

Abstract
The hepatopulmonary syndrome (HPS) results from intrapulmonary vasodilation in the setting of cirrhosis and portal hypertension. In experimental HPS, pulmonary endothelial endothelin B (ETB) receptor overexpression and increased circulating endothelin-1 (ET-1) contribute to vasodilation through enhanced endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO) production. In both experimental cirrhosis and prehepatic portal hypertension, ETB receptor overexpression correlates with increased vascular shear stress, a known modulator of ETB receptor expression. We investigated the mechanisms of pulmonary endothelial ETB receptor-mediated eNOS activation by ET-1 in vitro and in vivo. The effect of shear stress on ETB receptor expression was assessed in rat pulmonary microvascular endothelial cells (RPMVECs). The consequences of ETB receptor overexpression on ET-1-dependent ETB receptor-mediated eNOS activation were evaluated in RPMVECs and in prehepatic portal hypertensive animals exposed to exogenous ET-1. Laminar shear stress increased ETB receptor expression in RPMVECs without altering mRNA stability. Both shear-mediated and targeted overexpression of the ETB receptor enhanced ET-1-mediated ETB receptor-dependent eNOS activation in RPMVECs through Ca2+-mediated signaling pathways and independent of Akt activation. In prehepatic portal hypertensive animals relative to control, ET-1 administration also activated eNOS independent of Akt activation and triggered HPS. These findings support that increased pulmonary microvascular endothelial ETB receptor expression modulates ET-1-mediated eNOS activation, independent of Akt, and contributes to the development of HPS.
Keywords: endothelial nitric oxide synthase, Akt, shear stress
THE HEPATOPULMONARY SYNDROME (HPS) results when cirrhosis, hepatic injury, or portal hypertension triggers vasodilatation in the pulmonary microvasculature leading to the development of hypoxemia (5). This syndrome is found in 10–20% of patients with cirrhosis and has recently been recognized to significantly increase mortality (25, 28). The pathogenesis of intrapulmonary vasodilatation remains an area of active investigation. In humans, increased pulmonary nitric oxide (NO) production appears to be an important contributor to vasodilatation (2, 23, 24), but the stimuli and mechanisms that mediate NO overproduction in humans are unknown.
Common bile duct ligation (CBDL) in the rat is recognized as an animal model of HPS. In this model, the onset of HPS at 2 wk after biliary obstruction is also associated with increased NO synthase activity and pulmonary NO production (20, 32). In addition, hepatic production and release of endothelin-1 (ET-1) begins within 1 wk after CBDL and is followed by increased expression of the endothelin B (ETB) receptor in the pulmonary microvascular endothelium beginning 2 wk after CBDL (16). Both shear stress (3, 18) and ET-1 (17) may increase ETB receptor expression in vitro, and the increase in ETB receptor expression after CBDL occurs over the same time frame as the onset of a hyperdynamic circulation with increased vascular shear stress and in the setting of increased circulating ET-1 levels (16). ET-1 stimulation of the ETB receptor is hypothesized to activate endothelial NO synthase (eNOS), resulting in increased pulmonary NO production (13). This hypothesis is supported by the observation that administration of a selective ETB receptor antagonist to 2-wk CBDL animals significantly improves HPS (13). However, the signaling pathways through which ET-1-mediated ETB receptor activation occurs in the pulmonary microvascular endothelium and whether alterations in ETB receptor levels directly influence the magnitude of ET-1-induced eNOS activation are unknown.
Recently, a defect in ETB receptor-dependent, Akt-mediated eNOS activation in sinusoidal endothelial cells has been found to reduce hepatic microvascular endothelial NO production and exacerbate portal hypertension (15). Based on the finding that eNOS-derived NO production is increased rather than decreased in CBDL lung with HPS (13), we hypothesized that important differences in ET-1-dependent ETB receptor-mediated signaling may be present in pulmonary microvascular endothelium relative to that reported in sinusoidal endothelium and may contribute to susceptibility to HPS.
The aim of the current study was to investigate the mechanisms of pulmonary endothelial ETB receptor-mediated eNOS activation in experimental HPS. To accomplish this goal, we assessed the effects of shear stress and ET-1 on ETB receptor expression and evaluated the consequences of ETB receptor overexpression on ET-1-dependent, ETB receptor-mediated eNOS activation in rat pulmonary microvascular endothelial cells (RPMVECs). We then directly assessed if an increase in pulmonary endothelial ETB receptor levels in portal hypertensive animals sensitizes these animals to exogenous ET-1-mediated eNOS activation and HPS in vivo. Our findings reveal that shear stress and ET-1 increase ETB receptor expression in the pulmonary microvascular endothelium. Furthermore, ETB receptor overexpression results in enhanced ET-1-mediated eNOS activation through a calcium-mediated pathway independent of Akt activation in vitro and contributes to ET-1-mediated HPS in vivo.
MATERIALS AND METHODS
Reagents and antibodies
ETB receptor antagonist BQ-788 was obtained from Alexis (San Diego, CA). ET-1 peptide was from Peninsula Laboratories (Belmont, CA). Specific PLC (U-73122), inositol 1,4,5-trisphosphate (IP3) receptor (2-aminoethoxydiphenyl borate, 2-APB), and calmodulin (W-7) inhibitors and the Ca2+ chelator BAPTA-AM were purchased from Calbiochem (San Diego, CA). Pertussis toxin and wortmannin (Calbiochem) were used to evaluate the phosphatidylinositol 3 (PI3)-kinase/protein kinase B/Akt pathway. Actinomycin D was obtained from Sigma (St. Louis, MO). eNOS antibody was purchased from Transduction Laboratories. ETB receptor and GADPH antibodies were from Abcam (Cambridge, MA). Phospho-eNOS (Ser1177) and phospho-Akt (Ser473) antibodies were obtained from Cell Signaling (Beverly, MA).
Cell culture
RPMVECs were purchased from Vec Technologies (Rensselaer, NY). Cells were cultured in complete media MCDB-131 (Vec Technologies) at 37°C in presence of humidified 95% air and 5% CO2. Cell passages were between 2 and 10.
Shear stress
Laminar shear stress was applied to RPMVEC monolayers using a cone and plate system. When RPMVECs reached 80–90% confluence in 100-mm dishes (Falcon), cells were exposed to nonpulsatile, laminar shear stress at 15 dyn/cm2 to simulate levels intermediate between venous and arterial levels (18).
Adenoviral construction
The ETB receptor coding sequence was obtained from rat lung by RT-PCR using the following primers: forward 5′-ACGGGGATCCCACAGGAGCAAGCTGCAACATGCAATCGTCCGCAA-3′ and reverse 5′-CCCAAGCTTGCACACCTTTCCGCAAGCACG-3′. The fragments were ligated into the pShuttle-CMV vector after they were confirmed by direct sequencing (University of Alabama sequencing facility; Ref. 3). DNA isolated from selected viral clones was transfected into QBI-293A cells using the SuperFect Reagent (Qiagen, Valencia, CA) for large-scale proliferation (3). Virus was purified by standard CsCl gradient centrifugation and tittered by spectrophotometry at 260 nm, in which 1 unit of optical density at 260 nm equals 1.1 × 1012 viral particles (VP).
Adenoviral RPMVEC infection
RPMVECs were plated in six-well plates and allowed to reach ~80% confluence. Cells were infected with AdCMV-ETB receptor (1,000 VP/cell) for 1 h in 2% FBS infection media, and then serum concentration was increased to 10%. In preliminary studies, AdCMV-green fluorescent protein (AdCMVGFP) constructs were used to assess infection efficiency, and ~90% of RPMVECs were found to be infected.
Animal models
Male Sprague-Dawley rats (200–250 g; Charles River, Wilmington, MA) were used in all experiments. Portal vein ligation (PVL) was performed as previously described (6). ET-1 (3 ng/200 g body wt/h) or saline were administered to normal or PVL animals by miniosmotic pump for 2 wk as previously described (13). This dose of ET-1 resulted in a 25–30% increase in circulating ET-1 levels and did not increase systemic arterial pressure. Mean systemic arterial pressure (MSAP) and portal venous pressure (PVP) were measured directly. The presence of HPS was confirmed by measuring the alveolar-arterial oxygen gradient (AaPO2) by arterial blood gas on an ABL 520 analyzer (Radiometer America, Westlake, OH) as previously described (6). Lung homogenates were used for Western analysis as descried below.
Western blot analysis
The homogenates from lung or RPMVECs (from above) in RIPA buffer (1.0% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 50 mM Tris · HCl, 150 mM NaCl, 1 mM EDTA, 0.1 mM EGTA) containing protease inhibitors (1 μg/ml aprotinin, 50 μg/ml benzamidine HCl, 50 μg/ml leupeptin, 40 mg/ml PMSF, 1 mM sodium vanadate, 1 mM sodium fluoride) were centrifuged at 14,000 g for 15 min at 4°C. Equal amounts of protein were electrophoresed on SDS-PAGE gel and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Piscataway, NJ). Blots were incubated with primary antibody over night at 4°C, washed, incubated with secondary antibody conjugated with horseradish peroxidase, and then detected with enhanced chemiluminescence (Amersham). Autoradiographic signals were assessed using an Astra scanner (UMAX, Fremont, CA) and quantitated with ImagePC software (Scion, Frederick, MD). Signal intensity was shown to be a linear function of sample concentration over the range analyzed.
RNA analysis
Total RNA from RPMVECs exposed to various stimuli was extracted using the Ultraspec-II RNA isolation system (Biotecx, Houston, TX). The ProSTAR first-strand RT-PCR kit (Stratagene, La Jolla, CA) was used to generate cDNA. The primers for ETB receptor (forward 5′-agctggtgcccttcatacagaagg-3′ and reverse 5′-tgcacacctttccgcaagcacg-3′) or 18s rRNA (forward 5′-gaaacggctaccacatcc-3′ and reverse 5′-caccagacttgccctcca-3′) were obtained from Qiagen Operon (Alameda, CA). Autoradiographic signals were assessed as above.
NO measurements
NO amounts in cell culture media were measured by a colorimetric assay for determination of total nitrite using the manufacturers′ instructions (OXIS International, Portland, OR). Absorbance at 540 nm was measured, and net NO production was calculated. The lower limit of sensitivity of the assay is 0.25 μM.
Statistics
Data were analyzed using Student's t-test with Bonferroni correction for multiple comparisons between groups. Measurements are expressed as means ± SE. Statistical significance was designated as P < 0.05.
RESULTS
ETB receptor expression in RPMVECs
To test the hypothesis that shear stress and ET-1 directly modulate endothelial ETB receptor expression in the pulmonary microvasculature, we evaluated RPMVECs (Fig. 1). Exposure of RPMVECs to a level of shear stress expected in the setting of a hyperdynamic circulatory state (15 dyn/cm2) resulted in a significant twofold increase in ETB receptor mRNA and protein levels. In contrast, ET-1 (0.01 μM) had no significant effect on ETB receptor mRNA or protein levels. Since shear stress is established to modulate mRNA levels of a number of proteins through effects on mRNA stability, we also assessed shear effects on ETB receptor mRNA stability by examining the t1/2 of ETB receptor mRNA after stimulation by shear stress in the presence or absence of actinomycin D (100 ng/ml; Fig. 2). After peak ETB receptor mRNA induction by shear stress at 12 h, the addition of actinomycin D did not significantly alter the decline in mRNA levels over 8 h, supporting that mRNA stability is not significantly altered. Together, these findings support that shear stress increases ETB receptor transcription and expression.
Fig. 1.
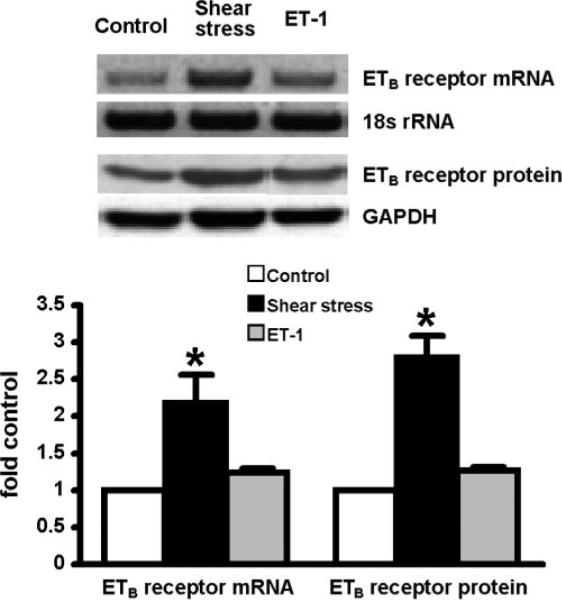
Effects of shear stress (15 dyn/cm2) and endothelin-1 (ET-1; 0.01 μM) on endothelin B (ETB) receptor expression in rat pulmonary microvascular endothelial cells (RPMVECs). Top shows representative RT-PCR for the ETB receptor mRNA (12 h) and immunoblots for ETB receptor protein (24 h). Bottom summarizes normalized ETB receptor mRNA and protein levels. Values are expressed as means ± SE (n = 5–7 for each group). *P < 0.05 compared with control cells.
Fig. 2.
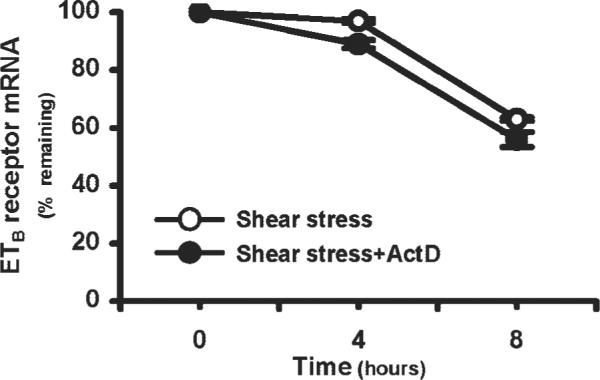
Expression of ETB receptor mRNA levels in RPMVECs incubated with actinomycin D (ActD; 100 ng/ml) for 4–8 h following peak ETB mRNA induction at 12 h by shear stress. mRNA levels are expressed as % of time 0 amounts remaining. Values are expressed as means ± SE (n = 5–7 for each group).
ETB receptor overexpression and ET-1-mediated eNOS activation in RPMVECs
To directly determine if increased ETB receptor protein levels in RPMVECs modulate ET-1-mediated eNOS activation, we pursued two approaches for increasing ETB receptor protein levels (Fig. 3). First, ETB receptor was overexpressed in RPMVECs using an AdCMV-ETB receptor construct. Second, ETB receptor levels were increased by exposing cells to shear stress. Subsequently, the effects of ET-1 on eNOS activation were evaluated. In control AdCMV-infected cells, ET-1 administration did not significantly influence eNOS activation assessed by phosphorylation of eNOS at Ser1177 (peNOS), likely due to low basal ETB receptor levels. In contrast, in RPMVECs infected with AdCMV-ETB receptor when ETB receptor levels increased 10.5-fold relative to control, exposure to ET-1 resulted in a significant fourfold increase in peNOS relative to AdCMV control infected cells. The increase in eNOS activation was associated with a significant increase in NO levels in cell culture media. The effects of ET-1 on both eNOS activation and NO production were completely inhibited by the selective ETB receptor antagonist BQ-788 (Fig. 3A). Similarly, in control RPMVECs not exposed to shear stress, ET-1 administration had no significant effect on peNOS levels. However, when ETB receptor levels were increased 2.5-fold relative to control by exposure to shear stress, ET-1 triggered a significant twofold increase in eNOS phosphorylation. This effect was also blocked by administration of BQ-788 (Fig. 3B).
Fig. 3.

Effects of adenovirus- (A) and shear stress- (B) mediated ETB receptor (ETB R) overexpression on endothelial nitric oxide synthase (eNOS) activation in response to exogenous ET-1 in RPMVECs. Top shows representative Western blots for ETB receptor expression, phosphorylation of eNOS at Ser1177 (peNOS), total eNOS from RPMVECs infected with AdCMV or AdCMV-ETB receptor, and cells under static conditions (control) or exposed to shear stress (15 dyn/cm2,24 h) in the absence or presence of ET-1 (0.01 μM) and BQ-788 (30 μM). Middle and bottom show the normalized peNOS/eNOS levels and nitrite amount in the media. Values are expressed as means ± SE (n = 4–5 for each group). *P < 0.05 relative to control or AdCMV treated RPMVECs. †P < 0.05 relative to ET-1 + AdCMV-ETB receptor transfected or ET-1 + shear stress-treated RPMVECs.
Signaling mechanisms of ET-1-mediated eNOS activation in RPMVECs
To define relevant signaling mechanisms, we explored two established pathways for ETB receptor-mediated signaling events, namely G protein-mediated Akt activation and activation of the PLC-calcium signaling pathway. Using AdCMV-ETB receptor-overexpressing RPMVECs stimulated with ET-1 (Fig. 4), we observed no change in phosphorylation of Akt at Ser473 (pAkt) relative to control. Moreover, neither the addition of wortmannin nor pertussis toxin to inhibit signaling through PI3K or Akt, respectively, blocked ET-1-mediated eNOS activation. In contrast, inhibitors of PLC/Ca2+ signaling including U-73122, BAPTA-AM, 2-APB, and W-7 all significantly decreased ET-1-mediated induction of peNOS levels, supporting an important role for PLC and Ca2+ signaling in ETB receptor-mediated events.
Fig. 4.
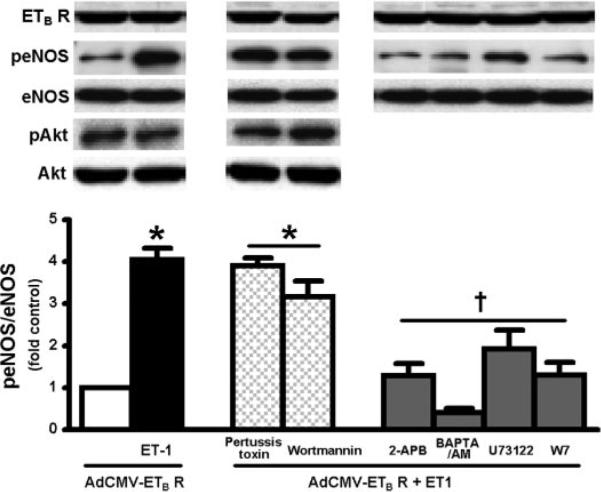
Effects of inhibition of Akt or Ca2+-mediated signaling pathways on ETB receptor-dependent eNOS activation by ET-1 in AdCMV-ETB receptor-infected RPMVECs. Top shows representative Western blots for ETB receptor expression, phosphorylation of eNOS at Ser1177 (peNOS) and Akt at Ser473 (pAkt) from RPMVECs stimulated with ET-1 (0.01 μM) for 5 min in the presence or absence of Akt inhibitors pertussis toxin (50 ng/ml) or wortmannin (500 nM), and Ca2+ inhibitors 2-aminoethoxydiphenyl borate (2-APB; 20 μM), BAPTA-AM (5 μM), U-73122 (2 μM), or calmodulin (W-7; 10 μM). Bottom summarizes normalized peNOS/eNOS levels. Values are expressed as means ± SE (n = 5–7 for each group). *P < 0.05 compared with AdCMV-ETB receptor-infected cells; †P < 0.05 compared with AdCMV-ETB receptor-infected cells activated by ET-1.
Effects of ET-1 administration on the development of HPS in normal and PVL animals
To directly test if ET-1 modulates lung eNOS activation and influences experimental HPS through the ETB receptor in vivo, ET-1 was administered intravenously by miniosmotic pump to normal or PVL animals for 2 wk (Table 1, Fig. 5). As we (16) previously reported, PVL animals developed significant portal hypertension, a hyperdynamic state, and an increase in pulmonary ETB receptor expression without the development of HPS. ET-1 infusion in normal animals did not alter PVP, MSAP, or arterial blood gases compared with vehicle-treated animals and was not associated with enhanced pulmonary eNOS activation. In contrast, ET-1-treated PVL animals developed a widened AaPO2, similar to changes observed in animals and humans with HPS. In addition, the development of HPS was associated with a significant increase in pulmonary eNOS phosphorylation in the absence of pulmonary Akt phosphorylation, similar to results in ET-1-treated ETB receptor-overexpressing RPMVECs.
Table 1.
MSAP, PVP, and arterial blood gases in PVL- and ET-1-treated animals
| Control | ET-1 | 2-wk PVL | 2-wk PVL + ET-1 | |
|---|---|---|---|---|
| MSAP, mmHg | 114.4±3.6 | 112.1±3.2 | 97.5±2.9* | 94.5±2.8* |
| PVP, mmHg | 7.5±1.1 | 8.3±0.9 | 13.7±1.2* | 12.5±1.0* |
| AaPO2, mmHg | 6.5±0.9 | 6.8±1.0 | 6.0±1.4 | 16.6±1.9* |
Values are means ± SE; n = 5–8 animals per group. MSAP, mean systemic arterial pressure; PVP, portal venous pressure; AaPO2, alveolar-arterial oxygen gradient; ET-1, endothelin-1; PVL, portal vein ligation.
P < 0.05 compared with control animals.
Fig. 5.

Effects of ET-1 (3 ng/200 g body wt/h) administration on pulmonary eNOS and Akt signaling activation in portal vein ligated (PVL) animals. Top shows representative Western blots of ETB receptor expression and phosphorylation of eNOS at Ser1177 (peNOS) and Akt at Ser473 (pAkt). Middle and bottom summarize peNOS/eNOS and pAkt/Akt levels expressed as fold control values. Values are expressed as means ± SE (n = 4–6 for each group). *P < 0.05 compared with control.
DISCUSSION
The current study was designed to explore the mechanisms and consequences of increased pulmonary microvascular endothelial ETB receptor expression in experimental HPS. In RPMVECs, we find that the direct application of laminar shear stress increases ETB receptor mRNA and protein levels, an event associated with increased mRNA expression. Furthermore, increased ETB receptor expression sensitizes RPMVECs to ET-1-mediated eNOS activation and NO production, an effect that occurs through a classic Ca2+-mediated signaling pathway, independent of Akt activation. Finally, in vivo administration of ET-1 in the setting of pulmonary endothelial ETB receptor overexpression resulting from prehepatic portal hypertension also activates eNOS in an Akt-independent fashion and triggers HPS. Together, these results support that pulmonary microvascular endothelial ETB receptor overexpression enhances ET-1-mediated eNOS activation independent of Akt both in vitro and in vivo and can trigger HPS. These findings are in line with the concept that increased vascular shear stress occurring in the setting of portal hypertension induces changes in the pulmonary microvascular endothelium that predispose to ET-1-mediated intrapulmonary vasodilatation.
The concept that increased pulmonary microvascular endothelial ETB receptor expression is an important event in the onset of experimental HPS derives from a number of observations. First, the increase in lung endothelial ETB receptor expression correlates with the onset of vasodilatation after CBDL (13). Second, although increased ETB receptor expression occurs in three different models of portal hypertension as the hyperdynamic state and increased shear stress develop, HPS occurs only when circulating ET-1 levels rise after CBDL or when exogenous ET-1 is infused in PVL animals (16). Third, pulmonary artery segments from portal hypertensive animals with increased endothelial ETB receptor expression exhibit enhanced ETB receptor-mediated, NO-dependent vasodilatation when exposed to ET-1 (13). Fourth, ETB receptor antagonists improve HPS in CBDL animals in vivo (13). Our current studies, using both a gene-targeting approach and application of shear stress, build on these results and directly demonstrate that rat pulmonary microvascular endothelial ETB receptor overexpression significantly enhances eNOS activation and NO production in response to physiologically relevant doses of ET-1 in vitro. The doses of ET-1 used in vivo increased circulating ET-1 levels by 25–30% and are in the range found in experimental HPS and in human cirrhosis where systemic vasoconstriction is absent (13). These observations support that increased ETB receptor expression alone is sufficient to sensitize the endothelium to ET-1-mediated NO production by activating eNOS. In our prior work (16), changes in endothelial ETB receptor levels were not found in the aorta of portal hypertensive animals, although whether they may occur in vascular beds and organs outside of the lung and directly influence vascular tone and perfusion in the setting of liver disease has not been studied.
ETB receptor stimulation mediates intracellular events, including eNOS activation, through classic G protein-coupled membrane receptor signaling pathways. In general, receptor activation catalyzes the exchange of GTP for GDP by the heterotrimeric G protein Gα-subunit followed by binding of GTP-bound Gα- to free Gβγ-subunits and activation of downstream pathways (7). In a number of cell types, ETB receptor activation stimulates PLC and a cascade ultimately leading to intracellular calcium release (1, 11), a known modulator of eNOS activation (21). In the current work, we assessed the contribution of Ca2+-mediated signaling pathways to ETB receptor-dependent eNOS activation in RPMVECs. Our results support that RPMVEC ETB receptor-dependent eNOS activation occurs through a PLC/IP3 Ca2+-dependent pathway based on the ability of inhibitors of PLC (U-73122), IP3 (2-APB), intracellular Ca2+ release (BAPTA-AM), and calmodulin (W-7) to significantly attenuate ET-1-mediated eNOS activation.
Phosphorylation/activation of eNOS may be mediated by a number of kinases including Akt, calmodulin-dependent kinase II, protein kinase A or G, and AMP-activated protein kinase (19). Recently, ETB receptor-mediated eNOS activation through G protein-βγ signaling to protein kinase B/Akt has been established in primary cultures of rat sinusoidal endothelial cells (14). Furthermore, inhibition of this signaling pathway mediated by G protein receptor kinase-2 overexpression has been shown to contribute to defective eNOS activation and increased portal pressure after CBDL (15). In the current study, ET-1-mediated eNOS activation was not associated with Akt phosphorylation and was not influenced by the addition of PI3-kinase and PKB/Akt inhibitors. Furthermore, infusion of ET-1 in vivo to prehepatic portal hypertensive animals with increased pulmonary microvascular ETB receptor levels triggered lung eNOS activation and HPS without altering lung Akt activation. Within the constraints of evaluating primary cell lines and using targeted gene overexpression of ETB receptors, our findings support that there are important differences in mechanisms of ETB receptor signaling between endothelial cells in different organs, which may be physiologically important. Defining the kinases involved in ET-1-mediated eNOS activation in the pulmonary endothelium is an important area for future study.
Endothelin receptors are members of the G protein-coupled membrane receptor family (4, 8, 9), and altered expression has been found in liver in experimental and human cirrhosis, particularly in activated stellate cells (12, 30, 31). In addition, portal hypertension is associated with the development of a hyperdynamic circulation that increases vascular shear stress, a recognized modulator of ETB receptor expression (3, 18). Our (16) prior finding that the increase in pulmonary endothelial ETB receptor levels in experimental models of cirrhosis and prehepatic portal hypertension correlates with the onset and severity of features of a hyperdynamic circulation is consistent with a role for increased shear stress in endothelial ETB receptor alterations. Our current in vitro studies provide direct evidence that increased laminar shear stress, in a physiologically relevant range, increases rat pulmonary endothelial ETB receptor expression. The molecular mechanisms that underlie shear-dependent increases in ETB receptor levels in the context of HPS remain to elucidated, although shear-mediated modulation of mRNA stability has been found to influence the levels of a number of endothelial proteins (10, 27, 29). Our findings here suggest that shear stress does not modulate ETB receptor mRNA stability in RPMVECs and support that effects on expression are operative. Other mediators, including ET-1 (17) and TNF-α (26), have been reported to increase ETB receptor expression, and each could contribute to changes in the pulmonary microvasculature in experimental HPS. Although our findings support that ET-1 alone does not significantly increase ETB receptor expression in vitro, defining all the factors that modulate pulmonary endothelial ETB receptor expression in the setting of HPS may be relevant to developing novel therapeutic strategies.
A key issue in experimental systems used to evaluate HPS is the relevance of findings to human disease. Our current results build on a series of studies and identify one mechanism for enhanced pulmonary microvascular sensitivity to ET-1-mediated vasodilatation in experimental HPS. The concept that vascular shear stress resulting from the onset of systemic hemodynamic alterations in the setting of portal hypertension may mediate this effect is consistent with the clinical finding that the great majority of patients with HPS have established portal hypertension (22). In addition, our results identify specific mechanisms and pathways to target in understanding the pathogenesis of human HPS. Finally, based on our current understanding of experimental HPS, among the important concepts to consider are whether pulmonary vascular endothelin signaling is altered and whether modulation of systemic hemodynamics or selective ETB receptor inhibition could have beneficial effects in human disease.
Acknowledgments
GRANTS This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-56804 and a Veterans Administration Merit Review grant to M. B. Fallon and National Heart, Lung, and Blood Institute Grant HL-70146 to R. P. Patel.
REFERENCES
- 1.Andreis PG, Tortorella C, Malendowicz LK, Nussdorfer GG. Endothelins stimulate aldosterone secretion from dispersed rat adrenal zona glomerulosa cells, acting through ETB receptors coupled with the phospholipase C-dependent signaling pathway. Peptides. 2001;22:117–122. doi: 10.1016/s0196-9781(00)00363-6. [DOI] [PubMed] [Google Scholar]
- 2.Brussino L, Bucca C, Morello M, Scappaticci E, Mauro M, Rolla G. Effect on dyspnoea and hypoxaemia of inhaled NG-nitro-L-arginine methyl ester in hepatopulmonary syndrome. Lancet. 2003;362:43–44. doi: 10.1016/S0140-6736(03)13807-X. [DOI] [PubMed] [Google Scholar]
- 3.Cahill PA, Hou MC, Hendrickson R, Wang YN, Zhang S, Redmond EM, Sitzman JV. Increased expression of endothelin receptors in the vasculature of portal hypertensive rats: role in splanchnic hemodynamics. Hepatology. 1998;28:396–403. doi: 10.1002/hep.510280216. [DOI] [PubMed] [Google Scholar]
- 4.Clement M, Albertini M. Differential release of prostacyclin and nitric oxide evoked from pulmonary and systemic vascular beds of the pig by endothelin-1. Prostaglandins Leukot Essent Fatty Acids. 1990;55:279–285. doi: 10.1016/s0952-3278(96)90009-5. [DOI] [PubMed] [Google Scholar]
- 5.Fallon M, Abrams G. Pulmonary dysfunction in chronic liver disease. Hepatology. 2000;32:859–865. doi: 10.1053/jhep.2000.7519. [DOI] [PubMed] [Google Scholar]
- 6.Fallon MB, Abrams GA, McGrath JW, Hou Z, Luo B. Common bile duct ligation in the rat: a model of intrapulmonary vasodilatation and hepatopulmonary syndrome. Am J Physiol Gastrointest Liver Physiol. 1997;272:G779–G784. doi: 10.1152/ajpgi.1997.272.4.G779. [DOI] [PubMed] [Google Scholar]
- 7.Gutkind J. Cell growth control by G protein-coupled receptors:from signal transduction to signal integration. Oncogene. 1998;17:1331–1342. doi: 10.1038/sj.onc.1202186. [DOI] [PubMed] [Google Scholar]
- 8.Haynes W, Webb D. Contribution of endogenous generation of endothelin-1 to basal vascular tone. Lancet. 1994;844:852–854. doi: 10.1016/s0140-6736(94)92827-4. [DOI] [PubMed] [Google Scholar]
- 9.Huggins JP, Pelton JT, Miller RC. The structure and specificity of endothelin receptors: their importance in physiology and medicine. Pharmacol Ther. 1993;59:55–123. doi: 10.1016/0163-7258(93)90041-b. [DOI] [PubMed] [Google Scholar]
- 10.Inoue H, Taba Y, Miwa Y, Yokota C, Miyagi M, Sasaguri T. Transcriptional and posttranscriptional regulation of cyclooxygenase-2 expression by fluid shear stress in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2002;22:1415–1420. doi: 10.1161/01.atv.0000028816.13582.13. [DOI] [PubMed] [Google Scholar]
- 11.Jaureguiberry MS, di Nunzio AS, Dattilo MA, Bianciotti LG, Vatta MS. Endothelin 1 and 3 enhance neuronal nitric oxide synthase activity through ETB receptors involving multiple signaling pathways in the rat anterior hypothalamus. Peptides. 2004;25:1133–1138. doi: 10.1016/j.peptides.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Leivas A, Jimenez W, Bruix J, Boix L, Bosch J, Arroyo V, Rivera F, Rodes J. Gene expression of endothelin-1 and ETA and ETB receptors in human cirrhosis: relationship with hepatic hemodynamics. J Vasc Res. 1998;35:186–193. doi: 10.1159/000025583. [DOI] [PubMed] [Google Scholar]
- 13.Ling Y, Zhang J, Luo B, Song D, Liu L, Tang L, Stockard C, Grizzle W, Ku D, Fallon MB. The role of endothelin-1 and the endothelin B receptor in the pathogenesis of experimental hepatopulmonary syndrome. Hepatology. 2004;39:1593–1602. doi: 10.1002/hep.20244. [DOI] [PubMed] [Google Scholar]
- 14.Liu S, Premont RT, Kontos CD, Huang J, Rockey DC. Endothelin-1 activates endothelial cell nitric oxide synthase (eNOS) via heterotrimeric G-protein beta gamma subunit signaling to protein kinase B/Akt. J Biol Chem. 2003;278:49929–49935. doi: 10.1074/jbc.M306930200. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med. 2005;11:952–958. doi: 10.1038/nm1289. [DOI] [PubMed] [Google Scholar]
- 16.Luo B, Liu L, Tang L, Zhang J, Stockard C, Grizzle W, Fallon M. Increased pulmonary vascular endothelin B receptor expression and responsiveness to endothelin-1 in cirrhotic and portal hypertensive rats: a potential mechanism in experimental hepatopulmonary syndrome. J Hepatol. 2003;38:556–563. doi: 10.1016/s0168-8278(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 17.Mallat A, Preaux AM, Gal CSL, Raufaste D, Gallois C, Brenner DA, Bradham C, Maclouf J, Iourgenko V, Fouassier L, Dhumeaux D, Mavier P, Lotersztajn S. Growth inhibitory properties of endothelin-1 in activated human hepatic stellate cells: a cyclic adenosine monophosphate-mediated pathway. Inhibition of both extracellular signal-regulated kinase and c-Jun kinase and upregulation of endothelin B receptors. J Clin Invest. 1996;98:2771–2778. doi: 10.1172/JCI119103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morawietz H, Talanow R, Szibor M, Rueckschloss U, Schubert A, Bartling B, Darmer D, Holtz J. Regulation of the endothelin system by shear stress in human endothelial cells. J Physiol. 2000;525:761–770. doi: 10.1111/j.1469-7793.2000.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Nunes H, Lebrec D, Mazmanian M, Capron F, Heller J, Tazi KA, Zerbib E, Dulmet E, Moreau R, Dinh-Xuan AT, Simonneau G, Herve P. Role of nitric oxide in hepatopulmonary syndrome in cirrhotic rats. Am J Respir Crit Care Med. 2001;164:879–885. doi: 10.1164/ajrccm.164.5.2009008. [DOI] [PubMed] [Google Scholar]
- 21.Pollock J, Forstermann U, Mitchell J, Warner T, Schmidt H, Nakane M, Murad F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci USA. 1991;88:10480–10484. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-Roisin R, Krowka MJ, Herve P, Fallon MB. ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD) Eur Respir J. 2004;24:861–880. doi: 10.1183/09031936.04.00010904. [DOI] [PubMed] [Google Scholar]
- 23.Rolla G, Brussino L, Colagrande P. Exhaled nitric oxide and impaired oxygenation in cirrhotic patients before and after liver transplantation. Ann Intern Med. 1998;129:375–378. doi: 10.7326/0003-4819-129-5-199809010-00005. [DOI] [PubMed] [Google Scholar]
- 24.Schenk P, Madl C, Rezale-Majd S, Lehr S, Muller C. Methylene blue improves the hepatopulmonary syndrome. Ann Intern Med. 2000;133:701–706. doi: 10.7326/0003-4819-133-9-200011070-00012. [DOI] [PubMed] [Google Scholar]
- 25.Schenk P, Schoniger-Hekele M, Fuhrmann V, Madl C, Silberhumer G, Muller C. Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology. 2003;125:1042–1052. doi: 10.1016/s0016-5085(03)01207-1. [DOI] [PubMed] [Google Scholar]
- 26.Smith PJ, Teichert-Kuliszewska K, Monge JC, Stewart DJ. Regulation of endothelin-B receptor mRNA expression in human endothelial cells by cytokines and growth factors. J Cardiovasc Pharmacol. 1998;31:S158–S160. doi: 10.1097/00005344-199800001-00045. [DOI] [PubMed] [Google Scholar]
- 27.Sokabe T, Yamamoto K, Ohura N, Nakatsuka H, Qin K, Obi S, Kamiya A, Ando J. Differential regulation of urokinase-type plasminogen activator expression by fluid shear stress in human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287:H2027–H2034. doi: 10.1152/ajpheart.00260.2004. [DOI] [PubMed] [Google Scholar]
- 28.Swanson K, Wiesner R, Krowka M. Natural history of hepatopulmonary syndrome: impact of liver transplantation. Hepatology. 2005;41:1122–1129. doi: 10.1002/hep.20658. [DOI] [PubMed] [Google Scholar]
- 29.Weber M, Hagedorn CH, Harrison DG, Searles CD. Laminar shear stress and 3′ polyadenylation of eNOS mRNA. Circ Res. 2005;96:1161–1168. doi: 10.1161/01.RES.0000170651.72198.fa. [DOI] [PubMed] [Google Scholar]
- 30.Yokomori H, Oda M, Ogi M, Kamegaya Y, Tsukada N, Nakamura M, Ishii H. Enhanced expression of endothelin receptor subtypes in cirrhotic rat liver. Liver. 2001;21:114–122. doi: 10.1034/j.1600-0676.2001.021002114.x. [DOI] [PubMed] [Google Scholar]
- 31.Yokomori H, Oda M, Yasogawa Y, Nishi Y, Ogi M, Takahashi M, Ishii H. Enhanced expression of endothelin B receptor at protein and gene levels in human cirrhotic liver. Am J Pathol. 2001;159:1353–1362. doi: 10.1016/S0002-9440(10)62522-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Ling Y, Luo B, Tang L, Stockard C, Grizzle WE, Fallon MB. Analysis of pulmonary heme oxygenase-1 and nitric oxide synthase alterations in experimental hepatopulmonary syndrome. Gastroenterology. 2003;125:1441–1451. doi: 10.1016/j.gastro.2003.07.005. [DOI] [PubMed] [Google Scholar]