

Abstract
The development of the head involves the interaction of several cell populations and coordination of cell signalling pathways, which when disrupted can cause defects such as facial clefts. This review concentrates on genetic contributions to facial clefts with and without cleft palate (CP). An overview of early palatal development with emphasis on muscle and bone development is blended with the effects of environmental insults and known genetic mutations that impact human palatal development. An extensive table of known genes in syndromic and non-syndromic CP, with or without cleft lip (CL), is provided. We have also included some genes that have been identified in environmental risk factors for CP/L. We include primary and review references on this topic.
Keywords: Cleft lip/palate, Human/murine palate genetics, Palate development
INTRODUCTION
Disturbances at any stage during palate development, e.g., defective palatal shelf growth, failed or delayed elevation, and blocked fusion, can result in cleft palate (CP)[1,2] with or without cleft lip (CL/P). As one of the most common congenital cranio-facial defects, CL/P occurs in approximately 1 per 750 live births in the United States[2,3] Clefts occur more frequently among Asians (about 1:400) and certain American Indians than Europeans or European descendants. Clefts are relatively less common among Africans and African Americans (about 1:1500).[4] Cleft lip and palate may not be life-threatening but many functions such as feeding, digestion, speech, middle-ear ventilation, hearing, respiration and facial and dental development can be disturbed because of the structures involved. These problems can also cause emotional, psycho-social, and educational difficulties. In addition, CP is an economic burden.
The aetiology of cleft lip with or without palate (CL/P) is theorized to be a combination of factors associated with genes and environment.[5,6] The advent of gene targeting technology and basic conventional techniques using animal models has led to the identification of genes associated with known and unknown etiologic factors. Characterization of the genomic sequences will greatly impact regulation of gene networks and pinpoint any variations in the different stages of craniofacial morphogenesis. In this article, emphasis is placed on different genes associated with the classifications of CL/P into syndromic [Table 1] and nonsyndromic [Table 2]. Each classification plays a significant role in understanding the molecular and genetic mechanisms affecting these types of craniofacial defects.[7–9] In addition to known genes there is strong evidence that several environmental factors (e.g., alcohol consumption, tobacco, and anti-convulsants) increase the risk of CL/P.[10,11] In contrast, several studies have shown that folic acid may have a protective effect on CL/P and neural tube defects.[12–16] Recent data from the National Birth Defect Prevention Network have indicated a decrease in neural tube defects from 5/10,000 to less than 2/10,000 after the fortification of the food supply with folic acid, indicating that this vitamin and the proteins that facilitate the uptake and metabolism of folic acid may be candidate genes in cranio-facial development.[14,17–20]
Table 1.
Syndromic genes associated with cleft lip and palate
| Syndrome | Clinical Features | Genes | Reference |
|---|---|---|---|
| Apert Syndrome (AS) | AD; high arched palate, bifid uvula, and cleft palate. | FGFR2 | 6, 114–118 |
| Bamforth-Lazarus | AR; hypothyroidism, athyroidal, CPO, choanal atresia, spiky hair. | FOXE1 | 6, 119, 120 |
| Syndrome (BLS) | |||
| Branchio-oculo facial syndrome (BOFS) | AD; pseudocleft of the upper lip resembling a poorly repaired cleft lip. | TFAP2A | 6, 121 |
| Down syndrome (DS) | Macroglossia, microstomia, atlantoaxial subluxation | duplication of portion of chromosome 21 | 122 |
| Ectrodactyly-ectodermal dysplasia-cleft syndrome (EEC) | AD; triad of ectrodactyly, ectodermal dysplasia, and facial clefting. | P63 | 6, 123, 124 |
| Fetal alcohol syndrome (FAS) | Disorder characterized by a pattern of minor facial anomalies, prenatal and postnatal growth retardation. | alcohol dehydrogenase 1B (ADH1B) | 125-128 |
| Goldenhar syndrome (GS) | Oculo auricular vertebral dysplasia; AD; incomplete development of the ear, nose, soft palate, lip, mandible. | Pericentric inversion of chromosome 9 | 129, 130 |
| Hereditary lymphoedema-distichiasis syndrome (HLD) | AD; lymphedema of the limbs, double rows of eyelashes, cardiac defects, and cleft palate. | FOXC mutations | 131 |
| Kallmann Syndrome (KS) | AR disorder; hypogonadotropic hypogonadism and anosmia | FGFR1 mutations | 6, 132, 133 |
| Margarita Island ectodermal dysplasia (ED4) | AR; unusual facies, dental anomalies, syndactyly, and cleft lip/cleft palate. | PVRL1 (nectin-1) mutation | 6, 134 |
| Pierre Robin | AD; triad of micrognathia, glossoptosis, and cleft palate. | Loci 2q24.1-33.3, 4q32qter, 11q2123.1, and 17q2124.325.1. | 135,136 |
| Sequence (PRS) | |||
| Smith–Lemli-Opitz | AR; defects in cholesterol biosynthesis, growth retardation, dysmorphic facial features including CLP/ CPO, postaxial polydactyly | DHCR | 6, 137, 138 |
| Syndrome (SLMOS) | |||
| Stickler Syndrome (SS) | |||
| AD; midface hypoplasia, micrognathia, Pierre Robin sequence, retinal detachment and early cataracts deafness, hypermobility of joints. | Col11A1, Col11A2, Col2A1 | 139,140 | |
| Treacher Collins (TC) | AD; craniofacial deformities such as downward slanting eyes, micrognathia, conductive hearing loss, underdeveloped zygoma. | Mutation in TCOF1 gene at chromosome 5q32-q33.1 | 141, 142 |
| van der Woude syndrome (VDWS) | AD; cleft lip palate, distinctive pits of the lower lips, or both. | IRF 6 (interferon regulatory factor 6) mutations | 6, 143 |
| Velocardiofacial | AD; cleft palate, heart defects, abnormal facial structure, and learning problems. | Chromosome 22q11 microdeletion | 144, 145 |
| Syndrome (VCFS) | |||
| Unnamed syndrome | CL/P and hereditary diffuse gastric cancer | CDH1 | 72 |
| Unnamed syndrome | Chromodomain helicase DNA-binding proteins; CL/P in Charge syndrome | CHD7 | 146, 147 |
| Unnamed syndrome | Bilateral CL/P, colobomas of the optic nerve and retina, agenesis of the corpus callosum. Dysphagia, reduced Oesophgeal peristalsis | PAX 9 | 6, 148 |
| Unnamed syndrome | X-linked mental retardation and CL/P | PH8 | 6, 149 |
| Unnamed syndrome | Holoprosencephaly 7, a spectrum of forebrain and midline anomalies and midline CL | PTCH | 6, 137, 150, 151 |
| Unnamed syndrome | CPO, craniofacial anomalies, osteoporosis, and cognitive defects | SATB2 | 6, 152 |
| Unnamed syndrome | Holoprosencephaly, a spectrum of anomalies ranging from severe (cyclopia) to subtle midline asymmetries. CL/P part of the spectrum | SHH | 6, 137 |
| Unnamed syndrome | Anomalies with most features of DiGeorge/velocardiofacial syndromes: CPO, thymus and parathyroid gland hypoplasia, vertebra, facial and cardiac outflow anomalies. | TBX1 | 6, 153 |
| Unnamed syndrome | X-linked CPO and ankyloglossia | TBX22 | 6, 51, 52 |
| Unnamed syndrome | Cardiovascular, craniofacial, skeletal, and cognitive alterations, bifid uvula and or/CPO | TGF Beta receptor | 6, 154 |
Table 2.
Non-syndromic genes: interaction effects of genes and environmental risk factors on oral clefts
| Gene | Functional Role | Risk Factor | Reference |
|---|---|---|---|
| Cytochrome P450 Proteins (CYP) CYPIA1, CYPIA2, CYPIB1 CYP2E1 | Highly polymorphic, having multiple functional alleles; Role in detoxification; metabolism of endogenous morphogens in the developing foetus. | Negative gene-smoking interaction effect | 155-157 |
| Epoxide Hydrolase (EPHX) | Class of proteins that catalyze the hydration of chemically reactive epoxides into their corresponding dihydrodiol products. | ||
| EPHX | Plays an important role in both the bioactivation and detoxification of exogenous chemicals such as PAHs, which are present in cigarette smoke. | Negative gene-smoking interaction effect | 155, 158 |
| EPHX1 Y113H | Variant of EPHX 1 found in the foetus and maternal smoking. | Positive gene-smoking interaction effect | 28, 159 |
| Glutathione Transferase Gene Family (GST) | Families of dimeric phase II enzymes that catalyze the conjugation of reduced glutathione with electrophilic groups of a wide variety of environmental agents. | ||
| GSTM1 | Major gene detoxifying PAHs and widely studied in many disorders and cancers. | Negative gene-smoking interaction effect | 160, 161 |
| GSTT1 | Expressed in a variety of tissues/organs such as erythrocytes, lung, kidney, brain, skeletal muscles, heart, and small intestine; elevated expression profile at the craniofacial regions during embryonic development. | Positive gene-smoking interaction effect | 162, 28, 157, 159 |
| GSTP1 | Major gene detoxifying PAHS; involvement in variety of disorders and cancers. Major enzyme involved in the inactivation of cigarette smoker's metabolites; most important isoform at the embryonic and early foetal developmental stages. | Positive gene-smoking interaction effect | 163, 28, 159 |
| GST A4 / GSTM3 | Two other types of GST gene family members. | Positive gene-smoking interaction effect | 28, 159 |
| Hypoxia-Induced Factor-1 (HIF1A) | Mechanism by which maternal smoking may affect embryonic development due to the production of carbon monoxide, which interferes with oxygen transfer to the placenta, or nicotine, which constricts the uterine wall resulting in hypoxia. | Positive gene-smoking interaction effect | 28, 159 |
| Arylamine N-Acetyltransferase gene Family | N-conjugation of arylamine by the action of N-acetyltransferases (NATs), UDP glucoronosyltransferases (UGTs), or sulfotransferases (SULTS) produces nontoxic compounds. | ||
| N-acetyltransferases1 (NAT 1) | Expressed in many tissues such as erythrocytes, bladder, lymphocytes, neural tissues, liver and intestines. | Negative gene-smoking interaction effect | 19, 164, 165 |
| N-acetyltransferases pseudogene, (NATP1) | Pseudogene identified, which is located at chromosome 8p23.1-8p21.3. | 19, 164, 165 | |
| N-acetyltransferases2 (NAT 2) | Expressed in the liver and epithelial cells of the intestine. | Positive gene-smoking interaction effect | 28, 157, 159 |
| Methylenetetrahydrofolate reductase (MTHFR) | Metabolism of folate by reducing methylenetrahydrofolate, primary donor for methionine synthesis. | Positive gene-smoking interaction effect | 166-172 |
| MTHFRC677T | Variant of methylenetetrahydrofolate reductase. | Negative gene-smoking interaction effect | |
| OTHER METABOLIC GENES | |||
| NAD(P)H quinine oxidoreductase (NQO1) | Flavoenzyme that catalyzes two electron reduction of quinine compounds to hydroquinone and is inducible by oxidative stress, dioxin, and PAHS found in cigarette smoke | Negative gene-smoking interaction effect | 28, 159 |
| SULT1A1 | Catalyzes transfer of the sulfonate group from the active sulfate to a substrate to form the respective sulfate or sulfamate ester. | Negative gene-smoking interaction effect | 28, 159 |
| UDP glycosyltransferases (UGTs) UGT1A7 variant | Catalyzes conjugation reactions where hydrophobic chemicals are transformed into water-soluble compounds. Potential maternal effects on embryonic development. | Positive gene-smoking interaction effect | 159, 173, 174 |
| DEVELOPMENTAL GENES FOR ORAL CLEFTS | |||
| Transforming Growth Factor A (TGF α) | Transmembrane protein expressed at the medial edge of the epithelium (MEE) of fusing palatal shelves. Its receptor epidermal growth factor (EGFR) is expressed in the degenerating MEE. | Positive gene-smoking interaction effect (smoking, alcohol drinking, vitamins) | 175-177 |
| Transforming growth Factor β-3 (TGF β3) | Regulator of many biological processes such as proliferation, differentiation, epithelial mesenchymal transformation and apoptosis. | Positive gene-smoking interaction effect (smoking, alcohol drinking) | 81, 176, 178 |
| Muscle Segment Homeobox1 (MSX1) | Transcriptional repressor important in craniofacial, limb, and nervous system development. | Positive gene-smoking interaction effect (smoking and alcohol drinking) | 176, 179, 180 |
| MSX2 | Similar to MSX1; rare cause of isolated cleft lip with or without cleft palate. | 179, 180 | |
| Acyl-CoA desaturase ACOD4 | Pericentric inversion disrupts a gene (ACOD4) on chromosome 4q21 that codes for a novel acyl-CoA desaturase enzyme that occurs in a single two-generation family with CL. | 181 | |
| Retinoic acid receptor (RAR) | Odds ratios for transmission of alleles at THRA1 were significant when ethnic group was included. | Negative gene-smoking interaction effect | 176 |
| CHD7 | Chromodomain helicase DNA-binding proteins. | 182 | |
| ESR1 | Ligand-activated TF estrogen receptor. | 183 | |
| FGF/ FGFR families FGF8 FGF3 FGF10 FGF18 FGFR1 FGFR2 FGFR3 | Expressed during craniofacial development and can rarely harbor mutations that result in human clefting syndromes. | 184 | |
| SPRY1/SPRY2 | Loss of function mutations in FGFR1 cause a syndromic form of clefting. | 185 | |
| TBX10 | Ectopically expressed in dancer cleft lip and palate mutant mice. | 185 | |
| GABRB3 | β3 subunit of GABA receptor CL/P. | 62, 186, 6 | |
| GLI2 | Mutations in GLi2 cause holoprosencephaly-like features with cleft lip and palate. | 185 | |
| ISGF3G | Similar to IRF6. | 185 | |
| OTHER CANDIDATE GENES | |||
| SKI, FOXE1, JAG2, LHX8 | Rare causes of isolated cleft lip with or without cleft palate | 185 | |
This review will concentrate on genetic contributions to facial clefts with/without cleft palate. We will begin with an overview of early palatal development, concentrate on muscle and bone development, and incorporate the effects of environmental insults and known genetic mutations that impact human palatal development.
EMBRYONIC PALATE DEVELOPMENT
The palatal structures are composed of the cranial neural crest (CNC)-derived mesenchyme and pharyngeal ectoderm.[21–24] Epithelia that cover the palatal shelves are regionally divided into oral, nasal and medial edge epithelia (MEE). The nasal and oral epithelia differentiate into pseudo-stratified and squamous epithelia, whereas MEE is removed from the fusion line [Figure 1].
Figure 1.

Schematic drawing showing coronal view of a normal palate shelf and key stages of mouse palatal development. At E12-E13 days in the mouse gestation, the palatal shelves grow downward along the tongue (t). At E13-E13.5 days, the palatal shelves become elevated above the tongue. At E14.5, the palatal shelves adhere to each other in the midline. After E15.5 days, the MES completely degrades, and the palate fuses
The secondary palate originates as an outgrowth of the maxillary prominences at approximately embryonic day 11.5 in the mouse (E11.5-m) [Figure 1] and post coital six weeks in humans (p.c.6wk-h). The palate shelves initially grow vertically along the sides of the tongue (E13.5-m; p.c.7wk-h) and then rise above the tongue as the latter drops in the oral cavity due to the forward and downward growth of the mandible (E14.0-m; p.c.8wks-h). With continued growth, the shelves appose at the midline (E14.5-m; p.c.10wks-h) and eventually fuse (E15.5-m; p.c.13wk-h).[25] Numerous genes similar in mice[26] and humans[25,27,28] are expressed [Table 1] during palatal development.
During fusion the epithelium covering the tip of the opposing palatal shelves, adheres, intercalates and thins into a single-layer midline epithelial seam (MES).[23] The disintegration of this seam results in the confluence of the palatal mesenchyme. Tremendous interest has arisen in cellular mechanisms underlying MES degradation. Epithelial-mesenchymal transition (EMT) is one of the proposed models that regulates medial edge epithelial (MEE) cell fate.[23,29–36] However, other mechanisms have been proposed, such as apoptosis,[37–40] in which all MEE cells are theorized to die during fusion. Alternatively, it is hypothesized by some researchers that MES cells disappear by migrating from the midline towards the nasal and oral epithelia.[41,42] Other investigators postulate that all events, including apoptosis, migration and EMT, may occur.[23,39,43] Interestingly, the fusion of the external surface of the bilateral maxillary processes with the naso-frontal prominence in the chick is similar to palatal fusion [Figure 2].[44] The outer periderm layer dies through apoptosis, and the lateral edge epithelium of the inter-maxillary segment of the naso-frontal process fuses with the medial edge epithelium of the external maxillary process to form a seam that transitions to a confluent mesenchyme [Figure 2].[44] Evidence supporting these theories, especially those involving EMT and apoptosis, will be presented and further discussed.
Figure 2.
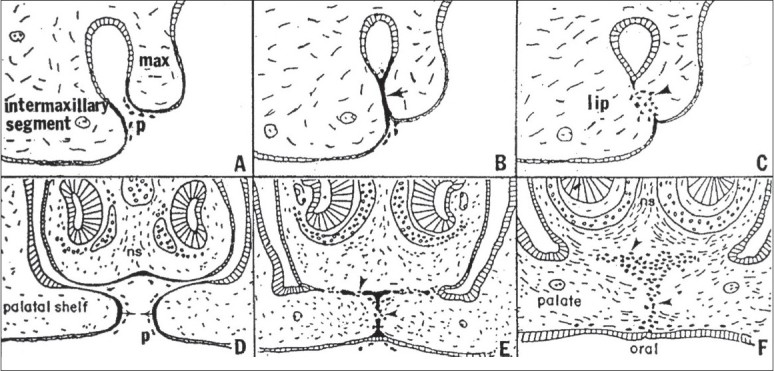
Comparison of the morphogenesis of the upper lip (A-C) with that of the palate (D-F). After the bilateral maxillary processes (max) fuse externally with the inter-maxillary segment, the resulting epithelial seam (arrow, B) gives rise to mesenchyme (arrowhead, C) to produce a confluent lip. At a later time, the palatal shelves arising internally from the maxillary processes fuse with each other (arrows, D) and with the nasal septum (ns) above them, creating an epithelial seam that transforms to mesenchyme (arrowheads, E) to produce the confluent palate (arrowheads, F). p, sloughed periderm cells. Reprinted with permission.[44]
MOLECULAR SIGNALLING EVENTS IN EMBRYONIC PALATAL DEVELOPMENT
As stated above, cleft palate with or without cleft lip is a complex trait caused by a combination of multiple genes and environmental factors.[5] Palatal shelf development defects will be divided into five categories for the purpose of this review:
Failure of palatal shelf formation
The failure of the palatal shelf formation is a rare severe defect. Recent studies have identified several molecular networks operating between the palatal shelf epithelium and mesenchyme during different steps of palatogenesis. These networks include signalling molecules and growth factors such as sonic hedgehog (Shh), members of the transforming growth factor β (TGfβ) super family, including bone morphogenetic proteins (Bmps) and Tgfβs, fibroblast growth factors (Fgfs) and their receptors (FgfR), effectors and targets.[25] Studies addressing the role of Fgf signalling during early palatal development by analyzing Fgf10 and FgfR2b mutants found altered cell proliferation within both mesenchyme and epithelium in the palatal shelves and increased apoptosis within the epithelium. It was reported that Fgf10 and FgfR2b mutations affected the initial development of palatal shelves, and the mouse pups had complete CP.[45] By signalling via its receptor, FgfR2b, in the palatal shelf epithelium, the mesenchymal derived Fgf10 supports epithelial proliferation and survival and also induces the expression of Shh within the epithelium. Shh, in turn, signals to the mesenchyme and stimulates cell proliferation.
In general, signalling activities are subject to tight spatio-temporal control, and, in many instances, too much or too little control is detrimental to the developing organ. This situation is well illustrated in anomalies caused by de-regulated hedgehog (hh) and Fgf signalling.[46,47] While Fgf10/FgfR2b activity plays a crucial role during palatogenesis, it appears to be subject to the tight spatio-temporal regulation shown in mice lacking Shox2. Shox2 mutant mice develop a very rare type of CP that may also be found in humans[48] the soft palate is intact, whereas the hard palate has a cleft. Abnormal proliferation and apoptosis are theorized to be the cause of the cleft. Surprisingly, a number of protagonists implicated in palatogenesis, including Msx1, Bmp4, Pax9, Lhx8, Osr2, Tgfβ3 and Jag 2, were expressed normally.[48] In contrast, Fgf10 and Fgfr2b were expressed at ectopic sites within the mesenchyme of the Shox2 mutant mice.[49] These studies emphasize the importance of the precise timing and determination of sites of signalling activities necessary for normal development. Mutation of activin-βA causes a severe facial primordial development defect, which may be responsible for the retardation of palatal shelf development and complete cleft palate. In addition, other genes, including Msx1, Lhx8, Shox2 and Osr2, assume important roles in the palatal shelf growth. The targeted mutation of these genes in mice generates CP, indicating the intrinsic requirement of these factors during palatogenesis.[49]
Fusion of the palatal shelf with the tongue or mandible
Under normal conditions, palatal shelves do not fuse with other oral structures. However, in mice that do not express Fgf10, the palatal shelf epithelium fuses with the tongue and mandible.[45] The loss of function mutations of Fgf10 results in anterior palatal shelf fusion with the tongue, whereas the middle and posterior palatal shelf regions adhere to the mandible, thus preventing the elevation of the palatal shelf.[50] There is a severe reduction of the expression of Jagged 2 (Jag2), thereby encoding a ligand for the Notch family receptors and ectopic Tgfβ3 production in the nasal epithelia of these mice. The analysis of Jag2 mutant embryos indicates that Jag2-Notch signalling prevents inappropriate palatal shelf adhesion to other oral epithelia through the control of oral epithelial differentiation. Another gene has also been associated with inappropriate adhesions. Mutations in TBX22 have been reported in families with X-linked cleft palate and ankyloglossia.[51–53]Tbx22 is expressed in the developing palate and tongue in mice, suggesting an important role in regulating tongue and palate development.
Failure of palatal elevation
Palatal shelf elevation is a rapid movement triggered by both intrinsic forces within the palatal shelves proper and by influences from other craniofacial and oral structures, including the movement of the tongue, and growth of the cranium and mandible.[1,54] The role of the extra-cellular matrix in palatal shelf elevation has been supported by some studies and is presently accepted as an important determinant of palatal shelf elevation.[55,56] Those studies[1] suggested that a progressive differential accumulation of glycosaminoglycans, primarily hyaluran in the palatal shelves, plays a role in their elevation.[55,56] Hyaluronan is a highly charged glycosaminoglycan that retains high amounts of water, forming hydrated gels leading to the expansion of the extracellular matrix. Other constituents of the palatal shelves including collagen fibers, vascularization, and the epithelial covering; the polarized alignment of the mesenchyme cells may also contribute to the intrinsic elevation force of the PS. Mutations of Pax9, Pitx1 or Osr2 can lead to failed palatal shelf elevation and cleft palate defect.[57–60] The cellular defect is associated with the CNC-derived palatal mesenchyme, suggesting the important functions of these transcription factors in regulating the fate of the CNC cells during palatogenesis.
Early studies attributed a role to neuro-transmitters during palatal shelf elevation.[1] At present, it is widely accepted that neuro-transmitter γ-aminobutyric acid (GABA) regulates not only neuronal activities but also cell migration, survival, proliferation and differentiation of neuronal and non-neuronal cells.[61–63] Terratological studies in rodents showed that GABA or GABA agonists generate CP by inhibiting palatal shelf elevation, whereas GABA antagonists stimulate the process.[64] The implication of GABA in palate development was demonstrated by genetic studies of mice lacking the β3 subunit of the GABA receptor that developed CP without other craniofacial malformations.[65]
Failure of palatal shelves to meet after elevation
Fusion of the opposing palatal shelves is an important step taking place through a sequence of events that includes the removal of the flat peridermal cells, contact and adhesion of the opposing MEE, which creates the MES, and the degeneration of the MES. The mesenchymal confluence thus forms at the midline.[22,23,34] Failure of shelf fusion is the most common type of cleft palate defect documented in animal studies. Mutations in Msx1 and Lhx8 and conditional inactivation of Tgfbr2 in CNC cells or Shh in the epithelium all result in retarded palatal shelf development.[45]
In many transgenic animals, the palatal shelves fail to meet at the midline because of hindrance by the tongue. This is usually associated with cases when the lower jaw does not move forward and downward during development, keeping the tongue between the palatal shelves. These secondary defects were evident in the Hand2 mutant mice in which the enhancer driving the expression of the gene in the pharyngeal arches was inactivated by targeted mutagenesis.[66] In these mice, the mandible did not grow properly, blocking the descent of the tongue, thus hindering palate fusion.[66,67]
Persistence of middle edge epithelium
Adhesion of the opposing MEE is an important event in both human and mouse embryos.[21,27,34,44,68] E-cadherin is expressed in the epithelia covering the fronto-nasal and medial nasal processes as well as during the different stages of palate development, including the epithelial islands, remnants of the MES.[69–71] Mutations of CDH1/E cadherin, which deletes the extracellular cadherin repeat domains required for cell-cell adhesion, have recently been associated with CL/P in families with hereditary diffuse cancer.[72] E-cadherins are known to form dimers, indicating that the mutant proteins may have trans-dominant negative effects over the normal proteins.[72]
Extensive efforts have been made to elucidate the role of Tgfβ3 during palatal fusion.[73–76] Adhesion of the MEE upon palatal shelf contact is a necessary step for fusion. TGfb3 is expressed in the MEE before and during fusion, and mediates MEE adhesion of the opposing palatal shelves through filopodia. E-cadherin is required for fusion, whereas filopodia seem to be crucial for proper alignment and guidance of cell sheets that are fated to fuse, but not for fusion itself.[77] Tgfβ3 is implicated in controlling the re-modelling of the extracellular matrix through regulation of the expression of the matrix metaloproteinases (Mmps) Mmp13, Mmp2 and the tissue inhibitor of metaloproteinase-2 (Timp).[78] Tgfβ3 signalling functions in the MEE by mediating the epithelial-masenchymal interactions leading to tissue changes that regulate palatal fusion. For example, EMT of the MES has been proposed as the major mechanism underlying the disappearance of the MES to generate mesenchyme continuity, thus preventing palatal clefts.[34] The establishment of the concept of EMT as the prevailing mechanism of MES disappearance led to studies attributing roles to different molecules, including Tgfβ3, Lef1, Smad, RhoA, phosphatidylinositol 3-kinase (PI-3 kinase), Mmps Twist and Snail.[22,33,79] In Tgfβ3 or Egfr mutant mice, there is an alteration of the fate of MEE cells.[80,81] In Tgfβ3 null mutant mice, MEE cells fail to undergo apoptosis and remain along the midline, preventing normal fusion.
OSSIFICATION OF THE PALATE
Palatal fusion signals the start of the ossification process in the anterior two-thirds of the palate to form the hard palatal tissues. This process entails the successful fusion of the three embryonic structures - lateral edges of the primary palate with the two anterior edges of the secondary palate. This process requires the synchronization of shelf movements together with the growth and withdrawal of the tongue and growth of the mandible and head.[82] Any form of disruption during the formative stages results in a pathological cleft. The same is true when ossification occurs too early. Sox9 is a gene controlling cartilage development and blocking the expression of Runx2, a transcription factor essential for osteoblast differentiation and bone formation associated with cleidocranial dysplasia. In Sox9 mutant, Runx2 expression is not repressed and ossification begins prematurely.[83] Since the palatal shelves are prematurely ossified, they cannot grow toward the midline and fail to fuse.
A wide range of studies on cranio-facial skeletal maturation has shown that the fusion of the palatal shelves along their length to form the mid-palatal (MP) suture occurs during the ossification of the maxillae and palatine bones before the mandibular condyle develops.[48,84,85] Ossification is observed where mesenchymal cells condense, the surrounding tissue vascularizes and the cells differentiate into osteoblasts that will form bone by mineral deposition. In this process, several growth and differentiation factors such as Bmps, core binding proteins (Cbf), Fgfs, and hedgehog (hh) proteins that interact with various signalling pathways to regulate the patterning of the undifferentiated mesenchyme, are involved. The Bmp-6 and the transcription factor Gli1 are also expressed during intra-membranous bone formation.[86,87] As in cranio-facial sutures, the MP and trans-palatal (TP) suture osteoblasts express Tgfβ1, 2 and 3, while the suture cells express primarily Tgfβ3.[88,89]
It has been established that cranial sutures are the growth sites for the neuro-cranium and that the dura mater provides the signalling molecules to regulate suture patency.[90] The MP and TP sutures have different morphology, so they are not in contact with the dura mater. Opperman's group hypothesized that these facial sutures are growth centres[88,89] and that the nasal capsular cartilage produces signalling molecules to regulate the fusion of MP and TP sutures [Figure 3].[89] They found that the nasal cartilage maintained the TP sutures as growth sites in experiments on rat palatal organ cultures (E20) with or without nasal cartilage. They theorized that the nasal cartilage may regulate mid-facial growth.[89]
Figure 3.
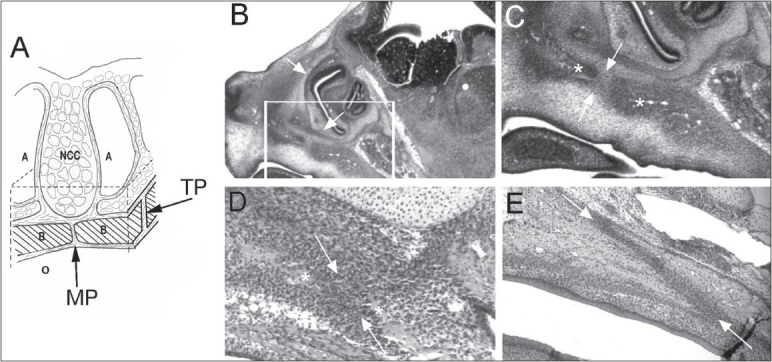
(A) Diagram showing the relationship between the NC cartilages and the transpalatal suture. Dotted lines indicate cut lines for removing the palate from the embryo and the NC cartilage from above the sutures. (B-E) Micrographs of parasagittal sections of foetal rat heads show the pre-natal development of TP sutures. (B) At E16, NC cartilages (arrows) can be seen directly above the presumptive TP suture region (in box). (C) High-power micrograph of the region in the box, showing the advancing palatal plate of the maxilla and horizontal plate of the palatal bone (asterisks) on either side of the presumptive TP suture (between arrows). (D) At E18, the advancing bone fronts (asterisks) begin to overlap one another, creating a highly cellular suture blastema (between arrows). (E) By E20, an elongated TP suture (between arrows) continues to form as the bone fronts proceed to overlap one another. A, airway; B, shelves of maxillary bones; MP, midpalatal suture; NCC, nasal capsular cartilage; O, oral cavity; TP, transpalatal suture. Reprinted with permission.[87]
Animal models have been developed to understand the aetiology and pathogenesis of orofacial clefts and the mechanisms of normal palatal ossification. The application of cyclic forces is an effective mechanical stimulus for the regulation of osteogenesis and osteoclastogenesis in the sutural growth of neonatal rats.[91] The process of tissue response and regeneration in the palato-maxillary suture under tensile forces was examined histologically and with fluorescence. A cyst-like zone appeared in the conjuncture of the bony front and the sutural connective tissue at the early stage of sutural expansion with increased proliferating osteoblasts and fibroblasts. New bone was deposited along the nasal septum and the front of the cyst until the new bone front formed and the suture restored its original morphology.[92]
The approach of utilizing MP suture expansion in mice has provided new insights into mechanical stress modulation as an important factor for the skeletal remodelling of bones and cartilage. The expansive force across the MP suture promotes both bone resorption through the activation of osteoclasts and bone formation through the increased proliferation and differentiation of the periosteal cells.[93] Similarly, the use of orthodontic wire expansion in growing rats showed that secondary cartilage can undergo chondrogenic and osteogenic differentiation in the maxillary arch. Interestingly, these induced changes were attributed to the alteration of the differentiation pathway of progenitor cells from chondroblastic to osteoblastic, in which many sutures temporarily form secondary cartilage during early development. Histological observations at days 7, 10, and 14 indicated that intra-membranous bone formation, which is partially recognized as mature bone,[94] occurred at the boundary between the pre-cartilaginous and cartilaginous cell layers where the calcified matrix was positive for osteocalcin antibody. The cellular events taking place at the MP suture cartilage in rat models as a result of expansion force have been observed as endochondral bone formation at the boundary between the maxillary bone and cartilage, whereas intra-membranous osteogenesis has appeared at the internal side of the cartilaginous layer.[95] To stimulate new bone formation in defective tissues, rat organ cultures with distracted palatal sutures were treated with Bmp-7 and Nell-1 for 8 days in vitro. The presence of Nell-1 increased chondrocyte hypertrophy and endochondral bone formation while Bmp-7 enhanced both chondrocyte proliferation and differentiation in the distracted palates of four-week-old male rats. This study indicates that Nell-1 is involved in the rapid osteoblast differentiation in palate sutures.[96] In another study, the application of TGF-β1 during the early stages of rat MP expansion induced rapid bone formation at the suture site.[97]
ORAL AND PALATAL MUSCULATURE AND RELATED DEFORMITIES
Overt CL/P encompasses a broad spectrum of defects, ranging from so-called microform clefts to complete unilateral or bilateral clefts of the lip and palate. The orbicularis oris (OO) muscle consists of numerous differently oriented strata of muscular fibres that surround the orifice of the mouth. At approximately seven weeks post-conception (p.c.) in humans, the two maxillary prominences fuse with the medial nasal prominence; however, lip fusion is not complete until the epithelial seam disappears through EMT and/or apoptosis[82] [Figure 2A–C]. By eight weeks p.c., a dense, continuous band of mesenchymal cells corresponding to the future OO muscle can be seen, with discernible OO muscle fibers present by 12 weeks.[98,104] The complete OO muscle architecture forms by 16 weeks. Any delay in fusion may result in sub-epithelial OO defects, such as the altered migration of the mesenchymal cells. Sub-epithelial (non-visible) defects of the orbicularis oris muscle represent the mildest form of cleft lip, and such defects are part of the phenotypic spectrum of CL/P. This defect usually is visualized as a ridge of tissue resembling a scar on the upper lip along the philtrum.[98]
Histological studies have demonstrated that such defects extend to the muscle fibres of the superior OO muscle. A method using high-resolution ultrasonography (USG) was developed to visualize the OO muscle non-invasively.[99] Significant differences in the defects of the OO are found in the first-degree relatives of CL/P individuals and controls. The OO muscle defect detected by ultrasound is consistent with the histological examination of cadavers.[99] Interestingly, the Bmp4 knockout mouse model shows bilateral cleft lip at E14.5, although this condition occurs at a rate of 22% after birth,[100] suggesting the initial cleft lip is rescued or healed in utero, leaving only the subepithelial OO defect. Potential mutations in BMP4 were found in two individuals with OO defects and none in the controls.[101] The strong evidence that OO discontinuities are indeed part of the phenotypic spectrum of CL/P provides an important clue for the clinical recurrence risk estimation for families with members affected with CL/P.
The mildest form of CP is termed a “submucosal cleft palate,” described as a bifid uvula, palatal muscle diastasis and a notch in the posterior surface of the hard palate.[102] Defects in the nasopharyngeal anatomy and/or physiology may lead to velopharyngeal incompetence (VPI). Although most VPI is caused by CP, the population prevalence of VPI due to other causes is estimated to be approximately 2.5%.[103] In such cases, VPI may be caused by submucosal muscular defects of the levator veli palatini or musculus uvulae. Most of the soft palate muscles are derived from myotome cells, which first invade pharyngeal arch 4 and then migrate to the palate, carrying their innervations from the vagus nerve. One muscle (tensor veli palatini) is derived from myotome cells that first invade arch 1 and are innervated by the trigeminal nerve.[104] In the mouse, the tensor veli palatine, levator veli palatini, medial pterygoid, and lateral pterygoid muscles are identified as myogenic fields as early as gestational day 15. The palatoglossus, palatopharyngeus, and musculus uvulae, however, are not clearly visible.[105] In principle, the presence of these anatomical features in unaffected individuals may signify an elevated risk for producing clefts in offspring.[106]
SUMO MODIFICATION OF SIGNALLING PATHWAYS IN PALATOGENESIS
The molecular understanding of NS CL/P is further complicated when one considers that large differences in penetrance often occur when the same mutations are placed on different mouse strains, indicating a potential role for both genetic and/or environmental modifiers in the pathogenesis of CL/P. Several lines of evidence point to the involvement of the small ubiquitin-like modifier (SUMO) posttranslational modification machinery.[107] A surprisingly specific role in oro-facial development has been revealed for protein modification by the SUMO, which might hint at a possible interaction with environmental factors. Small ubiquitin-related modifiers belong to the ubiquitin-related protein family, and SUMO proteins are ubiquitously expressed throughout the eukaryotic kingdom.[108] SUMO1 shows strong expression in the MEE of the secondary palate.[109] A translocation breakpoint interrupting SUMO1 was found in a patient with CLP.[109] The causative nature of the translocation defect has been confirmed in SUMO1-deficient mice having a distinct CP phenotype.[109] Furthermore, it was recently shown that mutations in TBX22 have a profound effect on its ability to be “sumoylated,” which is at least partially responsible for its loss of function.[110] Other SUMO targets include Smad4, Msx1, p63, Pax9, Eya1 and FGF signalling.[107] It seems likely that some of these factors may manifest through the disturbance of the SUMO pathway. De-stabilizing the normal balance of expression and activity for genes such as TBX22, MSX1, SATB2, and P63 during early pregnancy is likely to provide a high-risk environment for the occurrence of CL/P. Elucidating the relationship among environmental factors, the SUMO pathway, and the networks of craniofacial genes influenced by this post-transcriptional modification may be crucial to our understanding of the idiopathic forms of oro-facial clefts.
A-P GRADIENT OF MOLECULAR SIGNALLING IN PALATAL DEVELOPMENT
Multiple genes are critical for the development of the anterior region of the palate. Msx1, Bmp4, Bmp2, Fgf10, and Shox2 have restricted expression patterns in the anterior region of the palate.[45] In addition to the differential gene expression patterns along the A-P axis of the developing palate, there is also mesenchymal heterogeneity between the medial and lateral regions of the palatal shelf. The odd, skipped related genes Osr1 and Osr2 are expressed in a medial-lateral gradient in the palatal shelf. The mutation of the Osr2 gene results in the compromised development of the medial aspect of the palatal shelf and retards palatal shelf elevation.[60,111] The expression of Fgfr2 is focused on the medial aspect of the developing palatal shelf, suggesting a possible functional significance in regulating its development and elevation.
An important discovery has been the confirmation of genetic heterogeneity along the anterior-posterior and medial-lateral axes of the developing palate.[48] This heterogeneity may provide a differential regulatory mechanism for the fusion of the anterior vs. posterior region of the palate. MEE cells undergo apoptosis at different times during palatal fusion. It has been shown that the apoptosis of MEE cells is triggered by palatal shelf contact in the anterior region, whereas it is initiated before any contact between the opposing shelves in the posterior region.[38] This difference may be the result of dissimilar molecular signals in the palatal mesenchyme along the anteroposterior axis that instruct different fates to the palatal epithelium.[112] Recent studies have demonstrated that constant and reciprocal interactions between palatal epithelium and CNC-derived mesenchyme are responsible for setting up this genetic heterogeneity along the AP axis and are crucial for normal palatal development and fusion.[25,45,113] The specific gene expression patterns in the posterior region of the palatal mesenchyme are less understood. Fgfr2 is expressed in the epithelium, and the CNC-derived mesenchyme is found in the middle and posterior palate. FGF8 signalling selectively induces the expression of Pax9 in the posterior region of the palatal mesenchyme. The loss of Pax9 results in a palatal shelf development defect and a cleft palate[48,58]
CONCLUSION
It is clear from this and other review of literature that CL/P is caused by many factors, including both genes and environment. Gene targeting technology and basic conventional techniques using animal models led to the identification of genes associated with known and unknown aetiologic factors. In some cases, the human gene deficiency was identified first and replicated in an animal model, but in other cases, animal models led the way to understand gene/environment interactions. It is also clear from this extensive list of possible contributing genes that the molecular and cellular interactions associated with CL/P are not all understood. Fortunately, some subclinical changes in facial features may lead to a greater understanding of the gene/environment interactions in cranio-facial development.
Acknowledgments
We thank Dr. Lynne Opperman for discussions about palate ossification and for permitting the use of a figure from one of her publications on palatal suture development. We thank Jeanne Santa Cruz for editorial assistance. We also thank the publishers for permission to reprint figures from previous papers. The gene tables are an ongoing project.
Footnotes
Source of Support: Cleft Palate Foundation (LBR), NIH NIDCR U24 DE16472 (LBR, KKHS, WY, SSM), March of Dimes (KKHS) NIH NIDCR T32 DE 018380 (MS).
Conflict of Interest: None declared.
REFERENCES
- 1.Ferguson MW. Palate development. Develop. 1988;103:41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- 2.Christensen K, Juel K, Herskind AM, Murray JC. Long term follow up study of survival associated with cleft lip and palate at birth. BMJ. 2004;328:1405. doi: 10.1136/bmj.38106.559120.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fogh-Andersen P. Epidemiology and etiology of clefts. In: Bergsma D, editor. Birth defects: Original article series. Baltimore: Williams and Wilkins Co.; 1971. [PubMed] [Google Scholar]
- 4.Slavkin HC. Incidence of cleft lips, palates rising. J Am Dent Assoc. 1992;123:61–5. doi: 10.14219/jada.archive.1992.0297. [DOI] [PubMed] [Google Scholar]
- 5.Murray JC. Gene/environment causes of cleft lip and/or palate. [review] [122 refs] Clinical Genetics. 2002;61:248–56. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- 6.Gritli-Linde A. The etiopathogenesis of cleft lip and cleft palate usefulness and caveats of mouse models. Curr Top Dev Biol. 2008;84:37–138. doi: 10.1016/S0070-2153(08)00602-9. [DOI] [PubMed] [Google Scholar]
- 7.Gritli-Linde A. Molecular control of secondary palate development. Developmental Biology. 2007;301:309–26. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 8.Carinci F, Pezzetti F, Scapoli L, Martinelli M, Carinci P, Tognon M. Genetics of nonsyndromic cleft lip and palate: A review of international studies and data regarding the Italian population. Cleft Palate Craniofac J. 2000;37:33–40. doi: 10.1597/1545-1569_2000_037_0033_goncla_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 9.Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235:2353–75. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- 10.Wyszynski DF, Duffy DL, Beaty TH. Maternal cigarette smoking and oral clefts: A meta-analysis. Cleft Palate Craniofac J. 1997;34:206–10. doi: 10.1597/1545-1569_1997_034_0206_mcsaoc_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 11.Shaw GM, Lammer EJ, Zhu H, Baker MW, Neri E, Finnell RH. Maternal periconceptional vitamin use, genetic variation of infant reduced folate carrier (a80g), and risk of spina bifida. Am J Med Genet. 2002;108:1–6. doi: 10.1002/ajmg.10195. [DOI] [PubMed] [Google Scholar]
- 12.Boot MJ, Steegers-Theunissen RP, Poelmann RE, Van Iperen L, Lindemans J, Gittenberger-de Groot AC. Folic acid and homocysteine affect neural crest and neuroepithelial cell outgrowth and differentiation in vitro. Dev Dyn. 2003;227:301–8. doi: 10.1002/dvdy.10303. [DOI] [PubMed] [Google Scholar]
- 13.Briggs RM. Vitamin supplementation as a possible factor in the incidence of cleft lip/palate deformities in humans. Clin Plast Surg. 1976;3:647–52. [PubMed] [Google Scholar]
- 14.Finnell RH, Shaw GM, Lammer EJ, Brandl KL, Carmichael SL, Rosenquist TH. Gene-nutrient interactions: Importance of folates and retinoids during early embryogenesis. Toxicol Appl Pharmacol. 2004;198:75–85. doi: 10.1016/j.taap.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 15.Itikala PR, Watkins ML, Mulinare J, Moore CA, Liu Y. Maternal multivitamin use and orofacial clefts in offspring. Teratology. 2001;63:79–86. doi: 10.1002/1096-9926(200102)63:2<79::AID-TERA1013>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 16.Lammer EJ, Shaw GM, Iovannisci DM, Finnell RH. Periconceptional multivitamin intake during early pregnancy, genetic variation of acetyl-n-transferase 1 (nat1), and risk for orofacial clefts. Birth Defects Res A Clin Mol Teratol. 2004;70:846–52. doi: 10.1002/bdra.20081. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H, Curry S, Wen S, Wicker NJ, Shaw GM, Lammer EJ, et al. Are the betaine-homocysteine methyltransferase (bhmt and bhmt2) genes risk factors for spina bifida and orofacial clefts? Am J Med Genet A. 2005;135:274–7. doi: 10.1002/ajmg.a.30739. [DOI] [PubMed] [Google Scholar]
- 18.Tang LS, Santillano DR, Wlodarczyk BJ, Miranda RC, Finnell RH. Role of folbp1 in the regional regulation of apoptosis and cell proliferation in the developing neural tube and craniofacies. Am J Med Genet C Semin Med Genet. 2005;135:48–58. doi: 10.1002/ajmg.c.30053. [DOI] [PubMed] [Google Scholar]
- 19.Lammer EJ, Shaw GM, Iovannisci DM, Van Waes J, Finnell RH. Maternal smoking and the risk of orofacial clefts: Susceptibility with nat1 and nat2 polymorphisms. Epidemiology. 2004;15:150–6. doi: 10.1097/01.ede.0000112214.33432.cc. [DOI] [PubMed] [Google Scholar]
- 20.Shaw GM, Zhu H, Lammer EJ, Yang W, Finnell RH. Genetic variation of infant reduced folate carrier (a80g) and risk of orofacial and conotruncal heart defects. Am J Epidemiol. 2003;158:747–52. doi: 10.1093/aje/kwg189. [DOI] [PubMed] [Google Scholar]
- 21.Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 22.Kang P, Svoboda KK. Epithelial-mesenchymal transformation during craniofacial development. J Dent Res. 2005;84:678–90. doi: 10.1177/154405910508400801. [DOI] [PubMed] [Google Scholar]
- 23.Nawshad A. Palatal seam disintegration: To die or not to die? That is no longer the question. Dev Dyn. 2008;237:2643–56. doi: 10.1002/dvdy.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shuler CF. Programmed cell death and cell transformation in craniofacial development. Crit Rev Oral Biol Med. 1995;6:202–17. doi: 10.1177/10454411950060030301. [DOI] [PubMed] [Google Scholar]
- 25.Murray JC, Schutte BC. Cleft palate: Players, pathways, and pursuits. J Clin Invest. 2004;113:1676–8. doi: 10.1172/JCI22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu W, Ruest L, Svoboda K. Regulation of epithelial-mesenchymal transition in palatal fusion. Exp Biol Med (Maywood) 2009;234:483–91. doi: 10.3181/0812-MR-365. [DOI] [PubMed] [Google Scholar]
- 27.Britto JA, Evans RD, Hayward RD, Jones BM. Toward pathogenesis of Apert cleft palate: Fgf, fgfr, and tgf beta genes are differentially expressed in sequential stages of human palatal shelf fusion. Cleft Palate Craniofac J. 2002;39:332–40. doi: 10.1597/1545-1569_2002_039_0332_tpoacp_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 28.Shi M, Wehby GL, Murray JC. Review on genetic variants and maternal smoking in the etiology of oral clefts and other birth defects. Birth Defects Res C Embryo Today. 2008;84:16–29. doi: 10.1002/bdrc.20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shuler CF, Halpern DE, Guo Y, Sank AC. Medial edge epithelium fate traced by cell lineage analysis during epithelial-mesenchymal transformation in vivo. Dev Biol. 1992;154:318–30. doi: 10.1016/0012-1606(92)90071-n. [DOI] [PubMed] [Google Scholar]
- 30.Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 31.Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dyn. 1997;209:255–60. doi: 10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 32.Kang Y, Massague J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell. 2004;118:277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Nawshad A, Hay ED. Tgfbeta3 signaling activates transcription of the lef1 gene to induce epithelial mesenchymal transformation during mouse palate development. J Cell Biol. 2003;163:1291–301. doi: 10.1083/jcb.200306024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nawshad A, LaGamba D, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT) Arch Oral Biol. 2004;49:675–89. doi: 10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 35.Sun W, Vincent S, Settleman J, Johnson GL. Mek kinase 2 binds and activates protein kinase c-related kinase 2. Bifurcation of kinase regulatory pathways at the level of an mapk kinase kinase. J Biol Chem. 2000;275:24421–8. doi: 10.1074/jbc.M003148200. [DOI] [PubMed] [Google Scholar]
- 36.LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev Dyn. 2005;234:132–42. doi: 10.1002/dvdy.20489. [DOI] [PubMed] [Google Scholar]
- 37.Cuervo R, Covarrubias L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Develop. 2004;131:15–24. doi: 10.1242/dev.00907. [DOI] [PubMed] [Google Scholar]
- 38.Cuervo R, Valencia C, Chandraratna RA, Covarrubias L. Programmed cell death is required for palate shelf fusion and is regulated by retinoic acid. Dev Biol. 2002;245:145–56. doi: 10.1006/dbio.2002.0620. [DOI] [PubMed] [Google Scholar]
- 39.Mori C, Nakamura N, Okamoto Y, Osawa M, Shiota K. Cytochemical identification of programmed cell death in the fusing fetal mouse palate by specific labelling of DNA fragmentation. Anat Embryol (Berl) 1994;190:21–8. doi: 10.1007/BF00185843. [DOI] [PubMed] [Google Scholar]
- 40.Taniguchi K, Sato N, Uchiyama Y. Apoptosis and heterophagy of medial edge epithelial cells of the secondary palatine shelves during fusion. Arch Histol Cytol. 1995;58:191–203. doi: 10.1679/aohc.58.191. [DOI] [PubMed] [Google Scholar]
- 41.Carette MJ, Ferguson MW. The fate of medial edge epithelial cells during palatal fusion in vitro: An analysis by dii labelling and confocal microscopy. Development. 1992;114:379–88. doi: 10.1242/dev.114.2.379. [DOI] [PubMed] [Google Scholar]
- 42.Jin J-Z, Ding J. Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development. 2006;133:3341–7. doi: 10.1242/dev.02520. [DOI] [PubMed] [Google Scholar]
- 43.Martinez-Alvarez C, Blanco MJ, Perez R, Rabadan MA, Aparicio M, Resel E, et al. Snail family members and cell survival in physiological and pathological cleft palates. Dev Biol. 2004;265:207–18. doi: 10.1016/j.ydbio.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 44.Sun D, Baur S, Hay ED. Epithelial-mesenchymal transformation is the mechanism for fusion of the craniofacial primordia involved in morphogenesis of the chicken lip. Dev Biol. 2000;228:337–49. doi: 10.1006/dbio.2000.9946. [DOI] [PubMed] [Google Scholar]
- 45.Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, et al. Disruption of fgf10/fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. [see comment] J Clin Invest. 2004;113:1692–700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice R, Thesleff I, Rice DP. Regulation of Twist, Snail, and Id1 is conserved between the developing murine palate and tooth. Dev Dyn. 2005;234:28–35. doi: 10.1002/dvdy.20501. [DOI] [PubMed] [Google Scholar]
- 47.Nie X, Luukko K, Kettunen P. FGF signalling in craniofacial development and developmental disorders. Oral Dis. 2006;12:102–11. doi: 10.1111/j.1601-0825.2005.01176.x. [DOI] [PubMed] [Google Scholar]
- 48.Hilliard SA, Yu L, Gu S, Zhang Z, Chen YP. Regional regulation of palatal growth and patterning along the anterior-posterior axis in mice. J Anat. 2005;207:655–67. doi: 10.1111/j.1469-7580.2005.00474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu L, Gu S, Alappat S, Song Y, Yan M, Zhang X, et al. Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development. 2005;132:4397–406. doi: 10.1242/dev.02013. [DOI] [PubMed] [Google Scholar]
- 50.Alappat SR, Zhang Z, Suzuki K, Zhang X, Liu H, Jiang R, et al. The cellular and molecular etiology of the cleft secondary palate in fgf10 mutant mice. Dev Biol. 2005;277:102–13. doi: 10.1016/j.ydbio.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 51.Braybrook C, Doudney K, Marcano AC, Arnason A, Bjornsson A, Patton MA, et al. The t-box transcription factor gene tbx22 is mutated in x-linked cleft palate and ankyloglossia. Nat Genet. 2001;29:179–83. doi: 10.1038/ng730. [DOI] [PubMed] [Google Scholar]
- 52.Marcano AC, Doudney K, Braybrook C, Squires R, Patton MA, Lees MM, et al. Tbx22 mutations are a frequent cause of cleft palate. J Med Genet. 2004;41:68–74. doi: 10.1136/jmg.2003.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong FK, Hagg U. An update on the aetiology of orofacial clefts. Hong Kong Med J. 2004;10:331–6. [PubMed] [Google Scholar]
- 54.Ferguson MW. Palatal shelf elevation in the wistar rat fetus. J Anat. 1978;125:555–77. [PMC free article] [PubMed] [Google Scholar]
- 55.Brinkley LL, Morris-Wiman J, Brinkley LL, Morris-Wiman J. The role of extracellular matrices in palatal shelf closure. Curr Top Dev Biol. 1984;19:17–36. doi: 10.1016/s0070-2153(08)60393-2. [DOI] [PubMed] [Google Scholar]
- 56.Brinkley LL, Morris-Wiman J, Brinkley LL, Morris-Wiman J. Effects of chlorcyclizine-induced glycosaminoglycan alterations on patterns of hyaluronate distribution during morphogenesis of the mouse secondary palate. Development. 1987;100:637–40. doi: 10.1242/dev.100.4.637. [DOI] [PubMed] [Google Scholar]
- 57.Kist R, Greally E, Peters H. Derivation of a mouse model for conditional inactivation of pax9. Genesis. 2007;45:460–4. doi: 10.1002/dvg.20295. [DOI] [PubMed] [Google Scholar]
- 58.Peters H, Neubuser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998;12:2735–47. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szeto DP, Rodriguez-Esteban C, Ryan AK, O'Connell SM, Liu F, Kioussi C, et al. Role of the bicoid-related homeodomain factor pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes Dev. 1999;13:484–94. doi: 10.1101/gad.13.4.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao Y, Lan Y, Ovitt CE, Jiang R. Functional equivalence of the zinc finger transcription factors osr1 and osr2 in mouse development. Dev Biol. 2009;328:200–9. doi: 10.1016/j.ydbio.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wee EL, Zimmerman EF. Involvement of GABA in palate morphogenesis and its relation to diazepam teratogenesis in two mouse strains. Teratology. 1983;28:15–22. doi: 10.1002/tera.1420280104. [DOI] [PubMed] [Google Scholar]
- 62.Scapoli L, Martinelli M, Pezzetti F, Carinci F, Bodo M, Tognon M, et al. Linkage disequilibrium between gabrb3 gene and nonsyndromic familial cleft lip with or without cleft palate. Hum Genet. 2002;110:15–20. doi: 10.1007/s00439-001-0639-5. [DOI] [PubMed] [Google Scholar]
- 63.Varju P, Katarova Z, Madarasz E, Szabo G. GABA signalling during development: New data and old questions. Cell Tissue Res. 2001;305:239–46. doi: 10.1007/s004410100356. [DOI] [PubMed] [Google Scholar]
- 64.Ding R, Tsunekawa N, Obata K. Cleft palate by picrotoxin or 3-mp and palatal shelf elevation in GABA-deficient mice. Neurotoxicol Teratol. 2004;26:587–92. doi: 10.1016/j.ntt.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 65.Condie BG, Bain G, Gottlieb DI, Capecchi MR. Cleft palate in mice with a targeted mutation in the gamma-aminobutyric acid-producing enzyme glutamic acid decarboxylase 67. Proc Natl Acad Sci U S A. 1997;94:11451–5. doi: 10.1073/pnas.94.21.11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yanagisawa H, Clouthier DE, Richardson JA, Charite J, Olson EN. Targeted deletion of a branchial arch-specific enhancer reveals a role of hand in craniofacial development. Development. 2003;130:1069–78. doi: 10.1242/dev.00337. [DOI] [PubMed] [Google Scholar]
- 67.Barbosa AC, Funato N, Chapman S, McKee MD, Richardson JA, Olson EN, et al. Hand transcription factors cooperatively regulate development of the distal midline mesenchyme. Dev Biol. 2007;310:154–68. doi: 10.1016/j.ydbio.2007.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fitchett JE, Hay ED. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev Biol. 1989;131:455–74. doi: 10.1016/s0012-1606(89)80017-x. [DOI] [PubMed] [Google Scholar]
- 69.Montenegro MA, Rojas M, Dominguez S, Vergara A. Cytokeratin, vimentin and e-cadherin immunodetection in the embryonic palate in two strains of mice with different susceptibility to glucocorticoid-induced clefting. J Craniofac Genet Dev Biol. 2000;20:137–43. [PubMed] [Google Scholar]
- 70.Vaziri Sani F, Hallberg K, Harfe BD, McMahon AP, Linde A, Gritli-Linde A. Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev Biol. 2005;285:490–5. doi: 10.1016/j.ydbio.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 71.Yu W, Kamara H, Svoboda KH. The role of twist during palate development. Dev Dyn. 2008;237:2716–25. doi: 10.1002/dvdy.21627. [DOI] [PubMed] [Google Scholar]
- 72.Frebourg T, Oliveira C, Hochain P, Karam R, Manouvrier S, Graziadio C, et al. Cleft lip/palate and cdh1/e-cadherin mutations in families with hereditary diffuse gastric cancer. J Med Genet. 2006;43:138–42. doi: 10.1136/jmg.2005.031385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cui XM, Shiomi N, Chen J, Saito T, Yamamoto T, Ito Y, et al. Overexpression of smad2 in tgf-beta3-null mutant mice rescues cleft palate. Dev Biol. 2005;278:193–202. doi: 10.1016/j.ydbio.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 74.Fitzpatrick DR, Denhez F, Kondaiah P, Akhurst RJ. Differential expression of tgf beta isoforms in murine palatogenesis. Development. 1990;109:585–95. doi: 10.1242/dev.109.3.585. [DOI] [PubMed] [Google Scholar]
- 75.Pelton RW, Dickinson ME, Moses HL, Hogan BL. In situ hybridization analysis of tgf beta 3 rna expression during mouse development: Comparative studies with tgf beta 1 and beta 2. Development. 1990;110:609–20. doi: 10.1242/dev.110.2.609. [DOI] [PubMed] [Google Scholar]
- 76.Pelton RW, Hogan BL, Miller DA, Moses HL. Differential expression of genes encoding tgfs beta 1, beta 2, and beta 3 during murine palate formation. Dev Biol. 1990;141:456–60. doi: 10.1016/0012-1606(90)90401-4. [DOI] [PubMed] [Google Scholar]
- 77.Schock F, Perrimon N. Molecular mechanisms of epithelial morphogenesis. Annu Rev Cell Dev Biol. 2002;18:463–93. doi: 10.1146/annurev.cellbio.18.022602.131838. [DOI] [PubMed] [Google Scholar]
- 78.Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. Tgf-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell. 2001;12:1457–66. doi: 10.1091/mbc.12.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaartinen V, Haataja L, Nagy A, Heisterkamp N, Groffen J. TGFbeta3-induced activation of rhoa/rho-kinase pathway is necessary but not sufficient for epithelio-mesenchymal transdifferentiation: Implications for palatogenesis. Int J Mol Med. 2002;9:563–70. [PubMed] [Google Scholar]
- 80.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, et al. Abnormal lung development and cleft palate in mice lacking tgf-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–21. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 81.Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R, et al. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nature Genetics. 1999;22:69–73. doi: 10.1038/8773. [DOI] [PubMed] [Google Scholar]
- 82.Sperber G. Craniofacial development. Hamilton and London: BC Decker Inc; 2001. [Google Scholar]
- 83.Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci U S A. 2003;100:9360–5. doi: 10.1073/pnas.1631288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kjaer I. Human prenatal palatal shelf elevation related to craniofacial skeletal maturation. Eur J Orthod. 1992;14:26–30. doi: 10.1093/ejo/14.1.26. [DOI] [PubMed] [Google Scholar]
- 85.Okano J, Suzuki S, Shiota K. Regional heterogeneity in the developing palate: Morphological and molecular evidence for normal and abnormal palatogenesis. Congenit Anom (Kyoto) 2006;46:49–54. doi: 10.1111/j.1741-4520.2006.00103.x. [DOI] [PubMed] [Google Scholar]
- 86.Kerrigan JJ, Mansell JP, Sengupta A, Brown N, Sandy JR. Palatogenesis and potential mechanisms for clefting. J R Coll Surg Edinb. 2000;45:351–8. [PubMed] [Google Scholar]
- 87.Iwasaki M, Le AX, Helms JA. Expression of Indian hedgehog, bone morphogenetic protein 6 and gli during skeletal morphogenesis. 1997;69:197–202. doi: 10.1016/s0925-4773(97)00145-7. [DOI] [PubMed] [Google Scholar]
- 88.Adab K, Sayne JR, Carlson DS, Opperman LA. Tgf-beta1, tgf-beta2, tgf-beta3 and msx2 expression is elevated during frontonasal suture morphogenesis and during active postnatal facial growth. Orthod Craniofac Res. 2002;5:227–37. doi: 10.1034/j.1600-0544.2002.02227.x. [DOI] [PubMed] [Google Scholar]
- 89.Adab K, Sayne JR, Carlson DS, Opperman LA. Nasal capsular cartilage is required for rat transpalatal suture morphogenesis. Differentiation. 2003;71:496–505. doi: 10.1046/j.1432-0436.2003.7108003.x. [DOI] [PubMed] [Google Scholar]
- 90.Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219:472–85. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 91.Vij K, Mao JJ. Geometry and cell density of rat craniofacial sutures during early postnatal development and upon in vivo cyclic loading. Bone. 2006;38:722–30. doi: 10.1016/j.bone.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 92.Liu C, Song R, Song Y. A serial histological study on suture expansion osteogenesis for cleft palate closure. Zhonghua Zheng Xing Wai Ke Za Zhi. 2000;16:43–5. [PubMed] [Google Scholar]
- 93.Hou B, Fukai N, Olsen BR. Mechanical force-induced midpalatal suture remodeling in mice. Bone. 2007;40:1483–93. doi: 10.1016/j.bone.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takahashi I, Mizoguchi I, Nakamura M, Sasano Y, Saitoh S, Kagayama M, et al. Effects of expansive force on the differentiation of midpalatal suture cartilage in rats. Bone. 1996;18:341–8. doi: 10.1016/8756-3282(96)00012-9. [DOI] [PubMed] [Google Scholar]
- 95.Kobayashi ET, Hashimoto F, Kobayashi Y, Sakai E, Miyazaki Y, Kamiya T, et al. Force-induced rapid changes in cell fate at midpalatal suture cartilage of growing rats. J Dent Res. 1999;78:1495–504. doi: 10.1177/00220345990780090301. [DOI] [PubMed] [Google Scholar]
- 96.Cowan CM, Cheng S, Ting K, Soo C, Walder B, Wu B, et al. Nell-1 induced bone formation within the distracted intermaxillary suture. Bone. 2006;38:48–58. doi: 10.1016/j.bone.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 97.Sawada M, Shimizu N. Stimulation of bone formation in the expanding mid-palatal suture by transforming growth factor-beta 1 in the rat. Eur J Orthod. 1996;18:169–79. doi: 10.1093/ejo/18.2.169. [DOI] [PubMed] [Google Scholar]
- 98.Akita S, Hirano A. Surgical modifications for microform cleft lip repairs. J Craniofac Surg. 2005;16:1106–10. doi: 10.1097/01.scs.0000186309.10957.fd. [DOI] [PubMed] [Google Scholar]
- 99.Neiswanger K, Weinberg SM, Rogers CR, Brandon CA, Cooper ME, Bardi KM, et al. Orbicularis oris muscle defects as an expanded phenotypic feature in nonsyndromic cleft lip with or without cleft palate. Am J Med Genet A. 2007;143:1143–9. doi: 10.1002/ajmg.a.31760. [DOI] [PubMed] [Google Scholar]
- 100.Jiang R, Bush JO, Lidral AC. Development of the upper lip: Morphogenetic and molecular mechanisms. Dev Dyn. 2006;235:1152–66. doi: 10.1002/dvdy.20646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marazita ML. Subclinical features in non-syndromic cleft lip with or without cleft palate (cl/p): Review of the evidence that subepithelial orbicularis oris muscle defects are part of an expanded phenotype for cl/p. Orthod Craniofac Res. 2007;10:82–7. doi: 10.1111/j.1601-6343.2007.00386.x. [DOI] [PubMed] [Google Scholar]
- 102.Gosain AK, Conley SF, Marks S, Larson DL. Submucous cleft palate: Diagnostic methods and outcomes of surgical treatment. Plast Reconstr Surg. 1996;97:1497–509. doi: 10.1097/00006534-199606000-00032. [DOI] [PubMed] [Google Scholar]
- 103.Boorman JG, Varma S, Ogilvie CM. Velopharyngeal incompetence and chromosome 22q11 deletion. Lancet. 2001;357:774. doi: 10.1016/S0140-6736(00)04183-0. [DOI] [PubMed] [Google Scholar]
- 104.Sweeney L. Basic concepts in embryology: A student's survival guide. McGraw-Hill Professional; 1998. p. 443. [Google Scholar]
- 105.Trotman CA, Hou D, Burdi AR, Cohen SR, Carlson DS. Histomorphologic analysis of the soft palate musculature in prenatal cleft and noncleft a/jax mice. Cleft Palate Craniofac J. 1995;32:455–62. doi: 10.1597/1545-1569_1995_032_0455_haotsp_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 106.Weinberg SM, Neiswanger K, Martin RA, Mooney MP, Kane AA, Wenger SL, et al. The Pittsburgh oral-facial cleft study: Expanding the cleft phenotype. Background and justification. Cleft Palate Craniofac J. 2006;43:7–20. doi: 10.1597/04-122r1.1. [DOI] [PubMed] [Google Scholar]
- 107.Pauws E, Stanier P. Fgf signalling and sumo modification: New players in the aetiology of cleft lip and/or palate. Trends Genet. 2007;23:631–40. doi: 10.1016/j.tig.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 108.Zhang FP, Mikkonen L, Toppari J, Palvimo JJ, Thesleff I, Janne OA. Sumo-1 function is dispensable in normal mouse development. Mol Cell Biol. 2008;28:5381–90. doi: 10.1128/MCB.00651-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alkuraya FS, Saadi I, Lund JJ, Turbe-Doan A, Morton CC, Maas RL. Sumo1 haploinsufficiency leads to cleft lip and palate. Science. 2006;313:1751. doi: 10.1126/science.1128406. [DOI] [PubMed] [Google Scholar]
- 110.Andreou AM, Pauws E, Jones MC, Singh MK, Bussen M, Doudney K, et al. Tbx22 missense mutations found in patients with x-linked cleft palate affect DNA binding, sumoylation, and transcriptional repression. Am J Hum Genet. 2007;81:700–12. doi: 10.1086/521033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lan Y, Ovitt CE, Cho ES, Maltby KM, Wang Q, Jiang R. Odd-skipped related 2 (osr2) encodes a key intrinsic regulator of secondary palate growth and morphogenesis. Development. 2004;131:3207–16. doi: 10.1242/dev.01175. [DOI] [PubMed] [Google Scholar]
- 112.Ferguson MW, Honig LS, Slavkin HC. Differentiation of cultured palatal shelves from alligator, chick, and mouse embryos. Anat Rec. 1984;209:231–49. doi: 10.1002/ar.1092090210. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in msx1-deficient mice by transgenic bmp4 reveals a network of bmp and shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–46. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- 114.Kreiborg S, Cohen MM., Jr The oral manifestations of Apert syndrome. J Craniofac Genet Dev Biol. 1992;12:41–8. [PubMed] [Google Scholar]
- 115.Martelli H, Jr, Paranaiba LM, de Miranda RT, Orsi J, Jr, Coletta RD. Apert syndrome: Report of a case with emphasis on craniofacial and genetic features. Pediatr Dent. 2008;30:464–8. [PubMed] [Google Scholar]
- 116.Park WJ, Theda C, Maestri NE, Meyers GA, Fryburg JS, Dufresne C, et al. Analysis of phenotypic features and fgfr2 mutations in apert syndrome. Am J Hum Genet. 1995;57:321–8. [PMC free article] [PubMed] [Google Scholar]
- 117.Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, et al. Apert syndrome results from localized mutations of fgfr2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–72. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 118.Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, et al. Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet. 1996;13:48–53. doi: 10.1038/ng0596-48. [DOI] [PubMed] [Google Scholar]
- 119.Castanet M, Park SM, Smith A, Bost M, Leger J, Lyonnet S, et al. A novel loss-of-function mutation in ttf-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet. 2002;11:2051–9. doi: 10.1093/hmg/11.17.2051. [DOI] [PubMed] [Google Scholar]
- 120.Clifton-Bligh RJ, Wentworth JM, Heinz P, Crisp MS, John R, Lazarus JH, et al. Mutation of the gene encoding human ttf-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19:399–401. doi: 10.1038/1294. [DOI] [PubMed] [Google Scholar]
- 121.Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, et al. Tfap2a mutations result in branchio-oculo-facial syndrome. Am J Hum Genet. 2008;82:1171–7. doi: 10.1016/j.ajhg.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kallen B, Mastroiacovo P, Robert E. Major congenital malformations in Down syndrome. Am J Med Genet. 1996;65:160–6. doi: 10.1002/(SICI)1096-8628(19961016)65:2<160::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 123.Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of eec syndrome. Cell. 1999;99:143–53. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 124.McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the sam domain of p63. Hum Mol Genet. 2001;10:221–9. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- 125.Abel EL. Fetal alcohol syndrome: A cautionary note. Curr Pharm Des. 2006;12:1521–9. doi: 10.2174/138161206776389886. [DOI] [PubMed] [Google Scholar]
- 126.Green ML, Singh AV, Zhang Y, Nemeth KA, Sulik KK, Knudsen TB. Reprogramming of genetic networks during initiation of the fetal alcohol syndrome. Dev Dyn. 2007;236:613–31. doi: 10.1002/dvdy.21048. [DOI] [PubMed] [Google Scholar]
- 127.Seki M, Yoshida K, Kashimura M. [a case of fetal alcohol effects with orofacial cleft]. Nihon Arukoru Yakubutsu Igakkai Zasshi. 2005;40:137–43. [PubMed] [Google Scholar]
- 128.Wattendorf DJ, Muenke M. Fetal alcohol spectrum disorders. Am Fam Physician. 2005;72:279–82,85. [PubMed] [Google Scholar]
- 129.Kokavec R. Goldenhar syndrome with various clinical manifestations. Cleft Palate Craniofac J. 2006;43:628–34. doi: 10.1597/05-094. [DOI] [PubMed] [Google Scholar]
- 130.Vilkki SK, Hukki J, Nietosvaara Y, Hurmerinta K, Suominen E. Microvascular temporomandibular joint and mandibular ramus reconstruction in hemifacial microsomia. J Craniofac Surg. 2002;13:809–15. doi: 10.1097/00001665-200211000-00017. [DOI] [PubMed] [Google Scholar]
- 131.Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, et al. Mutations in foxc2 (mfh-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000;67:1382–8. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dode C, Fouveaut C, Mortier G, Janssens S, Bertherat J, Mahoudeau J, et al. Novel fgfr1 sequence variants in Kallmann syndrome, and genetic evidence that the fgfr1c isoform is required in olfactory bulb and palate morphogenesis. Hum Mutat. 2007;28:97–8. doi: 10.1002/humu.9470. [DOI] [PubMed] [Google Scholar]
- 133.Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, et al. Loss-of-function mutations in fgfr1 cause autosomal dominant Kallmann syndrome. [see comment] Nature Genetics. 2003;33:463–5. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 134.Suzuki K, Hu D, Bustos T, Zlotogora J, Richieri-Costa A, Helms JA, et al. Mutations of pvrl1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat Genet. 2000;25:427–30. doi: 10.1038/78119. [DOI] [PubMed] [Google Scholar]
- 135.Prows CA, Bender PL. Beyond Pierre Robin sequence. Neonatal Netw. 1999;18:13–9. doi: 10.1891/0730-0832.18.5.13. [DOI] [PubMed] [Google Scholar]
- 136.Cole A, Lynch P, Slator R. A new grading of Pierre Robin sequence. Cleft Palate Craniofac J. 2008;45:603–6. doi: 10.1597/07-129.1. [DOI] [PubMed] [Google Scholar]
- 137.Muenke M. The pit, the cleft and the web. Nat Genet. 2002;32:219–20. doi: 10.1038/ng1002-219. [DOI] [PubMed] [Google Scholar]
- 138.Wassif CA, Maslen C, Kachilele-Linjewile S, Lin D, Linck LM, Connor WE, et al. Mutations in the human sterol delta7-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome. Am J Hum Genet. 1998;63:55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Snead MP, Yates JR. Clinical and molecular genetics of Stickler syndrome. J Med Genet. 1999;36:353–9. [PMC free article] [PubMed] [Google Scholar]
- 140.Wilkin DJ, Mortier GR, Johnson CL, Jones MC, de Paepe A, Shohat M, et al. Correlation of linkage data with phenotype in eight families with Stickler syndrome. Am J Med Genet. 1998;80:121–7. [PubMed] [Google Scholar]
- 141.Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, et al. Tcof1/treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci USA. 2006;103:13403–8. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. The Treacher Collins syndrome (tcof1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci USA. 2004;101:10709–14. doi: 10.1073/pnas.0402492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, et al. Mutations in irf6 cause van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–9. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cuneo BF. 22q11.2 deletion syndrome: Digeorge, velocardiofacial, and conotruncal anomaly face syndromes. Curr Opin Pediatr. 2001;13:465–72. doi: 10.1097/00008480-200110000-00014. [DOI] [PubMed] [Google Scholar]
- 145.Moreno F, Zuazo E, Gonzalez S, Bereciartu P. 22q11 deletion syndrome: An expanding phenotype. Neurologia. 2009;24:69–71. [PubMed] [Google Scholar]
- 146.Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with chd7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43:211–7. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause charge syndrome. Nat Genet. 2004;36:955–7. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 148.Schuffenhauer S, Leifheit HJ, Lichtner P, Peters H, Murken J, Emmerich P. De novo deletion (14)(q11.2q13) including pax9: Clinical and molecular findings. J Med Genet. 1999;36:233–6. [PMC free article] [PubMed] [Google Scholar]
- 149.Abidi FE, Miano MG, Murray JC, Schwartz CE. A novel mutation in the phf8 gene is associated with x-linked mental retardation with cleft lip/cleft palate. Clin Genet. 2007;72:19–22. doi: 10.1111/j.1399-0004.2007.00817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, et al. Mutations in patched-1, the receptor for sonic hedgehog, are associated with holoprosencephaly. Hum Genet. 2002;110:297–301. doi: 10.1007/s00439-002-0695-5. [DOI] [PubMed] [Google Scholar]
- 151.Ribeiro LA, Murray JC, Richieri-Costa A. Ptch mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal mri. Am J Med Genet A. 2006;140:2584–6. doi: 10.1002/ajmg.a.31369. [DOI] [PubMed] [Google Scholar]
- 152.Leoyklang P, Suphapeetiporn K, Siriwan P, Desudchit T, Chaowanapanja P, Gahl WA, et al. Heterozygous nonsense mutation satb2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat. 2007;28:732–8. doi: 10.1002/humu.20515. [DOI] [PubMed] [Google Scholar]
- 153.Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of tbx1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–73. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- 154.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in tgfbr1 or tgfbr2. Nature Genetics. 2005;37:275–81. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 155.Hines RN, McCarver DG. The ontogeny of human drug-metabolizing enzymes: Phase I oxidative enzymes. J Pharmacol Exp Ther. 2002;300:355–60. doi: 10.1124/jpet.300.2.355. [DOI] [PubMed] [Google Scholar]
- 156.Stoilov I, Jansson I, Sarfarazi M, Schenkman JB. Roles of cytochrome p450 in development. Drug Metabol Drug Interact. 2001;18:33–55. doi: 10.1515/dmdi.2001.18.1.33. [DOI] [PubMed] [Google Scholar]
- 157.van Rooij IA, Wegerif MJ, Roelofs HM, Peters WH, Kuijpers-Jagtman AM, Zielhuis GA, et al. Smoking, genetic polymorphisms in biotransformation enzymes, and nonsyndromic oral clefting: A gene-environment interaction. Epidemiology. 2001;12:502–7. doi: 10.1097/00001648-200109000-00007. [DOI] [PubMed] [Google Scholar]
- 158.An S, Dickens MA, Bleu T, Hallmark OG, Goetzl EJ. Molecular cloning of the human edg2 protein and its identification as a functional cellular receptor for lysophosphatidic acid. Biochem Biophys Res Commun. 1997;231:619–22. doi: 10.1006/bbrc.1997.6150. [DOI] [PubMed] [Google Scholar]
- 159.Shi M, Christensen K, Weinberg CR, Romitti P, Bathum L, Lozada A, et al. Orofacial cleft risk is increased with maternal smoking and specific detoxification-gene variants. Am J Hum Genet. 2007;80:76–90. doi: 10.1086/510518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hartsfield JK, Jr, Hickman TA, Everett ET, Shaw GM, Lammer EJ, Finnell RA. Analysis of the ephx1 113 polymorphism and gstm1 homozygous null polymorphism and oral clefting associated with maternal smoking. Am J Med Genet. 2001;102:21–4. doi: 10.1002/1096-8628(20010722)102:1<21::aid-ajmg1409>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 161.Hozyasz KK, Mostowska A, Surowiec Z, Jagodzinski PP. [genetic polymorphisms of gstm1 and gstt1 in mothers of children with isolated cleft lip with or without cleft palate] Przegl Lek. 2005;62:1019–22. [PubMed] [Google Scholar]
- 162.Landi S. Mammalian class theta gst and differential susceptibility to carcinogens: A review. Mutat Res. 2000;463:247–83. doi: 10.1016/s1383-5742(00)00050-8. [DOI] [PubMed] [Google Scholar]
- 163.Raijmakers MT, Steegers EA, Peters WH. Glutathione s-transferases and thiol concentrations in embryonic and early fetal tissues. Hum Reprod. 2001;16:2445–50. doi: 10.1093/humrep/16.11.2445. [DOI] [PubMed] [Google Scholar]
- 164.Sim E, Payton M, Noble M, Minchin R. An update on genetic, structural and functional studies of arylamine n-acetyltransferases in eucaryotes and procaryotes. Hum Mol Genet. 2000;9:2435–41. doi: 10.1093/hmg/9.16.2435. [DOI] [PubMed] [Google Scholar]
- 165.Smelt VA, Upton A, Adjaye J, Payton MA, Boukouvala S, Johnson N, et al. Expression of arylamine n-acetyltransferases in pre-term placentas and in human pre-implantation embryos. Hum Mol Genet. 2000;9:1101–7. doi: 10.1093/hmg/9.7.1101. [DOI] [PubMed] [Google Scholar]
- 166.Martinelli M, Scapoli L, Pezzetti F, Carinci F, Carinci P, Stabellini G, et al. C677t variant form at the mthfr gene and cl/p: A risk factor for mothers? Am J Med Genet. 2001;98:357–60. doi: 10.1002/1096-8628(20010201)98:4<357::aid-ajmg1108>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 167.Blanton SH, Kolle BS, Hecht JT, Mulliken JB, Martin ER. No evidence supporting mthfr as a risk factor in the development of familial nsclp. Am J Med Genet. 2000;92:370–1. doi: 10.1002/1096-8628(20000619)92:5<370::aid-ajmg17>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 168.Botto LD, Yang Q. 5, 10-methylenetetrahydrofolate reductase gene variants and congenital anomalies: A huge review. Am J Epidemiol. 2000;151:862–77. doi: 10.1093/oxfordjournals.aje.a010290. [DOI] [PubMed] [Google Scholar]
- 169.Jugessur A, Wilcox AJ, Lie RT, Murray JC, Taylor JA, Ulvik A, et al. Exploring the effects of methylenetetrahydrofolate reductase gene variants c677t and a1298c on the risk of orofacial clefts in 261 Norwegian case-parent triads. Am J Epidemiol. 2003;157:1083–91. doi: 10.1093/aje/kwg097. [DOI] [PubMed] [Google Scholar]
- 170.Shaw GM, Rozen R, Finnell RH, Todoroff K, Lammer EJ. Infant c677t mutation in mthfr, maternal periconceptional vitamin use, and cleft lip. Am J Med Genet. 1998;80:196–8. doi: 10.1002/(sici)1096-8628(19981116)80:3<196::aid-ajmg2>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 171.Beaty TH, Wang H, Hetmanski JB, Fan YT, Zeiger JS, Liang KY, et al. A case-control study of nonsyndromic oral clefts in Maryland. Ann Epidemiol. 2001;11:434–42. doi: 10.1016/s1047-2797(01)00222-8. [DOI] [PubMed] [Google Scholar]
- 172.Pepe G, Camacho Vanegas O, Giusti B, Brunelli T, Marcucci R, Attanasio M, et al. Heterogeneity in world distribution of the thermolabile c677t mutation in 5, 10-methylenetetrahydrofolate reductase. Am J Hum Genet. 1998;63:917–20. doi: 10.1086/302015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Collier AC, Tingle MD, Paxton JW, Mitchell MD, Keelan JA. Metabolizing enzyme localization and activities in the first trimester human placenta: The effect of maternal and gestational age, smoking and alcohol consumption. Hum Reprod. 2002;17:2564–72. doi: 10.1093/humrep/17.10.2564. [DOI] [PubMed] [Google Scholar]
- 174.Shaw GM, Iovannisci DM, Yang W, Finnell RH, Carmichael SL, Cheng S, et al. Risks of human conotruncal heart defects associated with 32 single nucleotide polymorphisms of selected cardiovascular disease-related genes. Am J Med Genet A. 2005;138:21–6. doi: 10.1002/ajmg.a.30924. [DOI] [PubMed] [Google Scholar]
- 175.Hwang SJ, Beaty TH, Panny SR, Street NA, Joseph JM, Gordon S, et al. Association study of transforming growth factor alpha (tgf alpha) taqi polymorphism and oral clefts: Indication of gene-environment interaction in a population-based sample of infants with birth defects. Am J Epidemiol. 1995;141:629–36. doi: 10.1093/oxfordjournals.aje.a117478. [DOI] [PubMed] [Google Scholar]
- 176.Maestri NE, Beaty TH, Hetmanski J, Smith EA, McIntosh I, Wyszynski DF, et al. Application of transmission disequilibrium tests to nonsyndromic oral clefts: Including candidate genes and environmental exposures in the models. Am J Med Genet. 1997;73:337–44. doi: 10.1002/(sici)1096-8628(19971219)73:3<337::aid-ajmg21>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 177.Miettinen PJ, Perheentupa J, Otonkoski T, Lahteenmaki A, Panula P. Egf- and tgf-alpha-like peptides in human fetal gut. Pediatr Res. 1989;26:25–30. doi: 10.1203/00006450-198907000-00009. [DOI] [PubMed] [Google Scholar]
- 178.Hwang SJ. Association study of transforming growth factor alpha (tgf alpha) taqi polymorphism and oral clefts: Indication of gene-environment interaction in a population-based sample of infants with birth defects. Amer J Epidem. 1992;135:1000–11. doi: 10.1093/oxfordjournals.aje.a117478. [DOI] [PubMed] [Google Scholar]
- 179.Mitchell LE, Murray JC, O'Brien S, Christensen K. Evaluation of two putative susceptibility loci for oral clefts in the Danish population. American Journal of Epidemiology. 2001;153:1007–15. doi: 10.1093/aje/153.10.1007. [DOI] [PubMed] [Google Scholar]
- 180.Romitti PA, Lidral AC, Munger RG, Daack-Hirsch S, Burns TL, Murray JC. Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: Evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology. 1999;59:39–50. doi: 10.1002/(SICI)1096-9926(199901)59:1<39::AID-TERA9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 181.Beiraghi S, Zhou M, Talmadge CB, Went-Sumegi N, Davis JR, Huang D, et al. Identification and characterization of a novel gene disrupted by a pericentric inversion inv(4)(p13.1q21.1) in a family with cleft lip. Gene. 2003;309:11–21. doi: 10.1016/s0378-1119(03)00461-x. [DOI] [PubMed] [Google Scholar]
- 182.Felix TM, Hanshaw BC, Mueller R, Bitoun P, Murray JC. Chd7 gene and non-syndromic cleft lip and palate. Am J Med Genet A. 2006;140:2110–4. doi: 10.1002/ajmg.a.31308. [DOI] [PubMed] [Google Scholar]
- 183.Osoegawa K, Vessere GM, Utami KH, Mansilla MA, Johnson MK, Riley BM, et al. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridisation. J Med Genet. 2008;45:81–6. doi: 10.1136/jmg.2007.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, et al. Impaired fgf signaling contributes to cleft lip and palate. Proc Natl Acad Sci USA. 2007;104:4512–7. doi: 10.1073/pnas.0607956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vieira AR, Avila JR, Daack-Hirsch S, Dragan E, Felix TM, Rahimov F, et al. Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet. 2005;1:e64. doi: 10.1371/journal.pgen.0010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Inoue H, Kayano S, Aoki Y, Kure S, Yamada A, Hata A, et al. Association of the gabrb3 gene with nonsyndromic oral clefts. Cleft Palate Craniofac J. 2008;45:261–6. doi: 10.1597/06-142. [DOI] [PubMed] [Google Scholar]