

Abstract
The development of effective anti-tumor immune responses is normally constrained by low avidity, tumor-specific cytotoxic T lymphocytes (CTLs) which are unable to eradicate the tumor. Strategies to rescue anti-tumor activity of low avidity melanoma-specific CTLs in vivo may improve immunotherapy efficacy. To boost the in vivo effectiveness of low avidity CTLs we immunized mice bearing lung melanoma metastases with artificial Antigen Presenting Cells (aAPC), made by covalently coupling pepMHC-Ig dimers and B7.1-Ig molecules to magnetic beads. aAPC treatment induced significant tumor reduction in a mouse telomerase antigen system and complete tumor eradication in a mouse TRP-2 antigen system, when low avidity CTLs specific for these antigens were adoptively transferred. In addition, in an in vivo treatment model of subcutaneous melanoma, aAPC injection also augmented the activity of adoptively transferred CTLs and significantly delayed tumor growth. In vivo tumor clearance due to aAPC administration correlated with in situ proliferation of the transferred CTL. In vitro studies showed that aAPC effectively stimulated cytokine release, enhanced CTL-mediated lysis and TCR down-regulation in low avidity CTLs. Therefore, in vivo aAPC administration represents a potentially novel approach to improve cancer immunotherapy.
Keywords: artificial Antigen Presenting Cells (aAPC), melanoma, adoptive transfer, CTL affinity, costimulation
INTRODUCTION
Cancer vaccines have not significantly improved the clinical outcome and patient survival (1, 2), even though it is possible to detect tumor specific CTL in peptide-vaccinated patients. More encouraging results have been obtained, for both solid and hematological malignancies, in settings of adoptive immunotherapy by the administration of autologous tumor-specific CD8+ T cells expanded and activated in vitro (3, 4). Still, clinical success of adoptive immunotherapeutic approaches have been restricted to about 50% of patients, who show mostly partial rather than complete clinical responses (5).
Several factors clearly influence the challenge of an efficient immunotherapy: thymic selection, tumor-released inhibitory cytokines and chemokines, the presence of regulatory T cells, altered macrophage differentiation, and defects in DCs (6–10). Consistent with this idea, we have previously shown that vaccination with mouse TRP-2, a melanoma-associated self antigen (11), had only minor protective/therapeutic effect on B16 tumor challenge, due to an abortive immune response (12, 13). In this setting, TRP-2–specific CD8+ T lymphocytes, which proliferated after in vitro stimulation, efficiently recognized peptide-pulsed target cells but recognized B16 melanoma cells poorly (12, 13). This is probably due to reduced antigen- class I MHC complex expression on their surface (14).
Different approaches have been proposed to overcome the lack of tumoricidal activity of low avidity CD8+ T cells, such as the use of synthetic modified peptides to generate high avidity T cells (15–17), delivery of appropriate local “danger signals” inside the tumor microenvironment (18, 19), and injection of viral vectors expressing the costimulatory molecule B7.1 (CD80) into melanoma lesions (20). While these approaches have partially augmented the anti-tumor immune responses, they are not strong enough to result in complete tumor eradication.
To overcome issues of low avidity CTLs in tumor immunotherapy, we designed aAPC (see Suppl. Fig. 1) for in vivo administration. The aAPC are based on our previous work, where signal 1 (MHC-Ig) and signal 2 (anti-CD28) were coupled to magnetic beads and used to generate antigen-specific CTLs in vitro (21–23). Here we show that aAPC can also be used in vivo to augment the activity of adoptively transferred low avidity melanoma specific CTLs. This has been demonstrated in both lung metastasis models and in a subcutaneous treatment model. In all models, aAPC administration significantly augmented the in vivo anti-tumor activity of adoptively transferred CTLs leading to inhibition of tumor growth, in the telomerase-antigen specific lung model and subcutaneous melanoma model, or complete tumor clearance, in the TRP-2 antigen-specific lung metastasis model. This novel approach represents the first demonstration of an “off the shelf”, bead-based aAPC for systemic delivery of both antigen-specific and co-stimulatory signals to tumor-specific CTLs. aAPC administration can thus potentially be used to overcome current problems related to low avidity anti-tumor CTLs, therefore increasing the efficiency of the adoptive immunotherapy of cancer.
MATERIALS AND METHODS
Mice
Eight week-old female C57BL/6 (H-2b; B6) mice were purchased from Charles River Laboratories (Calco, Como, Italy). Procedures involving animals were in conformity with institutional guidelines.
Cell lines and CTL clones
MBL-2 is a leukemia cell line (H-2b) and B16Lu8 (hereafter referred to as B16) is a lung metastases forming melanoma cell line (H-2b) kindly provided by Dr. James C. Yang (NIH). B16Lu8 cells stably transfected with mouse B7.1 (B16.F1-mB7-1.32 hereafter referred to as B16-B7.1 cells) were a kind gift of P. Della Bona (Istituto Scientifico San Raffale, Milan, Italy).
TRP-2 specific CTL clones 8 and 24 were obtained by limiting dilution as described (12, 13). The m-TERT immunogenic peptide m-TERT198; VGRNFTNL restricted for H2-Kb was previously described (24). B6 mice were immunized against the mouse telomerase antigen and CTL lines were restimulated weekly with irradiated syngeneic splenocytes pulsed with m-TERT198.
Dimer and aAPC preparation
Soluble MHC-Ig fusion protein was derived as previously described (15, 25) and can be purchased under the brand name DimerX from BD. The Kb-Ig molecules were actively loaded either with the TRP-2180–181, SIY (SIYRYYGL, Commonwealth Biotechnologies, VA, USA), or m-TERT198 peptide (JPT Peptide Technologies, Berlin, Germany) (15). Briefly, Kb-Ig molecules were denaturated using an alkaline solution (150 mM NaCl; 15 mM Na2CO3 pH=11.5) in the presence of 75-fold molar excess of the relevant peptide for 15 min at RT, then neutralized with an equal volume of 250 mM Tris-HCl (pH=6.8) and incubated for 48 hours at 4 °C in a rotator. PepKb-Ig complexes were then used to generate aAPC as described ((21) see Supplemental Figure 1 Legend for details).
Tumor challenge and treatment
On day 0, B6 mice were injected i.v. with 105 B16 or B16-B7.1 tumor cells. On day 3, 5×106 TRP-2-specific or m-TERT-specific CD8+ T cells were adoptively transferred by i.v. injection. All mice were then treated i.p. with 30,000 IU IL-2, twice on day 3, 4, and 5. aAPC-treated mice received i.v. injections with 107 aAPC on day 4, 5, and 6. On day 14 all mice were sacrificed and pulmonary metastases were counted blindly.
In vivo proliferation
Both tumor-free and tumor-bearing B6 CD45.1+ (Ly5.1+) mice were injected with 5×106 CFSE-labeled (5 μg/ml; Invitrogen) clone 8 CTLs and treated as described above. On day 7, lungs, spleen and draining lymph nodes were removed and analyzed for expansion of the adoptively transferred, TRP-2-specific CTLs using anti-mouse CD8+-TriColor and anti-mouse CD45.2+-PE (Ly5.2; eBioscience San Diego, CA) mAbs. To prepare a single cell suspension, lungs from 3 mice were mechanically separated and then digested with an enzyme mixture (300 U/ml DNAse, 0,1% hyaluronidase, and 1% collagenase, all from Simga). Cell suspensions were enriched for viable cells by Ficoll centrifugation (Ficoll-Paque™ PLUS, Amersham Biosciences, Sweden). The samples were analyzed using a FACSCalibur flowcytometer (BD) in combination with Cellquest and Modfit software.
Enzyme-Linked Immunosorbent Assay
TRP-2-specific CTLs (105 cells) were stimulated for 24 hours in triplicate wells with an equal amount of target cells or beads. TERT-specific CD8+ T cells (105 cells) were incubated either with an equal amount of target tumor cells, with m-TERT-aAPC or with SIY aAPC at different T cell to aAPC ratios. T cell stimulation reached a plateau at 1:30 ratio. Supernatants were harvested and tested for the IFN-γ released in a sandwich enzyme-linked immunosorbent assay (ELISA, Endogen, Boston, MA)
TCR down regulation analysis
TRP-2-specific CTL clones 8 and 24 (105 cells/well) were incubated in a 96 well/plate (FALCON BD, Franklin Lakes, NJ, USA) in the presence of different stimuli: 106 RMA-S cells pulsed with increasing concentration of TRP-2 peptide, different amounts of cognate TRP-2 aAPC, or increasing concentrations of plate bound anti-mouse CD3 mAb. The plates were centrifuged to facilitate conjugate formation and incubated at 37°C. After 5h, cells were harvested and the amount of TCR down-regulation was measured by flowcytometry analysis using anti-mouse TCRαβ-PE mAb (Immunokontact, AMS Biotechonology Ldt-UK).
Effect of aAPC on s.c. tumor growth in vivo
B16 cells (5×105) were injected subcutaneously into thighs of 6–8 week old C57BL/6 female mice. When tumor area was ~10 mm2, mice were sublethally irradiated with 5 Gy to induce lymphopenia before adoptive cells transfer. After 6 hours, 5×106 hgp10025–33-specific CTL were injected i.v. 4–6 hours later, mice were injected i.v. with non-cognate ASN-aAPC or cognate hgp100-aAPC (107/mouse/injection) and treated i.p. with 30,000 IU of recombinant IL-2 twice a day. Injections of beads and IL-2 were repeated for three consecutive days. Tumor growth was monitored at 2–3 day intervals, using digital calipers, until tumor size was approximately 100mm2 at which point animals were euthanized.
Statistical analysis
Wilcoxon-Mann-Whitney test was used to examine the null hypothesis of rank identity between two sets of data. All p values presented are two-sided.
RESULTS
Low avidity antigen-specific CTL clones have poor in vitro and in vivo anti-tumor effects
Low avidity effector T cells can recognize peptide-pulsed target cells but often fail to recognize endogenous antigens on tumor cells (12, 13). We therefore analyzed the in vitro and in vivo activity of a high and low avidity CTL clones (clone 24 and clone 8) which are both specific for the cognate antigen, TRP-2180–188 presented by H-2Kb. As expected clone 8 produced significantly less IFN-γ than clone 24 when stimulated with B16 tumor cells (Fig. 1A) However when B16 tumor cells, transfected with the costimulatory molecule B7.1 (B16-B7.1), were used as targets, clone 8 produced large amounts of IFN-γ (Fig. 1A).
Figure 1. In vitro and in vivo activity of high and low avidity TRP-2-specific CTL clones.

(A) Antigen-specific IFN-γ release by TRP-2-specific CTLs was quantified by ELISA 24 h after stimulation with targets and antigens as indicated including MBL-2 cell or TRP-2 peptide pulsed MBL-2. (B) Number of pulmonary metastases (average ± SE; 2 experiments, n=10) counted on day 14 after the challenge with B16 tumor cells. Maximum number of resolvable metastases was 300.
We also found marked differences in vivo between clone 24 and clone 8. When analyzed in the B16 lung metastasis model (see M&M), only the high avidity CTL, clone 24, efficiently treated lung metastases while no significant difference was found between clone 8 treated and untreated mice (Fig. 1B). Therefore, clone 8 represents a classic low avidity anti-tumor CTL which is inefficient at eradicating tumors.
aAPC reverse in vivo inefficacy of the low avidity anti-tumor CTLs
To study the effect of aAPC, we tested the ability of aAPC to augment the efficacy of adoptively transferred, low avidity CTL clone 8. For these studies we generated aAPC in which signal 1, peptide-loaded Kb-Ig complexes, and signal 2, B7.1-Ig complexes, are covalently coupled to beads, see Suppl. Fig. 1 for schematic and details of all aAPC used for these studies. In mice previously injected with B16 tumor, we examined whether in vivo aAPC-activated clone 8 could eradicate lung metastases. As schematically shown in Figure 2A, on day 3 after B16 tumor cell injection, B6 mice received 5×106 TRP-2-specific CTLs and were injected i.v. on day 4, 5, and 6 with 107 aAPC/mouse and all mice were sacrificed 14 days later. Administration of cognate TRP-2 peptide-loaded aAPC led to complete eradication of the tumor by clone 8 which was otherwise ineffective (Fig. 2B).
Figure 2. Cognate TRP-2 aAPC treatment enhances in vivo anti-tumor activity of low avidity CTL clones.

(A) Schematic representation of the treatment protocol for aAPC administration. The treatment groups were: mice challenged with B16 melanoma and transferred with clone 8 alone or in combination with non-cognate SIY aAPC, signal 1 aAPC, signal 2 aAPC, or cognate TRP-2 aAPC; B6 mice transferred with clone 24 alone, mice that received only the tumor cells (no clone) and mice treated only with the cognate TRP-2 aAPC (no CTLs). (B) Therapeutic effectiveness of low avidity CTL clones 8 is dramatically enhanced by the treatment with cognate TRP-2-aAPC. Data result from the sum of two separated experiments (n=20). (C) In vivo stimulation of the anti-βgal CTL by cognate βgal-aAPC does not results in anti-metastatic activity (n=20).
To analyze the relevance of signal 1 and signal 2 on the aAPC, we tested the effect of administration of the following aAPC; 1) signal 1 aAPC, 2) signal 2 aAPC, and 3) non-cognate SIY aAPC (see Suppl. Figures 1 and 2). No anti-tumor effect was seen in mice treated with the signal 1 aAPC, signal 2 aAPC, or non-cognate SIY aAPC demonstrating that both cognate signal 1 and signal 2 were required for effective in vivo activation of low avidity CTLs (Fig. 2B). Additionally, no anti-tumor effect was seen in mice that were immunized with cognate TRP-2 aAPC only. These results indicate that aAPC provide more than basic costimulation to increase the efficiency of tumor immunotherapy.
To analyze the requirements for antigen-specific activation, a β-galactosidase-specific (β-gal) CTL clone was adoptively transferred to mice (Fig. 2A) and mice were injected with either β-gal or SIY aAPC (Fig. 2C). No effect was observed on metastasis formation, demonstrating that non-specific CTLs exert no anti-tumor activity.
aAPC induce in vivo proliferation of adoptively transferred TRP-2 specific CTLs
To further analyze the effect of aAPC, we evaluated in vivo proliferation of the low avidity CTLs. Tumor-bearing or tumor-free CD45.1+ B6 mice were adoptively transferred with 5×106 CFSE-labeled, CD45.2+ clone 8 cells. After 3 days, mice were immunized with either cognate TRP-2 aAPC or non-cognate SIY aAPC. Seven days later all mice were sacrificed and organs were collected and analyzed for the presence and expansion of CD45.2+/CD8+ cells. Transferred CTLs were found only in the lung, which could be related not only to the route of administration but, also, to the specific homing of the CTLs to the tumor site.
Administration of cognate TRP-2 aAPC induced significantly more proliferation of the transferred clone 8, than the non-cognate SIY aAPC in tumor-bearing mice as determined by CTL expansion (Fig. 3A) and CFSE dilution (Fig 3B). Interestingly, low avidity, TRP-2-specific clone 8 underwent more cell divisions in the presence of cognate TRP-2 aAPC in tumor-bearing mice (Fig. 3B). The majority (~80%) underwent 5 or more cell divisions and only 2.2% of the transferred CTLs did not proliferate. In contrast, significantly less expansion was observed in mice treated with non-cognate SIY aAPC (35.4% of the transferred CTLs remained undivided and only ~20% underwent more than 3 cell divisions). These data demonstrate that aAPC can be used to efficiently expand adoptively transferred CTLs in tumor-bearing mice. The reduced CTL proliferation in mice treated with the non-cognate SIY aAPC (Fig. 3A, and B) further confirms the finding that the B7.1 alone is not sufficient to induce anti-tumor activity or proliferation of the low avidity CTL clone.
Figure 3. In vivo proliferation of TRP-2-specific CTL clones after antigen-specific aAPC injection.

Tumor-free and B16 tumor-bearing CD45.1+ B6 mice, treated as described in Fig. 2A, were sacrificed on day 7 and the in vivo CD45.2+/CFSE/CD8+ T cells expansion in the lung was evaluated by FACS. (A) Absolute numbers of specific cells are presented. (B) Using Modfit software each division cycle of the transferred T cells was tracked and the percentage of undivided T cells for each treatment evaluated. (C) On day 7 and 14 following i.v. tumor injection, aAPC were separated from the lungs and quantified by FACS as described. To separate the aAPC, we prepared single cell suspensions from lungs, lysed the cells and separated the aAPC using a magnet (DYNAL). The amount of beads detected in the lung of tumor bearing mice was set to 100% and the amount in tumor-free mice calculated accordingly. Analysis was performed on a pool of 3 animals for each group.
Interestingly, TRP-2-specific CTL expansion triggered by cognate TRP-2 aAPC in the lung was more vigorous in tumor bearing mice, while only a limited expansion was observed in tumor-free mice (Fig. 3B, left panels). This effect might be related to the biodistribution of aAPC. Alternatively, it is also possible that, in the absence of tumor, activated T cells did not localize to the tumor site but distributed throughout the animal and were too dilute to be detected. To investigate whether the differences in proliferation between tumor-free and tumor-bearing mice were dependent on differences in the in vivo distribution of the aAPC, we injected tumor-bearing and tumor-free mice with aAPC and analyzed tissue distribution of aAPC in various organs. On day 7, we found ~40% fewer aAPC in the lungs of tumor-free mice than in the lungs of tumor-bearing mice and the absolute number of aAPC was further reduced by day 14 (Fig. 3C). In contrast, no differences were observed in the number of aAPC in other organs. Therefore, the presence of the tumor affects the biodistribution and persistence of aAPC, which may explain the increased T cell proliferation in lungs of tumor-bearing mice.
TRP-2-specific aAPC enhance in vitro activity of low avidity CTLs
To further analyze the effect of aAPC, we performed in vitro assays including effector cytokine secretion and TCR down-regulation assays on the TRP2-specifc clones. Both low and high avidity clones secreted IFN-γ at comparable levels after cognate TRP-2 aAPC stimulation (Fig. 4A). The amount of IFN-γ detected for non-cognate SIY aAPC, signal 1 and signal 2 aAPC was comparable to non-stimulated CTL clones (Fig. 4A). Only cognate TRP-2 aAPC induced significant effector functions in low avidity CTLs, clone 8. Analogously, anti-βgal CTLs were only stimulated to release IFN-γ by either βgal96–103 peptide-pulsed targets or βgal-aAPC, whereas there was no recognition even of B16-B7.1 melanoma targets (Fig. 4B).
Figure 4. Cognate TRP-2 aAPC enhance in vitro activity of low avidity CTL clones.

Specific IFN-γ release (pg/ml) quantified by ELISA after 24h of incubation of either the two anti-TRP-2 clones (A) or the anti-βgal clone (B). (C) TCR down-regulation of the two TRP-2-specific CTLs was evaluated after 5 hours of incubation with: RMA-S cells pulsed with increasing concentration of TRP-2180–188 peptide (left panel); increasing amount of anti-mouse CD3 bound to the plate (central panel); cognate TRP-2 vs. signal 1 aAPC (right panel). (D) Cognate TRP-2 aAPC do not reprogram low avidity CTLs to recognize wild type melanoma cells. Specific IFN-γ release (pg/ml) was quantified by ELISA assay using the supernatant from clones 8 and 24, 24 hours after incubation with cognate TRP-2 aAPC, non-cognate SIY aAPC, B16 wild type and B7-1-transfected B16 cells (Day 0). An aliquot of the same cells was cultured for 4 days with TRP-2 aAPC at 1:1 ratio (Day 4). After 4 days, specific IFN-γ release was quantified again by ELISA.
T cell activation was also evaluated by TCR down-regulation after stimulation with either, peptide-pulsed cells, plate bound anti-CD3 mAb, or aAPC. TCR down-regulation was more sensitive in the high avidity CTLs, clone 24, than in the low avidity CTLs, clone 8, when stimulated with peptide-pulsed target cells (Fig. 4C). The limited TCR down-regulation for clone 8 CTLs was not due to an overall impairment of the signaling machinery since stimulation with plate bound anti-CD3 induced strong TCR down-regulation in both clone 8 and clone 24 CTLs (Fig. 4C). Cognate TRP-2 aAPC efficiently induced similar TCR down-regulation in both CTL clones while signal 1 aAPC did not induce any TCR down-regulation (Fig. 4C). In summary, cognate TRP-2 aAPC are effective stimulators of activation of low avidity CTLs.
We determined whether low avidity CTLs stimulated with cognate aAPC in vitro might regain the capacity to recognize B16 tumors not expressing B7.1. Clone 8 and 24 CTL were tested immediately (Day 0, Fig. 4D) or 4 days following in vitro stimulation with cognate aAPC at 1:1 ratio (Day 4, Fig. 4D). Although on day 4 we detected a slightly higher reactivity in terms of IFN-γ released in culture upon stimulation, the functional response of clones 8 and 24 was not affected, i.e. clone 24 recognized efficiently both B16 melanoma and the B7.1 transfected variants whereas clone 8 only responded to B16-B7.1 cell stimulation (Fig. 4D). As expected, there was also no change in the overall CTL functional avidity as measured by peptide titrations (data not shown).
aAPC injection increases in vivo anti-tumor responses of low avidity, telomerase-specific CTLs
We analyzed the efficacy of aAPC in another clinically relevant system, a polyclonal mouse CTL line specific for mouse telomerase (TERT-CTL)(24). TERT-CTLs were greater than 95% CD8+ but only 36% antigen-specific, as determined by tetramer staining (data not shown).
TERT-CTLs also did not efficiently recognize B16 melanoma cells in vitro, but they did recognize peptide-pulsed target cells and B16-B7.1 targets (Fig. 5A). Incubation of TERT-CTLs with cognate m-TERT aAPC but not control aAPC induced significant release of IFN-γ, similar to the low avidity TRP-2-specific clone.
Figure 5. aAPC enhance the in vitro and in vivo activity of m-TERT specific CTLs.

(A) IFN-γ released by TERT specific CD8+ T cells (TERT-CTLs) was quantified by ELISA 24 h after stimulation with different targets, as indicated. (B) Number of metastases (average ± SE) counted on day 14 in the lung of B6 mice (n=10) treated as described in Fig.2A. Statistically relevant comparisons between experimental groups are indicated in the figure. (C) Varying numbers of mTERT-specific CTLs were incubated with 2×103, 51Cr-labeled B16 tumors either alone (inverted closed triangles) or in the presence of 2×105 SIY-aAPC (open triangles)- or mTERT-aAPC (closed squares). MBL2 tumor cells pulsed with mTERT198–205 peptide (closed circles) were used as positive controls and pulsed with β-gal peptide (open circles) negative controls. The graph shows data as mean ± SD of triplicate wells of a representative experiment. Significant differences (ANOVA, p<0.05) between the groups B16 plus mTERT-aAPC and B16 plus SIY-aAPC were found at all effector/target ratios.
To evaluate the aAPC-based in vivo stimulation of the adoptively transferred TERT-CTLs, we administered TERT peptide-loaded aAPC (see Fig. 2A schematic). While adoptive transfer of TERT-CTLs alone induced some tumor reduction (Fig. 5B), injection with cognate TERT-aAPC led to a significant decrease in tumor burden (p=0.0001). As expected, non-cognate aAPC did not reduce the tumor burden nor did TERT-aAPC alone, without adoptively transferred CTLs (Fig. 5B). Thus treatment with cognate TERT-specific aAPC also led to significant reduction in tumor burden in vivo.
One potential reason for incomplete tumor clearance could be epitope loss due to immunoselection by the adoptively transferred CTL. To examine this possibility, we isolated tumor after treatment with mTERT-specific CTL and aAPC and analyzed the ability of the mTERT-specific CTLs to recognize the tumor isolated from the various mice. There was no difference in recognition, by TERT-specific CTLs, of the tumors isolated from animals injected with cognate TERT-specific aAPC or non-cognate aAPC (Supplementary Figure 3). Thus alternative explanations including dosing of aAPC, activity of polyclonal TERT antigen-specific CTLs and level of TERT antigen on tumor cells may account for residual tumors in the TERT-system.
Mechanistically, we tested if aAPC directly stimulate low avidity CTL activity. For these studies, we used the low avidity mTERT-specific CTL and clone 8. CTL were incubated with 51Cr labeled B16 tumor cells, either alone or in the presence of cognate aAPC or non-cognate aAPC. Significant increases in CTL-mediated killing were seen in cognate aAPC-treated CTLs as compared to non-cognate SIY-aAPC (Fig. 5C). Similar results were obtained for clone 8 while no effect was seen on the high avidity clone 24 response (Supplemental Figure 4). Thus aAPC stimulation leads to enhanced killing by low avidity CTL toward tumors that otherwise would not be efficiently recognized.
aAPC administration reduces tumor growth in a subcutaneous tumor treatment model
To evaluate the impact of aAPC in a treatment model of subcutaneous tumors, we injected B16 tumor cells s.c. to induce solid tumors. Once tumors reached a size of ~10 mm2, hgp10025–33-specific CTLs were injected and mice were treated with either control or cognate aAPC. Administration of cognate aAPC led to a significant reduction in tumor growth (p=0.038) as compared to the control groups that were treated with either non-cognate aAPC or IL-2 alone (Fig. 6A). Kaplan-Meier survival analysis (Fig. 6B) also revealed statistically significant differences between cognate aAPC treatment and non-cognate aAPC (P=0.0046) and between cognate aAPC and animals receiving just CTLs alone and IL2 (P<0.001). In contrast there was no significant influence of treatment with non-cognate aAPC compared to untreated animals. Thus aAPC treatment is also effective in reducing growth of an established subcutaneous tumor.
Figure 6. Therapeutic activity of aAPC against subcutaneous tumors.
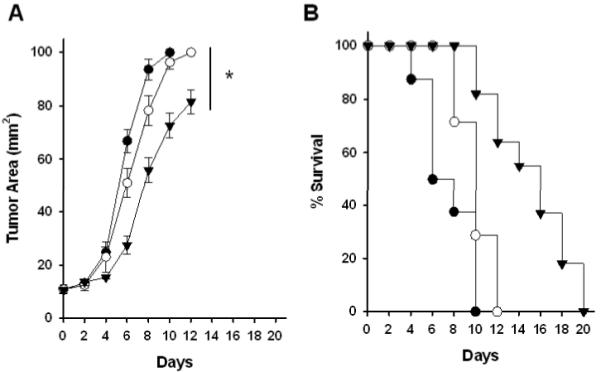
C57BL/6 mice were challenged subcutaneously with 5×105 B16 cells. When tumor area was ~10 mm2, mice were sublethally irradiated and 5×106 activated hgp10025–33-specific CTL were adoptively transferred/mouse. Mice were then injected i.v. with saline control (filled circles), Db-Ig-based aAPC loaded with noncognate ASN peptide (open circles), or cognate hgp10025–33-aAPC (inverted filled triangles) and analyzed as described. Tumors were measured blind using digital calipers and mice were euthanized when tumor area reached 100 mm2. Statistics: A, Mann-Whitney Test, P=0.038 between non-cognate ASN-aAPC and cognate hgp100-aAPC at day12; B, Kaplan-Meier analysis, cognate hgp100 aAPC vs non-cognate ASN-aAPC, P=0.0046; cognate hgp100-aAPC vs untreated, P<0.001; non-cognate ASN-aAPC vs untreated, P=0.104 (not significant).
Discussion
Many tumor-antigens are self-antigens. As a result it is not surprising that T cell anti-tumor immune responses are often of low avidity and fail to recognize endogenous antigens on tumor cells. Therefore, strategies are been developed to rescue the functional anti-tumor activity of low avidity CTL.
Using an adoptive transfer model, of low avidity TRP-2-specific CTLs in a B16 lung metastases melanoma model, we demonstrate that aAPC administration specifically induced in vivo CTL proliferation and CTL-mediated anti-tumor responses. Interestingly, CTL proliferation in the lung was more vigorous in lungs of tumor bearing mice which might be related to aAPC biodistribution, as the lungs of tumor-bearing mice retained more aAPC than tumor-free mice. Our results further show that only the cognate aAPC activate the melanoma-specific CTLs and lead to tumor clearance. While aAPC activation, of non-tumor specific β-gal-aAPC did not affect the metastatic tumor growth.
To further explore the potential of aAPC treatment, we studied aAPC mediated in vivo activation of low activity, m-TERT specific CTLs. aAPC administration also elicited a robust anti-tumor response confirming the value of the system in enhancing the therapeutic effectiveness of other low activity tumor-specific T cells. These results highlight the fact that the in vivo use of aAPC is efficient in enhancing an antigen-specific CTL response to another entirely different, yet clinically relevant antigen.
We also explored the generality of the effect of aAPC injection in a subcutaneous tumor treatment model. These studies demonstrated that aAPC treatment enhanced the activity of adoptively transferred CTLs and significantly delayed tumor growth of established subcutaneous tumors. In this system there are additional variables that may impact on the in vivo activity of aAPC. One to consider is the aAPC tissue distribution and the sites of CTL activation by the aAPC. The lungs are the first tissue bed where aAPC can lodge and activate CTLs while distribution in other tissues may be less efficient. Alternatively this finding may highlight the interest in adding targeting molecules that could help the aAPC home to either lymphoid organs or specific tissues
Approaches to targeting anti-tumor activity T cells are very broad. One previous approach has been to target antigen-specific CTLs using soluble MHC complexes, including soluble dimers and tetramers. Under some regimens soluble MHC appears to delete antigen-specific CTLs (26) while in others soluble dimers and/or tetramers alone activate CTL in vivo (27–29). In these previous reports, only high avidity CTLs were targeted, with no attention to anti-tumor activity of low avidity CTLs as we report here. Indeed, in our studies, aAPC made with only MHC-Ig dimers, signal 1 alone, were not effective. While there are many differences between the systems one possible reason for the requirement of both signal 1 and signal 2 could relate to the overall dose of antigen being delivered. To prepare aAPC we use only 1.5 μg per mouse (for 3×107 aAPC total dose) while much larger doses, up to 90 μg of tetramer per mouse were used previously (28). Another possibility is the role of endogenous antigen presenting cells, which may be different between the use of soluble MHC complexes and administration of aAPC beads. Overall either aAPC and/or soluble MHC complexes represent new approaches to antigen delivery that may each have valuable clinical applications in immunotherapy.
In recent trials, combination of myeloablation and adoptive immunotherapy with ex vivo expanded tumor-infiltrating lymphocytes (TILs) achieved objective clinical responses in about 50% of patients with metastatic melanomas (3, 5). Unfortunately, TILs can only be isolated and grown for a limited number of patients. To circumvent these limitations, high-avidity TCR, derived from TIL have been identified, cloned and transduced into PBMC of patients with cancer (30). However, there are still many problems that have so far limited a widespread clinical application. In this manuscript we show the feasibility of an alternative strategy aimed at rescuing the functional anti-tumor activity of low avidity T cells. This is the first report exploiting the use of an “off the shelf” aAPC injection as a novel approach for antigen-specific in vivo activation of tumor-specific CTL.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Joan Bieler for the editing and the critical review of the manuscript and Jonathon D. Benett, Carmela Mennuni, Luigi Aurisicchio, and Luigi Dolcetti for technical assistance. This work has been supported by grants from: Istituto Superiore Sanità - Alleanza contro il Cancro (project n. ACC8, V.B.) and Italy-US program (project n. 527/A/3A/1, V.B.); Italian Association for Cancer Research (AIRC; P.Z. and V.B.); the DOD Department of Defense grant PC 040972 (M.O.); the National Institution of Health grants RO1 CA108835, RO1 AI44129 and the DOD Department of Defense grant DAMD17-03-0370 (JPS).
Glossary
Abbreviation
- aAPC
artificial Antigen Presenting Cells
- pep-MHC-Ig
peptide-MHC-Ig
- TRP-2
Tyrosine Related Protein 2
- m-TERT
mouse Telomerase
REFERENCES
- 1.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mocellin S, Mandruzzato S, Bronte V, Lise M, Nitti D. Part I: Vaccines for solid tumours. Lancet Oncol. 2004;5:681–689. doi: 10.1016/S1470-2045(04)01610-9. [DOI] [PubMed] [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meidenbauer N, Marienhagen J, Laumer M, et al. Survival and tumor localization of adoptively transferred melan-a-specific T cells in melanoma patients. J Immunol. 2003;170:2161–2169. doi: 10.4049/jimmunol.170.4.2161. [DOI] [PubMed] [Google Scholar]
- 5.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 7.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature gr-1(+) myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 8.Lyman MA, Nugent CT, Marquardt KL, Biggs JA, Pamer EG, Sherman LA. The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J Immunol. 2005;174:2563–2572. doi: 10.4049/jimmunol.174.5.2563. [DOI] [PubMed] [Google Scholar]
- 9.Woo EY, Yeh H, Chu CS, et al. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168:4272–4276. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- 10.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bronte V, Apolloni E, Ronca R, et al. Genetic vaccination with “self” tyrosinase-related protein 2 causes melanoma eradication but not vitiligo. Cancer Res. 2000;60:253–258. [PMC free article] [PubMed] [Google Scholar]
- 13.De Palma R, Marigo I, Del Galdo F, et al. Therapeutic effectiveness of recombinant cancer vaccines is associated with a prevalent T-cell receptor alpha usage by melanoma-specific CD8+ T lymphocytes. Cancer Res. 2004;64:8068–8076. doi: 10.1158/0008-5472.CAN-04-0067. [DOI] [PubMed] [Google Scholar]
- 14.Seliger B, Ritz U, Abele R, et al. Immune escape of melanoma: first evidence of structural alterations in two distinct components of the MHC class I antigen processing pathway. Cancer Res. 2001;61:8647–8650. [PubMed] [Google Scholar]
- 15.Slansky JE, Rattis FM, Boyd LF, et al. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13:529–538. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 16.Romero P, Valmori D, Pittet MJ, et al. Antigenicity and immunogenicity of Melan-A/MART-1 derived peptides as targets for tumor reactive CTL in human melanoma. Immunol Rev. 2002;188:81–96. doi: 10.1034/j.1600-065x.2002.18808.x. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic tumor-associated peptide vaccine for the treatment of patients with metastatic melanoma. Nature Medicine. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 19.Yu P, Lee Y, Liu W, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. 2004;5:141–149. doi: 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- 20.Kaufman HL, Deraffele G, Mitcham J, et al. Targeting the local tumor microenvironment with vaccinia virus expressing B7.1 for the treatment of melanoma. J Clin Invest. 2005;115:1903–1912. doi: 10.1172/JCI24624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat Med. 2003;9:619–625. doi: 10.1038/nm869. [DOI] [PubMed] [Google Scholar]
- 22.Oelke M, Krueger C, Giuntoli RL, 2nd, Schneck JP. Artificial antigen-presenting cells: artificial solutions for real diseases. Trends Mol Med. 2005;11:412–420. doi: 10.1016/j.molmed.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Oelke M, Schneck JP. HLA-Ig-based artificial antigen-presenting cells: setting the terms of engagement. Clin Immunol. 2004;110:243–251. doi: 10.1016/j.clim.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 24.Mennuni C, Ugel S, Mori F, et al. Preventive vaccination with telomerase controls tumor growth in genetically engineered and carcinogen-induced mouse models of cancer. Cancer Res. 2008;68:9865–9874. doi: 10.1158/0008-5472.CAN-08-1603. [DOI] [PubMed] [Google Scholar]
- 25.Dal Porto J, Johansen TE, Catipovic B, et al. A soluble divalent class I major histocompatibility complex molecule inhibits alloreactive T cells at nanomolar concentrations. Proc Natl Acad Sci U S A. 1993;90:6671–6675. doi: 10.1073/pnas.90.14.6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Herrin SM, Slansky JE, Tang Q, et al. Antigen-specific blockade of T cells in vivo using dimeric MHC peptide. J Immunol. 2001;167:2555–2560. doi: 10.4049/jimmunol.167.5.2555. [DOI] [PubMed] [Google Scholar]
- 27.Cullen CM, Jameson SC, DeLay M, et al. A divalent major histocompatibility complex/IgG1 fusion protein induces antigen-specific T cell activation in vitro and in vivo. Cell Immunol. 1999;192:54–62. doi: 10.1006/cimm.1998.1434. [DOI] [PubMed] [Google Scholar]
- 28.Savage P, Millrain M, Dimakou S, Stebbing J, Dyson J. Expansion of CD8+ cytotoxic T cells in vitro and in vivo using MHC class I tetramers. Tumour Biol. 2007;28:70–76. doi: 10.1159/000099152. [DOI] [PubMed] [Google Scholar]
- 29.Carey B, DeLay M, Strasser JE, et al. A soluble divalent class I MHC/IgG1 fusion protein activates CD8+ T cells in vivo. Clin Immunol. 2005;116:65–76. doi: 10.1016/j.clim.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Johnson LA, Heemskerk B, Powell DJ, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.