

Abstract
Aims
Studies of arrhythmogenesis during ischemia have focused primarily on reentrant mechanisms manifested on the epicardial surface. The goal of this study was to use a physiologically-accurate model of acute regional ischemia phase 1A to determine the contribution of ischaemia-induced transmural electrophysiological heterogeneities to arrhythmogenesis following left anterior descending artery occlusion.
Methods and results
A slice through a geometrical model of the rabbit ventricles was extracted and a model of regional ischaemia developed. The model included a central ischaemic zone incorporating transmural gradients of IK(ATP) activation and [K+]o, surrounded by ischaemic border zones (BZs), with the degree of ischaemic effects varied to represent progression of ischaemia 2–10 min post-occlusion. Premature stimulation was applied over a range of coupling intervals to induce re-entry. The presence of ischaemic BZs and a transmural gradient in IK(ATP) activation provided the substrate for re-entrant arrhythmias. Increased dispersion of refractoriness and conduction velocity in the BZs with time post-occlusion led to a progressive increase in arrhythmogenesis. In the absence of a transmural gradient of IK(ATP) activation, re-entry was rarely sustained.
Conclusion
Knowledge of the mechanism by which specific electrophysiological heterogeneities underlie arrhythmogenesis during acute ischaemia could be useful in developing preventative treatments for patients at risk of coronary vascular disease.
Keywords: Ischaemia, Arrhythmogenesis, Transmural heterogeneites, Re-entry, Simulation
Introduction
Sudden cardiac death is the leading cause of adult mortality in the industrialized world, and its prevalence is increasing.1 It typically results from cardiac arrhythmias that arise following occlusion of a coronary vessel. The first 12 min following coronary occlusion are termed acute myocardial ischaemia phase 1A. Experimental studies using animal models indicate that lethal arrhythmias occur most frequently 4–7 min post-occlusion2–4 and that the arrhythmias are of re-entrant nature3,5,6 (see also Kléber7 for a review). A body of research8–12 has demonstrated that the most prominent electrophysiological changes in the heart during ischaemia phase 1A are as follows: extracellular potassium concentration ([K+]o) rises, metabolic inhibition leads to activation of the ATP-sensitive potassium current (IK(ATP)), and intracellular pH drops. Typically, only part of the heart is affected by the lack of blood due to coronary occlusion (regional ischaemia), and therefore ischaemic electrophysiological changes are spatially heterogeneous. Regional heterogeneities in [K+]o, IK(ATP), and pH result in increased dispersion of conduction velocity,3,5 of action potential duration (APD)6,13 and of effective refractory period (ERP).6,14–17 Experimental and theoretical studies have shown that the heterogeneous electrophysiological properties secondary to acute myocardial ischaemia provide the substrate for the initiation and sustenance of re-entry.6,13,15–18
However, studies of arrhythmogenesis in ischaemia have focused on re-entrant mechanisms manifested predominantly on the epicardium,6,13,14,17,19 and little is known about the contribution of transmural heterogeneities to arrhythmogenesis secondary to acute regional ischaemia. This is mostly due to limitations in the spatial resolution of plunge electrode recordings, limitations in depth penetration of optical mapping techniques, and the rapidity and complexity of electrophysiological changes that follow coronary occlusion.
Computer simulations offer the ability to examine the transmural electrophysiological activity at high resolution, and therefore have the potential to reveal the mechanisms underlying arrhythmogenesis under this pathological condition. We have previously developed models of acute global ischaemia in the heart20,21 and applied these models to investigate the effect of global ischaemia on defibrillation threshold. In the present study, we further develop our model of ischaemia to include regional heterogeneities associated with occlusion of a coronary artery. The goal of this study is to use a physiologically accurate computational model of acute regional ischaemia phase 1A to determine the contribution of ischaemia-induced transmural electrophysiological heterogeneities to arrhythmogenesis in the ventricles during the first 10 min following left anterior descending (LAD) coronary artery occlusion.
Methods
Tissue representation
We used a two-dimensional model with realistic representation of the electrophysiological heterogeneities secondary to acute coronary artery occlusion. The model geometry was an anterior–posterior cross-section through the equator of our model of the rabbit ventricles described previously,22,23 allowing for an effective examination of the contribution of transmural heterogeneities in ischaemia 1A to arrhythmia induction and maintenance. Electrical activity in the slice was modelled using the monodomain approach with isotropic conductivity of 0.1 mS/mm. Isotropic conductivities were used because inclusion of fibre directions would have been unrealistic if taken on a two-dimensional cross-section through the three-dimensional fibre field in the ventricles. Membrane kinetics were represented by a version of the Luo–Rudy dynamic model24,25 modified to account for alterations in membrane behaviour due to ischaemia as described below.
Representation of transmural electrophysiological heterogeneities secondary to acute regional ischaemia
In the two-dimensional cross-sectional geometry of the model, the ischaemic region was defined as the transmural area affected by the lack of blood flow following LAD occlusion (Figure 1), as described in experimental studies using rabbit hearts.26,27 The ischaemic substrate during the first 10 min post-occlusion was represented by changes in membrane dynamics due to hyperkalaemia, acidosis, and hypoxia, as done previously.12,17,20,21,28–30 Specifically, [K+]o was increased, whereas the maximum conductances of Na+ and L-type Ca2+ channels were decreased (via scaling factors SNa and SCaL) to represent inhibition by acidosis. In addition, activation of the IK(ATP) current caused by hypoxia was represented by increasing the fraction of K(ATP) channels opened (fATP).12
Figure 1.

(A) Distribution of ischaemic injury in the model, outlining the location of the ischaemic region (grey–white) and normal region (black). Electrode locations for pacing (square) and premature stimulation (circle) are marked. (B) Schematic profiles for the transmural distributions of [K+]o (top), fATP (middle), and SNa and SCaL (bottom). The horizontal axis indicates the transmural depth within the ventricular wall. The landmarks on this axis are, from left to right: epicardium (EPI, bottom left), epicardial normal zone (NZ), epicardial border zone (BZ), central ischaemic zone (CIZ), endocardial border zone (BZ), endocardial normal zone (NZ), and endocardium (ENDO, bottom right). The widths of the ‘BZ’ landmarks are based on the transmural distribution of [K+]o. Border zone widths for fATP and SCaL and SNa are shown as a percentage of [K+]o BZ width. The level of ischaemic injury is plotted on the vertical axes, from normal through the maximum level of injury for a given time post-occlusion.
Implementation of transmural heterogeneities in [K+]o, IK(ATP), and INa and ICa(L) conductivities in the model of regional ischaemia, as shown in Figure 1, was based on experimental data. Specifically, the central ischaemic zone (CIZ) was surrounded by lateral, endocardial, and epicardial border zones (BZs), which provided a transition in ischaemic parameters between the CIZ and the normal zone, as measured experimentally.26,31–37 In the absence of adequate data on the profile of ischaemic changes through the BZ, the transitions were assumed to be linear.17 In our model, the width of the lateral BZ for [K+]o was 5.5 mm, whereas epi- and endocardial [K+]o BZs were 1 mm wide, consistent with experimental data.31,32,34,36,38 In accordance with previous experimental and theoretical studies,17,31 BZs for the acidosis-induced inhibition of INa and ICa(L) were assumed to be half as wide as those of [K+]o (Figure 1A, bottom, BZs). The BZs of fATP were taken to be narrowest, at only 10% of the width of the [K+]o BZs, in agreement with experimental findings.17,39,40
Furthermore, experimental recordings show that during acute ischaemia, increase in [K+]o is faster in the subendocardium than in the subepicardium,31,40–43 and this results in transmural gradients in [K+]o within the central ischaemic region. Therefore, in our model, [K+]o was assumed to be highest in the subendocardium (Kmax) and decreased gradually with the distance to the subendocardium in the CIZ, reaching 60% of Kmax in the subepicardium (Ksubepi) of the CIZ.43 Similarly, fATP was increased linearly within the ischaemic region from the subendocardium to the subepicardium to represent the increased sensitivity of IK(ATP) in epicardial myocytes to changes in ATP and ADP concentrations.15,44 Subendocardial fATP was assumed to be 60% of subepicardial fATP (Figure 1A, middle, CIZ).15,45,46
Progression of time post-occlusion was simulated by linearly increasing the magnitude of the changes in [K+]o, INa, ICa(L), and fATP. Vulnerability to re-entry of the ischaemic substrate was evaluated in the time period 0–10 min following LAD occlusion at 2 min intervals. Table 1 presents the values for [K+]o,max, min SNa, min SCaL, and fATP,max for each level of ischaemia severity.
Table 1.
Central ischaemic zone parameters used in the model of ischaemia phase 1A
| Time P-O (min) | [K+]o,max (mM) | fATP,max (% activated) | min SNa, SCaL (% normal) |
|---|---|---|---|
| 0 | 5.0 | 0.11 | 100.0 |
| 2 | 7.4 | 0.28 | 95.0 |
| 4 | 9.8 | 0.38 | 90.0 |
| 6 | 12.2 | 0.45 | 85.0 |
| 8 | 14.6 | 0.54 | 80.0 |
| 10 | 17.0 | 0.63 | 75.0 |
For each time post-occlusion (time P-O), [K+]o,max (maximum extra-cellular potassium concentration), fATP,max (maximum fraction of activated K(ATP) channels), and the variables SCaL and SNa (scaling factors of INa and ICa(L) conductances) were incrementally varied toward their values at 10 min post-occlusion.
Analysing vulnerability to re-entry
Figure 1A illustrates the electrode locations used for stimulation of the model. To ensure a steady state in the ionic model, the tissue was paced with a train of nine square-wave transmembrane stimuli of duration 1 ms and strength 1 A/m2 via the pacing electrode in the left ventricular (LV) wall, at a basic cycle length of 300 ms. As most premature excitations (the trigger for arrhythmia induction) in ischaemia phase 1A originate in the subendocardial BZ5,47 (see Carmeliet8 for a review), in order to evaluate the arrhythmogenic propensity of the substrate, we chose for the site of premature stimulation a location in that region. Following a variable coupling interval, a square-wave premature stimulus of the same strength as the pacing stimulus was applied to the right ventricular (RV) subendocardium near the septum to initiate reentry.
A vulnerability grid, i.e. a two-dimensional grid that encompasses episodes of premature stimulation for all timings post-occlusion (0–10 min in 2 min steps) and all coupling intervals (range 155–205 ms in 5 ms steps), was constructed for the model of regional ischaemia. Each column referred to a time post-occlusion and each row corresponded to a coupling interval. Episodes of re-entrant activity within the vulnerability grid were classified as sustained and non-sustained according to the following criteria. Re-entries in which the re-entrant pathway was the same for three consecutive beats following the application of the premature stimulus were considered ‘sustained’. For re-entries in which the re-entrant pathway changed from beat to beat, computer simulations were continued either until the re-entry self-terminated, and was therefore considered ‘non-sustained’, or until the re-entrant wavefront entered into a repetitive pattern and the re-entry was therefore considered ‘sustained’.
It has been postulated that an epicardial BZ (which may or may not be present)26,33,41,48,49 and a gradient of IK(ATP) in the CIZ6,13,15,44,45,50 play key roles in arrhythmogenesis and maintenance of arrhythmia following coronary occlusion, although the cause–effect relationships remain unclear. In order to investigate the mechanisms by which these two phenomena augment the likelihood of re-entry during acute regional ischaemia, we constructed additional vulnerability grids with homogeneous ischaemic activation of IK(ATP), and without representation of the epicardial BZ. Overall, for the three vulnerability grids, more than 600 premature stimulation episodes were simulated, though for the sake of brevity only episodes over the range of coupling intervals for which re-entry was initiated are presented in Results.
Data analysis
To analyse the arrhythmogenic propensity of the regional ischaemia-induced electrophysiological substrate, local activation times, APD, and ERP were evaluated throughout the tissue. Local activation time was defined as the time at which the upstroke velocity of the action potential was maximal, relative to the last paced beat or to the application of the premature stimulus. Because of the wide range of action potential amplitudes and resting membrane potentials in the regionally ischaemic model, APD was computed as the time from local activation to the point of inflection (negative second derivative of transmembrane potential to positive second derivative) of the action potential repolarization phase.51 Spatial variation of the electrophysiological properties within the model precluded the determination of ERP by attempting to elicit a propagating wave. Instead, we determined the ERP by pacing the entire model simultaneously eight times at a cycle length of 300 ms, and then examining the local state of the h and j gates in INa. The time, relative to local activation time, at which the local product of the h and j gates exceeded 0.012 during recovery (this is the largest value of the product for which an action potential could not be elicited) was considered to be the ERP. Conduction velocities, where needed, were calculated by dividing the difference in isochrone times at a particular location by the distance between the isochrones.
Numerical aspects
Quality triangular meshing of the ventricular cross-section was done using the Triangle package,52 with an average edge length of 163 μm. The monodomain finite-element formulation was solved as described previously53 using a fixed time step of 1 μs.
Results
Characterization of the electrophysiological substrate during pacing
Figure 2 illustrates the effect of progression of regional ischaemia on activation (left column) and repolarization (middle column) maps, as well as on APD (right column) maps, for the last paced beat before the application of a premature stimulus. Each row corresponds to a time post-occlusion, indicated on the left of Figure 2. The activation maps in Figure 2 show that as ischaemia progresses, conduction in the ischaemic region briefly becomes faster (Figure 2; 2 and 4 vs. 0 min post-occlusion, compare green isochrones in the septum at t = 40–50 ms, red circles), due to supernormal conduction in the ischaemic tissue, as described by Shaw and Rudy28 and Rodríguez and Ferrero.21 With further progression of ischaemia, propagation in the ischaemic tissue becomes slower by 6 min post-occlusion (Figure 2, 6 vs. 0 min post-occlusion, leftward shift of green isochrones in the septum at t = 40–50 ms, red circle), and this results in changes in the activation sequence, with delayed activation (10 ms longer local activation times relative to those in normoxia) in the RV septal endocardial BZ and the CIZ. Slow conduction in ischaemic tissue is due to the lower availability of Na+ current, resulting from hyperkalemia-induced resting potential elevation, and to the decrease in the maximum conductance of the Na+ channel caused by acidosis.21,28 The latest activation time in the CIZ is extended from 40 ms in normoxia to 80 ms after 10 min post-occlusion.
Figure 2.

Activation and repolarization isochronal maps as well as action potential duration maps from 0 through 10 min post-occlusion in the regionally ischaemic model. Isochrones are 10 ms/colour. Locations of site at which action potential duration and effective refractory period were recorded for Figure 3 are marked in the 0 min action potential duration map. Red circles indicate the locations at which a shift in the isochrones occurs due to changes in conduction velocity at 2–6 min post-occlusion.
Changes in repolarization time become apparent soon after coronary occlusion (Figure 2, 2 and 4 min post-occlusion panels, middle column). These changes stem from alterations in activation times as explained above, and from APD shortening, as illustrated in the right panels of Figure 2. In normoxia (Figure 2, 0 min), the repolarization sequence follows the activation sequence, as APD is the same throughout the preparation. In regional ischaemia, however, APD becomes heterogeneously shorter in the ischaemic region, due mainly to the heterogeneous activation of IK(ATP).54 Shortening of APD in the ischaemic region results in a decrease of up to 90 ms in local repolarization times relative to those in normoxia. The shortest repolarization times always occur in the ischaemic portion of the LV mid-myocardium, where ischaemic tissue has the earliest local activation times. Shortest and longest repolarization times are 140 and 220 ms in normoxia and become 100 and 220 ms following 10 min post-occlusion.
Although APD in the ischaemic region becomes shorter with progression of time post-occlusion, elevated [K+]o leads to post-repolarization refractoriness. Figure 3 illustrates the time course of APD (solid lines) and of ERP, (broken lines) at three locations, one in the LV normal zone, another in the RV lateral BZ near the point of premature stimulation, and a third at the centre of the CIZ, all marked with arrows in the top right panel of Figure 2. In normoxia, APD and ERP are 106 and 117 ms, respectively, and therefore very similar, with ERP only 11 ms longer than APD. However, as acute ischaemia progresses, APD decreases monotonically from 2 to 10 min post-occlusion in both the CIZ and the BZ (from 96 to 10 ms and from 100 to 80 ms, respectively), whereas ERP decreases monotonically from 109 to 89 ms in the BZ but exhibits a non-monotonic behaviour in the CIZ: it decreases during the first 2–6 min post-occlusion from 104 to 92 ms, and then increases over the period 6–8 min post-occlusion, reaching 106 ms at 8 min post-occlusion, which indicates pronounced post-repolarization refractoriness. Progression of ischaemia to 10 min results in the prolongation of ERP beyond 300 ms in the CIZ.
Figure 3.

Action potential duration and effective refractory period at locations in the normal zone (squares), the lateral border zone (triangles), and the central ischaemic zone (diamonds, grey), as marked in Figure 2. Solid lines connect action potential duration data and dashed lines connect ERP data. The cross indicates conduction block beyond 8 min post-occlusion in the central ischaemic zone.
Vulnerability grids
Figure 4A–C shows the vulnerability grids (left) and total number of elicited re-entrant episodes (right) in the regionally ischaemic model, the regionally ischaemic model without a transmural gradient of fATP, and the regionally ischaemic model without an epicardial BZ, respectively. In all, for the chosen six times post-occlusion (0 min omitted from the figure), 63 re-entrant arrhythmias were initiated. Of these, 29 were sustained and 35 were non-sustained. In all three vulnerability grids, arrhythmias were initiated from 4 through 10 min post-occlusion. None were initiated at 0 or 2 min post-occlusion. However, significant differences in the number and type of re-entries were found between the three vulnerability grids.
Figure 4.

Left: vulnerability grids for the three cases considered ((A) regionally ischaemic model, (B) regionally ischaemic model without transmural gradient of IK(ATP) activation, (C) regionally ischaemic model without epicardial border zone). Rows correspond to coupling intervals in milliseconds. Columns correspond to time post-occlusion in minutes. Episodes (rectangles) are marked dark grey for sustained re-entry, light grey for non-sustained re-entry, or white for no re-entry. Right: total number of initiated re-entries by case. Each vertical bar is broken into sustained and non-sustained re-entry segments. Columns (A–C) correspond to the vulnerability grids denoted by the same letters.
In the regionally ischaemic model (Figure 4A), the vulnerable window (the range of coupling intervals over which re-entry, both sustained and non-sustained, is initiated for a given time post-occlusion) widens with the progression of ischaemia, from 10 ms at 4 min post-occlusion to 35 ms at 10 min post-occlusion, mainly due to a decrease in the shortest coupling interval at which re-entry is initiated. However, sustained re-entrant episodes are most common at 8 min post-occlusion. Although the vulnerable window becomes wider at 10 min post-occlusion, the ratio of sustained to non-sustained re-entrant episodes significantly decreases. Overall, 68% of the re-entrant episodes in this vulnerability grid are sustained (13 sustained vs. 6 non-sustained episodes).
Homogeneous IK(ATP) in the CIZ (Figure 4B) results in an overall increase in the number of episodes in which arrhythmia is initiated, and in an enlargement of the vulnerable windows for all stages of regional ischaemia. The vulnerable window increases from 25 ms at 4 min post-occlusion to 40 ms at 10 min post-occlusion. However, without a gradient of fATP in the CIZ, the majority of initiated re-entrant episodes are non-sustained. Only two sustained re-entries are initiated at coupling intervals of 160 and 155 ms, at 8 and 10 min post-occlusion, respectively. Thus, 7.7% of the re-entrant episodes in this vulnerability grid are sustained (2 sustained vs. 24 non-sustained episodes), much lower than when a transmural gradient of IK(ATP) is present in the CIZ.
In the absence of an epicardial BZ (Figure 4C), the vulnerable window width increases non-monotonically as ischaemia progresses from 4 to 10 min post-occlusion; it is 20 ms at 4 min post-occlusion, 25 ms at 6 min post-occlusion, 20 ms at 8 min post-occlusion, and 30 ms at 10 min post-occlusion. Although the number of re-entrant episodes (19) is the same regardless of whether an epicardial BZ is present, the absence of an epicardial BZ results in a shift of the range of the vulnerable window toward longer coupling intervals at 8–10 min post-occlusion. In the absence of an epicardial BZ, 74% of the re-entrant episodes in the vulnerability grid are classified as sustained (14 vs. 5 episodes), with only one additional re-entrant episode becoming sustained here as compared with the case with an epicardial BZ.
Overall, the most re-entrant episodes occur in the regionally ischaemic model which includes a transmural CIZ gradient of fATP, but does not include an epicardial BZ. The absence of an fATP gradient in the CIZ results in a significant decrease in the number of sustained re-entries, with almost all re-entries becoming non-sustained. The absence of an epicardial BZ leads to a shift in the range of coupling intervals over which re-entry is sustained, towards longer coupling intervals near the end of the 1A ischaemic period. In the next sections, we examine the mechanisms by which arrhythmias are generated by premature stimulation in the three models, representing different electrophysiological substrate scenarios in regional ischaemia phase 1A.
Analysis of mechanisms of arrhythmogenesis in the regionally ischaemic model
In this section, in order to investigate the mechanisms underlying the changes in vulnerable window duration and position within the vulnerability grid (Figure 4A) during the first 10 min of acute regional ischaemia, we examine the electrical activity following the application of premature stimuli at several timings post-occlusion in the regionally ischaemic model. Figure 5 shows the activation maps for the first three re-entrant cycles following premature stimulation at 4–10 min post-occlusion, at a coupling interval of 180 ms. All four cases are within the vulnerable window.
Figure 5.

Activation maps of the first three beats of re-entry at 4, 6, 8, and 10 min post-occlusion in the regionally ischaemic model following application of a premature stimulus at a coupling interval of 180 ms. White asterisks mark the location of the earliest activation for each beat. Black arrows outline the path of propagation. Flat line ends indicate termination of wavefront propagation. Red circle indicates the site of termination of propagation in the septal lateral border zone at 10 min post-occlusion. Dashed black line in Beat 1 at 4 min outlines the ischaemic region. Time increases from left to right, with each beat spanning ~120 ms. Time scales across the bottom indicate the time span of each beat. Isochrones are 10 ms apart in all maps. Black areas were not activated during the mapped beat.
At 4 min post-occlusion, propagation elicited by the premature stimulus is unidirectionally blocked by refractory tissue in the RV free wall, but proceeds through the LV and septal ischaemic tissue. Eventually, propagation re-enters through the RV free wall, once tissue there has recovered (Figure 5, 4 min, Beat 1). In the second re-entrant cycle, low availability of Na+ current following the first re-entrant beat results in slow propagation in the CIZ (Figure 5, 4 min, Beat 2, reduced isochrone spacing). Conduction block occurs in the LV lateral BZ 180 ms after application of the premature stimulus. Thus, the re-entrant wavefront propagates only through the septum, and then re-enters through both LV and RV free walls, resulting in a figure-of-eight re-entry. A similar pattern of re-entry is observed in the third re-entrant cycle (Figure 5, 4 min, Beat 3). Premature stimuli applied at a coupling interval of <180 ms result in bidirectional block of propagation because both the normal RV free wall and the ischaemic anterior region are refractory, and thus no arrhythmia is initiated. Conversely, premature stimuli applied at coupling intervals longer than 185 ms at 4 min post-occlusion result in propagation in all directions and again no re-entrant circuit is established.
As shown in Figure 5, the pattern of propagation in the preparation remains similar as ischaemia progresses from 4 to 6 and 8 min post-occlusion. The main difference is the amount of tissue in the ischaemic zone that experiences low excitability, alternans, and propagation block (black in activation maps for 6–8 min); this amount increases with time post-occlusion. However, the type of the re-entrant circuit remains the same, due to the presence of both endocardial and epicardial BZs, which sustain propagation through septal and LV tissue.
Increase in the width of the vulnerable window from 4 to 10 min post-occlusion is due to earlier recovery of tissue close to the site of premature stimulation (Figure 3), caused by shortening of APD and ERP in the ischaemic BZ (Figures 2 and 3). This results in wavefronts being elicited by premature stimuli at short coupling intervals; these wavefronts are able to traverse the epi- and endocardial BZs toward the LV and septum, but are blocked in the normal RV tissue. As a consequence, with the progression of acute ischaemia from 4 to 10 min post-occlusion, the vulnerable window expands towards shorter coupling intervals (Figure 4).
At 10 min post-occlusion, however, the majority of initiated re-entries are non-sustained. The activation pattern in the first beat following the premature stimulus is similar to that at 4, 6 and 8 min post-occlusion (Figure 5, 10 min, Beat 1). However, on the second re-entrant beat, conduction is blocked at the lateral BZ in the septum (red circle), and this leads to termination of the activity; arrhythmia is thus non-sustained. At 10 min post-occlusion, the lateral BZ tissue exhibits shorter APD than at 8 min post-occlusion (Figures 2 and 3). Consequently, at 10 min post-occlusion, tissue in the septal and LV lateral BZs repolarizes before the normal tissue just outside the ischaemic zone recovers excitability. Thus, propagation there is blocked and is unable to sustain re-entry. Therefore, short APD in the BZ results in increased likelihood of propagation block at 10 min post-occlusion, and decreased likelihood of establishment of sustained reentrant circuits.
Role of the transmural gradient in IK(ATP) activation in the initiation and sustenance of re-entry
As shown in Figure 4 (compare vulnerability grids in A vs. B and vertical bars), the absence of a gradient of fATP in the CIZ results in an overall increase in the number of re-entrant episodes in the vulnerability grid (from 19 to 26), but in a significant reduction in the number of sustained reentries (to only 2). Figure 6 illustrates the mechanisms underlying this change in vulnerability. The top row presents the data from the regionally ischaemic model, whereas the bottom row displays the data from the model lacking a transmural gradient of fATP, both at 6 min post-occlusion. Figure 6A shows the fATP, Figure 6B the APD maps during the last paced beat prior to premature stimulation, Figure 6C the activation maps of the first and second re-entrant beats following premature stimulation at a coupling interval of 175 ms, and Figure 6D the action potential traces at the black (subepicardial) and red (subendocardial) marker dots in Figure 6C.
Figure 6.

Top row: regionally ischaemic model with transmural gradient of IK(ATP) activation. Bottom row: regionally ischaemic model without transmural gradient of IK(ATP) activation. Data are from 6 min post-occlusion. The five columns are, from left to right: fraction of activated IK(ATP) channels (fATP); action potential duration in the last paced beat prior to application of the premature stimulus; activation map of the first beat following the application of a premature stimulus at a coupling interval of 175 ms; activation map of the second beat; and action potential traces from the locations marked by black and red circles in the fourth column. All isochrones are of 10 ms spacing. In the activation maps, white asterisks mark the first isochrone in the second cycle of re-entry. White ‘T’s mark locations of wavefront propagation block, whereas white arrows indicate that the wavefront continued to propagate. Black areas were not activated during the mapped period.
Both in the presence and in the absence of a transmural gradient of fATP, the premature stimulus elicits a propagating wave that proceeds through the septum and LV free wall, and eventually re-enters through the RV free wall, as shown in the left column of Figure 6C. In the second re-entrant cycle (right column of Figure 6C), both in the presence and in the absence of a transmural gradient of fATP, the re-entrant wavefront proceeds through the RV endocardial and LV epicardial BZs. The wavefront that traverses the epicardial BZ is eventually blocked, in both cases, at the LV BZ, when it encounters the normal tissue that is still refractory following the first re-entrant beat (right column of Figure 6C). Propagation then proceeds only through the LV endocardial BZ, where it is affected by the degree of IK(ATP) activation, which is different in the two cases. The consequences of this effect, which eventually determine the outcome of premature stimulation, are analysed in Figure 6D and discussed next.
In the presence of a transmural gradient of fATP, propagation through the endocardial BZ is slightly slower than through the epicardial BZ. This is due to the fact that APD during the first re-entrant beat is longer in the endocardium than in the epicardium, resulting in a shorter diastolic interval, and thus in lower availability of Na+ current in the endocardium than in the epicardium. As a consequence, the wavefront propagating through the RV endocardium reaches the lateral BZ in the septum after the normal tissue in its vicinity has recovered. Propagation proceeds towards the RV and LV, and sustained figure-of-eight re-entry is established.
In contrast, in the absence of a transmural gradient of fATP, IK(ATP) and thus APD are similar in the epicardial and endocardial BZs (Figure 6B), although slight differences exist due to greater [K+]o in the subendocardium. Similar APD in the endocardial and epicardial BZs results also in similar diastolic intervals preceding the second cycle of activation (Figure 6D), and therefore in a similar availability of Na+ current. Consequently, in the absence of a transmural gradient of fATP, conduction velocity in the endocardial BZ is similar to that in the epicardial BZ (Figure 6C, right column) and faster than that in the endocardial BZ in the presence of a transmural gradient of fATP. Therefore, in the absence of a transmural gradient of fATP, wavefront propagation through the endocardial BZ reaches the normal tissue proximal to the lateral BZ in the septum 10 ms earlier than in the presence of such gradient, allowing less time for the normal tissue close to the BZ to recover, and resulting in propagation block 190 ms after the premature stimulation, and thus in non-sustained arrhythmia. Therefore, the presence of a transmural gradient of fATP in the CIZ results in an increased dispersion of refractoriness and conduction velocity in the ischaemic region, and thereby increases the likelihood of sustained re-entry.
Role of the epicardial border zone in vulnerability to re-entry
In this section, we examine the role of the epicardial BZ in modifying vulnerability to re-entry in regional ischaemia by analyzing the activity before and after the application of a premature stimulus in the presence and absence of an epicardial BZ. As shown in Figure 4C, the absence of an epicardial BZ results in a shift in the range of coupling intervals, over which sustained re-entry is initiated, toward long coupling intervals at 8 and 10 min post-occlusion. The mechanisms underlying this effect are illustrated in Figure 7, which presents the activation maps of the last paced beat (Figure 7A) before premature stimulation, and the first and second beats of re-entrant activity (Figure 7B) following the premature stimulus, at 10 min post-occlusion, in the presence (top) and absence (bottom) of an epicardial BZ. In Figure 7B, the coupling interval at which the premature stimulus is applied is 190 ms.
Figure 7.
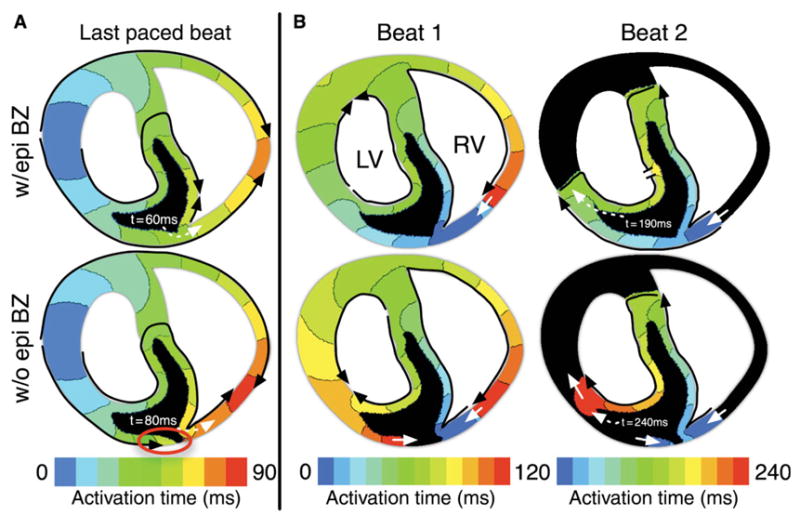
(A) Activation maps for the last paced beat 10 min post-occlusion with (w/epi BZ) and without (w/o epi BZ) an epicardial border zone. Black arrows outline the conduction pathway. Activation time isochrones are in the range from 0 to 90 ms following the application of a pacing stimulus, 10 ms/colour. Black areas were not activated during the mapping period. Red circle indicates the site of conduction block. Dashed white arrows indicate the time at which the site of premature stimulation was activated by the paced wavefront. (B) Activation maps from the first and second reentrant beats (Beats 1 and 2) with and without an epicardial border zone. Activation isochrones range from 0 to 240 ms (120/beat) and are spaced at 10 ms/colour. Black areas were not activated during the mapped period. Solid white arrows indicate continued propagation from one beat to the next. Dashed white arrows indicate the time at which the wavefront reached the left ventricular lateral border zone in the second re-entrant beat. Flat line ends rather than arrowheads (Beat 2, left ventricular endocardium) indicate termination of wavefront propagation. Coupling interval is 190 ms in all maps.
At 10 min post-occlusion, the majority of the CIZ is inexcitable (black in activation maps). In the presence of an epicardial BZ, both epicardial and endocardial BZs provide a pathway for propagation through the ischaemic region (Figure 7A, w/epi BZ, black arrows). In the absence of an epicardial BZ, however, epicardial tissue experiences prolonged refractoriness and low excitability caused by ischaemia-induced decrease in the availability of Na+ current. Thus, in the absence of an epicardial BZ, propagation elicited by the pacing stimulus proceeds through the endocardial BZ, but is blocked in the CIZ, and eventually fails in the subepicardium, resulting in a change in the propagation pattern compared with when the epicardial BZ is present (Figure 7A, compare top vs. bottom panels). As a result of this change in conduction pathway, the activation time at the site of premature stimulation increases from 60 ms when the epicardial BZ is present to 80 ms when it is absent. Long activation times at the site of premature stimulation also result in long repolarization times. Therefore, premature stimuli applied at short coupling intervals in the absence of an epicardial BZ result in propagation block because tissue at the site of premature stimulation is still refractory. This explains the shift in the vulnerable window towards long CIs in this case.
Our results also show that a greater number of arrhythmias are sustained at 10 min post-occlusion in the absence than in the presence of an epicardial BZ. Figure 7B illustrates the mechanisms underlying this phenomenon. In the presence of an epicardial BZ, a premature stimulus applied at a coupling interval of 190 ms elicits propagation through the epicardial BZ as well as the RV endocardial BZ. Propagation continues through the LV free wall and the septum, and eventually re-enters through the RV free wall (Figure 7B, w/epi BZ, Beat 1). In the second beat (Figure 7B, w/epi BZ, Beat 2), however, propagation proceeds through the BZs but is blocked in the normal regions of the LV free wall and septum, regions that are still refractory following the first re-entrant beat. Thus the arrhythmia is not sustained.
In contrast, in the absence of an epicardial BZ, propagation elicited by the premature stimulus initially proceeds only through the RV endocardial border zone, as the epicardial tissue is still refractory. Eventually, the wavefront re-enters through the RV free wall and retrogradely through the epicardium in the central ischaemic region (Figure 7B, w/o epi BZ, Beat 1). Whereas the latter wavefront eventually dies out due to low excitability in the CIZ (Figure 7B, w/o epi BZ, Beat 2), the wavefront that re-enters through the RV free wall propagates around the CIZ and through the RV and LV endocardial BZs in the second re-entrant circuit. The pathway of propagation in the absence of the epicardial BZ is therefore longer than when the zone is present, increasing the period of time over which the tissue in the LV free wall is able to recover before the re-entrant wavefront reaches it. This increase in recovery time allows re-entry to continue over a longer range of coupling intervals, resulting in a greater number of sustained re-entries at 10 min post-occlusion.
Discussion
In this study, a two-dimensional model of regional ischaemia in the rabbit ventricles was developed to investigate the contribution of ischaemia-induced transmural electro-physiological heterogeneities to arrhythmogenesis in the first 10 min following LAD occlusion. Our results show that the number of arrhythmias induced by premature stimulation increases from 4 to 10 min post-occlusion. Increased vulnerability to arrhythmia induction in regional ischaemia is caused by increased dispersion of refractoriness and conduction velocity in the ischaemic BZs. The absence of a transmural gradient of IK(ATP) activation in the central ischaemic region drastically reduces the number of sustained re-entries induced. In contrast, the absence of an epicardial BZ has little effect on the number of re-entries initiated; however, it results in a shift of the vulnerable window towards longer coupling intervals at the 8–10 min post-occlusion period. The specific mechanisms underlying these effects are analysed in detail in the following sections.
Changes in electrophysiological properties in the model of acute regional ischaemia
Previous studies have shown that changes in myocardial electrophysiological behaviour during ischaemia phase 1A mainly result from the impact of hyperkalaemia, acidosis, and hypoxia on ionic concentrations and currents.8,28 However, the time course and spatial distribution of these changes depend on a variety of factors, including the degree of coronary flow reduction, animal species, pacing rate, recording site, and characteristics of the experimental setup, such as the medium surrounding the preparation. This study focuses on arrhythmogenesis in the rabbit heart subsequent to complete occlusion of the LAD; thus, changes in [K+]o, maximum conductances of Na+ and L-type Ca2+ channels, and IK(ATP) activation incorporated in the model are specific to the rabbit heart during the first 10 min of no-flow ischaemia. Accordingly, changes in electrophysiological properties in our rabbit model are consistent with the experimental results as discussed below.
Following coronary occlusion, acute ischaemia-induced activation of IK(ATP) and increased [K+]o result in APD shortening in the ischaemic zone. Our simulations show that heterogeneities in IK(ATP) and [K+]o lead to heterogeneous APD shortening. Action potential duration shortening is greatest in the CIZ, where mean APD is 84.2±2.1% of its normal value (mean± standard deviation, n = 6336) at 2 min post-occlusion and 44.2±27.0% of its normal value at 10 min post-occlusion. Although there is normal tissue just beneath the epi- and endocardial surfaces, electrotonic effects resulting from coupling to the CIZ lead to ischaemia-induced changes in electrophysiological properties even in these regions. In our simulations, APD was shorter in the epicardial layer of the ischaemic zone (ranging from 91% of normal APD at 2 min post-occlusion to 73% of normal APD at 10 min post-occlusion) than in the endocardial layer (ranging from 94% of normal APD at 2 min post-occlusion to 76% of normal APD at 10 min post-occlusion), due to the greater degree of IK(ATP) activation in the epicardium than in the endocardium. However, at 10 min post-occlusion, high [K+]o in the endocardium results in slightly shorter APD on the endocardium (56% of normal) than in the epicardium (59% of normal). Shortening of APD in our regionally ischaemic model is in agreement with the experimental recordings. In several studies,9,55–58 APD was recorded from rabbit RV endocardium during global no-flow ischaemia at various pacing rates. Experimental data from the globally ischaemic endocardium paced at a cycle length of 400 ms, which are similar to ours (cycle length of 300 ms), indicate that APD was shortened to 59% of the normal over the first 10 min post-occlusion.9 Action potential duration shortening on the epicardial surface of the rabbit heart has been also observed by Behrens et al.13 (to 77% of normal after 10 min) and Cheng et al.59 (73% after 30 min), although these reductions in APD were less than those in our study, as these studies induced reduced-flow rather than no-flow ischaemia.
Activation delay (i.e. local activation time in ischaemia minus local activation time in normoxia) in our model resulted from a combination of reduced conduction velocity and transient conduction block in the ischaemic zone. As shown in Figure 2, in regional ischaemia in the presence of an epicardial BZ, activation delay at the epicardium was minimal, ranging from −1 ms during supernormal conduction (2–4 min post-occlusion) to 5 ms at 10 min post-occlusion. However, tissue in the CIZ experienced more extensive activation delay, reaching a maximum of 31 ms at 8 min post-occlusion–the time at which the preparation was most prone to sustained re-entries. In the absence of an epicardial BZ, the greatest delay was at the surface of the epicardium, rather than in the CIZ. At the epicardium, maximum activation delay ranged from −2 ms during supernormal conduction at 2 ms post-occlusion to 25 ms at 10 min post-occlusion. Both activation delay and the number of sustained re-entries peaked at 10 min post-occlusion in the absence of an epicardial BZ. Although the maximum activation delay is highly dependent on the geometrical characteristics of a given preparation, the coincidence of peak activation delay and peak arrhythmogenesis is common across a variety of preparations. Kaplinsky et al.3 reported a peak in both epicardial activation delay and arrhythmogenesis at 5 min post-occlusion in the dog. Likewise, Fleet et al.4 observed, in pigs, a peak in activation delay at 5 min post-occlusion and a mean time to onset of ventricular fibrillation of 4.6 min. The alignment of the time of peak activation delay and peak arrhythmogenesis both in the present study and in experimental studies supports the validity of our model of arrhythmogenesis in ischaemia and emphasizes the important role that ischaemia-induced transmural heterogeneities play in the pro-arrhythmic ischaemic substrate.
Elevated resting potential is a hallmark of myocardial ischaemia, caused primarily by extracellular accumulation of potassium. Although volumes of experimental data exist on the time course of potassium accumulation in ischaemia, few studies have actually quantified the degree to which resting potential is elevated, and to our knowledge, only one has provided quantification of ischaemia-induced elevation of resting potential in a preparation comparable to our model. In that study,60 the resting potential in the rabbit RV subendocardium (depth <500 μm) was found to be elevated by 17.9 mV after 11.5 min of no-flow ischaemia. Their preparation was paced at a cycle length of 350–400 ms, comparable to the cycle length of 300 ms used in our model. We observed resting potential elevation in the subendocardium (~100 μm depth), from −88.3 to −72.0 mV after 10 min of simulated no-flow ischaemia, a net elevation of 16.3 mV. Accounting for the additional 1.5 min elapsed in the experimental recordings (no data were provided at 10 min post-occlusion), this is an excellent match to our data. Other studies have quantified elevation of resting potential in rabbit papillary muscle61 and guinea pig62 and are not directly comparable to our findings, although they agree qualitatively with our results. In animal models, the rate of increase in [K+]o depends on pacing rate. It should be noted that [K+]o is not updated dynamically in our model and is therefore not affected by pacing rate. As such, the agreement discussed above is a confirmation that we have chosen adequate levels of [K+]o for the pacing rate used in our simulations.
Role of transmural electrophysiological heterogeneities in vulnerability to re-entry in regional ischaemia
Our simulation results show that the number of arrhythmias induced by premature stimulation increased progressively between 4 and 10 min following LAD occlusion. Progressive widening of the vulnerable window over the time course of acute regional ischaemia stemmed from increased dispersion of refractoriness in the lateral BZ. Specifically, early recovery of tissue in the ischaemic BZ allowed propagation elicited by early premature stimuli to enter the ischaemic region, whereas normal tissue adjacent to the BZ was still refractory. This resulted in unidirectional block and re-entry initiation at shorter coupling intervals in regional ischaemia than in normoxia. Thus, our simulation results show that an increased dispersion of refractoriness across the lateral BZ is a key factor in the initiation of re-entrant activity, and thus in the extension of the vulnerable window duration in acute regional ischaemia. Experimental studies have shown that spontaneous initiation of re-entry in ischaemia typically originates close to the ischaemic BZs.5,63 Our simulation results demonstrate that increased dispersion of refractoriness in the BZ increases the likelihood that premature stimuli applied there would result in re-entry. Thus, it may be the case that only premature activations occurring close to the BZ result in re-entry, whereas those occurring at other locations die out because they do not encounter a unidirectional block.
Our simulation results show that the number of sustained re-entrant episodes in our two-dimensional model progressively increased until 8 min post-occlusion, and then decreased at 10 min post-occlusion. A biphasic change in the incidence of sustained ventricular arrhythmias over the first 10 min following occlusion of the LAD has also been observed experimentally in pigs and dogs.3,4,64 To our knowledge, no data exist regarding the time course of sustained ventricular arrhythmias in rabbits during ischaemia phase 1A. Smith et al.64 found that the number of arrhythmic events clearly peaked at 6–8 min post-occlusion in pigs, as in our simulations. Kaplinsky et al.3 observed in dogs a rapid increase in the incidence of ventricular arrhythmias from 0 to 5 min post-occlusion, and a slower decline over the period from 5 to 10 min post-occlusion. Similarly, Fleet et al.4 found that in pigs the onset of ventricular fibrillation occurred in the range of 2.8–6.0 min post-occlusion, and on average 4.6 min post-occlusion, also consistent with our results.
Sustained re-entry in our model was typically anatomical, circling one or both of the ventricular cavities (Figure 5). The type of re-entry initially varied between beats, but settled to a figure-of-eight re-entry, with one rotor around each ventricular cavity and a common pathway in the septum (Figure 5). If block occurred in both the septal and LV lateral BZs, re-entry was non-sustained (Figure 5, 10 min). Data from high-resolution transmural electrical recordings in studies of acute regional ischaemia are not abundant; we are aware of one study in which such mapping was attempted. Pogwizd and Corr5 mapped re-entry in the cat at 4–5 min post-occlusion. They observed extensive activation delay in the ischaemic zone, as we documented at 6 min post-occlusion (Figure 5, 6 min, Beat 2), with re-entry driven by conduction around the LV.
Beginning at 6 min post-occlusion, portions of the CIZ were activated or inactivated on alternating beats in our simulations (Figure 5); it is a common finding in experimental studies and has been noted to precede the initiation of arrhythmias.14 Our results show, however, that alternans in the CIZ is not directly involved in the establishment of a reentrant circuit, but it is an indicator of increased severity of electrophysiological changes with the progression of ischaemia, and particularly of increased dispersion of refractoriness and conduction velocity, which is key in re-entry initiation and sustenance. Although the aforementioned patterns of re-entry are dependent on the site of stimulation in our pacing protocol as well as on the geometry of the preparation used, the mechanisms underlying re-entry would likely be the same with another protocol—the specific pathways might be different, but we expect block at the lateral BZs to be critical to both the initiation (unidirectional block) and termination (in the non-sustained episodes) of re-entry.
Our simulation results also show that a transmural gradient of IK(ATP) in the central ischaemic region is key to the sustenance of re-entry. Reduced dispersion of APD and conduction velocity in the absence of a transmural fATP gradient results in conduction block following the first re-entrant beat at both LV and septal lateral BZs, and the majority (92%) of re-entrant arrhythmias initiated are thus non-sustained (Figure 4B). Although a transmural gradient of fATP in the central ischaemic region exists physiologically in the heart, these results suggest that pharmacological interventions that reduce the differences in IK(ATP) throughout the ischaemic region may be useful in preventing sustained arrhythmias during acute ischaemia.
There is experimental evidence of the presence of an epicardial BZ following more than 30 min of ischaemia65 and in infarction,38 but no such BZ has been directly reported in early acute ischaemia. Its presence or absence may depend on the medium surrounding the heart ex vivo, or on collateral vascularization. Collateral myocardial vascularization is known to vary extensively between species27 and even between specimens from the same species. The canine model, in particular, is well perfused by collateral circulation (as much as 20% during ischaemia22), which may produce (or enhance the extent of) an epicardial BZ in acute regional ischaemia. The human heart does not normally exhibit such extensive collateral vascularization. Nonetheless, a large number of studies of acute regional ischaemia have favoured canine models.3,6,27,33,34,37,39,41,42,47,66–71 Furthermore, rabbits, another common animal model in electrophysiology studies, and the basis of our ventricular model, have been found in some cases to have increased collateral vascularization, particularly as they age (Igor Efimov, personal communication). Thus, young rabbits may have no epicardial BZ, whereas one might be present in older specimens.
In this study, the presence of an epicardial BZ had little effect on the number of sustained re-entries induced, but its absence resulted in a shift of the vulnerable window toward long coupling intervals at 8–10 min post-occlusion. This shift occurred because at that time, conduction through the ischaemic zone was restricted to the subendocardial BZs, with no activity on the epicardium. Thus, in experiments that only image the epicardium, re-entry in ischaemia in the absence of an epicardial BZ may be misinterpreted as spontaneous activation.
Limitations
The model used in this study is two-dimensional rather than three-dimensional. Although the model geometry does not represent that of the three-dimensional ventricles, the use of a two-dimensional geometry was necessary to decrease the complexity of the model, in order to enable dissection of the mechanisms of arrhythmogenesis. Additionally, this study assumed that the tissue was isotropic. Inclusion of tissue anisotropy might be expected to alter somewhat conduction pathway and conduction velocity in the model. However, as the mechanisms of arrhythmogenesis uncovered in this study stem primarily from heterogeneous changes in APD and ERP, we expect that the main findings of this study would remain valid in an anisotropic model. Finally, although many changes in membrane kinetics occur during acute ischaemia,8 increase in [K+]o and IK(ATP), and acidosis-induced reduction of Na+ and L-type Ca++ channel conductances, as incorporated in our model, have been shown to reproduce the behaviour of acutely ischaemic tissue.28 Despite these limitations, the present study provides new insight into the role of transmural electrophysiological heterogeneities in acute regional ischaemia.
Acknowledgments
The authors extend their thanks to Dr Felipe Aguel for implementation of the ionic model and to Dr Jonathan R. Shewchuk for his public distribution of the Triangle meshing program.
Funding
This study was supported in part by AHA pre-doctoral fellowship #0615280B to B. T., by an NSF award CBET-0601935 to N.T. and by the EPSRC-funded Integrative Biology E-Science pilot project (GR/S72 023/01) to B.R.
Footnotes
Conflict of interest: none declared.
References
- 1.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–51. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 2.Scherlag BJ, el-Sherif N, Hope R, Lazzara R. Characterization and localization of ventricular arrhythmias resulting from myocardial ischemia and infarction. Circ Res. 1974;35:372–83. doi: 10.1161/01.res.35.3.372. [DOI] [PubMed] [Google Scholar]
- 3.Kaplinsky E, Ogawa S, Balke CW, Dreifus LS. Two periods of early ventricular arrhythmia in the canine acute myocardial infarction model. Circulation. 1979;60:397–403. doi: 10.1161/01.cir.60.2.397. [DOI] [PubMed] [Google Scholar]
- 4.Fleet WF, Johnson TA, Cascio WE, Shen J, Engle CL, Martin DG, et al. Marked activation delay caused by ischemia initiated after regional K+ elevation in in situ pig hearts. Circulation. 1994;90:3009–17. doi: 10.1161/01.cir.90.6.3009. [DOI] [PubMed] [Google Scholar]
- 5.Pogwizd SM, Corr PB. Reentrant and nonreentrant mechanisms contribute to arrhythmogenesis during early myocardial ischemia: results using three-dimensional mapping. Circ Res. 1987;61:352–71. doi: 10.1161/01.res.61.3.352. [DOI] [PubMed] [Google Scholar]
- 6.Kuo CS, Munakata K, Reddy CP, Surawicz B. Characteristics and possible mechanism of ventricular arrhythmia dependent on the dispersion of action potential durations. Circulation. 1983;67:1356–67. doi: 10.1161/01.cir.67.6.1356. [DOI] [PubMed] [Google Scholar]
- 7.Kléber AG. Conduction of the impulse in the ischemic myocardium–implications for malignant ventricular arrhythmias. Experientia. 1987;43:1056–61. doi: 10.1007/BF01956039. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. 1999;79:917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- 9.Weiss JN, Venkatesh N, Lamp ST. ATP-sensitive K+ channels and cellular K+ loss in hypoxic and ischaemic mammalian ventricle. J Physiol. 1992;447:649–73. doi: 10.1113/jphysiol.1992.sp019022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kagiyama Y, Hill JL, Gettes LS. Interaction of acidosis and increased extracellular potassium on action potential characteristics and conduction in guinea pig ventricular muscle. Circ Res. 1982;51:614–23. doi: 10.1161/01.res.51.5.614. [DOI] [PubMed] [Google Scholar]
- 11.Sato R, Noma A, Kurachi Y, Irisawa H. Effects of intracellular acidification on membrane currents in ventricular cells of the guinea pig. Circ Res. 1985;57:553–61. doi: 10.1161/01.res.57.4.553. [DOI] [PubMed] [Google Scholar]
- 12.Ferrero JM, Sáiz J, Ferrero JM, Thakor NV. Simulation of action potentials from metabolically impaired cardiac myocytes. Role of ATP-sensitive K+ current. Circ Res. 1996;79:208–21. doi: 10.1161/01.res.79.2.208. [DOI] [PubMed] [Google Scholar]
- 13.Behrens S, Li C, Franz MR. Effects of myocardial ischemia on ventricular fibrillation inducibility and defibrillation efficacy. J Am Coll Cardiol. 1997;29:817–24. doi: 10.1016/s0735-1097(96)00571-2. [DOI] [PubMed] [Google Scholar]
- 14.Downar E, Janse MJ, Durrer D. The effect of acute coronary artery occlusion on subepicardial transmembrane potentials in the intact porcine heart. Circulation. 1977;56:217–24. doi: 10.1161/01.cir.56.2.217. [DOI] [PubMed] [Google Scholar]
- 15.Kimura S, Bassett AL, Kohya T, Kozlovskis PL, Myerburg RJ. Simultaneous recording of action potentials from endocardium and epicardium during ischemia in the isolated cat ventricle: relation of temporal electrophysiologic heterogeneities to arrhythmias. Circulation. 1986;74:401–9. doi: 10.1161/01.cir.74.2.401. [DOI] [PubMed] [Google Scholar]
- 16.Avitall B. Computer simulation of ventricular tachyarrhythmias during coronary artery ligation and release. J Electrocardiol. 1979;12:17–22. doi: 10.1016/s0022-0736(79)80040-0. [DOI] [PubMed] [Google Scholar]
- 17.Ferrero JM, Jr, Trénor B, Rodríguez B, Sáiz J. Electrical activity and reentry during acute regional myocardial ischemia: insights from simulations. Int J Bif Chaos. 2003;13:1–13. [Google Scholar]
- 18.Seigneuric RG, Chassé JL, Auger PM, Bardou AL. Role of the dispersion of refractoriness on cardiac reentries. Math Biosci. 1999;157:253–67. doi: 10.1016/s0025-5564(98)10085-8. [DOI] [PubMed] [Google Scholar]
- 19.Zaitsev AV, Guha PK, Sarmast F, Kolli A, Berenfeld O, Pertsov AM, et al. Wavebreak formation during ventricular fibrillation in the isolated, regionally ischemic pig heart. Circ Res. 2003;92:546–53. doi: 10.1161/01.RES.0000061917.23107.F7. [DOI] [PubMed] [Google Scholar]
- 20.Rodríguez B, Tice BM, Eason JC, Aguel F, Ferrero JM, Trayanova N. Effect of acute global ischemia on the upper limit of vulnerability: a simulation study. Am J Physiol Heart Circ Physiol. 2004;286:H2078–H2088. doi: 10.1152/ajpheart.01175.2003. [DOI] [PubMed] [Google Scholar]
- 21.Rodríguez B, Tice BM, Eason JC, Aguel F, Trayanova N. Cardiac vulnerability to electric shocks during phase 1A of acute global ischemia. Heart Rhythm. 2004;1:695–703. doi: 10.1016/j.hrthm.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Trayanova N, Eason J. Shock-induced arrhythmogenesis in the myocardium. Chaos. 2002;12:962–72. doi: 10.1063/1.1483955. [DOI] [PubMed] [Google Scholar]
- 23.Rodríguez B, Trayanova N. Upper limit of vulnerability in a defibrillation model of the rabbit ventricles. J Electrocardiol. 2003;36:51–6. doi: 10.1016/j.jelectrocard.2003.09.066. [DOI] [PubMed] [Google Scholar]
- 24.Luo C, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–96. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 25.Zeng J, Laurita K, Rosenbaum D, Rudy Y. Two components of the delayed rectifier K+ current in ventricular myocytes of the guinea pig type: theoretical formulation and their role in repolarization. Circ Res. 1995;77:140–52. doi: 10.1161/01.res.77.1.140. [DOI] [PubMed] [Google Scholar]
- 26.Di Rocco RJ, Bauer AA, Pirro JP, Kuczynski BL, Belnavis L, Chan YW, et al. Delineation of the border zone of ischemic rabbit myocardium by a technetium-labeled nitroimidazole. Nucl Med Biol. 1997;24:201–7. doi: 10.1016/0969-8051(95)02035-7. [DOI] [PubMed] [Google Scholar]
- 27.Harken AH, Simson MB, Haselgrove J, Wetstein L, Harden WR, Barlow CH. Early ischemia after complete coronary ligation in the rabbit, dog, pig, and monkey. Am J Physiol. 1981;241:H202–H210. doi: 10.1152/ajpheart.1981.241.2.H202. [DOI] [PubMed] [Google Scholar]
- 28.Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia. A mechanistic investigation of action potential conduction and conduction failure. Circ Res. 1997;80:124–38. doi: 10.1161/01.res.80.1.124. [DOI] [PubMed] [Google Scholar]
- 29.Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia: a theoretical study of altered cell excitability and action potential duration. Cardiovasc Res. 1997;35:256–72. doi: 10.1016/s0008-6363(97)00093-x. [DOI] [PubMed] [Google Scholar]
- 30.Ferrero JM, Trénor B, Saiz J, Rodriguez B. Simulation of figure-of-eight reentry during acute inhomogeneous myocardial ischemia: role of ATP-sensitive potassium current. Comput Cardiol. 2002:251–4. [Google Scholar]
- 31.Coronel R, Fiolet JW, Wilms-Schopman FJ, Schaapherder AF, Johnson TA, Gettes LS, et al. Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation. 1988;77:1125–38. doi: 10.1161/01.cir.77.5.1125. [DOI] [PubMed] [Google Scholar]
- 32.Coronel R, Wilms-Schopman FJ, Opthof T, van Capelle FJ, Janse MJ. Injury current and gradients of diastolic stimulation threshold, TQ potential, and extracellular potassium concentration during acute regional ischemia in the isolated perfused pig heart. Circ Res. 1991;68:1241–9. doi: 10.1161/01.res.68.5.1241. [DOI] [PubMed] [Google Scholar]
- 33.Factor SM, Okun EM, Kirk ES. The histological lateral border of acute canine myocardial infarction. A function of microcirculation. Circ Res. 1981;48:640–9. doi: 10.1161/01.res.48.5.640. [DOI] [PubMed] [Google Scholar]
- 34.Hearse JD, Opie LH, Katzeff IE, Lubbe WF, Van der Werff TJ, Peisach M, et al. Characterization of the ‘border zone’ in acute regional ischemia in the dog. Am J Cardiol. 1977;40:716–26. doi: 10.1016/0002-9149(77)90187-4. [DOI] [PubMed] [Google Scholar]
- 35.Jones D, Sohla A, Klein G. Internal cardiac defibrillation threshold: effects of acute ischemia. Pacing Clin Electrophysiol. 1986;9:322–31. doi: 10.1111/j.1540-8159.1986.tb04487.x. [DOI] [PubMed] [Google Scholar]
- 36.Kléber AG, Janse MJ, van Capelle FJ, Durrer D. Mechanism and time course of S-T and T-Q segment changes during acute regional myocardial ischemia in the pig heart determined by extracellular and intracellular recordings. Circ Res. 1978;42:603–13. doi: 10.1161/01.res.42.5.603. [DOI] [PubMed] [Google Scholar]
- 37.Kralios AC, Leonard MT, Anderson FL, Kralios FA. Prevention of extracellular K+ inhomogeneity across the ischemic border by coronary venous obstruction in the dog: salutary antiarrhythmic effects of enhanced myocardial hydration. J Mol Cell Cardiol. 1994;26:1349–56. doi: 10.1006/jmcc.1994.1153. [DOI] [PubMed] [Google Scholar]
- 38.Schaapherder AF, Schumacher CA, Coronel R, Fiolet JW. Transmural inhomogeneity of extracellular [K+] and pH and myocardial energy metabolism in the isolated rat heart during acute global ischemia; dependence on gaseous environment. Basic Res Cardiol. 1990;85:33–44. doi: 10.1007/BF01907012. [DOI] [PubMed] [Google Scholar]
- 39.Harken AH, Barlow CH, Harden WR, Chance B. Two and three dimensional display of myocardial ischemic ‘border zone’ in dogs. Am J Cardiol. 1978;42:954–9. doi: 10.1016/0002-9149(78)90681-1. [DOI] [PubMed] [Google Scholar]
- 40.Hearse DJ, Yellon DM. The ‘border zone’ in evolving myocardial infarction: controversy or confusion? Am J Cardiol. 1981;47:1321–34. doi: 10.1016/0002-9149(81)90266-6. [DOI] [PubMed] [Google Scholar]
- 41.Cohn PF, Kirk ES, Downey JM, Sonnenblick EH, Gorlin R. Autoradiographic evaluation of myocardial collateral circulation in the canine heart. Cardiovasc Res. 1973;7:181–5. doi: 10.1093/cvr/7.2.181. [DOI] [PubMed] [Google Scholar]
- 42.Hirzel HO, Sonnenblick EH, Kirk ES. Absence of a lateral border zone of intermediate creatine phosphokinase depletion surrounding a central infarct 24 hours after acute coronary occlusion in the dog. Circ Res. 1977;41:673–83. doi: 10.1161/01.res.41.5.673. [DOI] [PubMed] [Google Scholar]
- 43.Hill JL, Gettes LS. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation. 1980;61:768–78. doi: 10.1161/01.cir.61.4.768. [DOI] [PubMed] [Google Scholar]
- 44.Kimura S, Bassett AL, Furukawa T, Cuevas J, Myerburg RJ. Electrophysiological properties and responses to simulated ischemia in cat ventricular myocytes of endocardial and epicardial origin. Circ Res. 1990;66:469–77. doi: 10.1161/01.res.66.2.469. [DOI] [PubMed] [Google Scholar]
- 45.Furukawa T, Kimura S, Furukawa N, Bassett AL, Myerburg RJ. Role of cardiac ATP-regulated potassium channels in differential responses of endocardial and epicardial cells to ischemia. Circ Res. 1991;68:1693–702. doi: 10.1161/01.res.68.6.1693. [DOI] [PubMed] [Google Scholar]
- 46.Gilmour RF, Zipes DP. Different electrophysiological responses of canine endocardium and epicardium to combined hyperkalemia, hypoxia, and acidosis. Circ Res. 1980;46:814–25. doi: 10.1161/01.res.46.6.814. [DOI] [PubMed] [Google Scholar]
- 47.Kaplinsky E, Ogawa S, Michelson EL, Dreifus LS. Instantaneous and delayed ventricular arrhythmias after reperfusion of acutely ischemic myocardium: evidence for multiple mechanisms. Circulation. 1981;63:333–40. doi: 10.1161/01.cir.63.2.333. [DOI] [PubMed] [Google Scholar]
- 48.Li L, Nikolski V, Wallick DW, Efimov IR, Cheng Y. Mechanisms of enhanced shock-induced arrhythmogenesis in the rabbit heart with healed myocardial infarction. Am J Physiol Heart Circ Physiol. 2005;289:H1054–H1068. doi: 10.1152/ajpheart.01253.2004. [DOI] [PubMed] [Google Scholar]
- 49.Brooks H, Al-Sadir J, Schwartz J, Rich B, Harper P, Resnekov L. Biventricular dynamics during quantitated anteroseptal infarction in the porcine heart. Am J Cardiol. 1975;36:765–75. doi: 10.1016/0002-9149(75)90458-0. [DOI] [PubMed] [Google Scholar]
- 50.Kimura S, Bassett AL, Furukawa T, Furukawa N, Myerburg RJ. Differences in the effect of metabolic inhibition on action potentials and calcium currents in endocardial and epicardial cells. Circulation. 1991;84:768–77. doi: 10.1161/01.cir.84.2.768. [DOI] [PubMed] [Google Scholar]
- 51.Efimov IR, Huang DT, Rendt JM, Salama G. Optical mapping of repolarization and refractoriness from intact hearts. Circulation. 1994;90:1469–80. doi: 10.1161/01.cir.90.3.1469. [DOI] [PubMed] [Google Scholar]
- 52.Shewchuk JR. Delaunay refinement algorithms for triangular mesh generation. Comput Geom. 2002;22:21–74. [Google Scholar]
- 53.Vigmond EJ, Aguel F, Trayanova NA. Computational techniques for solving the bidomain equations in three dimensions. IEEE Trans Biomed Eng. 2002;49:1260–9. doi: 10.1109/TBME.2002.804597. [DOI] [PubMed] [Google Scholar]
- 54.Rodríguez B, Ferrero JM, Trénor B. Mechanistic investigation of extracellular K+ accumulation during acute myocardial ischemia: a simulation study. Am J Physiol Heart Circ Physiol. 2002;283:H490–500. doi: 10.1152/ajpheart.00625.2001. [DOI] [PubMed] [Google Scholar]
- 55.Weiss J, Shine KI. [K+]o accumulation and electrophysiological alterations during early myocardial ischemia. Am J Physiol. 1982;243:H318–327. doi: 10.1152/ajpheart.1982.243.2.H318. [DOI] [PubMed] [Google Scholar]
- 56.Weiss J, Shine KI. Effects of heart rate on extracellular [K+] accumulation during myocardial ischemia. Am J Physiol. 1986;250:H982–991. doi: 10.1152/ajpheart.1986.250.6.H982. [DOI] [PubMed] [Google Scholar]
- 57.Kanda A, Watanabe I, Williams ML, Engle CL, Li S, Koch GG, et al. Unanticipated lessening of the rise in extracellular potassium during ischemia by pinacidil. Circulation. 1997;95:1937–44. doi: 10.1161/01.cir.95.7.1937. [DOI] [PubMed] [Google Scholar]
- 58.Yan GX, Yamada KA, Kléber AG, McHowat J, Corr PB. Dissociation between cellular K+ loss, reduction in repolarization time, and tissue ATP levels during myocardial hypoxia and ischemia. Circ Res. 1993;72:560–70. doi: 10.1161/01.res.72.3.560. [DOI] [PubMed] [Google Scholar]
- 59.Cheng Y, Mowrey KA, Nikolski V, Tchou PJ, Efimov IR. Mechanisms of shock-induced arrhythmogenesis during acute global ischemia. Am J Physiol Heart Circ Physiol. 2002;282:H2141–2151. doi: 10.1152/ajpheart.00561.2001. [DOI] [PubMed] [Google Scholar]
- 60.Wilensky RL, Tranum-Jensen J, Coronel R, Wilde AA, Fiolet JW, Janse MJ. The subendocardial border zone during acute ischemia of the rabbit heart: an electrophysiologic, metabolic, and morphologic correlative study. Circulation. 1986;74:1137–46. doi: 10.1161/01.cir.74.5.1137. [DOI] [PubMed] [Google Scholar]
- 61.Kléber AG, Riegger CB, Janse MJ. Electrical uncoupling and increase of extracellular resistance after induction of ischemia in isolated, arterially perfused rabbit papillary muscle. Circ Res. 1987;61:271–9. doi: 10.1161/01.res.61.2.271. [DOI] [PubMed] [Google Scholar]
- 62.Kléber AG. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ Res. 1983;52:442–50. doi: 10.1161/01.res.52.4.442. [DOI] [PubMed] [Google Scholar]
- 63.Janse MJ, van Capelle FJ, Morsink H, Kléber AG, Wilms-Schopman F, Cardinal R, et al. Flow of ‘injury’ current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts. Evidence for two different arrhythmogenic mechanisms. Circ Res. 1980;47:151–65. doi: 10.1161/01.res.47.2.151. [DOI] [PubMed] [Google Scholar]
- 64.Smith WT, Fleet WF, Johnson TA, Engle CL, Cascio WE. The Ib phase of ventricular arrhythmias in ischemic in situ porcine heart is related to changes in cell-to-cell electrical coupling. Experimental Cardiology Group, University of North Carolina. Circulation. 1995;92:3051–60. doi: 10.1161/01.cir.92.10.3051. [DOI] [PubMed] [Google Scholar]
- 65.de Groot JR, Wilms-Schopman FJ, Opthof T, Remme CA, Coronel R. Late ventricular arrhythmias during acute regional ischemia in the isolated blood perfused pig heart. Role of electrical cellular coupling. Cardiovasc Res. 2001;50:362–72. doi: 10.1016/s0008-6363(01)00222-x. [DOI] [PubMed] [Google Scholar]
- 66.Babbs CF, Whistler SJ, Yim GK, Tacker WA, Geddes LA. Dependence of defibrillation threshold upon extracellular/intracellular K+ concentrations. J Electrocardiol. 1980;13:73–8. doi: 10.1016/s0022-0736(80)80013-6. [DOI] [PubMed] [Google Scholar]
- 67.Becker LC, Ferreira R, Thomas M. Mapping of left ventricular blood flow with radioactive microspheres in experimental coronary artery occlusion. Cardiovasc Res. 1973;7:391–400. doi: 10.1093/cvr/7.3.391. [DOI] [PubMed] [Google Scholar]
- 68.Cox JL, McLaughlin VW, Flowers NC, Horan LG. The ischemic zone surrounding acute myocardial infarction. Its morphology as detected by dehydrogenase staining. Am Heart J. 1968;76:650–9. doi: 10.1016/0002-8703(68)90164-6. [DOI] [PubMed] [Google Scholar]
- 69.Lie JT, Pairolero PC, Holley KE, McCall JT, Thompson HK, Titus JL. Time course and zonal variations of ischemia-induced myocardial cationic electrolyte derangements. Circulation. 1975;51:860–6. doi: 10.1161/01.cir.51.5.860. [DOI] [PubMed] [Google Scholar]
- 70.Marcus ML, Kerber RE, Ehrhardt J, Abboud FM. Three dimensional geometry of acutely ischemic myocardium. Circulation. 1975;52:254–63. doi: 10.1161/01.cir.52.2.254. [DOI] [PubMed] [Google Scholar]
- 71.Lazzara R, el-Sherif N, Scherlag BJ. Early and late effects of coronary artery occlusion on canine Purkinje fibers. Circ Res. 1974;35:391–9. doi: 10.1161/01.res.35.3.391. [DOI] [PubMed] [Google Scholar]