

Abstract
Treatment of mice bearing established ovarian tumors with T cells expressing chimeric NKG2D receptors (chNKG2D) develop protective host immune responses to tumor antigens. In this study, the mechanisms that chNKG2D T cells require to induce host immunity against ovarian tumors and which of the host immune cells are involved in tumor elimination were determined. Treatment with chNKG2D T cells led to a sustained, increased IFNγ production by host NK, CD4+, and CD8+ T cells in the spleen and at the tumor site and this continued for many weeks after T cell injection. Tumor antigen presentation was enhanced in chNKG2D T cell treated mice, and there were greater numbers of tumor-specific T cells at the tumor site and in draining lymph nodes after treatment with chNKG2D T cells. The increase in host cell cytokine secretion and antigen presentation was dependent on chNKG2D T cell-derived perforin, IFNγ, and GM-CSF. Host immune mechanisms were involved in tumor elimination because inhibition of tumor growth was limited in mice that lacked perforin, IFNγ, NK cells, or T and B cells (Rag1−/−). There was no role for host-derived GM-CSF or CD1-dependent NKT cells, as mice deficient in these were able to clear tumors as well as treated wildtype B6 mice. In summary, chNKG2D T cells required both cytotoxicity and cytokine secretion as well as the participation of host immune cells for development of a host anti-tumor immune response and complete efficacy.
Keywords: Tumor immunity, NK cells, T cells, Cytotoxic
INTRODUCTION
Adoptive T cell transfer has the potential to provide effective specific therapy for cancer. It has been found that prior leukodepletion of patients and long-term survival of the transferred T cells may enhance anti-tumor efficacy (1, 2). However, antigen escape variants and downregulation of MHC class I can ultimately lead to evasion of immune response and eventual tumor progression in patients (3). Development of a host immune response that targets a variety of tumor antigens may help prevent the outgrowth of antigen-loss tumor variants and improve clinical outcome.
One benefit of using T cell therapies for treatment of cancer is that T cells can specifically target tumor cells with limited cross-reaction with normal tissues. T cells recognize tumor antigens as peptides associated with MHC molecules, or T cells can be engineered to recognize antigens directly through chimeric receptors. Many current immunotherapy trials target a variety of tumor associated antigens including melanoma antigens, cancer testes antigens, p53, Her-2 neu, and α-folate receptor through use of T cells expressing specific receptors for tumor antigens (3, 4). Another potential tumor specific target are the ligands for the NKG2D receptor. NKG2D is an activating receptor expressed on NK cells, CD8+ T cells, and subsets of other T cells (5). The ligands for this receptor are expressed on many different types of carcinomas and hematopoetic tumors, and are not expressed on the cell surface of most normal tissues (6, 7).
Induction of host cell production of interferon γ (IFNγ) at the ovarian tumor site may be one way to increase anti-tumor immunity. Treating ovarian cancer patients with a combination of IFNγ and chemotherapy has shown some benefits and IFNγ has many anti-tumor mechanisms, including activating antigen presenting cells, increasing antigen presentation by tumor cells, and having cytostatic effects on tumor cells directly (8–10). In addition, increasing the activation of host antigen-presenting cells to induce host T cell and NK cell responses to tumor antigens may also improve cancer therapy (11).
Previously, we reported on a chimeric NKG2D receptor, which consists of the NKG2D receptor fused to the cytoplasmic region of the CD3ζ chain (12–16). Treatment of mice bearing established ovarian cancer with chNKG2D T cells led to long-term, tumor-free survival (16). These tumor-surviving mice had developed host CD4 and CD8 T cell memory responses to tumor antigens, and this immune response was protective against tumor rechallenge (16). This indicates that chNKG2D T cell therapy induces the development of host immune responses to tumor antigens. In this study, we determined the mechanisms whereby chNKG2D T cells activate host immune responses to tumor antigens, examining host cell IFNγ production, antigen presentation, and trafficking of tumor-specific T cells. We also determined the requirement of host molecules in the development of the host immune response and tumor elimination.
MATERIALS AND METHODS
Mice
Female C57BL/6 (B6) and B6-LY5.2/Cr(CD45.1+) mice were purchased from the National Cancer Institute (Frederick, MD). B6.Rag1−/− mice were bred and maintained at Dartmouth Medical School.C57BL/6 interferon-γ deficient mice B6.129S7-Ifngtm1Ts/J (IFN-γ−/−), interferon-γ receptor 1 deficient mice B6.129S7-IfngR1tm1Agt/J (IFN-γR−/−), perforin deficient mice C57BL/6-Prf1tm1Sdz/J (Pfp−/−), and C57BL/6-TgTcraTcrb1100Mjb/J (OT-I) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). GM-CSF deficient mice (GM-CSF−/−) on a C57BL/6 background were kindly provided by Dr. Jeff Whitsett of the University of Cincinnati, and B6.CD1−/− mice were kindly provided by Dr. Mark Exley (Harvard University, Boston, MA). Mice used were between 7 to 12 weeks of age at the start of the experiments. All animal work was performed in the Dartmouth Medical School Animal Facility in accordance with Institutional guidelines.
Injection of ID8-GFP cells and treatment of mice with genetically modified T cells
Mouse spleen cells were stimulated with ConA for 18 hours (1μg/ml) and transduced as previously described (13, 14). Two days after transduction, T cells were selected in media containing G418 (0.5 mg/mL) and 25 U/mL recombinant human (rHu) IL-2 for three days. Viable cells were isolated using Histopaque-1083 (Sigma, St Louis, MO) and expanded for two days without G418 (13, 14). ID8-GFP cells (2 ×106) were injected intraperitoneally (i.p.) into B6 mice or mice deficient in IFNγ, GM-CSF, perforin, Rag1, or CD1. Mice were treated i.p. with wtNKG2D or chNKG2D T cells (5 × 106) one week after tumor injection. Mice were sacrificed and a peritoneal wash was performed using 10ml PBS. The number of solid tumors on the peritoneal wall was counted. Red blood cells in the peritoneal washes were lysed with ACK lysis buffer (0.15M NH4Cl, 1mM KHCO3, 0.1mM EDTA, pH 7.3), the number of cells was counted, and percent GFP+ cells was determined by flow cytometry. Absolute number of tumor cells in the peritoneal washes was determined by multiplying percent GFP+ cells by the number of cells in the peritoneal wash.
In vivo NK cell depletion
To deplete NK cells, 200 μg of anti-NK1.1 (PK136) or control mouse γ globulin (Jackson ImmunoResearch Laboratories) was injected into mice i.p. two days before and three days after T cell injection (day +5 and day +10 relative to tumor cell injection).
Cytokine secretion and intracellular cytokine detection by flow cytometry
Peritoneal wash cells (106) from tumor-bearing wtNKG2D or chNKG2D T cell treated mice were cultured in 48-well plates in complete media. Twenty-four hour cell-free conditioned media were assayed for IFNγ by ELISA using mouse Duoset ELISA kits (R&D Systems) and for nitric oxide using Griess’s Reagent for nitrite (Sigma) according to manufacturers’ protocols. Seventy-two hour conditioned media were assayed for additional cytokines using multiplex analysis (Bio-Rad) that was performed by the Immune Monitoring Laboratory of the Norris Cotton Cancer Center (Lebanon, NH). For intracellular staining, peritoneal wash cells (106) or spleen cells (2.5 × 106) were cultured in complete media for 24 hours. During the last five hours of culture, 10μg/ml brefeldin A was added to the wells (Sigma). Cells were then incubated with FcR block and stained with FITC-conjugated anti-CD8β (clone CT-CD8β), APC-conjugated anti-NK1.1 (clone PK136), or APC-conjugated anti-CD45.1 (clone A20), and biotin-conjugated anti-CD3 (clone eBio500A2), with a PE-Cy5.5 conjugated-streptavidin secondary. Cells were fixed with 1% paraformaldehyde, permeabilized with 0.1% saponin, and stained with PE-conjugated anti-IFNγ (clone XMG12), or PE-conjugated anti-rat IgG1 isotype control. All antibodies were purchased from eBioscience (San Diego, CA). Cell fluorescence was monitored using a FACSCalibur cytometer (Becton Dickinson, San Jose, CA).
In vitro tumor antigen presentation assay
ID8-GFP-tOva cells were made by retroviral transduction of ID8-GFP cells with a vector containing a truncated OVA gene where nucleotides encoding aa 1–40 (the leader peptide) were removed (17–19). ID8-GFP-tOva or ID8-GFP cells (2 ×106) were injected i.p. into B6 mice, and mice were treated with wtNKG2D or chNKG2D T cells (5 × 106) i.p. after seven days (d+7). Seven days after T cell injection (day +14), spleen cells and mediastinal lymph node cells were isolated. CD8+ OT-I T cells were purified from spleen and lymph node cells using magnetic bead selection (Miltenyi Biotec) and FITC-conjugated anti-CD8β Abs according to the manufacturer’s instructions and purity was > 95% for OT-I T cells (CD8β+). CFSE-labeled OT-I T cells (5 × 104) were cultured with spleen and lymph node cells (105) and proliferation of T cells was determined by flow cytometry after 72 hrs. For peptide pulsing, spleen and lymph node cells were incubated with OVA257–264 peptide (10−9 – 10 −11 M) for 2 hours at 37°C, and the cells were washed three times to remove non-bound peptides before culture with OT-I T cells.
In vivo OT-I T cell trafficking and survival
ID8-GFP-tOva or ID8-GFP cells (2 ×106) were injected i.p. into B6 mice, and after seven days mice were treated i.p. with 5 × 106 wtNKG2D or chNKG2D T cells. At the same time as T cell injection, purified, CFSE-labeled OT-I T cells (2 × 106) that were congenically marked with both Ly5.1 and Ly5.2 were injected i.v. Four days after transfer of OT-I T cells, a peritoneal wash was performed, spleen cells and mediastinal lymph node cells were processed, and the presence of Ly5.1+/Ly5.2+ CFSE-labeled OT-I T cells was determined by flow cytometry.
Statistical analysis
Differences between groups were analyzed using the Student’s t test or ANOVA using Prism software (GraphPad Software, San Diego, CA). Values of p<0.05 were considered significant.
RESULTS
Treatment of tumor bearing mice with chNKG2D T cells induces cytokine secretion from host immune cells
Treatment of mice bearing an established ovarian tumor burden with chNKG2D T cells led to long-term, tumor-free survival and the induction of host memory responses to tumor antigens (16). However, the mechanism of how chNKG2D T cells induce systemic immune responses against tumor antigens is unclear. To study the development of the host immune response, ID8-GFP ovarian tumor cells were injected i.p. into mice and were treated one week later with T cells expressing chNKG2D receptors or wtNKG2D receptors i.p. One, three, and seven days after T cell injection, spleen cells from treated mice were cultured with media, and the supernatants were analyzed for IFNγ production (Figure 1). Spleen cells from mice treated with chNKG2D T cells secreted significantly more IFNγ compared to mice treated with wtNKG2D T cells. The increase in IFNγ secretion from spleen cells began three days after T cell treatment with a peak at seven days after chNKG2D T cell injection. Similar data were found when mice were treated with chNKG2D T cells five weeks after tumor inoculation (data not shown). To determine which cell types secreted IFNγ, intracellular cytokine staining was performed (Figure 1B and Supplementary Figure 1). ChNKG2D T cells were not a source of IFNγ as these T cells did not traffic to the spleen after i.p. injection, as shown by the lack of Ly5.1+cells in the spleen at any time-point analyzed (Figure 1B). There was an increase in the production of IFNγ by host NK cells, CD8+ and CD4+ T cells three and seven days after chNKG2D T cell injection, indicating that treatment with chNKG2D T cells led to an increased number of host cells that secreted IFNγ.
Figure 1. Host NK cells and T cells have increased IFNγ production after treatment with chNKG2D T cells.

One, three, and seven days after T cell injection, spleen cells from wtNKG2D (white bars) or chNKG2D T cell (black bars) treated tumor bearing mice were cultured in media for 24 hours. (A) Cell-free supernatants were assayed for IFNγ or (B) cells were assayed for IFNγ production by intracellular staining. Cells were gated on either CD3+Ly5.1+, CD8+CD3+, CD4+CD3+, or NK1.1+CD3− as indicated. The average of each group (n=4) is shown as percent IFNγ+ cells + SD. Treatment with chNKG2D T cells significantly increased IFNγ secretion compared to control treated mice (*-p<0.05). Data are representative of at least 5 separate experiments.
In mice with an established tumor burden, a single treatment with chNKG2D T cells led to a significantly reduced tumor burden compared to mice treated with wtNKG2D T cells (14). The kinetics of the host IFNγ response were analyzed to determine if the induced host immune response was sustained for long periods of time. The increased IFNγ secretion in chNKG2D T cell treated mice was maintained up to ten weeks after T cell injection, and both lymphocytes at the tumor site and in the spleen were producing IFNγ (Figure 2A). This response decreased over time with the peak IFNγ response occurring seven days after T cell injection. Host cells from the peritoneal cavity also secreted a significant amount of nitric oxide for at least four weeks after chNKG2D T cell injection. Spleen cells did not secrete nitric oxide at any timepoint analyzed (data not shown). Similar to earlier timepoints, host NK, CD8+, and CD4+ T cells from chNKG2D T cell treated mice produced more IFNγ compared to mice treated with wtNKG2D T cells, and this response decreased over time.
Figure 2. Tumor bearing mice treated with chNKG2D T cells have a sustained IFNγ response from host cells.

(A) ID8-GFP cells were injected i.p. and mice were treated with wtNKG2D T cells (white bars) or chNKG2D T cells (black bars) one week later. Three, five, eight, or eleven weeks after tumor cell injection, peritoneal wash cells and spleen cells from T cell treated or naive mice (grey bars) were cultured in media for 24 hours. Cell-free supernatants were collected and assayed for IFNγ or for nitric oxide. (B) Intracellular staining for IFNγ was performed on peritoneal wash cells and spleen cells cultured as described in (A). Cells were gated on either CD8+CD3+, CD4+CD3+, or NK1.1+CD3− as indicated. The average of each group (n=4) is shown + SD. Treatment with chNKG2D T cells significantly increased IFNγ and nitric oxide secretion compared to control treated mice (*-p<0.05). Data are representative of 2 separate experiments.
As the secretion of additional cytokines may be affected by chNKG2D T cell treatment, cytokine production by peritoneal and spleen cells from wtNKG2D or chNKG2D T cell-treated mice was analyzed (Table I). Seven weeks after chNKG2D T cell injection, cells isolated from the tumor site and the spleen secreted elevated amounts of many cytokines, including IL-2, GM-CSF, CCL3, and CCL4. Additionally, there was a decrease in KC and CCL2 in mice treated with chNKG2D T cells. Together, these data demonstrate that treatment of tumor-bearing mice with chNKG2D T cells induced a sustained host immune response and proinflammatory cytokine production from NK and T cells at the tumor site and in the spleen.
Table I.
Cytokine secretion eight weeks after tumor injection
| Cytokine | Spleen cells | Peritoneal cells | ||
|---|---|---|---|---|
| WT | CH | WT | CH | |
| IL-2 | 1596a | 5096* | 1033 | 3077* |
| IL-3 | 438 | 2805* | 129 | 2532* |
| IL-4 | 41 | 100* | 8 | 171* |
| IL-5 | 21 | 551* | 181 | 757b* |
| IL-9 | 58 | 154* | 98 | 179* |
| IL-13 | 116 | 648* | 185 | 1190* |
| GM-CSF | 269 | 1242* | 131 | 890* |
| CCL3 | 892 | 2530* | 233 | 377* |
| CCL4 | 6146 | 10765* | 167 | 1294* |
| Eotaxin | 336 | 631* | 506 | 696* |
| TNFα | 43 | 107* | 88 | 160* |
| KC | 437 | 70* | 4393 | 1484* |
| CCL2 | 6158b | 1479* | 6158b | 6158b |
WT- wtNKG2D T cell treated mice, CH- chNKG2D T cell treated mice
- Values shown as pg/ml and are an average of three mice.
- Values were above detection limit of assay.
- Significantly different than wtNKG2D T cell treated mice, (p<0.05).
Induction of IFNγ production from host cells requires chNKG2D T cell-derived cytokines and perforin
It has been shown that chNKG2D T cell-derived IFNγ, GM-CSF, and perforin are essential for complete anti-tumor efficacy (16). The requirement for cytokine secretion and tumor lysis may be partially due to their involvement in inducing a systemic host immune response to tumor antigens. Tumor-bearing mice were treated with chNKG2D T cells derived from wildtype B6 mice or from mice deficient in IFNγ, GM-CSF, or perforin. ChNKG2D T cell-derived IFNγ, GM-CSF, and perforin were all required for a complete induction of an IFNγ response, as shown by decreased IFNγ secretion from spleen cells both three and seven days after treatment with chNKG2D T cells deficient in any of these effector molecules (Figure 3A). As chNKG2D T cell-derived IFNγ could be acting directly on the tumor cells or on the host immune cells, mice deficient in the IFNγR were used as hosts to determine if host cell responsiveness to IFNγ was required. An increase in IFNγ secretion was not observed after chNKG2D T cell treatment in mice deficient in the IFNγR, indicating that host cells needed to respond to the IFNγ secreted by the chNKG2D T cells for the induction of the host immune response (Figure 3B).
Figure 3. Induction of host IFNγ production requires chNKG2D T cell- derived cytokines and perforin.

(A) Spleen cells from tumor bearing mice treated with B6-derived wtNKG2D (white bars) or chNKG2D T cell (black bars), or chNKG2D T cells derived from GM-CSF- (hashed bars), IFNγ-(striped bars), or perforin-deficient mice (grey bars) or (B) spleen cells from mice deficient in IFNγR1 treated with wtNKG2D (hashed bars) or chNKG2D (striped bars) T cells were cultured in media for 24 hours. Cell-free supernatants were collected and assayed for IFNγ by (A, B) ELISA or (C) intracellular staining. Cells were gated on either NK1.1+ CD3−, CD8+CD3+, or CD4+CD3+ as indicated. The average of each group (n=4) is shown + SEM. Treatment with chNKG2D T cells significantly increased IFNγ secretion compared to wtNKG2D T cell treated mice (*-p<0.05). Treatment with chNKG2D T cells deficient in effector molecules secreted significantly less IFNγ compared to chNKG2D T cell treated mice (§-p<0.05). Data are representative of at least 2 separate experiments.
Intracellular staining was performed on spleen cells to determine which host cells were affected by chNKG2D T cell-derived molecules. ChNKG2D T cell-derived GM-CSF was required for the induction of host NK cell and CD4+ T cell IFNγ production, while chNKG2D T cell-derived IFNγ and perforin were required for the induction of host CD8+ and CD4+ T cell responses, as shown by decreased percent of IFNγ+ cells from mice treated with T cells deficient in these molecules (Figure 3C). Thus, chNKG2D T cell-derived cytokines and perforin were involved in the induction of a systemic host anti-tumor response.
Treatment with chNKG2D T cells increased tumor antigen presentation and tumor-specific T cell infiltration at the tumor site
Treatment with chNKG2D T cells may increase host immune responses to ID8 tumor cells by increasing antigen presentation and therefore the activation of host T cells. To test this, mice were injected with ID8-GFP cells that expressed a truncated OVA gene (ID8-GFP-tOva) that is not secreted and therefore remains an intracellular tumor antigen (18, 19). After seven days of tumor growth, mice were treated with either PBS, wtNKG2D, or chNKG2D T cells. One week after T cell injection, spleen cells and draining lymph node cells were isolated and cultured with CFSE-labeled OT-I T cells, which recognize the tumor specific Ova peptide presented by MHC class I molecules. After three days of culture, OT-I T cells proliferated more when cultured with spleen or lymph node cells from chNKG2D T cell treated mice compared to mice treated with wtNKG2D T cells or with PBS (Figure 4A). Spleen and lymph node cells from wtNKG2D and chNKG2D T cell treated mice had similar numbers of antigen presenting cells including CD11c+ dendritic cells and macrophages and these antigen presenting cells expressed similar amounts of MHC class I, MHC class II, CD80, and CD86 (data not shown). This suggests that spleen and lymph node cells from the chNKG2D T cell treated mice were presenting more antigen to the OT-I T cells. To determine if the increase in OT-I T cell proliferation was dependent on specific antigen, similar experiments were performed with mice bearing ID8-GFP tumors that did not express Ova. OT-I T cells proliferated more when cultured with spleen and lymph node cells from chNKG2D T cell treated mice than with cells from wtNKG2D or PBS-treated mice, but the increased proliferation was modest compared to OT-I T cells that were cultured with cells from chNKG2D T cell treated mice bearing tumors that expressed Ova (Figure 4). In addition, lymph node and spleen cells from chNKG2D T cell-, wtNKG2D T cell-, and PBS-treated mice were pulsed with increasing concentrations of Ova peptides which induced proliferation of OT-I T cells (Figure 4C and data not shown). This indicates that chNKG2D T cells induced T cell proliferation because of increased antigen presentation and other additional factors, likely cytokine secretion, from spleen and lymph node cells. The increase in tumor antigen presentation was dependent on chNKG2D T cell-derived GM-CSF, IFNγ, and perforin as cells isolated from mice treated with chNKG2D T cell deficient in one of these molecules did not have increased antigen presentation by cells taken from the draining lymph node or spleen (Figure 4D).
Figure 4. Treatment with chNKG2D T cells increases tumor antigen presentation and requires chNKG2D T cell expression of GM-CSF, IFNγ, and perforin for antigen presentation.

(A) ID8-GFP-tOva or (B) ID8-GFP cells were injected i.p. and mice were treated with wtNKG2D T cells (white bars), chNKG2D T cells (black bars), or PBS (grey bars) one week later. Seven days after T cell injection, cells isolated from the mediastinal lymph node and spleen were cultured with CFSE-labeled OT-I T cells. OT-I T cell proliferation was measured after 72 hours of culture. (C) Lymph node cells isolated from ID8-GFP bearing mice treated as described in figure 4B were pulsed with different concentrations of Ova peptide as indicated, and were cultured with CFSE-labeled OT-I T cells. OT-I T cell proliferation was measured after 72 hours of culture. (D) ID8-GFP-tOva cells were injected i.p. and mice were treated with wtNKG2D T cells or chNKG2D T cells from B6 mice or chNKG2D T cells from GM-CSF- (hashed bars), IFNγ-(striped bars), or perforin-deficient mice (grey bars) one week later. Seven days after T cell injection, cells from the mediastinal lymph node and spleen were cultured as described in (A). The average of each group + SD is shown (n=4). Treatment with chNKG2D T cells significantly increased tumor antigen specific T cell proliferation compared to wtNKG2D T cell treated mice (*-p<0.05). Treatment with chNKG2D T cells deficient in effector molecules had significantly less T cell proliferation compared to chNKG2D T cell treated mice (§-p<0.05). Data are representative of 2 separate experiments.
To address how chNKG2D T cell treatment affects tumor antigen-specific T cells in vivo, wtNKG2D or chNKG2D T cells were injected i.p. into ID8-GFP-tOva bearing mice and concurrently mice were injected with CFSE-labeled OT-I T cells i.v. Three days after OT-I T cell transfer, the trafficking of the tumor antigen-specific T cells was measured. A higher number of OT-I T cells were found in the draining lymph node and at the tumor site in the chNKG2D T cell treated mice compared to mice treated with wtNKG2D T cells or PBS, demonstrating that chNKG2D T cells also increased in vivo trafficking and/or survival of tumor antigen-specific T cells at the tumor site and in draining lymph nodes (Figure 5). To determine if this T cell recruitment was antigen-specific, trafficking of OT-I T cells was measured in mice bearing ID8-GFP tumors that did not express Ova. Compared to mice treated with wtNKG2D T cells or PBS, treatment with chNKG2D T cells did not increase trafficking of OT-I T cells to the tumor draining lymph node, indicating that the increase in OT-I T cells at the lymph node was antigen-dependent. While an increased number of OT-I T cells was found in the peritoneal cavity in mice that received chNKG2D T cells, this increase was not as great as seen in mice bearing tumors expressing Ova. This suggests that chNKG2D T cells increased T cell trafficking to the tumor site in both antigen-dependent and antigen-independent manners, possibly through an alteration in chemokine production, as shown in Table I.
Figure 5. ChNKG2D T cell treatment increases T cell trafficking to the tumor site.
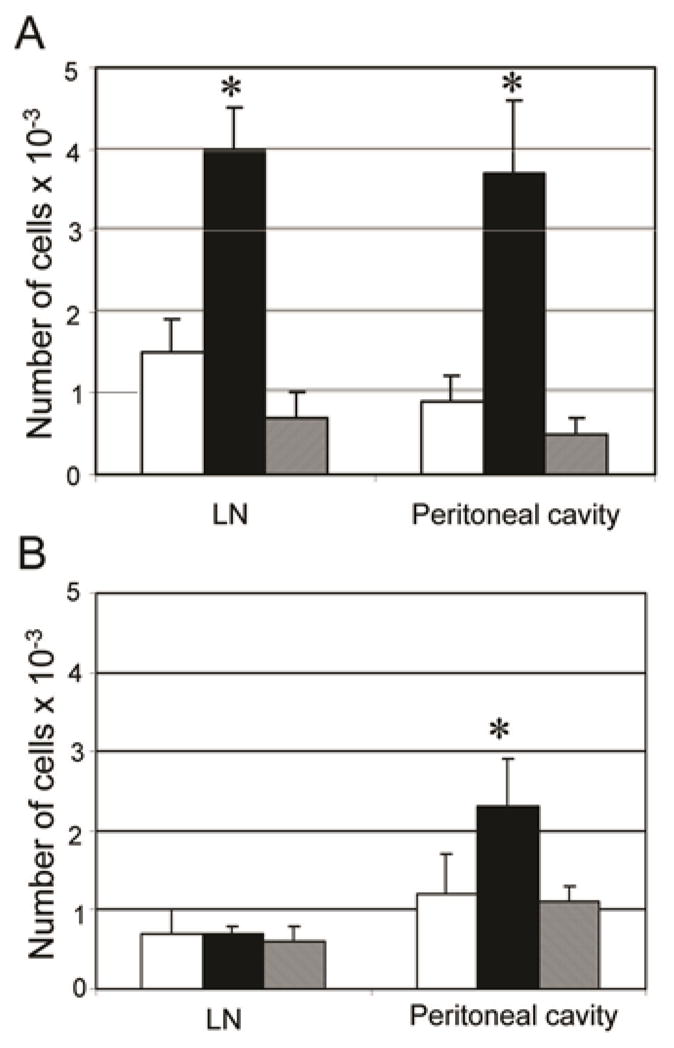
(A) ID8-GFP-tOva cells or (B) ID8-GFP cells were injected i.p. One week later, mice were treated i.p. with wtNKG2D T cells (white bars), chNKG2D T cells (black bars), or PBS (grey bars), and CFSE-labeled OT-I T cells were transferred i.v. Absolute number of OT-I T cells in the mediastinal lymph node and peritoneal cavity was determined three days after T cell injection. The average of each group (n=4) is shown + SD. Treatment with chNKG2D T cells significantly increased OT-I T cell trafficking in vivo compared to control treated mice (*-p<0.05). Data are representative of 2 independent experiments.
ChNKG2D T cells require host lymphocytes and IFNγ and perforin from host cells for in vivo efficacy
While chNKG2D T cells induced a systemic host immune response to tumor antigens, the requirements of host components for tumor elimination were unknown. To investigate the role of the host immune system in this response, wildtype B6 mice or mice deficient in perforin, GM-CSF, or IFNγ were injected with ID8 tumor cells and treated one week later with wtNKG2D or chNKG2D T cells. Wildtype B6 mice treated with chNKG2D T cells had a significantly reduced number of tumor cells in the peritoneal cavity and solid tumors compared to mice treated with wtNKG2D T cells after eight weeks (Figure 6). Mice deficient in IFNγ had no reduction in tumor burden after chNKG2D T cell treatment, demonstrating that host cell-derived IFNγ was essential for chNKG2D T cell anti-tumor efficacy. Mice deficient in perforin had a reduced tumor burden after chNKG2D T cell treatment, but not as low as recipient mice that expressed perforin, indicating that perforin expression by host cells was also required for complete anti-tumor efficacy. While chNKG2D T cell-derived GM-CSF was required for reducing the tumor burden, chNKG2D T cells were able to completely reduce the tumor burden in mice that could not produce GM-CSF, demonstrating that the host did not require expression of GM-CSF for tumor elimination.
Figure 6. ChNKG2D T cells require host cell-derived perforin and IFN γ for in vivo efficacy.

ID8-GFP cells were injected i.p. into (A) B6 or B6.IFNγ−/− mice or (B) B6, B6.Perforin−/−, or B6.GM-CSF−/− mice. After seven days, mice received wtNKG2D (WT) or chNKG2D (CH) T cells i.p. Tumor burden was determined after 8 weeks by measuring the absolute number of GFP+ cells in the peritoneal wash or number of solid tumors on the peritoneum. Each data point represents an individual and the average of each group is shown. Data are from at least two separate experiments. * p<0.05- compared with WT T cell treatment. § p<0.05, compared with CH T cell treatment.
Many cell types in the mouse express perforin and IFNγ, including T cells and NK cells. To examine the requirement of different host lymphocytes, tumor-bearing Rag1 deficient mice (lacking T cells and B cells) were treated with wtNKG2D or chNKG2D T cells. ChNKG2D T cells significantly reduced the tumor burden in Rag1−/− mice, but not as effectively as in mice with intact T and B cells, indicating a role for both chNKG2D T cells and host lymphocytes in anti-tumor efficacy (Figure 7A). Rag1−/− mice treated with wtNKG2D T cells had lower tumor burden than B6 mice, which may reflect the enhanced NK cell activity present in these mice (20). A similar effect was observed if NK cells were depleted, showing that host NK cells were involved in decreasing the tumor burden after chNKG2D T cell injection (Figure 7B). Host CD1-dependent NKT cells were not required for anti-tumor efficacy because chNKG2D T cells were able to reduce the tumor burden in CD1−/− hosts to the same extent as in wildtype B6 mice (Figure 7C). Overall, chNKG2D T cell reduction of an established tumor burden required components of the host immune system for best efficacy, including host-derived perforin and host lymphocytes.
Figure 7. ChNKG2D T cells require host immune cells for in vivo efficacy.

ID8-GFP cells were injected i.p. into (A) B6 or B6.Rag1−/− mice or (B) B6 mice injected with anti-NK1.1 mAbs (PK136) or control mouse gamma globulin (msgg) or (C) B6 or B6.CD1−/− mice. After seven days, mice received wtNKG2D (WT) or chNKG2D (CH) T cells i.p. Tumor burden was determined after 8 weeks by measuring the absolute number of GFP+ cells in the peritoneal wash or number of solid tumors on the peritoneum. Each data point represents an individual and the average of each group is shown. Data are from two separate experiments. * p<0.05, compared with WT T cell treatment. § p<0.05, compared with CH T cell treatment.
DISCUSSION
Transfer of chNKG2D T cells to mice bearing ovarian cancer led to the induction of a host immune response against the tumor. This included an increase in host NK cell, CD8+ and CD4+ T cell responses, and these responses were sustained for up to ten weeks after chNKG2D T cell injection. ChNKG2D T cell treatment also increased antigen presentation in tumor draining lymph nodes and in the spleen, and increased tumor-specific T cell homing to the lymph nodes and tumor site in vivo. The induction of the host immune response and antigen presentation by chNKG2D T cells required their production of IFNγ, GM-CSF, and perforin. Complete anti-tumor efficacy also required host-cell derived IFNγ and perforin, and host lymphocytes.
Currently there are many T cell therapy approaches being developed for cancer treatment. In some of these studies, it was found that a non-myeloablative chemotherapy regimen and radiation treatment prior to transfer of tumor-specific T cells enhanced anti-tumor efficacy (1, 21, 22). A possible reason for this is that depleting a patient of immune cells prior to T cell transfer decreases competition for antigen presenting cells and homeostatic cytokines such as IL-15, which may allow for the transferred T cells to survive long-term (23–25). Also, these pretreatment regimens may affect regulatory cells, such as myeloid derived suppressor cells and regulatory T cells which may decrease the anti-tumor responses of the transferred T cells (23, 26). While many of the current T cell therapies aim to induce the survival of their transferred T cells, this study shows that although chNKG2D T cells do not live long-term after injection, they instead induce long-lived anti-tumor immune responses in the host. This induction of a host immune response may be beneficial for many reasons. If the tumor cells decrease the expression of NKG2D ligands or if there were some tumor cells present in a patient that did not express NKG2D ligands, then the tumor cells may be able to escape direct recognition by the transferred T cells. However, the induction of a host immune response to tumor antigens may result in epitope spreading and the development of immunity against multiple tumor antigens. Thus, chNKG2D T cells may induce a response to ligand negative tumor cells through induction of host anti-tumor immune responses. Furthermore, the fact that the chNKG2D T cells do not survive long-term may be also good for safety reasons. Some patients that have tumor-antigen specific T cells that survive long-term show signs of autoimmunity due to antigen expression on normal tissues (27). While the ligands for NKG2D are not expressed on most normal tissues, cell surface expression can be upregulated in some situations, such as after infection with certain microbes (28, 29). This could be potentially dangerous if chNKG2D T cells survived long-term as these T cells may then recognize and attack normal tissues if NKG2D ligand expression was expressed at the cell surface.
Components of the endogenous immune system may also be involved in the reduction of ovarian tumor growth. In Figure 1, intracellular staining showed that although chNKG2D T cells increased the NK cell secretion of IFNγ, NK cells in wtNKG2D T cell treated mice also produced IFNγ, indicating that the host responded to the tumor, but this response was not sufficient for tumor elimination. ChNKG2D T cells increased host IFNγ production three days after T cell injection, which suggests that the chNKG2D T cell response may enhance an ongoing host response. In addition, wtNKG2D T cell-treated mice that were deficient in IFNγ or NK cell depleted had a somewhat increased tumor burden compared to B6 mice. This indicates that these host mechanisms may play a role in controlling tumor growth, however these clearly cannot control tumor growth by themselves. ChNKG2D T cells likely act together with host immune cells to overcome local immune suppression and lead to tumor elimination.
Although the role of host immune components in T cell adoptive therapies is not well understood, the data presented here demonstrate that host cells play an important role in anti-tumor efficacy of the transferred T cells. ChNKG2D T cell treatment induced the sustained secretion of many proinflammatory cytokines even ten weeks after chNKG2D T cell injection. While three doses of chNKG2D T cells leads to tumor-free survival, one dose of chNKG2D T cells may not completely eliminate the tumor (14, 16). Therefore host immune cells may be continually responding to a low level of residual tumor present after a single chNKG2D T cell treatment, as tested here. Without host cell-derived perforin or lymphocytes, the anti-tumor efficacy of chNKG2D T cells was decreased. Thus the chNKG2D T cells did not act alone to reduce the tumor burden, but instead they elicited the host immune system to respond against the tumor. Consequently, chNKG2D T cell therapy provides both passive immunity and also has a tumor vaccine-like effect in supporting the induction of active host anti-tumor immunity.
ChNKG2D T cells increased tumor antigen presentation in both the draining lymph node and spleen of tumor bearing mice. The increase in antigen presentation depended not only on chNKG2D T cell derived perforin, which may be acting to lyse tumor cells thus releasing more antigen for antigen presenting cells, but also chNKG2D T cell derived GM-CSF and IFNγ. These cytokines may mature dendritic cells at the tumor site and cause them to traffic to the draining lymph node to initiate host T cell responses (30). An increased number of tumor antigen-specific T cells were found in the draining lymph node and at the tumor site in chNKG2D T cell treated mice compared to mice treated with wtNKG2D T cells or PBS. This could be due to a combination of increased proliferation, survival, or trafficking of tumor antigen-specific T cells in chNKG2D T cell treated mice.
Understanding the mechanisms that transferred T cells use to decrease tumor burden increases our understanding of how to improve on T cell immunotherapies for cancer. This study shows that transfer of chNKG2D T cells was able to initiate the induction of a host immune response in a mouse model of ovarian cancer. The longevity of the immune response to tumor antigens did not require the survival of chNKG2D T cells, but instead induced a proinflammatory environment through cytokine secretion and increased tumor antigen presentation to induce an effective host-dependent anti-tumor response.
Supplementary Material
Acknowledgments
The authors wish to thank Gary Ward and Alice Givan at the Englert Cell Analysis Laboratory for assistance with flow cytometry (Norris Cotton Cancer Center, Lebanon, NH), the Immune Monitoring Laboratory for assistance in luminex analysis (Norris Cotton Cancer Center, Lebanon, NH), and the Animal Resource Center at Dartmouth Medical School for help with the animal studies.
Grant Support: This study was supported in part by grants from the Department of Microbiology and Immunology and the National Institutes of Health (T32 AI07363, CA130911). AB was supported by a John H. Copenhaver, Jr., and William H. Thomas, M.D., 1952 Fellowship from the Dartmouth Graduate Office. The contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
Publisher's Disclaimer: “This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.”
References
- 1.Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, Restifo NP. Increased intensity lymphodepletion and adoptive immunotherapy--how far can we go? Nat Clin Pract Oncol. 2006;3:668–681. doi: 10.1038/ncponc0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June CH. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leen AM, Rooney CM, Foster AE. Improving T Cell Therapy for Cancer. Annu Rev Immunol. 2006 doi: 10.1146/annurev.immunol.25.022106.141527. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sentman CL, Barber MA, Barber A, Zhang T. NK cell receptors as tools for cancer immunotherapy. Advances in Cancer Research. 2006;95:249–292. doi: 10.1016/S0065-230X(06)95007-6. [DOI] [PubMed] [Google Scholar]
- 6.Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene. 2008;27:5944–5958. doi: 10.1038/onc.2008.272. [DOI] [PubMed] [Google Scholar]
- 7.Bryceson YT, Ljunggren HG. Tumor cell recognition by the NK cell activating receptor NKG2D. Eur J Immunol. 2008;38:2957–2961. doi: 10.1002/eji.200838833. [DOI] [PubMed] [Google Scholar]
- 8.Blankenstein T, Qin Z. The role of IFN-gamma in tumor transplantation immunity and inhibition of chemical carcinogenesis. Curr Opin Immunol. 2003;15:148–154. doi: 10.1016/s0952-7915(03)00007-4. [DOI] [PubMed] [Google Scholar]
- 9.Marth C, Windbichler GH, Hausmaninger H, Petru E, Estermann K, Pelzer A, Mueller-Holzner E. Interferon-gamma in combination with carboplatin and paclitaxel as a safe and effective first-line treatment option for advanced ovarian cancer: results of a phase I/II study. Int J Gynecol Cancer. 2006;16:1522–1528. doi: 10.1111/j.1525-1438.2006.00622.x. [DOI] [PubMed] [Google Scholar]
- 10.Pujade-Lauraine E, Guastalla JP, Colombo N, Devillier P, Francois E, Fumoleau P, Monnier A, Nooy M, Mignot L, Bugat R, Marques C, Mousseau M, Netter G, Maloisel F, Larbaoui S, Brandely M. Intraperitoneal recombinant interferon gamma in ovarian cancer patients with residual disease at second-look laparotomy. J Clin Oncol. 1996;14:343–350. doi: 10.1200/JCO.1996.14.2.343. [DOI] [PubMed] [Google Scholar]
- 11.Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet. 2008;371:771–783. doi: 10.1016/S0140-6736(08)60241-X. [DOI] [PubMed] [Google Scholar]
- 12.Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106:1544–1551. doi: 10.1182/blood-2004-11-4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Research. 2006;66:5927–5933. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 14.Barber A, Zhang T, DeMars LR, Conejo-Garcia J, Roby KF, Sentman CL. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007;67:5003–5008. doi: 10.1158/0008-5472.CAN-06-4047. [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Barber A, Sentman CL. Chimeric NKG2D Modified T Cells Inhibit Systemic T-Cell Lymphoma Growth in a Manner Involving Multiple Cytokines and Cytotoxic Pathways. Cancer Res. 2007;67:11029–11036. doi: 10.1158/0008-5472.CAN-07-2251. [DOI] [PubMed] [Google Scholar]
- 16.Barber A, Zhang T, Sentman CL. Immunotherapy with chimeric NKG2D receptors leads to long-term tumor-free survival and development of host anti-tumor immunity in murine ovarian cancer. J Immunol. 2008;180:72–78. doi: 10.4049/jimmunol.180.1.72. [DOI] [PubMed] [Google Scholar]
- 17.Huang B, Zhao J, Li H, He KL, Chen Y, Chen SH, Mayer L, Unkeless JC, Xiong H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005;65:5009–5014. doi: 10.1158/0008-5472.CAN-05-0784. [DOI] [PubMed] [Google Scholar]
- 18.Barber MA, Zhang T, Gagne BA, Sentman CL. NK cells negatively regulate antigen presentation and tumor-specific CTLs in a syngeneic lymphoma model. J Immunol. 2007;178:6140–6147. doi: 10.4049/jimmunol.178.10.6140. [DOI] [PubMed] [Google Scholar]
- 19.Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, Benencia F, Stan RV, Keler T, Sarobe P, Sentman CL, Conejo-Garcia JR. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res. 2008;68:7684–7691. doi: 10.1158/0008-5472.CAN-08-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grundy MA, Sentman CL. Immunodeficient mice have elevated numbers of NK cells in non-lymphoid tissues. Exp Cell Res. 2006;312:3920–3926. doi: 10.1016/j.yexcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 21.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams KM, Gress RE. Immune reconstitution and implications for immunotherapy following haematopoietic stem cell transplantation. Best Pract Res Clin Haematol. 2008;21:579–596. doi: 10.1016/j.beha.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tey SK, Bollard CM, Heslop HE. Adoptive T-cell transfer in cancer immunotherapy. Immunol Cell Biol. 2006;84:281–289. doi: 10.1111/j.1440-1711.2006.01441.x. [DOI] [PubMed] [Google Scholar]
- 27.Uchi H, Stan R, Turk MJ, Engelhorn ME, Rizzuto GA, Goldberg SM, Wolchok JD, Houghton AN. Unraveling the complex relationship between cancer immunity and autoimmunity: lessons from melanoma and vitiligo. Adv Immunol. 2006;90:215–241. doi: 10.1016/S0065-2776(06)90006-6. [DOI] [PubMed] [Google Scholar]
- 28.Mistry AR, O’Callaghan CA. Regulation of ligands for the activating receptor NKG2D. Immunology. 2007;121:439–447. doi: 10.1111/j.1365-2567.2007.02652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borchers MT, Harris NL, Wesselkamper SC, Zhang S, Chen Y, Young L, Lau GW. The NKG2D-activating receptor mediates pulmonary clearance of Pseudomonas aeruginosa. Infect Immun. 2006;74:2578–2586. doi: 10.1128/IAI.74.5.2578-2586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrantini M, Capone I, Belardelli F. Dendritic cells and cytokines in immune rejection of cancer. Cytokine Growth Factor Rev. 2008;19:93–107. doi: 10.1016/j.cytogfr.2007.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.