

Abstract
Aneuploidy and extensive chromosomal rearrangements are common in human tumors. The role of DNA damage response proteins p53 and p21CIP1/WAF1 in aneugenesis and clastogenesis was investigated in telomerase immortalized diploid human fibroblasts using siRNA suppression of p53 and p21CIP1/WAF1. Cells were exposed to the environmental carcinogen sodium arsenite (15 and 20 µM), and the induction of micronuclei (MN) was evaluated in binucleated cells using the cytokinesis-block assay. To determine whether MN resulted from missegregation of chromosomes or from chromosomal fragments, we used a fluorescent in situ hybridization with a centromeric DNA probe. Micronuclei were predominantly of clastogenic origin in control cells regardless of p53 or p21CIP1/WAF1 expression. MN with centromere signals in cells transfected with NSC siRNA or Mock increased 30% after arsenite exposure, indicating that arsenite induced aneuploidy in the tGM24 cells. Although suppression of p53 increased the fraction of arsenite-treated cells with MN, it caused a decrease in the fraction of with centeromeric DNA. Suppression of p21CIP1/WAF1 like p53 suppression decreased the fraction of with centromeric DNA. Our results suggest that cells lacking normal p53 function cannot become aneuploid because they die by mitotic arrest-associated apoptosis, whereas cells with normal p53 function that are able to exit from mitotic arrest can become aneuploid. Furthermore our current results support this role for p21CIP1/WAF1. Since suppression of p21CIP1/WAF1 caused a decrease in aneuploidy induced by arsenite suggesting that p21CIP1/WAF1 plays a role in mitotic exit.
Keywords: aneuploidy, arsenite, p21CIP1/WAF1, p53
1. Introduction
Aneuploidy and extensive chromosomal rearrangements are frequently found in human tumors (reviewed in [1]). Aneuploidy arises from chromosomal missegregation whereas chromosomal rearrangements arise from clastogenic induction of cycles of chromosome breakage and rejoining. Although aneuploidy may arise spontaneously, certain types of environmental agents induce aneuploidy by induction of chromosome loss or gain during cell division in somatic and germ cells [2]. Likewise, chromosome breakage arises from DNA strand breakage often caused directly or indirectly by environmental genotoxic agents. Arsenic in the trivalent form (arsenite) causes both aneuploidy [3, 4] and clastogenesis [5]. Arsenic induces a mitotic delay and aneuploidy in diploid human fibroblasts and peripheral blood lymphocytes [3, 4]. Both aneuploidy and clastogenesis have been proposed as mechanisms of arsenic-carcinogenesis [6].
Accurate chromosome segregation during mitosis is critical in the prevention of aneuploidy. In fact, loss of mitotic checkpoint, abnormal centrosome amplification and defects in the kinetochore-microtubule attachment are events that disrupt chromosome segregation and contribute to aneuploidogenesis [7]. The spindle assembly checkpoint, activated immediately upon entry into mitosis, is the main mitotic checkpoint controlling mitotic progression [8] and ensures proper alignment of the chromosomes at the metaphase plate before segregation. The major regulator of the spindle assembly checkpoint is the mitotic checkpoint complex composed of MAD2, BUB1 and MAD3/BUBR1 [9]. However, it is likely that regulation may occur by other pathways as ATM and p21CIP1/WAF1 which suppress aneuploidy in a genome instability background [10]; p53 may also play a role in suppressing aneuploidy [11] by regulating mitotic progression either directly or indirectly through p21CIP1/WAF1 (aka CDKN1A) [12].
Induction of p53 by mitotic checkpoint activation is essential to the conservation of normal ploidy caused by mitotic failure. In addition, p53 deficiencies induce insufficient mitotic arrest, compromise apoptosis, and can cause aneuploidy [13]. Apoptosis is an important defense mechanism to prevent the survival of aneuploid cells. Cells that proceed through a defective mitosis and slip through the arrest often undergo apoptosis in the ensuing pseudo-G1 phase. Paradoxically, p53 expression allows exit from arsenite-induced mitotic arrest in a p21CIP1/WAF1-dependent manner. p21CIP1/WAF1 is a cyclin dependent kinase inhibitor involved in both G1 and G2 checkpoints. Expression of p21CIP1/WAF1 is suggested to play a role in release of cells from mitotic arrest induced by paclitaxel [14] as well as by arsenite [12].
Arsenite is an aneugenic agent and arsenite–induced apoptosis has been reported to be associated with activation of spindle checkpoint [15–17]. Chronic exposure to inorganic arsenic from drinking water has been associated with diabetes mellitus, cardiovascular disorders, and a variety of skin and internal organ cancers [6, 18, 19]. Paradoxically, arsenic trioxide has also been used as an effective chemotherapeutic agent in the treatment of patients with acute promyelocytic leukemia, particularly in patients showing resistance to therapy with all-trans-retinoic acid or other chemotherapeutic drugs [20]. Trivalent inorganic arsenic compounds, such as arsenic trioxide and arsenite, kill cancer cells by induction of apoptosis consequent to mitotic disruption [15, 21–29].
Disruption of mitosis can be caused by interference with spindle function, destabilization of centrosomes, or disruption of mitotic spindles. Arsenite induces G2 and mitotic arrest in cell lines that lack functional p53 [28, 30, 31]. We have previously demonstrated that arsenite induces accumulation of mitotic cells with non-functional p53, but not in p53-expressing cells. In addition, the p53-deficient cells underwent apoptosis, whereas most of p53-expressing cells exited from M phase [32]. Furthermore, p53-dependent p21CIP1/WAF1 induction, releases cells from mitotic arrest and prevents the arrest-associated apoptosis [12]. Arsenic-induced mitotic arrest associated apoptosis is directly related to the functional activation of spindle checkpoint [15, 17] and can be prevented by inhibition of the cyclin dependent kinase activity by roscovitine [15].
In the present study, we investigated the effect of suppression of p53 and p21CIP1/WAF1 on chemical-induced aneuploidy and clastogenesis in telomerase immortalized diploid human fibroblasts (tGM24 cells). Only cells that recover from mitotic disruption can become aneuploid, whereas the cells that do not recover will suffer mitotic arrest-associated apoptosis (aka mitotic catastrophe). Elucidation of this recovery process might provide information relevant to the mechanisms of both arsenic carcinogenesis and chemotherapy by mitotic disruption.
Here we tested the effect of siRNA suppression of p53 and p21CIP1/WAF1 expression on genotoxicant-induced aneuploidy and clastogenesis in telomerase immortalized diploid human fibroblasts which are genetically stable cells. We compared chromosomal effects of arsenite with the microtubule disruptor colcemid and the DNA crosslinking agent mitomycin C.
2. Materials and methods
Chemicals
Fresh working aqueous solutions of sodium arsenite (NaAsO2, Sigma, CAS 1327-53-3) were prepared on the day of treatment and filter prior to use. Colcemid (Gibco, CAS 477-30-5), an aneugenic agent and mitomycin C (Sigma, CAS 50-07-7), a DNA crosslinking agent, were used as positive controls of both clastogenic and aneugenic effects. Sterile water was used as the solvent for all chemicals (stock solutions). Cytochalasin B (Cyt B; Sigma, CAS 14930-96-2) was dissolved in DMSO (1 mg/ml) and kept at−20°C.
Cell culture
Telomerase-immortalized diploid human fibroblasts (HTERT-GM00024, “tGM24”) were obtained on Material Transfer Agreement with Geron Inc. as described previously [33]. tGM24 cells were grown in Eagle's minimal essential medium containing Earle's salts (Gibco-BRL) supplemented with non-essential amino acids (0.1 mM), L-glutamine (2 mM), penicillin (100 units/ml), streptomycin (100 µg/mL) and fetal bovine serum (10%, Hyclone). The cells were incubated at 37 °C, with 5% CO2, and 90% relative humidity.
Arsenite toxicity
tGM24 cells were treated with NaAsO2 at 5–50 µM for 24, 48, 72 and 144 h. Cultures were monitored with an inverted phase-contrast microscope and cell viability by AlamarBlue fluorescence assay as described [12].
Knock-down of p53 and p21CIP1/WAF1 by siRNA transfection
RNA interference was carried out as described previously [12]. Briefly, cells were harvested from growing cultures and transfected with p53 and p21CIP1/WAF1 Smartpool siRNAs (Dharmacon, Lafayette, CO). Mock transfections were performed without any siRNA. Nonspecific control transfections were performed with 2 µg of non-specific control (NSC) siRNA. Specific transfections were performed with 2 µg of TP53 siRNA or p21CIP1/WAF1 siRNA; siRNA was transfected into the cells using Lipofectamine 2000 (Invitrogen). At 24 h post–transfection, the cells were treated for 24 h. Specific suppression of p53 and p21CIP1/WAF1 was demonstrated in tGM24 cells by Western blot analysis of samples 72 h after induction by UV radiation.
Western blot analysis
Suppression of p53 and p21CIP1/WAF1 was confirmed by western blot analysis performed described previously [12]. Membranes were stained with Ponceau stain to determine even gel loading and transfer to the membrane.
Cytokinesis block micronucleus assay
The micronucleus assay was performed as described previously [34]. Briefly, 0.4×106 cells were seeded onto a cover slip in 60 mm dishes. After 24 h of culture, cells were exposed to NaAsO2 (15 and 20 µM), colcemid (1 µM) or mitomycin C (1 µM) and also to cytochalasin B (3 µg/ml final concentration). After 24 h of exposure, the cells were harvested. Cells on the cover slips were fixed. Air-dried cover glasses were stained with Wright´s colorant and mounted on slides for microscopic evaluation.
The frequency of micronuclei (MN) was determined in 1000 binucleated cells per treatment group. Cell proliferation kinetics were analyzed by determining the frequency of mononucleated (Mono), binucleated (Bi) and polynucleated (Poly) cells, corresponding to 0, 1, and 2 or more in vitro cell divisions, respectively. Cytostatic activity was determined by calculating the percentage of binucleated cells. Three independent experiments were analyzed.
Fluorescence in situ hybridization (FISH) with a pancentromeric DNA probe
The chromosomal composition of MN was assessed by fluorescence in situ hybridization (FISH) technique to distinguish between aneugenic and clastogenic origin of the MN. For FISH analysis, samples were selected in which the quality was confirmed by observation after standard Wright´s staining. The FISH analysis using commercial human pancentromeric probes directly labelled with Cy3 (Cambio UK) was carried out according to the manufacturer´s protocol with slight modifications. Briefly, the cells were treated with RNase (1 mg/ml) at 3700B0C for 15 min and proteinase K (50 ng/ml) at 37°C for 5 min. DNA was denatured with 70% formamide/2XSSC for 2 min at 70°C. The DNA probe was denatured for 10 min at 85°C and was added to the slide under reduced light. The slides were covered with a cover slip and sealed with rubber cement. Following an overnight hybridization at 37°C in a humidified box, slides were washed in 50% formamide/2XSSC for 5 min at 37°C. The cells were then counterstained with DAPI in antifade. One thousand binucleated cells with well-preserved cytoplasm were examined using a fluorescent microscope equipped with a double-band pass filter for visualization of Cy3 (red) and DAPI (blue) signals and single-band pass filter for visualization of DAPI signals alone. If one or several red spots (Cy3-labelled centromeres) were observed inside MN, the MN was classified as a MN with centromere signals or positive. While a MN without signals was classified as MN negative.
Statistical analysis
Normality of data was evaluated by the Kolmogorov–Smirnov test. The difference between groups for multiple variables was assessed with one-way analysis of variance (ANOVA) followed by Tukey´s post-test. The Fisher´s exact test was applied for comparisons between MN+ and MN− frequencies. Differences were considered significant at p<0.05.
3. Results
We sought to examine the role of p53 and p21CIP1/WAF1 in suppressing aneuploidy and clastogenesis induced by arsenic in cells that were genetically stable. Thus, we chose to use a telomerase immortalized diploid human fibroblast cell line, tGM24. Like their non-immortalized forbears, the tGM24 cells are genetically stable and exhibit normal DNA repair functions [33]. The sensitivity to sodium arsenite (NaAsO2) was evaluated in the tGM24 cells.
Cells treated with 0–50 µM were examined for signs of cytotoxicity at 24, 48, 72 and 144 h (Figure 1). Cell viability assay suggested that dose and time-dependent cytotoxicity was occurring because the apparent concentration - dependent decrease in AlamarBlue fluorescence increased with increasing time of incubation (Figure 1A). However, when cells were observed at NaAsO2 concentrations up to 20 µM using phase contrast microscopy, it became apparent that the principle effect was a slowing of cell growth as indicated by the NaAsO2 - concentration-dependent delay of increase in cell density (Figure 1B). Arsenite-treated cells had a similar morphology to non-treated cells at 5, 10 and 20 µM for 24 h. However, after 48 h when compared with non-treated cells, a decrease of growth was observed with concentrations up to 20 µM. It was noted that the cells grow more slowly but they did not die. Morphological impacts due to cytotoxicity were evident in cultures exposed at NaAsO2 concentrations ≥ 30 µM. These results were essentially identical to those obtained in similar experiments conducted with non-immortalized GM24 cells [28].
Fig. 1.

Analysis of arsenite-induced sensitivity in tGM24 fibroblasts. Cells were treated with the indicated concentrations of arsenite for 24 to 144 h. A) Cell viability assay using AlamarBlue. Data are reported arbitrary fluorescence units above the blank versus time. B) Following the exposure, morphological changes were monitored with a phase-contrast microscope.
In order to establish the changes in the cell proliferation in relation to functional status of p53 and p21CIP/WAF1, we analyzed the cell proliferation kinetics in tGM24 cells after silencing with the corresponding siRNAs. Suppression of p53 and p21CIP/WAF1 was confirmed by Western blot (Figure 2). Effects on cell proliferation were evaluated by the scoring of mono, bi and polynucleated cells (Figure 3A, B). Suppression of p53 produced a small non-significant increase in the percentage of binucleated cells (Figure 3C). In contrast suppression of p21CIP/WAF1 induced a significant decrease in the percentage of binucleated cells (Figure 3C), indicating a slowing of cell proliferation and suggesting that p21CIP/WAF1 may play a role in mitotic exit. NaAsO2 treatment decreased the percent binucleated Mock and NSC transfected cells (Fgiure 3C) indicating a slowing of cell proliferation. Only cells treated with 15 µM arsenite had a significant increase in cell proliferation when p53 was suppressed (Figure 3C).
Fig. 2.

Confirmation of p53 and p21CIP/WAF1 knockdown. Western blot analysis of p53 and p21CIP/WAF1 in tGM24 fibroblasts unexposed and exposed to UV radiation. Uniformity of loading and transfer were confirmed by staining the membrane with Ponceau Red.
Fig. 3.
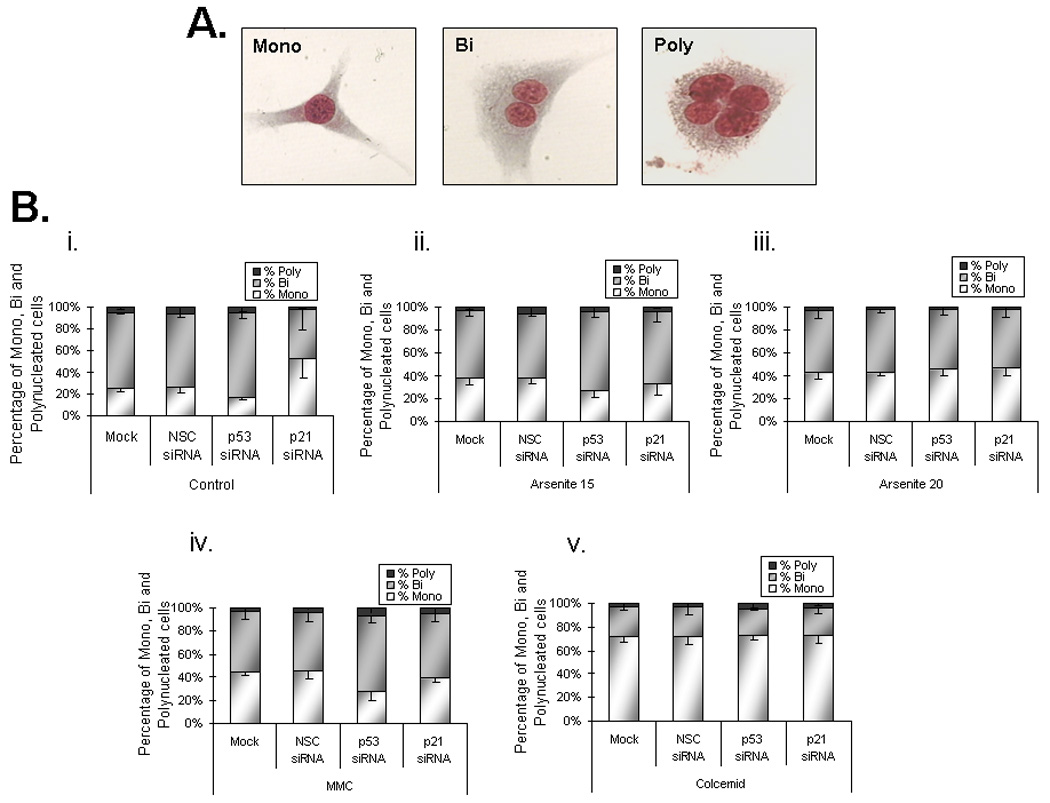

Effects on cell proliferation kinetics by suppression of p53 and p21CIP1/WAF1 in tGM24 fibroblasts. A) Representative images of mononuclear, binuclear and polynuclear cells are shown. B) Analysis of mononucleated (Mono), binucleated (Bi) and polynucleated (Poly) cells. C) Analysis of the cytostatic effect. Y-axis indicates the percentage of binucleated cells (BN) graphed as bars. Cells were incubated with sodium arsenite (15 and 20 µM), mitomycin C and colcemid for 24 h. Knock-down of p53 and p21CIP1/WAF1 by siRNA transfection was performed as indicated (Materials and methods). Data represent the mean ± S.D. calculated from three independent experiments. *p ≤ 0.05 with respect to corresponding control cells; **p ≤ 0.05 with respect to each treatment as indicated by brackets (ANOVA test).
Suppression of p21CIP/WAF1 did not alter cell proliferation in tGM24 cells treated with 15 or 20 µM of arsenite (Figure 3C). It should be noted that mitomycin C and colcemid significantly impair cell proliferation kinetics as indicated by the large increases in percent mononucleated cells (Figures 3Biv, 3Bv,) and deceases in binucleated cells (Figure 3C) compared to controls (Figure 3Bi). Although siRNA knockdown of p53 and p21CIP/WAF1 did not impact proliferation of tGM24 cells treated with colcemid, knockdown of p53, but not p21CIP1/WAF1, increased proliferation of cells treated with mitomycin C suggesting that cell cycle arrest caused by mitomycin C was p53 dependent but p21CIP1/WAF1 independent.
The effects of p53 and p21CIP1/WAF1 suppression on MN induction were tested using the cytokinesis block assay (Figure 4) in tGM24 cells. Addition of cytochalasin B leads to binucleated cells due to inhibition of cytokinesis but not of karyokinesis (Figure 4A). Suppression of p21WAF1/CIP1, but not p53, caused a significant increase in MN in control cells (Figure 4B). MN were induced by NaAsO2 and colcemid independent of the suppression of p53 or p21CIP1/WAF1 and the induction showed NaAsO2 dose response in Mock, NSC and p53 siRNA transfected cells. MN induction was further increased by p53 suppression with all given treatments. Suppression of p21CIP1/WAF1 significantly further increased MN in cells treated with colcemid but not mitomycin C or NaAsO2.
Fig. 4.

Effects on cell proliferation kinetics by suppression of p53 and p21CIP1/WAF1 in tGM24 fibroblasts. A) Representative images of mononuclear, binuclear and polynuclear cells are shown. B) Analysis of mononucleated (Mono), binucleated (Bi) and polynucleated (Poly) cells. C) Analysis of the cytostatic effect. Y-axis indicates the percentage of binucleated cells (BN) graphed as bars. Cells were incubated with sodium arsenite (15 and 20 µM), mitomycin C and colcemid for 24 h. Knock-down of p53 and p21CIP1/WAF1 by siRNA transfection was performed as indicated (Materials and methods). Data represent the mean ± S.D. calculated from three independent experiments. *p ≤ 0.05 with respect to corresponding control cells; **p ≤ 0.05 with respect to each treatment as indicated by brackets (ANOVA test).
To determine whether MN resulted from missegregation of chromosomes or from chromosomal fragments, we used fluorescent in situ hybridization (FISH) with a centromeric DNA probe (Figure 5). MN that are positive for centromeric DNA (MN+, Figure 5B) are derived from whole chromosomes whereas MN without centromeric DNA (MN−, Figure 5C) are derived from chromosome fragments. Although suppression of p21 in control cells increased the percentage of cells with MN (Figure 4B), it did not change the fraction of MN+ in control cells (Figure 6). Likewise, suppressing p53 did not change the ratio of MN+/MN− in control cells.
Fig. 5.

Fluorescence in situ hybridization (FISH) images of centromeric DNA probe hybridization in tGM24 cells. Only cells with well-preserved cytoplasm were examined under DAPI filter (left panels) and centromeric regions in the same cells with the filter for visualization of Cy3 (merged image in right panels). A) Typical binucleated cell. B) Binucleated cell with a signal in MN (MN+; arrows). C) Binucleated cell without a centromeric signal (MN−; arrows).
Fig. 6.
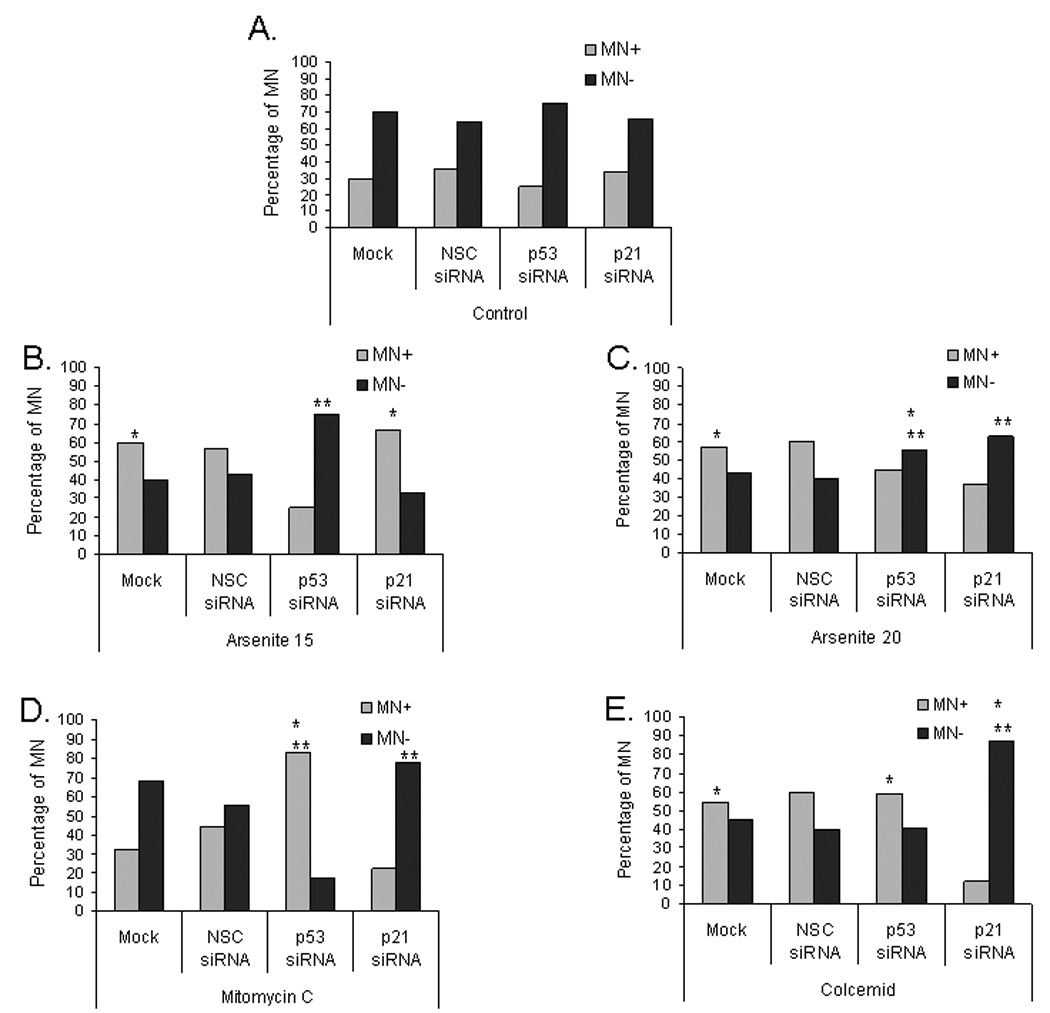
Suppression of p53 and p21CIP1/WAF1 reduces arsenite-induced aneuploidy. MN frequency was evaluated in tGM24 fibroblasts treated with sodium arsenite (15 and 20 µM), mitomycin C and colcemid for 24 h. Knock-down of p53 and p21CIP1/WAF1 by siRNA transfection was performed as indicated (Materials and methods). Data show the percentage of cells containing MN centromere positive (MN+) or MN centromere negative (MN−). *p ≤ 0.05 with respect to corresponding control cells; **p ≤ 0.05 with respect to each treatment as indicated by brackets (Fisher´s exact test).
MN+ cells transfected with NSC siRNA or Mock increased 30% after arsenite exposure (both 15 and 20 µM), indicating that arsenite induced aneuploidy in the tGM24 cells. Similar results were observed in colcemid treated cells. Although suppression of p53 increased the fraction of arsenite-treated cells with MN (Figure 4B), it caused a decrease in the fraction of MN+ cells treated with 15 µM NaAsO2 (Figure 6). This change in MN+/MN− ratio also was significant but not as great in cells treated with 20 µM NaAsO2 (Figure 6). Thus, when the MN+/MN− ratio was evaluated in 15 µM NaAsO2 treated cells, the suppression of p21 had no effect on the MN+/MN− ratio, but 20 µM NaAsO2 treated cells did show a significant effect on the MN+/MN− ratio and decreased the fraction of MN+ similar to p53 suppression. Suppression of p21CIP1/WAF1 dramatically suppressed the percentage of MN+ in colcemid treated cells (Figure 6) while increasing the total fraction of cells that had MN (Figure 4). Surprisingly, p53 suppression resulted in an increase in the percentage of aneuploid MN which showed centromere signals in mitomycin C-treated tGM24 cells.
4. Discussion
The induction of cytogenetic alterations by arsenite is probably not due to its direct damage effect on DNA. Arsenite is a genotoxic agent that affects cell division and the mitotic spindle apparatus, resulting in the loss or gain of whole chromosomes, thereby inducing aneuploidy. In the current study, we show that the suppression of p53 is associated with a reduction of aneuploidy induced by arsenite in telomerase immortalized diploid human fibroblasts. Arsenic induces mitotic arrest and subsequent apoptosis in cancer cell lines and p53 expression has previously been shown to affect the ability of cells to escape from mitotic arrest induced by arsenic [12, 31, 32]. Our results suggest that cells lacking normal p53 function cannot become aneuploid because they die by mitotic arrest-associated apoptosis, whereas cells with normal p53 function that are able to exit from mitotic arrest can become aneuploid. Furthermore our current results support this role for p21CIP1/WAF1. Since suppression of p21CIP1/WAF1 caused a decrease in aneuploidy induced by NaAsO2 (20 µM) suggesting that p21CIP1/WAF1 plays a role in exit from NaAsO2 – induced mitotic delay (Figure 7). These observations are consistent with the role of p53 induced p21CIP/WAF1 in allowing cells arrested in mitosis by arsenite to exit the block [12]. Thus, cells that lack p21CIP1/WAF1 and don’t divide cannot become aneuploid. On the other hand, cells not exposed to genotoxic agents and lacking p21CIP1/WAF1 exhibited an increase in MN−, indicating an increase in clastogenesis.
Fig. 7.

Schematic of proposed role of p21CIP1/WAF induction in exit from NaAsO2-induced mitotic delay and the effect on aneuploidy. Cyclin B is normally targeted for proteasomal degradation by polyubiquitinylation by the anaphase promoting complex/cyclosome (APC/C) and thus CDK1 activity is decreased allowing mitotic exit. NaAsO2 induces mitotic delay or mitotic arrest by stabilization of cyclin B and maintenance of CDK1 activity [12,16] most likely a result of inhibition of APC/C. Thus, inhibition of CDK1 activity is essential to exit from NaAsO2 – induced mitotic arrest [15]. Cells unable to exit mitosis suffer mitotic catastrophe and die by apoptosis [12], whereas the cells that recover from mitotic disruption can become aneuploid, NaAsO2 induces p53 in mitotically arrested cells, which in turn induces the cyclin dependent kinase inhibitor p21CIP1/WAF1 which inhibits CDK1 activity allowing mitotic exit and avoidance of apoptosis.
Aberrations in the centrosome duplication cycle result in the formation of monopolar or multipolar spindles, leading to aneuploidy (reviewed in [1]). There is evidence linking p53 and centrosome amplification [7,11]. Centrosome amplification in tumors and cell lines has been linked to numerous genetic aberrations, including the loss of the tumor suppressor protein p53 and the deregulated expression of its regulators (Mdm2) and downstream targets (p21CIP/WAF1, Gadd45) [35]. Our data indicate that arsenite-induced aneuploidy is clearly reduced in tGM24 cells when p53 was suppressed. These results are further supported by the findings that arsenite induces centrosome amplification prior to the induction of mitotic arrest and apoptosis [21, 36]. In addition to arsenite-induced abnormal centrosome amplification, subsequent mitotic arrest has been associated with HSP70, a protein involved in organization of the spindle apparatus [21].
It should be noted that it is known that p53 plays a key role in the mitotic checkpoint. We found that the suppression of p53 inhibits the arsenite-induced aneuploidy. Our findings are supported by the role of p53 in mitotic checkpoint activation. Induction and/or activation of p53 seems to be essential for protecting cells against the development of abnormal levels of chromosomal ploidy that would be induced should the cells escape the checkpoint [11].
MN frequency was induced in cells treated with sodium arsenite, mitomycin C and colcemid. These results are consistent with the induction of DNA damage in cells treated with genotoxic agents [34]. Since the formation of MN were evaluated in cells with one cell division, it could indicate that in presence of DNA damage, the cells can ultimately undergo an aberrant mitosis and enter the ensuing G1. More importantly, a higher induction of MN was observed in cells treated with all the chemicals when p53 was suppressed. As compromised function of p53 is linked to centrosome amplification, it is possible that the micronuclei formation is due to centrosome amplification during prolonged mitotic arrest induced by arsenic.
Current data show that the p21CIP/WAF1 suppression causes a decrease in nuclear proliferation index in control cells suggesting that p21CIP/WAF1 may play a role in mitotic exit by inhibiting the cyclin B dependent kinase CDK1 (CDC2). It has been shown that suppression of p21CIP/WAF1 increased the occurrence of mitotic catastrophe (apoptotic mitotic death) in arsenite treated TR9-7 cells [12]. Also when A375 melanoma cells arrested in mitosis by arsenite were treated with roscovitine (a chemical inhibitor of CDK1), they exited from arsenite induced mitotic arrest [15]. Considered together, the release from mitotic arrest by both p21CIP1/WAF1 and roscovitine suggest that inhibition of CDK1 activity is essential to release from mitotic arrest induced by arsenite. Data obtained with p21CIP/WAF1 suppression in control cells support the hypothesis that inhibition of CDK1 by p21CIP/WAF1 is involved in the mitotic exit network (Figure 7). Furthermore, we know that in p53 deficient cells, p21CIP/WAF1 is still expressed although not as high as in cells with functional p53. It may be that the mitotic arrest is not as complete in p53 deficient cells as it is in p21CIP/WAF1 suppressed cells. Hence, the micronuclei formation may be dependent on partial mitotic progression that is less likely in p21 CIP/WAF1 suppressed cells.
In a recent study, Yu et al [37] examined the gene expression changes in response to arsenite using a microarray-based analysis. Although arsenite induces apoptosis and cell cycle arrest in a dose-dependent manner in both wildtype and p53 deficient mouse embryonic fibroblasts, arsenite induces differential gene pathways leading to cell cycle arrest and apoptosis in p53+/+ cells versus p53−/− cells [37]. Yu et al. revealed also that arsenite induced p53-dependent gene expression alterations in DNA damage and cell cycle regulation genes in p53+/+ cells, while in the p53−/− cells, arsenite induced a significant up-regulation of apoptosis-related genes (Noxa) and down-regulation of genes involved in the regulation of immune modulation.
The present study demonstrates a direct correlation between the arsenite-induced aneuploidy and the suppression of p53 and p21CIP/WAF1. Increase in MN frequency could be consequence of increases in abnormal centrosome number which give rise to aneuploidy by altering the number of spindle poles. Considering our findings we propose that p53 expression in the presence of arsenite might accelerate the chromosomal instability and aneuploidy in p53+/+ cells. However, given that p53 is only a part of the regulatory mechanism that maintains p53 functions, future studies are needed to better understand arsenic-induced aneuploidy.
Acknowledgements
This work was supported by NIEHS grants R01ES011314 to JCS and P30ES014443, and by grants from CONACYT and PAPIIT-DGAPA (UNAM) to AMS. We appreciate the support of L. Brito.
Reference List
- 1.Pihan GA, Doxsey SJ. The mitotic machinery as a source of genetic instability in cancer. Semin. Cancer Biol. 1999;9:289–302. doi: 10.1006/scbi.1999.0131. [DOI] [PubMed] [Google Scholar]
- 2.Aardema MJ, Albertini S, Arni P, Henderson LM, Kirsch-Volders M, Mackay JM, Sarrif AM, Stringer DA, Taalman RD. Aneuploidy: a report of an ECETOC task force. Mutat. Res. 1998;410:3–79. doi: 10.1016/s1383-5742(97)00029-x. [DOI] [PubMed] [Google Scholar]
- 3.Vega L, Gonsebatt ME, Ostrosky-Wegman P. Aneugenic effect of sodium arsenite on human lymphocytes in vitro: an individual susceptibility effect detected. Mutat. Res. 1995;334:365–373. doi: 10.1016/0165-1161(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 4.Yih LH, Ho IC, Lee TC. Sodium arsenite disturbs mitosis and induces chromosome loss in human fibroblasts. Cancer Res. 1997;57:5051–5059. [PubMed] [Google Scholar]
- 5.Chakraborty T, De M. Clastogenic effects of inorganic arsenic salts on human chromosomes in vitro. Drug Chem. Toxicol. 2009;32:169–173. doi: 10.1080/01480540802594509. [DOI] [PubMed] [Google Scholar]
- 6.National Research Council. Arsenic in Drinking Water: 2001 Update. Washington DC: National Academy Press; 2001. [PubMed] [Google Scholar]
- 7.Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230:6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 8.Lew DJ, Burke DJ. The spindle assembly and spindle position checkpoints. Annu. Rev. Genet. 2003;37:251–282. doi: 10.1146/annurev.genet.37.042203.120656. [DOI] [PubMed] [Google Scholar]
- 9.Tan AL, Rida PC, Surana U. Essential tension and constructive destruction: the spindle checkpoint and its regulatory links with mitotic exit. Biochem.J. 2005;386:1–13. doi: 10.1042/BJ20041415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen KC, Heng H, Wang Y, Lu S, Liu G, Deng CX, Brooks SC, Wang YA. ATM and p21 cooperate to suppress aneuploidy and subsequent tumor development. Cancer Res. 2005;65:8747–8753. doi: 10.1158/0008-5472.CAN-05-1471. [DOI] [PubMed] [Google Scholar]
- 11.Duensing A, Duensing S. Guilt by association? p53 and the development of aneuploidy in cancer. Biochem. Biophys. Res. Commun. 2005;331:694–700. doi: 10.1016/j.bbrc.2005.03.157. [DOI] [PubMed] [Google Scholar]
- 12.Taylor BF, McNeely SC, Miller HL, Lehmann GM, McCabe MJ, Jr, States JC. p53 suppression of arsenite-induced mitotic catastrophe is mediated by p21CIP1/WAF1. J. Pharmacol. Exp. Ther. 2006;318:142–151. doi: 10.1124/jpet.106.103077. [DOI] [PubMed] [Google Scholar]
- 13.Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol. Cell Biol. 1998;18:1055–1064. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barboule N, Chadebech P, Baldin V, Vidal S, Valette A. Involvement of p21 in mitotic exit after paclitaxel treatment in MCF-7 breast adenocarcinoma cell line. Oncogene. 1997;15:2867–2875. doi: 10.1038/sj.onc.1201469. [DOI] [PubMed] [Google Scholar]
- 15.McNeely SC, Taylor BF, States JC. Mitotic arrest-associated apoptosis induced by sodium arsenite in A375 melanoma cells is BUBR1-dependent. Toxicol. Appl. Pharmacol. 2008;231:61–67. doi: 10.1016/j.taap.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNeely SC, Belshoff AC, Taylor BF, Fan TW, McCabe MJ, Jr, Pinhas AR, States JC. Sensitivity to sodium arsenite in human melanoma cells depends upon susceptibility to arsenite-induced mitotic arrest. Toxicol. Appl. Pharmacol. 2008;229:252–261. doi: 10.1016/j.taap.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu YC, Yen WY, Yih LH. Requirement of a functional spindle checkpoint for arsenite-induced apoptosis. J. Cell Biochem. 2008;105:678–687. doi: 10.1002/jcb.21861. [DOI] [PubMed] [Google Scholar]
- 18.Coronado-Gonzalez JA, Del Razo LM, Garcia-Vargas G, Sanmiguel-Salazar F, Escobedo-de la Pena J. Inorganic arsenic exposure and type 2 diabetes mellitus in Mexico. Environ. Res. 2007;104:383–389. doi: 10.1016/j.envres.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 19.States JC, Srivastava S, Chen Y, Barchowsky A. Arsenic and cardiovascular disease. Toxicol. Sci. 2009;107:312–323. doi: 10.1093/toxsci/kfn236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou WC, Dang CV. Acute promyelocytic leukemia: recent advances in therapy and molecular basis of response to arsenic therapies. Curr. Opin. Hematol. 2005;12:1–6. doi: 10.1097/01.moh.0000148552.93303.45. [DOI] [PubMed] [Google Scholar]
- 21.Taylor BF, McNeely SC, Miller HL, States JC. Arsenite-induced mitotic death involves stress response and is independent of tubulin polymerization. Toxicol. Appl. Pharmacol. 2008;230:235–246. doi: 10.1016/j.taap.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen ZY, Shen J, Cai WJ, Hong C, Zheng MH. The alteration of mitochondria is an early event of arsenic trioxide induced apoptosis in esophageal carcinoma cells. Int.J.Mol.Med. 2000;5:155–158. doi: 10.3892/ijmm.5.2.155. [DOI] [PubMed] [Google Scholar]
- 23.Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou L, Huang Y, Zhang JW, Xiong SM, Chen SJ, Wang ZY, Chen Z, Chen GQ. Arsenic trioxide-induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling pathways in acute promyelocytic leukemia. Leukemia. 2000;14:262–270. doi: 10.1038/sj.leu.2401650. [DOI] [PubMed] [Google Scholar]
- 24.Uslu R, Sanli UA, Sezgin C, Karabulut B, Terzioglu E, Omay SB, Goker E. Arsenic trioxide-mediated cytotoxicity and apoptosis in prostate and ovarian carcinoma cell lines. Clin. Cancer Res. 2000;6:4957–4964. [PubMed] [Google Scholar]
- 25.Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, Kakizuka A. Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen-independent prostate cancer. Cancer Res. 2001;61:5432–5440. [PubMed] [Google Scholar]
- 26.Yu J, Qian H, Li Y, Wang Y, Zhang X, Liang X, Fu M, Lin C. Arsenic trioxide (As2O3) reduces the invasive and metastatic properties of cervical cancer cells in vitro and in vivo. Gynecol. Oncol. 2007;106:400–406. doi: 10.1016/j.ygyno.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 27.Huang S, Huang CF, Lee T. Induction of mitosis-mediated apoptosis by sodium arsenite in HeLa S3 cells. Biochem.Pharmacol. 2000;60:771–780. doi: 10.1016/s0006-2952(00)00397-x. [DOI] [PubMed] [Google Scholar]
- 28.States JC, Reiners JJ, Jr, Pounds JG, Kaplan DJ, Beauerle BD, McNeely SC, Mathieu P, McCabe MJ., Jr Arsenite disrupts mitosis and induces apoptosis in SV40-transformed human skin fibroblasts. Toxicol. Appl. Pharmacol. 2002;180:83–91. doi: 10.1006/taap.2002.9376. [DOI] [PubMed] [Google Scholar]
- 29.Cai X, Yu Y, Huang Y, Zhang L, Jia PM, Zhao Q, Chen Z, Tong JH, Dai W, Chen GQ. Arsenic trioxide-induced mitotic arrest and apoptosis in acute promyelocytic leukemia cells. Leukemia. 2003;17:1333–1337. doi: 10.1038/sj.leu.2402983. [DOI] [PubMed] [Google Scholar]
- 30.McCabe MJ, Jr, Singh KP, Reddy SA, Chelladurai B, Pounds JG, Reiners JJ, Jr, States JC. Sensitivity of myelomonocytic leukemia cells to arsenite-induced cell cycle disruption, apoptosis, and enhanced differentiation is dependent on the inter-relationship between arsenic concentration, duration of treatment, and cell cycle phase. J. Pharmacol. Exp. Ther. 2000;295:724–733. [PubMed] [Google Scholar]
- 31.Yih LH, Hsueh SW, Luu WS, Chiu TH, Lee TC. Arsenite induces prominent mitotic arrest via inhibition of G2 checkpoint activation in CGL-2 cells. Carcinogenesis. 2005;26:53–63. doi: 10.1093/carcin/bgh295. [DOI] [PubMed] [Google Scholar]
- 32.McNeely SC, Xu X, Taylor BF, Zacharias W, McCabe MJ, Jr, States JC. Exit from arsenite-induced mitotic arrest is p53 dependent. Environ. Health Perspect. 2006;114:1401–1406. doi: 10.1289/ehp.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porter PC, Clark DR, McDaniel LD, McGregor WG, States JC. Telomerase-immortalized human fibroblasts retain UV-induced mutagenesis and p53-mediated DNA damage responses. DNA Repair (Amst) 2006;5:61–70. doi: 10.1016/j.dnarep.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Salazar AM, Sordo M, Ostrosky-Wegman P. Relationship between micronuclei formation and p53 induction. Mutat Res. 2009;672:124–128. doi: 10.1016/j.mrgentox.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 35.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- 36.Yih LH, Tseng YY, Wu YC, Lee TC. Induction of centrosome amplification during arsenite-induced mitotic arrest in CGL-2 cells. Cancer Res. 2006;66:2098–2106. doi: 10.1158/0008-5472.CAN-05-2308. [DOI] [PubMed] [Google Scholar]
- 37.Yu X, Robinson JF, Gribble E, Hong SW, Sidhu JS, Faustman EM. Gene expression profiling analysis reveals arsenic-induced cell cycle arrest and apoptosis in p53-proficient and p53-deficient cells through differential gene pathways. Toxicol. Appl. Pharmacol. 2008;233:389–403. doi: 10.1016/j.taap.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]