplasm. The leading genome end may penetrate the outer envelope of the cell through a conduit formed by the ejected core proteins, but the majority of the DNA must remain stably inside the head for at least two hours during infection by T7 virions containing gp16-CΔ7-F19.
Penetrating the cytoplasmic membrane
Any channel across the cytoplasmic membrane formed by virions containing gp16-CΔ7-F19 must be closed because infected cells continue to grow and divide normally. The phage genome itself could block the channel, preventing collapse of the membrane potential, and the physiological defect of an infection by the mutant phage may simply be the failure to translocate any DNA into the cell. Alternatively, the mutant gp16 may only partially traverse the cytoplasmic membrane, thereby failing to make an aqueous connection that would allow genome transfer between the phage capsid and the cell cytoplasm. However, it seems unlikely that the highly charged C terminus of wild-type gp16 is necessary to penetrate the phospholipid bilayer, although the positive charges may be important in anchoring the protein on the membrane. Perhaps a complete channel does transiently form but the lack of positive charges on the C terminus of gp16-CΔ7-F19 causes the extended tail of the phage to retract onto the periplasmic face of the membrane. That no genome internalization occurs after infection by virions containing gp16-CΔ7-F19 is consistent with either possibility, as even a productive infection by T7 does not lead to a transient drop in the membrane potential (Kuhn and Kellenberger, 1985; E. Ramanculov and I. J. M., unpublished data).
Suppressors of the defect caused by replacing the last seven residues of gp16 with non-specific sequences alter gene 15. The changes in gp15 are located in the C-terminal quarter of the protein and their isolation may be more of a response to an altered gp16 sequence near residue 1310 than to the loss of the positively charged C-terminus. It seems possible that the gp15 suppressor mutants stabilize the C-terminal α-helix of gp16 (Fig. 1B), a function normally governed by the native C-terminal charged residues. Although the gene 15 mutants restore infectivity to virions containing a 1312 or 1313 residue gp16 (gp16-CΔ6 and gp16-CΔ5), suppression is more efficient with virions containing a 1309 residue gp16 (gp16-CΔ9 and gp16-CΔ11-F2). Both phage yields and plaque sizes are comparable to those obtained with gp16-CΔ7-F19, whose use allowed isolation of the suppressors, as well as to those obtained after complementation of T7 Δ16 by wild-type gp16.
Does gp15 escort DNA from the virion into the cell?
The 747 residue gp15 is also a potential candidate to escort the leading end of the phage genome into the cell. Seventeen of its C-terminal 51 amino acids are Arg/Lys (7 Asp/Glu residues), which could bind to the leading end of the genome. However, plaque-forming ability is fully maintained when 12 positive (and three negative) charges are removed by truncating gp15 to 713 residues (I. J. M., unpublished observations), and thus a positively charged C terminus may not be essential. Larger truncations of gp15 result in a loss of infectivity although the precise defect has not been determined. The gene 15 suppressor mutations affect residues far from the non-essential C terminus and they may simply alter the structure of gp15 such that it retains function in the presence of a mutant (or wild-type) gp16. Presumably, the region of gp15 that includes amino acids 560 to 614 normally interacts with the C terminus of gp16. At present, we cannot determine which of gp15 or gp16, or whether both, escort the leading end of the T7 genome into the cell. However, both proteins clearly function together in the overall T7 DNA internalization translocation process, and they are jointly responsible for transcription-independent genome translocation.
Gp15 and gp16 actively translocate the T7 genome into an infected cell
The majority of the T7 genome is actively transported into the cell, at rates characteristic of the different enzymes used (García and Molineux, 1995; 1999; Kemp et al., 2004). However, in order to allow the various enzymes to access their recognition sites, the leading end of the genome must first be internalized. We have shown here that the C terminus of gp16 is essential for this process but, in its absence, substitutions in gp15 can fully compensate. The exact mechanism by which gp15 and gp16 function in genome translocation has not yet been established, but at 30°C the two proteins translocate DNA into the infected cell at ~ 70 bp/sec. This mode of DNA translocation must be an active, energy-requiring process as the rate of internalization is constant across the entire phage genome. Neither a passive mechanism resulting from forces resulting from the closely packed DNA inside the capsid, nor entropic changes in DNA as the genome enters the cell, can account for the kinetics observed. We suggest that both gp15 and gp16 cooperate to form a molecular motor that utilizes the membrane potential of the cell (Kemp et al., 2004) to ratchet the T7 genome from the capsid into the infected cell.
Phage DNA ejection in vitro
The above results using T7 in vivo are in stark contrast to ejection of T5 DNA ejection in vitro, which occurs spontaneously after addition of the FhuA receptor. Individual molecules are ejected in a stepwise fashion, attaining speeds of up to 75 kb/sec between distinct pauses, which may correspond to the defined nicks in the T5 genome (Mangenot et al., 2005). The pauses become longer as more of the genome is ejected. A simpler picture emerges from similar studies on λ: ejection of individual genomes occurs at speeds > 60 kb/sec but again the rate is not constant, slowing significantly before the capsid is empty (Grayson et al., 2007). It was concluded from both studies that forces associated with the closely packed DNA in the phage head are responsible for the rapid initial ejection phase.
The internal pressure in various phage virions has been estimated from single molecule DNA packaging studies to be at least 60 atmospheres (atm) (Fuller et al., 2007; Smith et al., 2000). In comparison, suppression of genome ejection in vitro using concentrated PEG solutions suggests that the internal pressure is lower: 15–20 atm for λ or T5, and somewhat less than 50 atm for the Bacillus subtilis phage SPP1 (Grayson et al., 2006; Leforestier et al., 2008; São-José et al., 2007). As the genomes of all tailed phages, including T7, are packaged at comparable densities (~500 mg/ml), pressure has often been suggested to provide the driving force for phage DNA ejection into the cell. However, forces internal to the virion are necessarily insufficient for transport of a complete genome into a living cell (Molineux, 2001, 2006b). Furthermore, the internal forces available for DNA ejection may be considerably lower than commonly assumed. Disrupting particles of several phages at the instant of deposition on a carbon grid resulted in electron micrographs where the phage genomes still maintained a compact structure and conserved the internal organization seen in mature particles (Richards et al., 1973). Compact DNA structures would not be seen if the packaged genome were exerting substantial forces on the capsid inner wall prior to its disruption.
Phage DNA ejection in vivo
The kinetics of T7 genome internalization in vivo are not consistent with any forces internal to the infecting virion controlling the process. Although the general mechanism of T7 genome internalization into the infected cell is applicable to members of this phage family (perhaps also N4; Molineux, 2006b), internal virion forces have not yet been shown to cause genome ejection of any phage into an infected cell. The rate of DNA internalization between markers on the SP82 genome – the only other direct measurement of phage genome transfer rates in vivo - also show a temperature-dependence that could be fitted to Arrhenius kinetics (McAllister, 1970). Those data are inconsistent with internal virion forces ejecting the genome into the cell. Furthermore, stripping the capsid from a T5 particle after first-step genome transfer had occurred does not prevent the remaining naked 110 kb from being internalized by the cell at a rate not substantially different from an unperturbed infection (Labedan and Legault-Demare, 1973). This may be the clearest example that forces internal to a phage virion cannot be required for efficient genome uptake by an infected cell.
MATERIALS AND METHODS
Phages, plasmids and bacteria
T7 Δ16 contains a unique EcoRI site replacing T7 DNA #30577–34551 inclusive; the deletion removes the entire coding sequence of gene 16. The complementing plasmids pPK69 and pPK70, which differ only in the orientation of gene 16, relative to vector promoters, contain T7 DNA #30577–34551 flanked by EcoRI sites in the plasmid pWSK129 (Wang and Kushner, 1991). No significant difference in complementing activity was noted when gene 16 is expressed from the vector T7 (pPK69) or T3 and lacP promoters (pPK70). Derivatives of pPK69 or pPK70 are listed in Table 5. T7 strains were grown and titered at 30°C on IJ1133 [K-12 ΔlacX74thi Δ(mcrC-mrr)102∷Tn10], using complementing plasmids as required. Phages were concentrated from lysates by precipitation with polyethylene glycol or by ultracentrifugation, followed by isopycnic density gradient centrifugation in CsCl. Visible phage bands or the corresponding region of the gradient were removed by syringe through the side of the centrifuge tube, dialyzed against 10 mM tris-HCl pH 7.6, 10 mM MgCl2, 1 M NaCl; phage concentrations were determined by titer and/or measurements of A260 and A280. The protease-deficient strain HM130 [K-12 ΔlacX74 galE galK thi-1 rpsL ΔphoA(PvuII) ΔompT degP41(ΔPstI)∷ΩKnR Δtsp3∷ΩKnReda51∷Tn10 ptr-32∷ΩCmR] (Meerman and Georgiou, 1994) was used in some experiments where T7 was eluted from the surface of infected cells. Genome segment swaps were performed by ligating appropriate restriction enzyme fragments and transfecting into CaCl2-treated competent IJ1126 [K-12 recB21 recC22 sbcA5 endA gal thi supL Δ(mcrC-mrr)102∷Tn10] or its T7 gene 1-containing derivative IJ1127.
Table 5.
Gene 16 plasmids.
| Plasmid | Gene16 | Common nameb | Parent vector and description |
|---|---|---|---|
| pPK69 | wild-type | pWSK129; T7 #30577-34551 inserted at EcoRI site | |
| pPK70 | wild-type | pWSK129; T7 #30577-34551 inserted at EcoRI site | |
| pCYC5 | wild-type | pWKS30; T7 #30577-34551 inserted at EcoRI site | |
| pCYC7 | wild-type | pPK70; NotI-BamHI, fill-in (ΔXbaI-SpeI) | |
| pCYC9 | wild-type | pWSK29; T7 #30577-34551 inserted at EcoRI site | |
| pCYC9.19 | 16-CΔ7-F19 | pCYC9; HpaI-EcoRV deletion | |
| pCYC9.9 | 16-CΔ7-F9 | pCYC9.19; HindIII (fill-in) - ApaI (blunt) | |
| pCYC9.7 | 16-CΔ7-F7 | pCYC9.19; HindIII (fill-in) - KpnI (blunt) | |
| pCYC10 | wild-type | pCYC9; NotI-BamHI, fill-in (ΔXbaI-SpeI) | |
| pCYC10.19 | 16-CΔ7-F19 | pCYC10; HpaI-EcoRV deletion | |
| pCYC10 (I754T) |
I754T | pCYC10 - BstBI-XbaI swap from T7 16-I754T DNA | |
| pCYC10.19 (I754T) |
16-I754T, CΔ7-F19 |
pCYC10.19 - BstBI-XbaI swap from T7 16-I754T DNA | |
| pCYC51 | R1317am | 16-CΔ2 | pCYC5; HpaI-HindIII swap 5′-AACTTGAGGGAGCGTTAGAAATAAGAATTC TTGAACTCCCTCGCAATCTTTATTCTTAAGTCGA-5′ |
| pCYC52 | E1315am | 16-CΔ4 | pCYC5; HpaI-HindIII swap 5′-AACTTGAGGTAGCGTAGGAAATAAGAATTC TTGAACTCCATCGCATCCTTTATTCTTAAGTCGA-5′ |
| pCYC53 | R1314am | 16-CΔ5 | pCYC5; HpaI-HindIII swap 5′-AACTTGTAGGAGCGTAGGAAATAAGAATTC TTGAACATCCTCGCATCCTTTATTCTTAAGTCGA-5′ |
| pCYC54 | L1313am | 16-CΔ6 | pCYC5; HpaI-HindIII swap 5′-AACTAGAGGGAGCGTAGGAAATAAGAATTC TTGATCTCCCTCGCATCCTTTATTCTTAAGTCGA-5′ |
| pCYC71 | G1310am | 16-CΔ9 | pCYC7; XbaI-HpaI swap with PCR product using F: 5′-ACCGTACACATCTAGAGAGGa R: 5′-CTCCCTCAAGTTAACCTAGTTCGCCTCATAAATC |
| pCYC72 | E1307am | 16-CΔ12 | pCYC7; XbaI-HpaI swap with PCR product using F: 5′-ACCGTACACATCTAGAGAGGa R: 5′-CTCAAGTTAACACCGTTCGCCTAATAAATCTTCAACAC |
| pCYC73 | Q1300am | 16-CΔ19 | pCYC7; XbaI-HpaI swap with PCR product using F: 5′-ACCGTACACATCTAGAGAGGa R: 5′-CTCAAGTTAACACCGTTCGCCTCATAAATCTTCAACACAAGCTATTGAGTCAATG |
| pCYC75 | 16-CΔ11-F2 | pCYC7; mutant PCR product from construction of 16-CΔ9 | |
PCR products (T7 DNA template) were cut with XbaI/HpaI (underlined) and cloned into corresponding sites of pCYC7
Name applicable in Su- cells only.
Preparation of 35S-labeled T7
BL21 or BL21(pCYC10.19) were grown at 30°C in M9 glucose media containing all amino acids except methionine and cysteine, and were then infected with T7 or T7 Δ16 at a multiplicity of five. After 10 min, 70 μCi/ml carrier-free 35S-Met and – Cys (EXPRESS Protein Labeling Mix; New England Nuclear) was added, and the culture was then incubated until lysis. Labeled phage particles were harvested and purified by isopycnic density gradient centrifugation in CsCl. The phage band was isolated and its concentration estimated by absorption, using 4 × 1011 particles per A260.
Assays of ejected virion proteins after infection
IJ1133 or the protease-deficient strain HM130 were grown in LB media at 30°C to a density of 5 × 108 cells /ml. 10 min before infection at a multiplicity of 8–10 35S-labeled phage/cell, cells were treated with 200 μg/ml rifampicin to prevent phage development, in particular transcription mediated genome internalization (García and Molineux, 1995, 1996). After 10 min of infection, cells were collected by centrifugation and washed five times, using vortex agitation, with water plus 100 μg/ml rifampicin at 0°C. This procedure releases most virion proteins (from both reversibly and irreversibly adsorbed phage) from the surface of the cell, but bacteria retain those virion proteins that have penetrated the cell envelope (Israel, 1977; Kemp et al., 2005). The five aqueous eluates were combined, lyophilized, resuspended, and the sample was then subjected to SDS-PAGE. A portion of the cell pellet was saved for SDS-PAGE, the remainder was lysed in a French press and the membrane fraction isolated by centrifugation at 100,000 × g for 2 hr. The membrane pellet was then extracted with 0.25% Sarkosyl in the absence of divalent ions for 2 hr at 4°C. Sarkosyl selectively solubilizes cytoplasmic membrane proteins, outer membrane proteins remaining insoluble (Filip et al., 1973). Insoluble and soluble fractions were then subjected to SDS-PAGE, and the radioactive T7 proteins were selectively visualized using a phosphorimager.
Sucrose gradient centrifugation was also employed to separate outer and inner membranes (Osborn et al., 1972). Infected, washed cells were converted into spheroplasts by lysozyme treatment. Spheroplasts were lysed by a single passage through a French press at 16,000 psi, separated from unlysed cells by a brief centrifugation, and layered on a discontinuous 30 –55% sucrose gradient. Inner and outer membranes were separated by centrifugation at 120,000 × g for 14 hr at 4°C, gradient fractions were collected from the tube bottom. Portions were taken for scintillation counting in order to detect radioactive proteins, and for assays of NADH oxidase activity as an inner membrane marker. The remaining part of the gradient fractions was analyzed by SDS-PAGE. Radioactive T7 proteins were detected by phosphorimagery. The outer membrane protein OmpA was visualized by immunoblotting using anti-OmpA antibody and a secondary antibody coupled to alkaline phosphatase.
Lysis curves
Exponential phase cells IJ1133 cells, growing in LB media at 30°C, were infected at time zero using a multiplicity of five particles per cell. Apparent absorption of the cultures was determined using a Klett-Summerson colorimeter.
Assays of genome internalization
DNA entry experiments measuring the kinetics of genome internalization were performed essentially as described by García and Molineux (1995). Briefly, IJ1133 cells carrying pTP166, a plasmid overexpressing dam methylase, were grown at 30°C to a density of 2 × 108/ml. Ten minutes before infection cells were centrifuged and resuspended in the same medium containing 200 mg/ml chloramphenicol or 200 mg/ml chloramphenicol and 500 mg/ml rifampicin. Cells were infected at a multiplicity of 0.4 using phage that had been grown on IJ922 [K-12 ΔlacX74 supE44 galK2 galT22 mcrA rfbD1 mcrB1hsdS3dam-13∷Tn9] containing a gene 16 plasmid. At various times portions of the culture were treated with a phenol/ethanol killing solution, cells were harvested by centrifugation and treated with proteinase K. DNA was isolated by successive phenol-chloroform and phenol extractions, taking care to achieve a constant efficiency of recovery between samples. Variable recovery leads to lane-to-lane variation in band intensities on the Southern blot. After digested with either Sau3A I or Dpn I, DNA was electrophoresed through agarose and was then Southern hybridized using a probe of randomly primed T7 DNA. DNA was visualized using a phosphorimager and band intensities were determined using Quantity One (BioRad) software. The intensity of each Dpn I band was divided by the intensity of the corresponding Sau3A I band; the ratio corresponds to the fraction of a particular DNA fragment that has entered the cell and been methylated by Dam. This process normalizes for variations in the efficiencies of transfer of different-sized fragments to the membrane and for the hybridization step. The initial slope of Dpn I to Sau3A I band intensities versus time was used to estimate the time when ~50% of the maximum ratio is reached. This time is when a particular fragment is deemed to have entered the cell. The maximum Dpn I to Sau3A I band intensity ratio varies in different experiments because the fraction of phage particles that adsorb to cells and initiate genome internalization during a short infection in liquid varies with different preparations.
ACKNOWLEDGEMENTS
We are grateful to Bill Robins, who independently quantified the Southern hybridization data in order to confirm rates of genome entry. We thank Whitney Yin for constructive criticisms on the manuscript. This work was supported by NIH grant GM32095 to IJM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agirrezabala X, Martin-Benito J, Caston JR, Miranda R, Valpuesta JM, Carrascosa JL. Maturation of phage T7 involves structural modification of both shell and inner core components. EMBO J. 2005a;24:3820–3829. doi: 10.1038/sj.emboj.7600840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agirrezabala X, Martín-Benito J, Valle M, González JM, Valencia A, Valpuesta JM, Carrascosa JL. Structure of the connector of bacteriophage T7 at 8 Å resolution: structural homologies of a basic component of a DNA translocating machinery. J. Mol. Biol. 2005;347:895–902. doi: 10.1016/j.jmb.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Anand VS, Patel SS. Transient state kinetics of transcription elongation by T7 RNA polymerase. J. Biol. Chem. 2006;281:35677–35685. doi: 10.1074/jbc.M608180200. [DOI] [PubMed] [Google Scholar]
- Anderson CW, Eigner J. Breakdown and exclusion of superinfecting T-even bacteriophage in Escherichia coli. J. Virol. 1971;8:869–886. doi: 10.1128/jvi.8.6.869-886.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer ME. Adsorption of bacteriophages to adhesions between wall and membrane of Escherichia coli. J. Virol. 1968;2:346–356. doi: 10.1128/jvi.2.4.346-356.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattoraj DK, Inman RB. Location of DNA ends in P2, 186, P4, and lambda bacteriophage heads. J. Mol. Biol. 1974;87:11–22. doi: 10.1016/0022-2836(74)90556-7. [DOI] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deléage G. NPS@: Network Protein Sequence Analysis. Trends Biochem, Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- Cuervo A, Vaney M-C, Antson AA, Tavares P, Oliveira L. Structural rearrangements between portal protein subunits are essential for viral DNA translocation. J. Biol. Chem. 2007;282:18907–18913. doi: 10.1074/jbc.M701808200. [DOI] [PubMed] [Google Scholar]
- Filip C, Fletcher G, Wulff JL, Earhart CF. Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J. Bacteriol. 1973;115:717–722. doi: 10.1128/jb.115.3.717-722.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DN, Raymer DM, Rickgauer JP, Robertson RM, Catalano CE, Anderson DL, Grimes S, Smith DE. Measurements of single DNA molecule packaging dynamics in bacteriophage λ reveal high forces, high motor processivity, and capsid transformations. J. Mol. Biol. 2007;373:1113–1122. doi: 10.1016/j.jmb.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García LR, Molineux IJ. Rate of translocation of bacteriophage T7 DNA across the membranes of Escherichia coli. J. Bacteriol. 1995;177:4066–4076. doi: 10.1128/jb.177.14.4066-4076.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García LR, Molineux IJ. Transcription-independent DNA translocation of bacteriophage T7 DNA into Escherichia coli. J. Bacteriol. 1996;178:6921–6929. doi: 10.1128/jb.178.23.6921-6929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García LR, Molineux IJ. Translocation and cleavage of bacteriophage T7 DNA by the type I restriction enzyme EcoKI in vivo. Proc. Natl. Acad. Sci USA. 1999;96:12430–12435. doi: 10.1073/pnas.96.22.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson P, Evilevitch A, Inamdar MM, Purohit PK, Gelbart WM, Knobler CM, Phillips R. The effect of genome length on ejection forces in bacteriophage lambda. Virology. 2006;348:430–436. doi: 10.1016/j.virol.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson P, Han L, Winther T, Phillips R. Real-time observations of single bacteriophage lambda DNA ejections in vitro. Proc. Natl. Acad. Sci. USA. 2007;104:14652–14657. doi: 10.1073/pnas.0703274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel V. E proteins of bacteriophage P22. J. Virol. 1977;23:91–97. doi: 10.1128/jvi.23.1.91-97.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Chang J, Weigele P, King J, Chiu W. Structure of epsilon15 bacteriophage reveals genome organization and DNA packaging/injection apparatus. Nature. 2006;439:612–616. doi: 10.1038/nature04487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp P, García LR, Molineux IJ. Changes in bacteriophage T7 virion structure at the initiation of infection. Virology. 2005;340:310–317. doi: 10.1016/j.virol.2005.06.039. [DOI] [PubMed] [Google Scholar]
- Kemp P, Gupta M, Molineux IJ. Bacteriophage T7 DNA ejection into cells is initiated by an enzyme-like mechanism. Mol. Microbiol. 2004;53:1251–1265. doi: 10.1111/j.1365-2958.2004.04204.x. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Kellenberger E. Productive phage infection in Escherichia coli with reduced internal levels of the major cations. J. Bacteriol. 1985;163:906–912. doi: 10.1128/jb.163.3.906-912.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labedan B, Legault-Demare J. Penetration into host cells of naked partially injected (post FST) DNA of bacteriophage T5. J. Virol. 1973;12:226–229. doi: 10.1128/jvi.12.2.226-229.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science. 2006;312:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- Lavigne R, Briers Y, Hertveldt K, Robben J, Volckaert G. Identification and characterization of a highly thermostable bacteriophage lysozyme. Cell. Mol. Life Sci. 2004;61:2753–2759. doi: 10.1007/s00018-004-4301-y. [DOI] [PubMed] [Google Scholar]
- Leforestier A, Braselès S, de Frutos M, Raspaud E, Letellier L, Tavares P, Livolant F. Bacteriophage T5 DNA ejection under pressure. J. Mol. Biol. 2008;384:730–739. doi: 10.1016/j.jmb.2008.09.035. [DOI] [PubMed] [Google Scholar]
- Leiman PG, Battisti AJ, Bowman VD, Stummeyer K, Mühlenhoff M, Gerardy-Schahn R, Scholl D, Molineux IJ. The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J. Mol. Biol. 2007;371:836–849. doi: 10.1016/j.jmb.2007.05.083. [DOI] [PubMed] [Google Scholar]
- Lhuiller S, Gallopin M, Gilquin B, Brasilès S, Lancelot N, Letellier G, Gilles M, Dethan G, Orlova EV, Couprie J, Tavares P, Zinn-Justin S. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA-gating. Proc. Natl. Acad. Sci. USA. 2009;106:8507–8512. doi: 10.1073/pnas.0812407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangenot S, Hochrein M, Radler J, Letellier L. Real-time imaging of DNA ejection from single phage particles. Curr. Biol. 2005;15:430–435. doi: 10.1016/j.cub.2004.12.080. [DOI] [PubMed] [Google Scholar]
- McAllister WT. Bacteriophage infection: which end of the SP82G genome goes in first? J. Virol. 1970;5:194–198. doi: 10.1128/jvi.5.2.194-198.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerman HJ, Georgiou G. Construction and characterization of a set of E. coli strains deficient in all known loci affecting the proteolytic stability of secreted recombinant proteins. Bio/Technology (NY) 1994;12:1107–1110. doi: 10.1038/nbt1194-1107. [DOI] [PubMed] [Google Scholar]
- Moak M, Molineux IJ. Role of the gp16 lytic transglycosylase motif in bacteriophage T7 virions at the initiation of infection. Mol. Microbiol. 2000;37:345–355. doi: 10.1046/j.1365-2958.2000.01995.x. [DOI] [PubMed] [Google Scholar]
- Moak M, Molineux IJ. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol. Microbiol. 2004;51:1169–1183. doi: 10.1046/j.1365-2958.2003.03894.x. [DOI] [PubMed] [Google Scholar]
- Moffatt BA, Studier FW. Entry of bacteriophage T7 DNA into the cell and escape from host restriction. J. Bacteriol. 1988;170:2095–2105. doi: 10.1128/jb.170.5.2095-2105.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineux IJ. No syringes please, ejection of T7 DNA from the virion is enzyme-driven. Mol. Microbiol. 2001;40:1–8. doi: 10.1046/j.1365-2958.2001.02357.x. [DOI] [PubMed] [Google Scholar]
- Molineux IJ. The T7 group. In: Calendar R, editor. The Bacteriophages. Oxford University Press; New York: 2006a. pp. 277–301. [Google Scholar]
- Molineux IJ. Fifty-three years since Hershey and Chase; much ado about pressure but which pressure is it? Virology. 2006b;344:221–229. doi: 10.1016/j.virol.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Osborn MJ, Gander JE, Parisi E, Carson J. Mechanism of assembly of the outer membrane of Salmonella typhimurium. J. Biol. Chem. 1972;247:3962–3972. [PubMed] [Google Scholar]
- Plisson C, White HE, Auzat I, Zafarani A, São-José C, Tavares P, Orlova EV. Structure of the bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 2007;26:3720–3728. doi: 10.1038/sj.emboj.7601786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards KE, Williams RC, Calendar R. Mode of DNA packaging within bacteriophage heads. J. Mol. Biol. 1973;78:255–259. doi: 10.1016/0022-2836(73)90114-9. [DOI] [PubMed] [Google Scholar]
- Roeder GS, Sadowski PD. Bacteriophage T7 morphogenesis: phage-related particles in cells infected with wild-type and mutant T7 phage. Virology. 1977;76:263–285. doi: 10.1016/0042-6822(77)90302-6. [DOI] [PubMed] [Google Scholar]
- Saigo K. Tail-DNA connection and chromosome structure in bacteriophage T5. Virology. 1975;68:154–165. doi: 10.1016/0042-6822(75)90157-9. [DOI] [PubMed] [Google Scholar]
- Saigo K, Uchida H. Connection of the right-hand terminus of DNA to the proximal end of the tail in bacteriophage lambda. Virology. 1974;61:524–536. doi: 10.1016/0042-6822(74)90287-6. [DOI] [PubMed] [Google Scholar]
- São-José C, de Frutos M, Raspaud E, Santos MA, Tavares P. Pressure built by DNA packing inside virions: enough to drive DNA ejection in vitro, largely insufficient for delivery into the bacterial cytoplasm. J. Mol. Biol. 2007;374:346–355. doi: 10.1016/j.jmb.2007.09.045. [DOI] [PubMed] [Google Scholar]
- Serwer P. Internal proteins of bacteriophage T7. J. Mol. Biol. 1976;107:271–291. doi: 10.1016/s0022-2836(76)80005-8. [DOI] [PubMed] [Google Scholar]
- Serwer P. Observation of DNA by negative staining: phage T7 DNA-capsid complexes. Ninth International Congress on Electron Microscopy. 1978;II:228–229. [Google Scholar]
- Serwer P, Wright ET, Hakala KW, Weintraub ST. Evidence for bacteriophage T7 tail extension during DNA injection. BMC Research Notes. 2008;1:36. doi: 10.1186/1756-0500-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C. The bacteriophage ø29 portal motor can package DNA against a large internal force. Nature. 2001;413:748–752. doi: 10.1038/35099581. [DOI] [PubMed] [Google Scholar]
- Struthers-Schlinke JS, Robins WP, Kemp P, Molineux IJ. The internal head protein gp16 of bacteriophage T7 controls DNA ejection from the virion. J. Mol. Biol. 2000;301:35–45. doi: 10.1006/jmbi.2000.3940. [DOI] [PubMed] [Google Scholar]
- Tang J, Olson N, Jardine PJ, Grimes S, Anderson DL, Baker TS. DNA poised for release in bacteriophage ø29. Structure. 2008;16:935–943. doi: 10.1016/j.str.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JO. Chemical linkage of the tail to the right-hand end of bacteriophage lambda DNA. J. Mol. Biol. 1974;87:1–9. doi: 10.1016/0022-2836(74)90555-5. [DOI] [PubMed] [Google Scholar]
- Vogel U, Jensen KF. The RNA chain elongation rate in Escherichia coli depends on the growth rate. J. Bacteriol. 1994;176:2807–2813. doi: 10.1128/jb.176.10.2807-2813.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Kushner SR. Construction of versatile low-copy-number vectors for cloning, sequencing, and gene expression in Escherichia coli. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- Xiang Y, Morais MC, Battisti AJ, Grimes S, Jardine PJ, Anderson DL, Rossmann MG. Structural changes of bacteriophage ø29 upon DNA packaging and release. EMBO J. 2006;25:5229–5239. doi: 10.1038/sj.emboj.7601386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavriev SK, Shemyakin MF. RNA polymerase-dependent mechanism for the stepwise T7 phage DNA transport from the virion into E. coli. Nucleic Acids Res. 1982;10:1635–1652. doi: 10.1093/nar/10.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]