

Abstract
SAR exploration of the 2,4-diamino-6,7-dimethoxyquinazoline template led to the discovery of 8 (UNC0224) as a potent and selective G9a inhibitor. A high resolution X-ray crystal structure of the G9a-8 complex, the first co-crystal structure of G9a with a small molecule inhibitor, was obtained. The co-crystal structure validated our binding hypothesis and will enable structure-based design of novel inhibitors. 8 is a useful tool for investigating the biology of G9a and its roles in chromatin remodeling.
Multicellular organisms have evolved elaborate mechanisms to enable differential and cell-type specific expression of genes. Epigenetics refers to these heritable changes in how the genome is accessed in different cell-types and during development and differentiation. This capability permits specialization of function between cells even though each cell contains the same genome. Over the last decade, the cellular machinery that creates these heritable changes has been the subject of intense scientific investigation as there is no area of biology or for that matter no area of human health, where epigenetics may not play a fundamental role.1
The template upon which the epigenome is written is chromatin – the complex of histone proteins, RNA and DNA that efficiently package the genome in an appropriately accessible state within each cell. The state of chromatin, and therefore access to the genetic code, is mainly regulated by covalent and reversible PTMs to histone proteins and DNA, and the recognition of these marks by other proteins and protein complexes. The PTMs of histones and DNA include: histone lysine methylation, arginine methylation, lysine acetylation, sumoylation, ADP-ribosylation, ubiquitination, glycosylation and phosphorylation, and DNA methylation.2 Given the wide-spread importance of chromatin regulation to cell biology, the enzymes that produce these modifications (the ‘writers’), the proteins that recognize them (the ‘readers’), and the enzymes that remove them (the ‘erasers’) are critical targets for manipulation in order to further understand the histone code3,4 and its role in human disease. Indeed, small molecule histone de-acetylase inhibitors5 and DNA methyltransferase inhibitors6 have already proven useful in the treatment of cancer.
Histone lysine methylation refers to covalent methylation of histone lysine tails to produce mono-,di-, or trimethylated states. Among a myriad of PTMs, histone lysine methylation catalyzed by histone lysine methyltransferases (HMTs) has received great attention because of its essential function in many biological processes including gene expression and transcriptional regulation, heterochromatin formation, and X-chromosome inactivation.7 It is therefore considered to be one of the most significant PTMs of histones. Since the first HMT was characterized in 20008, more than 50 human histone methyltransferases have been identified.9 Growing evidence suggests that HMTs play important roles in the development of various human diseases including cancer.10,11 For example, G9a, a H3K9 methyltransferase also known as EHMT2, is overexpressed in human cancers and knockdown of G9a inhibits cancer cell growth.12,13
Despite the tremendous progress made in identifying new HMTs, only two small molecule HMT inhibitors14–16, which are not SAM-related analogs, have been reported since the first HMT was characterized in 2000.8 Therefore, creating multiple, high quality small molecule HMT inhibitors as research tools for studying the biological function of HMTs is urgently needed.
In this letter, we report the design and synthesis of novel compounds to explore the 2,4-diamino-6,7-dimethoxyquinazoline template, and biological evaluation of these compounds that led to the discovery of 8 (UNC0224) as a potent and selective G9a inhibitor. In addition, we disclose a high resolution (1.7 Ǻ) X-ray crystal structure of the G9a-8 complex, the first co-crystal structure of G9a with a small molecule inhibitor.
The only previously reported small molecule inhibitor of G9a in the literature is 2,4-diamino-6,7-dimethoxy quinazoline 2a (BIX-01294)15,17 (Fig. 1), which also inhibited GLP (also known as EHMT1). GLP is another H3K9 methyltransferase that shares 80% sequence identity with G9a in their respective SET domains. Because no SAR has been reported for this quinazoline scaffold, we decided to explore multiple regions of this template to elucidate the SAR and improve potency and selectivity as part of our efforts to create multiple chemical probes for epigenetic targets and make these probes available to the research community without restrictions on their use.
Figure 1.

An efficient two-step synthetic sequence was developed to explore the 4-amino and 2-amino regions of the quinazoline scaffold (Scheme 1). Displacing the 4-chloro of commercially available 2,4-dichloro-6,7-dimethoxyquinazoline (1) with the first set of amines at room temperature, followed by displacement of the 2-chloro with the second set of amines under microwave heating conditions, yielded the desired 2,4-diamino-6,7-dimethoxyquinazolines 2 in good yields. Using this efficient synthesis, we rapidly prepared the compounds listed in Table 1 and Table 2. These compounds were evaluated in two orthogonal and complementary biochemical assays18: (1) Thioglo-based G9a inhibitory assay for monitoring the conversion of SAM to SAH;19 and (2) G9a AlphaScreen for the detection of methylated histone peptides.20
Scheme 1.

Synthesis of 2,4-diamino-6,7-dimethoxy quinazolines 2a
a (a) R’ amines, DMF, DIEA, rt; (b) R” amines, i-PrOH, 4 M HCl/dioxane, microwave, 160 °C.
Table 1.
SAR of 4-amino moiety.
 | |||
|---|---|---|---|
| Compound ID | R’ | G9a IC50 (µM) |
|
| Thioglo Assay |
Alpha- Screen |
||
|
2a
(BIX-01294) |
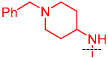 |
0.11 | 0.29 |
| 2b | 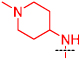 |
0.33 | 0.23 |
| 2c | 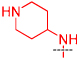 |
< 30% inhibition at 1 µM |
> 10 |
| 2d |  |
< 30% inhibition at 1 µM |
> 10 |
| 2e |  |
< 30% inhibition at 1 µM |
> 10 |
| 2f | < 30% inhibition at 1 µM |
5.1 | |
| 2g | < 30% inhibition at 1 µM |
5.8 | |
Table 2.
SAR of 2-amino moiety.
 | |||
|---|---|---|---|
| Compound ID | R” | G9a IC50 (µM) |
|
| Thioglo Assay |
Alpha- Screen |
||
| 2b | 0.33 | 0.23 | |
| 2h | 0.68 | 0.20 | |
| 2i | 0.55 | 0.51 | |
| 2j | 1.6 | 0.81 | |
| 2k | 0.91 | 6.5 | |
| 2l | 1.1 | 0.90 | |
| 2m | < 30% inhibition at 1 µM |
9.1 | |
The 4-amino moiety of the quinazoline scaffold was first explored (Table 1). Replacing the 1-benzyl piperidin-4-yl-amino group (2a) with 1-methyl piperidin-4-yl-amino (2b) resulted in no potency loss. This result is consistent with the X-ray crystal structure of the GLP-2a complex as the benzyl group of 2a was outside the peptide binding groove and did not make any interactions.17 This key SAR finding allowed us to reduce the molecular weight and lipophilicity of this chemical series. On the other hand, replacing the 1-methyl piperidin-4-yl-amino (2b) with piperidin-4-yl-amino (2c), tetrahydropyran-4-yl-amino (2d), or cyclohexylamino (2e) led to significant potency loss – indicating that an alkylated nitrogen is important for inhibitory activity. Analogs containing a smaller amino group such as cyclopropylamino (2f) and isopropylamino (2g) were also significantly less potent compared to 2a and 2b. In general, assay results from the Thioglo-based assay and AlphaScreen are consistent. However, the AlphaScreen appears to be more sensitive for weakly active compounds. The sensitivity of AlphaScreen for weakly active compounds is likely due to the assay detecting amplified signal and to the use of a lower concentration of peptide substrate.
The 2-amino moiety of the quinazoline scaffold was investigated next. In general, modifications to the 2-amino region were well tolerated (Table 2). The methyl homopiperazine (2b) could be replaced with the methyl piperazine (2h) and piperidine (2i) without significant potency loss. Analogs containing morpholine (2j), diethylamine (2k), or dimethylamine (2l) had moderate potency. The 2-choloro analog 2m had poor potency in both assays.
Having established initial SAR for the 2- and 4- amino regions, we next explored the 7-methoxy moiety. The X-ray crystal structure of the GLP-2a complex revealed that 2a occupied the histone peptide binding site and did not interact with the narrow lysine binding channel.17 We hypothesized that adding a 7-aminoalkoxy side chain to the quinazoline scaffold would make new interactions with the lysine binding channel while the rest of molecule maintained interactions with the peptide binding grove. Thus, we designed compound 8, which possesses a 7-dimethylaminopropoxy chain and also combines the best 2- and 4-amino moieties identified previously. Synthesis of 8 is outlined in Scheme 2. Benzyl protection of commercially available 2-methoxy-4-cyanophenol (3), followed by nitration, and subsequent reduction of the nitro group, produced aniline 4. Aniline 4 was then converted to quinazolinedione 5 via formation of methyl carbamate and subsequent saponification of the cyano group and ring closure. POCl3 treatment of intermediate 5 resulted in 2,4-dichloro quinazoline 6, which underwent two consecutive chloro displacement reactions to yield 2,4-diamino quinazoline 7. Debenzylation of intermediate 7, followed by Mitsunobu reaction with 3-(dimethylamino)propan-1-ol, produced the desired compound 8.
Scheme 2.

Synthesis of compound 8a
a (a) BnBr, K2CO3, DMF, rt; (b) HNO3, Ac2O, 0 °C to rt; (c) Fe dust, NH4Cl, i-PrOH-H2O, reflux, 67% over 3 steps; d) methyl chloroformate, DIEA, DMF-DCM, 0 °C to rt; (e) NaOH, H2O2, EtOH, reflux, 70% over 2 steps; (f) N,N-diethylaniline, POCl3, reflux, 59%; (g) 4-amino-1-methylpiperidine, DIEA, THF, rt; (h) 1-methylhomopiperazine, HCl, i-PrOH, 160 °C, microwave, 82% over 2 steps; (i) Pd/C, H2, EtOH, rt; (j) 3-(dimethylamino)propan-1-ol, PPh3, DIAD, THF, 0 °C to rt, 46% over 2 steps.
We were pleased to find that 8 was a potent G9a inhibitor with an IC50 of 15 nM, 7 times more potent compared to 2a (IC50 = 106 nM), in the G9a Thioglo assay (Figure 2). Although 8 (IC50 = 289 nM) had similar potency compared to 2a (IC50 = 290 nM) in the G9a AlphaScreen (likely due to 8 reaching the IC50 limit of the G9a AlphaScreen), the high potency of 8 was confirmed in several secondary assays. In ITC experiments that measure the binding affinity of a small molecule to the G9a protein21, 8 (Kd = 23 ± 8 nM (n = 2)) has about 5-fold higher binding affinity compared to 2a (Kd = 130 ± 18 nM (n = 2)) (Figure 3). 8 also displaced fluorescein labeled 15-mer H3 peptide (1–15) better than 2a in a G9a FP assay (Figure 4). In addition, 8 stabilized G9a better compared to 2a in DSF experiments.22 These results together strongly suggest that 8 is a significantly more potent G9a inhibitor compared to 2a.
Figure 2.

Full concentration response curves of 8 (○) (IC50 = 15 ± 10 nM) and 2a (●) (IC50 = 106 ± 20 nM) in the G9a Thioglo assay.
Figure 3.

8 showed higher binding affinity to G9a compared to 2a in an ITC experiment.
Figure 4.

8 (●) displaced fluorescein labeled 15-mer H3 peptide (1–15) better than 2a (○) (unlabeled 25-mer H3 peptide (1–25) (▼) used as a positive control).
Although 8 also potently inhibited GLP with an IC50 of 20 nM and 58 nM in the Thioglo assay and AlphaScreen, respectively, 8 was more than 1000-fold selective for G9a over SET7/9 (a H3K4 HMT) and SET8/PreSET7 (a H4K20 HMT) in Thioglo-based biochemical assays. In addition, 8 was clean (less than 20% inhibitions at 1 µM) against a broad range of G-protein coupled receptors, ion channels, and transporters in a 30-target selectivity panel (tested by MDS Pharma Services) except hitting muscarinic M2 receptor at 82% inhibition at 1 µM and histamine H1 receptor at 31% inhibition at 1 µM.
A high resolution (1.7 Ǻ) X-ray crystal structure of the G9a-8 complex, the first crystal structure of a G9a-small molecule inhibitor complex, was obtained. As shown in Figure 5, we were pleased to find that the 7-dimethylamino propoxy side chain of 8 indeed occupied the lysine binding channel of G9a nicely, thus validating our binding hypothesis. The higher potency of 8 compared to 2a can be explained by these additional interactions between the 7-dimethylamino propoxy side chain and the lysine binding channel and these interactions were absent in the GLP-2a complex. Other key features include: (1) the distal nitrogen of the homopiperazine interacts with Asp1074; (2) 4-amino group interacts with Asp1083; and (3) the bulk of 8 occupies the histone peptide binding site. The inhibitor-enzyme interactions revealed by this high resolution co-crystal structure will enable future structure-based design of novel HMT inhibitors.
Figure 5.

X-ray crystal structure of the G9a-8 complex (PDB code 3K5K). Compound 8 is in light and dark blue. The superposed histone backbone trace and the methylated lysine side chain are in magenta.
In conclusion, compound 8, a potent and selective inhibitor of histone lysine methyltransferase G9a, was discovered via SAR exploration and structure-based design. The first X-ray crystal structure of G9a with a small molecule inhibitor was obtained. This high resolution co-crystal structure of the G9a-8 complex validated our binding hypothesis and will enable structure-based design of novel inhibitors. 8 is a potentially valuable small molecule tool for the research community to investigate the biology of G9a and its roles in chromatin regulation.
Supplementary Material
Acknowledgement
We thank Dr. Yizhou Dong and Professor K.H. Lee for HRMS and HPLC support.
Abbreviations
- PTMs
post-translational modifications
- HMT
histone lysine methyltransferase
- EHMT2
euchromatic histone lysine methyltransferase 2
- H3K9
histone 3 lysine 9
- SAM
S-adenosyl-L-methionine
- SAR
structure activity relationships
- GLP
G9a like protein
- EHMT1
euchromatic histone lysine methyltransferase 1
- SET
suppressor of variegation 3–9, enhancer of zeste, and trithorax
- SAH
S-adenosyl-L-homocysteine
- AlphaScreen
amplified luminescence proximity homogeneous assay
- ITC
isothermal titration calorimetry
- FP
fluorescence polarization
- DSF
differential scanning fluorimetry
Footnotes
The coordinates and structure factors of UNC0224 co-crystallized with G9a have been deposited in the Protein Data Bank (www.pdb.org, PDB code 3K5K).
Supporting Information Available: procedures and characterization data for all compounds. Procedures for biochemical assays. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 2.Gelato KA, Fischle W. Role of histone modifications in defining chromatin structure and function. Biol Chem. 2008;389:353–363. doi: 10.1515/BC.2008.048. [DOI] [PubMed] [Google Scholar]
- 3.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 4.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 5.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 6.Lyko F, Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J Natl Cancer Inst. 2005;97:1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- 7.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 8.Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 9.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Spannhoff A, Sippl W, Jung M. Cancer treatment of the future: inhibitors of histone methyltransferases. Int J Biochem Cell Biol. 2009;41:4–11. doi: 10.1016/j.biocel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 11.Fog CK, Jensen KT, Lund AH. Chromatin-modifying proteins in cancer. APMIS. 2007;115:1060–1089. doi: 10.1111/j.1600-0463.2007.apm_776.xml.x. [DOI] [PubMed] [Google Scholar]
- 12.McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006;66:3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 13.Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, Haddad BR, Issa JP. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat Chem Biol. 2005;1:143–145. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- 15.Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 16.Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The G9a Thioglo assay IC50 values in this paper are the average of at least duplicate assay runs with standard deviation (SD) within 1 fold unless noted otherwise. The G9a AlphaScreen IC50 values in this paper are the average of at least duplicate assay runs with standard error measurement (SEM) < 35%. The observed potency difference for 2a (BIX-01294) in our G9a Thioglo and AlphaScreen assays (Table 1) versus the previously reported IC50 values (shown in Figure 1) is due to using different assays.
- 19.Collazo E, Couture JF, Bulfer S, Trievel RC. A coupled fluorescent assay for histone methyltransferases. Anal Biochem. 2005;342:86–92. doi: 10.1016/j.ab.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Quinn AM, Allali-Hassani A, Vedadi M, Simeonov A. A Chemiluminescence-based Method for Identification of Histone Lysine Methyltransferase Inhibitors. Molecular BioSystems. doi: 10.1039/b921912a. submitted. G9a AlphaScreen assay protocol is included in Supporting Information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Min J, Allali-Hassani A, Nady N, Qi C, Ouyang H, Liu Y, MacKenzie F, Vedadi M, Arrowsmith CH. L3MBTL1 recognition of mono- and dimethylated histones. Nat Struct Mol Biol. 2007;14:1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- 22.DSF graphs of 8 (UNC0224) and 2a are included in Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.