

Abstract
We report on the current state of in vivo amyloid imaging. While this technique is less than a decade old, a wealth of information is emerging as the initial clinical studies are reported. Imaging of patients with Alzheimer's Disease (AD) have allowed for quantitative exploration of the natural history of amyloid deposition and it's relationship to neurodegeneration. Amyloid imaging also shows significant promise in differential diagnosis of mild cognitive impairment or atypical dementias. However, amyloid detection may be of greatest utility in healthy elderly in whom amyloid imaging has confirmed prior autopsy reports of a significant percentage of asymptomatic adults with Alzheimer's pathology. Understanding the relationship between this pathology and future cognitive status has significant implications for the application of disease modifying medications in the ‘pre-clinical’ phase of disease. Given the considerable clinical experience compared to other tracers, the current review focuses on the literature involving Pittsburgh Compound-B (PiB) PET.
I. Introduction
Alzheimer's disease (AD) is the most common form of dementia and it is currently estimated that 5.3 million people in the United States are afflicted with this disease. In addition to the significant emotional and social impact on patients and caregivers, close to $150 billion per year is spent on the care needs of this population. Due to changing demographics and enhanced longevity, it is anticipated that up to 16 million people will have AD by the year 2050. Given these staggering numbers, considerable resources have been focused on the development of disease modifying medications, which are designed to directly delay the pathophysiologic process of the disease.
A parallel research goal has been the search for biomarkers which allow for early detection of AD and the ability to accurately chart disease progression. The first desire follows from the notion that interventions are felt to most likely be effective early in the course of the disease and less likely to reverse the brain injury already sustained. The ultimate hope is for a biomarker that allows for “diagnosis” of the underlying pathological process prior to the onset of symptoms, which would be the ideal clinical scenario for initiation of a disease modifying medication. The second motivation is based on the need in clinical trials for more specific and less variable markers of progression than the currently used clinical measures, which would allow for accurate assessment of efficacy in potentially smaller cohorts of patients. In addition to these roles, biomarkers also have the potential to provide invaluable information about the pathophysiology and course of the disease.
Amyloid imaging, which provides an in vivo measurement of one of the hallmark pathological features of AD – fibrillar Aβ plaques – has great potential to fulfill these goals. A number of different PET ligands have displayed affinity for amyloid plaques (e.g. [18F]-FDDNP 1 and [11C]-SB13 2), but the thioflavin-T derivative N-methyl [11C] 2-(4′-methylaminophenyl)-6-hydroxybenzothiazole, or Pittsburgh Compound-B (PiB), has been the most widely studied 3. Since the initial human studies performed in Sweden, PiB PET imaging has been utilized at numerous sites across the United States and world in thousands of subjects. Along with its inclusion in the Alzheimer's Disease Neuroimaging Initiative (ADNI), PiB PET has effectively become the benchmark for amyloid imaging. As such, the current review will focus on this ligand 4.
In addition to the ex vivo evidence supporting PiB's capacity to bind to fibrillar Aβ 5, 6, PiB PET imaging in patients with AD (as described below), has revealed a distribution of binding consistent with pathological descriptions of the topography of amyloid deposition in AD 3, 7. Further, high levels of PiB uptake have been found to be associated with low CSF concentrations of CSF Aβ1-42, a biomarker associated with AD pathology, in both patients with AD and healthy controls 8. More directly, autopsy data in a patient who had a PiB PET scan 10 months prior to death revealed a high correlation between regional in vivo PiB uptake and plaque load 9. An additional autopsy study of a patient with a clinical diagnosis of dementia with Lewy Bodies and significant amyloid angiopathy also displayed a strong correlation with in vivo imaging 10. Recently, Leinonen and colleagues reported on 10 patients who underwent biopsy and intracranial pressure (ICP) monitoring as part of their work-up for normal pressure hydrocephalus and then subsequently had PiB PET imaging. Five of the 10 patients had evidence of significant amyloid pathology on biopsy 11. While no “false positives” were reported, one patient with a reasonable degree of amyloid pathology detected by immunohistochemistry (although minimal plaques on Bielschowski silver stain) did not display elevated PiB binding. This case may be the first example in humans that PiB PET may produce false negative results, which could be related to ultrastructural aspects of the amyloid pathology 12. Nonetheless, this study still demonstrated a high overall correlation between Aβ pathology and detection by PiB PET.
II. PiB PET Imaging in AD
The first cohort of patients with AD who underwent PiB PET imaging was reported in 2004 by Klunk and colleagues 3. Sixteen patients with mild-moderate clinical AD were compared to age-matched and young controls. Increased PiB retention was found in regions commonly associated with amyloid deposition in pathological studies, including frontal cortex, lateral temporal and parietal cortex, anterior and posterior cingulate, precuneus, and striatum 7, 13 (see Figure 1 and Table). Further, areas in which there is little fibrillar amyloid deposition in AD, such as subcortical white matter and cerebellum, were not significantly different in PiB uptake between the patients and controls (Figure 2). Importantly, consistent with pathological studies, medial temporal regions tended to have less PiB signal than neocortical association areas.
Figure 1.
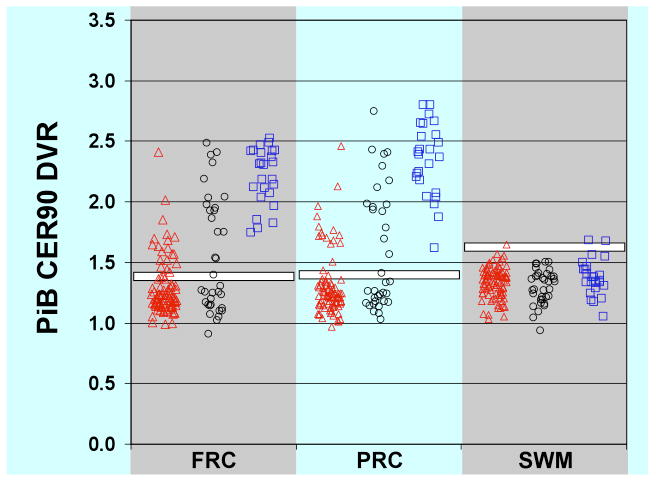
PiB retention by region in healthy controls, patients with mild cognitive impairment, and patients with Alzheimer's disease. PiB distribution volume ratio (DVR) values based on 90 minutes of scanning with cerebellum as reference quantified for frontal cortex (FRC), precuneus (PRC), and subcortical white matter (SWM). The white bars represent University of Pittsburgh cut-off values for dichotomous determination of “amyloid-positive” versus “amyloid-negative” scans. Red triangles: healthy controls; black circles: mild cognitive impairment patients; blue squares: Alzheimer's disease patients.
Table 1.
Mean DVR values for PiB uptake by region
| HC PiB-neg. | HC PiB-pos. | MCI PiB-neg | MCI PiB-pos. | AD | |
| ACG | 1.2 | 1.7 | 1.2 | 2.2 | 2.4 |
| FRC | 1.2 | 1.6 | 1.2 | 2.0 | 2.2 |
| LTC | 1.1 | 1.4 | 1.1 | 1.8 | 2.0 |
| MTC | 1.0 | 1.2 | 1.1 | 1.3 | 1.3 |
| PAR | 1.2 | 1.5 | 1.2 | 1.9 | 2.0 |
| PRC | 1.2 | 1.7 | 1.2 | 2.0 | 2.4 |
| SWM | 1.3 | 1.4 | 1.3 | 1.3 | 1.4 |
| PON | 1.5 | 1.5 | 1.4 | 1.5 | 1.5 |
Note: Regions associated with amyloid plaques on autopsy display higher PiB DVR values in PiB-positive individuals than PiB-negative ones, but not in other regions (e.g. SWM, PON); PiB-pos.: PiB binding above University of Pittsburgh cut-off; PiB-neg.: PiB binding below University of Pittsburgh cut-off; DVR: distribution volume ratio with cerebellum as reference; ACG: anterior cingulate gyrus; FRC: frontal cortex; LTC: lateral temporal cortex; MTC: medial temporal cortex; PAR: parietal cortex; PRC: precuneus; SWM: subcortical white matter; PON: pons
Figure 2.

Axial and mid-sagittal PiB PET images of amyloid-positive (NC+) and amyloid-negative (NC-) healthy controls and patients with mild cognitive impairment (MCI+ and MCI-, respectively) compared with a typical patient with Alzheimer's disease. Images are quantified by distribution volume ratio (DVR).
Since this report, a number of studies have similarly demonstrated robust differences of regional PiB uptake in AD patients relative to healthy controls 3, 14-16. However, not all patients with clinically diagnosed AD have been reported to have levels of PiB binding which differ from controls 3, 16, 17. Presumably, these cases reflect misdiagnosis although without pathological confirmation, insensitivity of the technique to amyloid deposition cannot be ruled out in these cases. Nonetheless, this finding is completely consistent with the imperfect specificity of the clinical diagnosis of AD 18. Indeed, the three reported PiB negative patients by Klunk and colleagues all had an atypical course with high Mini-Mental Status Exam scores (MMSE) and stable cognitive status over the two to four years prior to the study. Post-mortem analysis of such patients will ultimately establish whether these cases truly represent misdiagnosis or false-negative results.
In addition to a potential role in confirmation of diagnosis, amyloid imaging also has the potential to provide significant insight into role of Aβ in the pathophysiology and clinical manifestations of AD. For example, the natural history of amyloid plaque deposition can be evaluated in individual AD patients. Previously, the rate of such deposition has been assessed through comparison of autopsy specimens for patients at different stages of disease. A couple of studies have now reported longitudinal data on PiB binding in AD patients 17, 19. Engler et al. noted essentially no change in PiB levels regardless of disease progression after approximately two years. However, cognitive function and FDG PET metabolism appeared to decline. These authors suggested that amyloid deposition may reach a plateau by the time of clinical AD despite continued neurodegenerative changes signaled by alterations in brain metabolism and cognition. In distinction, Jack and colleagues reported a small, but slow longitudinal increase in PiB uptake regardless of cognitive status. Further, while no overall statistical effect was found, many of the patients reported by Engler et al. displayed some small increase in PiB levels 20. Whatever the case, it does appear that these changes are small at most, which suggests that anti-amyloid interventions must demonstrate more than a stabilizing effect on amyloid levels in order to be effective. This finding is also consistent with the notion from autopsy studies that amyloid plaque burden only weakly correlates with cognition and clinical status relative to other markers of disease burden, such as neurofibrillary tangle burden or synapse loss 21, 22.
Nonetheless, combined studies of fluorodeoxyglucose (FDG) and PiB PET have reported inverse correlations between degree of PiB uptake and parietal hypometabolism 3, 16, 19. However, despite the high levels of PiB binding in the frontal cortex of AD patients, frontal hypometabolism is relatively uncommon in the mild stages of disease. This contrast with a strong inverse correlation in more posterior regions suggests that the relationship between amyloid plaque deposition and metabolism is complex.
Imaging with PiB PET has also begun to allow for assessing the role of brain reserve or compensatory activity in modifying the relationship between AD pathology and phenotype. For example, it has been frequently reported that education may be protective against the risk of developing clinical AD 23. Autopsy work has suggested that patients with equivalent loads of AD pathology may display different degrees of cognitive impairment with more highly educated patients displaying the least impairment; alternatively, highly educated AD patients appear to require more pathology to reach a certain level of cognitive decline relative to their lower educated counterparts 24, 25. As in vivo confirmation of these findings, Kemppainen et al. reported higher levels of PiB uptake in high compared to low educated AD patients matched for disease severity 26. Recent work has also started to examine the relationship of regional PiB binding and cortical recruitment during cognitive tasks with functional imaging 27.
III. PiB PET in Non-AD Dementias
The use of PiB PET has been explored in other dementias with implications for the utility of this method for differential diagnosis. For example, several reports have revealed lower levels of PiB binding in patients with Parkinson's Disease (PD) and Parkinson's Disease Dementia (PDD) 28, 29. However, patients with Dementia with Lewy Bodies (DLB) frequently have elevated levels of PiB uptake above that of controls and occasionally equivalent to AD 15. This result is not unexpected, as the presence of significant Aβ pathology is a frequent finding in DLB with less than 20% of such patients having purely Lewy Body pathology 30. While cortical PiB binding is generally quantitatively less than that seen in AD, Rowe and colleagues reported an acceleration in the time from symptom onset to the full spectrum of DLB in those with the highest levels of PiB uptake suggesting a modifying role of Aβ in the pathophysiology of DLB. While the overlap in PiB binding between AD and DLB limits the ability of this technique to clearly differentiate these conditions, it does provide potentially important information about the underlying contribution of AD pathology to such patients which may have implications for treatment approaches.
The potential differential diagnostic utility of PiB PET may be best demonstrated by studies of patients with Frontotemporal Dementia (FTD) spectrum disorders, including Semantic Dementia (SD), Progressive Nonfluent Aphasia (PNFA), and Corticobasal Degeneration (CBD), as well as other forms of atypical focal dementias (e.g. Posterior Cortical Atrophy; PCA). These clinical phenotypes are all associated with a proportion of patients who have primarily AD pathology (Alladi et al., 2007). For example, Alladi et al. reported that 40% of patients with PNFA had AD pathology at autopsy rather than the spectrum of pathology traditionally associated with FTD (e.g. tau or TDP-43 pathology). Consistent with clinicopathologic series, studies of patients with FTD has revealed a significant minority of patients who display elevated levels of PiB binding 31, 32. For example, Rabinovici et al. (2007) designated four out of 12 FTD patients as “PiB-positive” based on visual inspection of the scans. In a later series, this group described PiB PET findings on 15 patients with either primary progressive aphasia or semantic dementia. Six of these patients had uptake above control level, including all of the patients with so-called logopenic aphasia (four patients), a subtype of PNFA with a high frequency of AD pathology on autopsy studies 33. Interestingly, these patients did not have asymmetric PiB binding despite the left hemisphere symptoms and FDG findings, which is also consistent with pathological studies with regard to amyloid plaque distribution in such cases. While the possibility of concomitant non-AD pathology remains in these patients, the results are entirely consistent with clinicopathologic series and suggest that amyloid imaging may be critical in differentiating the underlying etiology of such cases with greater accuracy than can be accomplished based on the clinical phenotype. This would obviously have important implications for the use of disease specific interventions in such cases. Additionally, early identification of these atypical presentations of AD may allow for further insight into the potential mechanisms driving these abnormal presentations.
IV. PiB Imaging in Mild Cognitive Impairment
As mentioned earlier, one of the major goals of AD research over the past decade is the early identification of AD patients before fully manifesting dementia symptoms. In some sense, the diagnostic construct of Mild Cognitive Impairment (MCI) can be thought of as a clinical attempt to accomplish this goal. While this designation has been operationalized in a number of different ways, the essence of the definition is the presence of cognitive decline in at least one domain of cognitive function (e.g. memory or language) to an insufficient extent to result in obvious functional impairment or to merit the diagnosis of dementia 34. As such, it is often conceptualized as a transitional stage between normal aging and dementia. In support of this notion, patients with amnestic-MCI (a-MCI), the most studied subtype, have an annual conversion rate of 5-15%/year with the majority developing clinical AD 34. Nonetheless, not all patients convert to dementia even after several years, and when they do, AD is not the only possible etiology 35. Indeed, even in patients classified as converters to AD, pathologic diagnosis may not conform 36. Further, conversion rates vary depending on the criteria applied or the population studied. For example, relatively frequent “reversion to normal” rates have been reported in epidemiologic studies suggesting instability of the diagnosis 37.
Given this prognostic and pathologic uncertainty, there has been considerable effort in establishing biomarkers that provide this information. Establishing the underlying pathology at this stage of disease has important implications for potential early medicinal intervention. Further, such specification could play an important role in the development of AD-specific disease modifying therapeutics by potentially allowing for MCI clinical trials to only incorporate patients with AD, rather than the heterogeneous population produced by a purely clinical designation. Indeed, application of the current MCI criteria limits the power of detecting an effect for an AD-specific drug, as a significant proportion of patients in the active and placebo arms of such studies will not have underlying AD pathology. While volumetric MRI (e.g. hippocampal volume), FDG PET, cerebrospinal fluid (CSF) Aβ and tau, and certain psychometrics (e.g. delayed recall) all have been shown to enhance prediction of clinical conversion to AD 38-41, it remains unclear whether these methods offer adequate sensitivity and specificity in detection of AD pathology.
Amyloid imaging, which provides a direct measure of AD pathology, may be ideal in this regard. There have now been several reported studies of PiB PET imaging in patients with MCI 14, 15, 42-45. In studies that have operationalized a cut-off of PiB uptake to designate “amyloid-positive” versus “amyloid-negative” patients, there has generally been a rate of 50-65% who were amyloid-positive. While application of a dichotomous label to a continuous variable may seem arbitrary, there is logic to the notion that any evidence of amyloid pathology is abnormal. Nonetheless, determination of these cutoffs have varied across laboratories and no clear consensus has been reached upon what constitutes a clearly abnormal PiB PET scan. Interestingly, this issue tends to be less of a concern for patients with MCI than controls (discussed below), as the distribution of PiB binding tends to be largely dichotomous. With some exceptions, MCI patients with elevated PiB binding tend to have levels of uptake similar to that of patients with clinical AD (see Figure 2), suggesting that there may be a relatively asymptomatic phase of amyloid plaque accumulation before the manifestation of significant cognitive symptoms.
Consistent with the likelihood that the amyloid-positive MCI patients represent the early symptomatic phase of Alzheimer's, these patients tend to have features characteristic of this diagnosis. For example, Forsberg and colleagues (2007) reported a correlation between PiB binding and decreased CSF Aβ and increased CSF tau, the pattern typical seen with AD. Amyloid-positive patients also have been reported to have poorer memory than their amyloid-negative counterparts in some 42, 43, 45, but not all 44, studies. Our group described smaller hippocampal volumes in amyloid-positive patients 45. As all of these characteristics are also predictive of clinical conversion to AD in longitudinal studies, these findings are consistent with the notion that MCI patients with elevated PiB binding have early AD as the etiology for their cognitive impairment and will eventually qualify for a diagnosis of clinical AD by current criteria.
The strongest evidence in this regard will come from longitudinal follow-up. While the data on conversion in MCI patients who underwent PiB imaging is limited, a few groups have reported outcomes consistent with the expectation that those with elevated PiB binding are likely to convert to clinical AD 42, 45, 46. Forsberg et al. reported the first follow-up data on 21 MCI patients. While the follow-up period was short (mean approximately 8 months), 7 out of the 21 patients converted, all with elevated PiB uptake at the AD level, and, overall, the converters had a higher level of PiB binding than the non-converters. In our MCI cohort, patients were followed for a mean of approximately 21 months 45. Of those patients designated amyloid-positive based on PiB imaging, 38% (5/13) converted to clinical AD. None of the amyloid-negative patients converted, and, in fact, three “reverted to normal” on follow-up. Thus, PiB imaging had significant prognostic value. However, further follow-up data will be needed in this and other cohorts to confirm that all or most MCI patients with elevated PiB uptake convert to AD and that those who are amyloid-negative remain stable or develop non-AD dementias.
V. PiB Imaging in Healthy Controls
Potentially the greatest utility of amyloid imaging will come in the detection of early AD pathology in pre-symptomatic individuals. It is well-know from neuropathological studies that a significant proportion of cognitively normal older adults display significant AD pathology 47. Likewise, several groups have now reported similar proportions (20-30%) of healthy controls who have elevated PiB binding 17, 43, 48; see Figure 1 for an example of an amyloid-positive control). The ability to follow these patients longitudinally allows for potentially answering one critical question unanswered by the autopsy studies: would these asymptomatic individuals with AD pathology have developed clinical AD if they had lived long enough?
A couple of additional questions are being addressed in this population. One is whether cognitively normal subjects with elevated amyloid, as measured by PiB, display any subtle cognitive changes compared to their amyloid-negative counterparts. Thus far, the data have been mixed. A couple of studies did not detect significant cognitive differences between healthy controls with and without increased levels of PiB binding 44, 48. However, Pike and colleagues found that PiB uptake correlated with episodic memory performance 43. Given the relatively small numbers of subjects in these studies and the potential for individual variability in cognitive performance, it is perhaps not surprising that these results have been somewhat inconsistent. Interestingly, in a recent study PiB PET scans were performed in a group of healthy elderly individuals who had been followed with psychometric testing for up to 10 years prior to the study 49. This group was then divided into subjects who displayed some degree of cognitive decline over that period versus those who did not. Overall, the decliners were much more likely to display elevated PiB uptake, which suggests that the term “asymptomatic” may be relative. Along these lines, a recent report found evidence of cortical thinning in cognitively normal elderly with elevated PiB binding supporting the notion that despite being classified as normal, these individuals are already beginning to display neurodegenerative brain changes 50.
Another issue that may be addressed in control populations is the natural history of amyloid plaque deposition over time, complementing what is known about such changes in symptomatic patients (MCI and AD). As noted above, patients with MCI tend to have a similar degree of PiB uptake as AD patients, suggesting that the accumulation of amyloid plaques occurs prior to the clearly symptomatic stages of disease. Indeed, data from PiB imaging in healthy controls have supported this contention. While some controls do have levels of PiB uptake similar to that seen in AD patients, these subjects tend to be the exception rather than the rule. Unlike the somewhat dichotomous distribution of MCI, the degree of PiB binding is more continuous in cognitively normal controls, spanning the clearly amyloid-negative range to the higher levels seen in MCI and AD 48 (see Figure 2 and Table).
These findings have led some to suggest that amyloid slowly accumulates over an asymptomatic stage although the rapidity of this increase remains to be determined (Aizenstein et al., 2008; Jack et al., 2009). While there may be some subtle evidence of cognitive decline and neurodegeneration (e.g. neuronal or synaptic loss as measured by volumetric MRI), these individuals appear normal. However, once a threshold of amyloid deposition has been reached, more obvious clinical symptoms appear (i.e. MCI). At that point, while further amyloid accumulation appears to be slow 17, if at all 19, cognition continues to decline concordant with other markers of neurodegenerative (e.g. hippocampal volume) or metabolic change (FDG PET). Indeed, these latter measures appear to more closely track the cognitive manifestations of the disease 16, 17. It should, again, not be surprising that once patients are symptomatic that PiB binding would only weakly correlate with cognition, as pathologically studies have frequently reported higher correlations of cognition with neurofibrillary tangles and synaptic loss than with amyloid plaques 21, 22. Whatever the case, this relatively asymptomatic stage of amyloid accumulation may prove ideal for the application of disease modifying therapies prior to the potentially irreversible development of significant neuronal and synaptic loss.
VI. Conclusion
A great deal of information has been acquired about the course and clinical manifestations of Aβ pathology from amyloid imaging. As we appear to be about to enter the era of more etiologically specific interventions, rather than the current symptomatic ones, techniques that allow for visualization of underlying pathology will be invaluable. Given that such treatments will likely be associated with some degree of toxicity, determination of patients with the appropriate underlying pathological substrate will be important. From this perspective, PiB PET has displayed great potential in the differential diagnosis of dementia and MCI. Further, amyloid imaging in healthy controls may offer the possibility of ‘pre-symptomatic’ diagnosis and the institution of early preventative measures. This technique will also likely play an important role in the development of these potential disease-specific treatments by assuring that study populations in such clinical trials have underlying AD pathology and as a biomarker for measures of efficacy.
Nonetheless, there is still much to be learned from and about this imaging modality. It is a certainty that additional ligands and, perhaps, imaging platforms (e.g. MRI) will be used more frequently in the future, and it is likely that some form of amyloid imaging will play a role in clinical care. In order for more widespread clinical use, tracers will need to be labeled with radionuclides with a longer half-life than carbon-11 (20 minutes), which is currently used in PiB studies and requires an on-site cyclotron for production. Several promising agents with fluorine-18 (110 minute half-life) are in development (e.g. AV-45 and 3′-F-PiB), but will need to be validated in post-mortem tissue and, perhaps, relative to the performance of carbon-11 PiB. Whatever the case, amyloid imaging will continue to play an essential role in our understanding of this devastating disease.
Contributor Information
David A. Wolk, Department of Neurology, Penn Memory Center, University of Pennsylvania, 3615 Ralston House, Philadelphia, PA 19104, (p) 215 662-7810, (f) 215 662-7812, david.wolk@uphs.upenn.edu
William E. Klunk, Department of Psychiatry, Alzheimer's Disease Research Center, University of Pittsburgh, 200 Lothrop Street, Pittsburgh, PA 15213, (p) 412 692-2700, klunkwe@upmc.edu
References
- 1.Small GW, Kepe V, Ercoli LM, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–63. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 2.Verhoeff NP, Wilson AA, Takeshita S, et al. In-vivo imaging of Alzheimer disease beta-amyloid with [11C]SB-13 PET. Am J Geriatr Psychiatry. 2004;12:584–95. doi: 10.1176/appi.ajgp.12.6.584. [DOI] [PubMed] [Google Scholar]
- 3.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 4.Mueller SG, Weiner MW, Thal LJ, et al. The Alzheimer's disease neuroimaging initiative. Neuroimaging Clin N Am. 2005;15:869–77. xi–xii. doi: 10.1016/j.nic.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klunk WE, Lopresti BJ, Ikonomovic MD, et al. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer's disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klunk WE, Wang Y, Huang GF, et al. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23:2086–92. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 8.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]; • Important paper describing the relationship between PiB binding and the CSF biomarker Aβ1-42. Elevated PiB uptake in controls appeared to explain abnormally low Aβ1-42 in healthy controls.
- 9.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131:1630–45. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First report of comparison between in vivo PiB uptake and post-mortem measures of amyloid deposition in a patient with Alzheimer's disease. This work found a high regional correlation between pre- and post-mortem measures, supporting the efficacy of in vivo PiB for quantification of amyloid pathology.
- 10.Bacskai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431–4. doi: 10.1001/archneur.64.3.431. [DOI] [PubMed] [Google Scholar]
- 11.Leinonen V, Alafuzoff I, Aalto S, et al. Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh Compound B. Arch Neurol. 2008;65:1304–9. doi: 10.1001/archneur.65.10.noc80013. [DOI] [PubMed] [Google Scholar]
- 12.Klunk WE. Biopsy support for the validity of Pittsburgh compound B positron emission tomography with a twist. Arch Neurol. 2008;65:1281–3. doi: 10.1001/archneur.65.10.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 14.Kemppainen NM, Aalto S, Wilson IA, et al. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology. 2007;68:1603–6. doi: 10.1212/01.wnl.0000260969.94695.56. [DOI] [PubMed] [Google Scholar]
- 15.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 16.Edison P, Archer HA, Hinz R, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–8. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- 17.Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009 doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides description of longitudinal changes in PiB levels across different populations (controls, MCI, and AD). Demonstrates the complementary role of structural measures for disease progression and PiB for disease classification.
- 18.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–46. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 19.Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129:2856–66. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 20.Klunk WE, Mathis CA, Price JC, Lopresti BJ, DeKosky ST. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain. 2006;129:2805–7. doi: 10.1093/brain/awl281. [DOI] [PubMed] [Google Scholar]
- 21.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 22.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 23.Stern Y, Gurland B, Tatemichi TK, Tang MX, Wilder D, Mayeux R. Influence of education and occupation on the incidence of Alzheimer's disease. JAMA. 1994;271:1004–10. [PubMed] [Google Scholar]
- 24.Roe CM, Xiong C, Miller JP, Morris JC. Education and Alzheimer disease without dementia: support for the cognitive reserve hypothesis. Neurology. 2007;68:223–8. doi: 10.1212/01.wnl.0000251303.50459.8a. [DOI] [PubMed] [Google Scholar]
- 25.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Education modifies the association of amyloid but not tangles with cognitive function. Neurology. 2005;65:953–5. doi: 10.1212/01.wnl.0000176286.17192.69. [DOI] [PubMed] [Google Scholar]
- 26.Kemppainen NM, Aalto S, Karrasch M, et al. Cognitive reserve hypothesis: Pittsburgh Compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer's disease. Ann Neurol. 2008;63:112–8. doi: 10.1002/ana.21212. [DOI] [PubMed] [Google Scholar]
- 27.Nelissen N, Vandenbulcke M, Fannes K, et al. Abeta amyloid deposition in the language system and how the brain responds. Brain. 2007;130:2055–69. doi: 10.1093/brain/awm133. [DOI] [PubMed] [Google Scholar]
- 28.Johansson A, Savitcheva I, Forsberg A, et al. [(11)C]-PIB imaging in patients with Parkinson's disease: preliminary results. Parkinsonism Relat Disord. 2008;14:345–7. doi: 10.1016/j.parkreldis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Maetzler W, Reimold M, Liepelt I, et al. [11C]PIB binding in Parkinson's disease dementia. Neuroimage. 2008;39:1027–33. doi: 10.1016/j.neuroimage.2007.09.072. [DOI] [PubMed] [Google Scholar]
- 30.McKeith I, Mintzer J, Aarsland D, et al. Dementia with Lewy bodies. Lancet Neurol. 2004;3:19–28. doi: 10.1016/s1474-4422(03)00619-7. [DOI] [PubMed] [Google Scholar]
- 31.Rabinovici GD, Furst AJ, O'Neil JP, et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–12. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- 32.Rabinovici GD, Jagust WJ, Furst AJ, et al. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64:388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Excellent paper reporting on PiB binding in primary progressive aphasia. This work demonstrates the potential utility of amyloid imaging in atypical dementia patients in which a significant proportion represent abnormal presentations of AD. Additionally, they note dissociations between regional FDG and PiB measures with a greater correlation of the former with phenotype, suggesting these modalities provide complementary information.
- 33.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol. 2008;63:709–19. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen RC. Mild cognitive impairment as a diagnostic entity. Journal of Internal Medicine. 2004;256:183–94. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 35.Visser PJ, Kester A, Jolles J, Verhey F. Ten-year risk of dementia in subjects with mild cognitive impairment. Neurology. 2006;67:1201–7. doi: 10.1212/01.wnl.0000238517.59286.c5. [DOI] [PubMed] [Google Scholar]
- 36.Jicha GA, Parisi JE, Dickson DW, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63:674–81. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- 37.Larrieu S, Letenneur L, Orgogozo JM, et al. Incidence and outcome of mild cognitive impairment in a population-based prospective cohort. Neurology. 2002;59:1594–9. doi: 10.1212/01.wnl.0000034176.07159.f8. [DOI] [PubMed] [Google Scholar]
- 38.Fleisher AS, Sowell BB, Taylor C, Gamst AC, Petersen RC, Thal LJ. Clinical predictors of progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology. 2007;68:1588–95. doi: 10.1212/01.wnl.0000258542.58725.4c. [DOI] [PubMed] [Google Scholar]
- 39.Jack CR, Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999;52:1397–403. doi: 10.1212/wnl.52.7.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–34. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 41.Drzezga A, Grimmer T, Riemenschneider M, et al. Prediction of individual clinical outcome in MCI by means of genetic assessment and (18)F-FDG PET. J Nucl Med. 2005;46:1625–32. [PubMed] [Google Scholar]
- 42.Forsberg A, Engler H, Almkvist O, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29:1456–65. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]; • First study to report on longitudinal outcome of patients with MCI with respect to PiB PET imaging. As expected, they found that converters had higher levels of PiB retention than non-converters.
- 43.Pike KE, Savage G, Villemagne VL, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130:2837–44. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]; • Represents one of the first reports to carefully examine the relationship between PiB uptake and cognition in healthy controls. While this study did find a relationship with episodic memory, subsequent studies have not and this issue remains an active area of research.
- 44.Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–80. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolk DA, Price JC, Saxton JA, et al. Amyloid imaging in Mild Cognitive Impairment subtypes. Annals of Neurology. 2009 doi: 10.1002/ana.21598. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koivunen J, Pirttila T, Kemppainen N, et al. PET amyloid ligand [11C]PIB uptake and cerebrospinal fluid beta-amyloid in mild cognitive impairment. Dement Geriatr Cogn Disord. 2008;26:378–83. doi: 10.1159/000163927. [DOI] [PubMed] [Google Scholar]
- 47.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 48.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–17. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• In addition to reporting on the cognitive manifestation of elevated PiB uptake in healthy controls, this manuscript establishes a methodology for determination of cut-offs for dichotomous determination of amyloid status.
- 49.Villemagne VL, Pike KE, Darby D, et al. Abeta deposits in older non-demented individuals with cognitive decline are indicative of preclinical Alzheimer's disease. Neuropsychologia. 2008;46:1688–97. doi: 10.1016/j.neuropsychologia.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009;19:497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]