

Abstract
Phor21-βCG(ala), a 36-amino acid peptide comprised of a lytic peptide (Phor21) conjugated to a modified 15-amino acid segment of the β-chain of chorionic gonadotropin (βCG(ala)), selectively kills cancer cells that over-express luteinizing hormone/chorionic gonadotropin (LH/CG) receptors by disrupting cellular membrane structure. These studies were designed to further characterize its in-vitro inhibition and in-vivo destruction of prostate cancer cells, biostability and pharmacokinetics to determine its pharmacokinetic and pharmacodynamic profile. Inhibitory effects of Phor21-βCG(ala) were tested in PC-3 and Caco-2 cells as well as in nude mice bearing PC-3 cells transfected with the luciferase gene (PC-3.luc). Plasma stability, protease hydrolysis and pharmacokinetics of Phor21-βCG(ala) were measured by using liquid chromatography mass spectrometry (LC/MS/MS). Phor21-βCG(ala) selectively inhibited proliferation in-vitro and in-vivo metastases of PC-3 cells. Phor21-βCG(ala) was relatively stable in mouse, rat, dog and human plasma. Its degradation was partially due to protease hydrolysis and thermodynamic catalysis. Intravenous administration of Phor21-βCG(ala) showed its blood Cmax and AUC0→∞ around the in-vitro effective levels. In the tested rodents, Phor21-βCG(ala) displayed a moderate volume of distribution at steady state (Vdss) and slow clearance (Cl) in the rodents. In conclusion, Phor21-βCG(ala) displayed promising in-vitro and in-vivo anti-cancer activity with favourable pharmacokinetics, and may offer a novel approach to metastatic cancer chemotherapy.
Introduction
Contrary to popular belief and despite the expenditure of massive amounts of research funds, there has been no great improvement in the survival rates of patients with distant metastatic cancer since 1973 (Jemal et al 2003). Many chemotherapeutic drugs now in use are effective only against rapidly dividing cells and are thus ineffective against dormant or slow-growing metastatic cells. This sobering fact has led to attempts to develop more specific treatments by targeting hormone receptors expressed by the cancer cells and to develop drugs that are able to seek out and destroy distant and dormant metastatic cells. Remarkable success has recently been attained in developing drugs that target luteinizing hormone/chorionic gonadotropin (LH/CG) receptors expressed by prostate, breast, ovarian and testicular cancer cells (Hansel 2005; Hansel et al 2007b).
Among these drugs is a class of peptide molecules developed by us (Hansel et al 2001; Leuschner et al 2001; Bodek et al 2003) based on the use of membrane-disrupting peptides that are abundant in nature. Tumour cells are usually 50 times more sensitive to these membrane-disrupting peptides than normal cells (Johnstone et al 2000). Luteinizing hormone releasing hormone (LHRH) and beta chorionic gonadotropin (βCG) receptors on membranes of prostate, breast, ovarian and testicular cancer cells are up-regulated by oestrogens and follicle-stimulating hormone (FSH) and thus are more easily recognized by the targeting segment of these membrane-disrupting peptides, and become more vulnerable to these peptides (Leuschner & Hansel 2004). To date, a number of these peptides have been synthesized and their activities have been tested by us (Leuschner & Hansel 2005; Hansel et al 2007b). Structurally, they consist of a membrane-disrupting lytic peptide (e.g. Hecate, phor14, or phor21) conjugated to a 15-amino acid segment of the beta chain of chorionic gonadotropin to form Hecate-βCG (Zaleska et al 2003, 2004), Phor14-βCG or Phor21-βCG(ala). The βCG segment binds the membrane-disrupting peptides to the cancer cell membrane, resulting in changes in cell membrane electrochemical potentials and subsequent cell death. Because these lytic peptides are specifically bound by the targeting segment (βCG or LHRH) they destroy both primary and metastatic cells and their effects are independent of cell proliferation. In addition, they are not antigenic because they are relatively small and metabolized without side effects, except for inhibition of spermatogenesis and ovulation.
Hecate is a 23-amino-acid amphipathic lytic peptide and Phor14 is a 14-amino-acid peptide. We showed that both Hecate-βCG and Phor14-βCG were active in-vitro and in-vivo against human breast, prostate and ovarian cancer cells (Gawronska et al 2002; Leuschner et al 2003a, b). Because the doses of Hecate-βCG and Phor14-βCG required for prostate and breast cancer cell treatment were relatively high (8–10 and 12–24 mg kg−1, respectively), an additional compound, Phor21-βCG(ala) (NSC 731443), was recently developed. Phor21-βCG(ala) consists of the lytic peptide Phor21 (three heptads of the amino-acid sequence KFAKFAK) linked to 15 amino acids (81–95) of the βCG, in which the cysteines were replaced by alanines (Phor21-βCG(ala); Figure 1).
Figure 1.
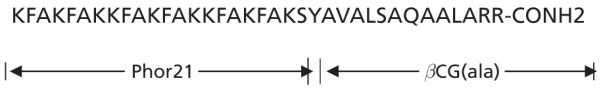
Structure of Phor21-βCG(ala).
In early preclinical studies, Phor21-βCG(ala) was shown to be effective in reducing primary tumour weight in nude mice bearing human tumour xenografts at much lower dose levels than Hecate-βCG or Phor14-βCG (Hansel et al 2007a). It reduced breast and prostate metastatic cells in bones and lymph nodes to nearly undetectable levels (Hansel et al 2007a, b). Similar to the actions of Hecate-βCG and Phor14-βCG, Phor21-βCG(ala) had adverse effects on ovarian follicle growth and corpus luteum formation in mice; however, no histopathological lesions were observed in other tissues (Leuschner & Hansel 2005). The lytic peptide conjugates have the potential to selectively destroy metastatic cancer cells that express high levels of LH/CG. A comprehensive understanding of the molecule’s characteristics, including its in-vitro inhibition of growth of human prostate cancer cells, its destruction of prostate metastases in animal models and its biostability and pharmacokinetic profile in animals, is important for further development of Phor21-βCG(ala). As of this writing, however, no pharmacokinetics and little cytotoxicity data have been reported for Phor21-βCG(ala). To this end, in-vitro studies were conducted to characterize Phor21-βCG(ala) with regard to cytotoxicity using human prostate PC-3 cells that express LH/CG receptors and Caco-2 adenocarcinoma cells that do not. In-vivo pharmacokinetic studies in mice and rats, and studies of the effects of Phor21-βCG(ala) on disseminated luciferase transfected PC-3 cells (PC-3.luc) implanted in nude mice were also conducted to facilitate future development of Phor21-βCG(ala).
Materials and Methods
Test compound and animal care
Phor21-βCG(ala) (MW 4010; Lot No. 03TR1211) was synthesized by SynPep Corporation (Dublin, CA, USA) and maintained at 5°C. The stated purity of the compound, as determined by HPLC analysis, was 95.0%. All animals used in these investigations were handled in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996) and the Guidelines of the US Department of Agriculture (Animal Welfare Act; Public Law 99-198). The institutional animal care and use committee reviewed and approved all animal use procedures based on the above regulations, including use of appropriate species, quality and number of animals, avoidance or minimization of discomfort, distress and pain of animals in concert with sound science, use of appropriate anaesthesia and euthanasia.
Cytotoxicity assay
Human Caco-2 and prostate PC-3 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and grown under standard cell culture conditions in RPMI-1640 medium containing 5% fetal bovine serum and 2 mm glutamine (complete medium). Cell cultures were passaged twice weekly by harvesting and suspending in fresh complete medium. The LH/CG receptor levels were determined according to our established method (Leuschner et al 2003a, b). The cell count was determined with a Coulter Model Z1 cell counter and viability was measured with propidium iodide staining followed by analysis on a Beckman Coulter EPICS XL flow cytometer.
Tissue culture cluster plates (96-well) were seeded at a density of 30 000 cells per well in 200-μL volumes. The wells were rinsed with complete medium before plating to ensure uniform cell attachment. The plates were incubated for 72 h until the cells reached 90% confluence, which was confirmed by microscopic inspection. The complete medium was removed and replaced with 100 μL of a 2× concentrated dosing solution to obtain final concentrations of Phor21-βCG(ala) in the range 0.06–125 μm. Eight wells per concentration were treated. Each cluster plate also contained 8 wells each of a medium control (no cells, background) and an untreated control. After dosing was completed, the plates were incubated at 37°C with 5% CO2 and 95% humidity for 6 h. Viable cell number was measured using the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) (Promega, Madison, WI, USA) cell viability assay (Jia et al 2003). The assay involved incubating cells for 4 h with MTS and an electron-coupling reagent (phenazine ethosulfate). MTS was bio-reduced by viable cells into a formazan that was soluble in tissue culture medium. The absorbance of formazan in each monolayer was measured at 490 nm on a microplate reader.
The data were transferred and processed in an Excel spreadsheet program to calculate the mean and standard deviation for viable cell number as estimated by optical density values. The potency of Phor21-βCG(ala) to reduce viable cell number was analysed using Prism 4 software (GraphPad Software, Inc., San Diego, CA, USA) and expressed as an IC50 value (50% inhibitory concentration).
Mice injected with PC-3.luc and treated with Phor21-βCG(ala)
The study tested the hypothesis that Phor21-βCG(ala) was able to destroy disseminated cells derived from prostate cancer xenografts in nude mice. Briefly, the PC-3 cells were transfected with exogenous DNA by lipofection using the plasmid pRC/CMV-luc containing the Photinus pyralis luciferase gene and an antibiotic resistance gene under the transcription control of the cytomegalovirus promoter (Rubio et al 1998, 2000). The stably transfected PC-3.luc cells were selected and only the clones with the highest expression of luciferase gene were used and characterized. The passage numbers of PC-3 cells used in this study were 26–28.
To assess the ability of Phor21-βCG(ala) to inhibit growth and development of PC-3.luc cells, the transfected cells suspended in phosphate-buffered saline were injected into the thigh muscles of male athymic Balb/c nude mice, as described by Rubio et al (1998, 2000), which were then treated with Phor21-βCG(ala). The nude mice were purchased at 4 weeks of age from Harlan Sprague Dawley (Indianapolis, IN, USA) and were housed in autoclaved cages fitted with high efficiency filter-tops and with autoclaved bedding. The mice were fed with irradiated Purina Chow (5350) and allowed free access to autoclaved water. After quarantine, the sexually mature mice, 6 weeks of age and 25–30 g, were inoculated with a suspension of 1 × 106 PC-3.luc cells into the thigh muscle using a 27-gauge needle (Rubio et al 1998, 2000). Body weights were determined weekly and at necropsy.
The mice were allotted into 4 groups (n = 8 per group) and were treated according to the following protocol: Group 1, treated on days 1, 2 and 3 after tumour cell injection and necropsied on day 4; Group 2, treated on days 7, 8 and 9 after tumour cell injection and necropsied on day 10; Group 3, treated on days 14, 15 and 16 after tumour cell injection and necropsied on day 17; and Group 4, treated on days 21, 22 and 23 after tumour cell injection and necropsied on day 24. Treatment consisted of tail-vein injections of saline (control) or 0.8 mg kg−1 of Phor21-βCG(ala). At necropsy the mice were weighed. Their hind legs were removed, weighed, frozen in liquid nitrogen and stored at −80°C until further analysis.
Luciferase-positive cells were determined in the clear supernatants of whole homogenates of each hind leg prepared in fresh reporter lysis buffer (Promega, Madison, WI, USA) using the luciferase assay kit following the manufacturer’s procedures. Standard curves were prepared from homogenates of legs removed from non-tumour-bearing mice spiked with known numbers of PC-3.luc cells. The number of luciferase-positive cells per gram of leg tissue was calculated from the difference in the values of relative light units from legs from control mice without tumours and from mice with tumour cells and divided by the slope of the standard curve, as described by Rubio et al (1998, 2000).
Stability of Phor21-βCG(ala) in plasma
Sample preparation for stability of Phor21-βCG(ala) in plasma was similar to that previously described (Jia et al 2005a). Briefly, Phor21-βCG(ala) was added to mouse, rat, dog or human plasma at a final concentration of 10 μg mL−1. Samples were then maintained in a 37°C water bath for 0.25, 0.5, 1, 2, 4 and 6 h. The samples were also maintained at 5 and −20°C (freezer) for a certain period of time to determine proper handling or shipping conditions for internal and external collaborative studies. Phor21-βCG(ala) concentrations in individual samples were measured at the beginning (time 0) and end of each incubation period using liquid chromatography tandem mass spectrometry (LC/MS/MS).
To test the hypothesis that proteases play a role in hydrolysing the peptide drugs, Phor21-βCG(ala) (10 μg mL−1) was incubated with two typical proteases, trypsin and chymotrypsin, respectively, at 37°C in phosphate-buffered saline (pH 7.4) for 6 h, and the samples were separated and analysed by the LC/MS/MS method. The MS Digest Program (Protein Prospector, San Francisco, CA, USA) was used to predict the possible hydrolysed fragments.
Analytical method for Phor21-βCG(ala)
The HPLC system consisted of a Perkin-Elmer (Foster City, CA, USA) series 200 autosampler and two series 200 micropumps. Separation of Phor21-βCG(ala) and the internal standard (Phor14-βCG) was achieved using a Betabasic C18 HPLC column (150 × 2 mm i.d., 5 μm particle size) (ThermoElectron, Bellefonte, PA, USA) protected by a Betabasic C18 guard cartridge (ThermoElectron). A gradient elution profile, delivered at a flow rate of 0.3 mL min−1, consisted of de-ionized water and acetonitrile each with 0.1% trifluoroacetic acid (TFA); 20% acetonitrile was held for 0.5 min and then increased over a 9.5-min period to 60% acetonitrile, and then decreased back to 20% acetonitrile and re-equilibrated for 3 min.
The elutes were analysed on a PE Sciex (Concord, Canada) API 3000 LC/MS/MS equipped with an electrospray ionization source operated at 5 KV and 450°C (Jia et al 2005b). High-purity nitrogen was used as the curtain gas while hydrocarbon-free air was used for nebulization. The ring and orifice potentials were set at 80 and 350 V, respectively. The instrument was operated in the positive-ion mode using nitrogen as the collision gas and employing multiple reaction monitoring detection. Multiple ion transitions as a result of different mass to charge (m/z) ratios produced by electrospray ionization of Phor21-βCG(ala) were summed using a dwell time of 200 ms for each transition (M+4H)+4, (M+5H)+5 and (M+6H)+6 at m/z 1003.7, 803.2 and 669.5, respectively. Quantitation was based on the formation of the fragment ion at m/z 129.3. Instrument control and quantitation were performed using Analyst 1.4 (Applied Biosystems, Foster City, CA, USA).
Blood sample preparation and extraction of Phor21-βCG(ala)
Plasma samples (100 μL) used in the in-vitro and in-vivo studies were fortified with 10 μL of a 50 μg mL−1 internal standard spiking stock, vortexed and then mixed with 400 μL of 1 m HCl to make the final concentration 1 μg mL−1. Control plasma samples, spiked with known amounts of Phor21-βCG(ala), were similarly fortified with the internal standard and mixed with 4 volumes of 1 m HCl. Red blood cell (RBC) samples were lysed in 4 volumes of 1 m HCl and then fortified with the internal standard, at the same final concentration as added to plasma samples (1 μg mL−1). For extraction, 500 μL of each plasma or RBC sample were loaded into individual wells of a preconditioned OASIS HLB (10 mg) 96-well solid-phase extraction plate (Waters Corp., Milford, MA, USA). Preconditioning of the wells consisted of eluting 500 μL of acetonitrile through each well followed by 500 μL of 2% ammonium hydroxide. After the samples were loaded, each well was washed with 500 μL of a 10% acetonitrile solution containing 0.1% TFA and eluted with 75 μL of a 60% acetonitrile solution containing 0.1% TFA, followed by an additional 75 μL of 0.1% TFA. A 125-μL volume of the eluate was injected into the LC/MS/MS.
Pharmacokinetics of Phor21-βCG(ala) in mice and rats
Male C57BL/6 mice were procured from Charles River Laboratories (Kingston, NY, USA). The mice were approximately 11 weeks old and weighed 24.3 ± 0.7 g on the day of dosing. Male Fischer rats were obtained from Charles River Laboratories (Portage, MI, USA); each rat was procured with an indwelling jugular vein catheter. The rats were approximately 10 weeks old and weighed 201 ± 8 g on the day of dosing.
Dose solutions of Phor 21-βCG(ala) were prepared in 0.9% sterile saline. Each mouse was given a single intravenous dose, via a tail vein, of 0.8 or 8 mg kg−1 (2.4 or 24 mg m−2) of Phor21-βCG(ala) in a dose volume of 10 mL kg−1. Each rat was given a single intravenous dose of 6 mg kg−1 (36 mg m−2) of Phor21-βCG(ala) by injection into a tail vein in a dose volume of 5 mL kg−1.
At each of the following times after administration of either dose level of Phor21-βCG(ala), three mice were anaesthetized with CO2–O2 and approximately 0.6 mL of blood was collected from the retro-orbital plexus of each mouse: 2, 5, 15 and 30 min and 1, 2, 4 and 8 h. Blood samples were collected from the jugular vein catheter of un-anaesthetized rats (n = 4 per time point) at 2, 5, 15, 30 min and 1, 2, 4 and 8 h after dosing. Blood samples collected into EDTA-containing tubes were immediately placed on ice and then centrifuged to separate plasma and RBCs, both of which were stored at −70°C before LC/MS/MS analysis.
The plasma concentration data were subjected to pharmacokinetic analysis using WinNonlin (Professional Version 4.1; Pharsight Corp., Mountain View, CA, USA). The area under the plasma concentration versus time curve (AUC) was calculated from time 0 to infinity.
Statistical analysis
All results were expressed as mean ± s.d. unless otherwise noted. To analyse experimental results, the Student’s t-test or one-way analysis of variance was applied to compare the means of two or more groups simultaneously and examine significance of differences, and P < 0.05 was considered to be statistically significant.
Results
Cytotoxicity of Phor21-βCG(ala)
In-vitro studies were conducted to determine the difference in cytotoxicity of Phor21-βCG(ala) against the human prostate PC-3 cells and Caco-2 adenocarcinoma cells. The former expressed significant numbers of the LH/CG receptor and the latter did not. Phor21-βCG(ala) reduced viable cell number in both cell lines in a dose-dependent manner. The PC-3 cell line was more sensitive to Phor21-βCG(ala) with an IC50 value of 5.5 μm compared with the human Caco-2 cell line, which displayed an IC50 of 52.2 μm (Figure 2). Since the cytotoxic effect of a test drug can also be dependent on time of cell exposure to the drug (Jia et al 2003), we found that longer exposure of the Caco-2 cells to Phor21-βCG(ala) up to 72 h increased the cells’ sensitivity to the peptide.
Figure 2.
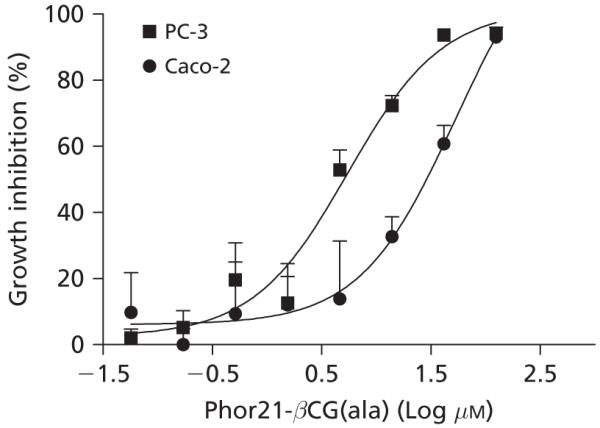
Growth inhibitory activity of Phor21-βCG(ala) against human prostate PC-3 and Caco-2 adenocarcinoma cell lines following the 6-h treatment. Data are means ± s.e., n = 8 per concentration.
Treatment of nude mice with Phor21-βCG(ala)
Luciferase-positive cell determinations were used as a measure of live PC-3.luc tumour cells. Characterization of the PC-3 cells demonstrated that neither the receptor binding capacity nor the EC50 values (concentration producing 50% of the maximum response) of the PC-3 cells were significantly affected by transfection with the luciferase gene: EC50 values for the transfected cells were 5.6 ± 1.3 μm compared with 6.1 ± 1.6 μm for the wild type cells (P > 0.05). Figure 3 shows the numbers of luciferase-positive cells in the dissected legs of mice treated with Phor21-βCG(ala) or saline on days 1–3, 7–9, 14–16 and 21–23 and necropsied on days 4, 10, 17 and 24, respectively, after tumour cell injection. Luciferase-positive cells were significantly decreased in all treatment groups compared with saline control (P < 0.004, n = 8 per group), suggesting destruction of disseminated PC-3.luc cells seeded in the muscle. In contrast, saline controls had large numbers of PC-3.luc cells, indicating live tumour cells at the time of necropsy. There were no significant differences in luciferase-positive cells among either saline-treated control mice or among the treated mice. Body weights remained stable during the entire study in both treated and control mice and there were no significant differences among the treated groups, suggesting that the dose of Phor21-βCG(ala) was adequate to destroy the disseminated tumour cells without producing toxicity. As expected, no primary vascularized tumours were visible in this experiment because the primary vascularized tumours require 20 days to develop after intramuscular injection of PC-3 cells (Rubio et al 1998, 2000).
Figure 3.
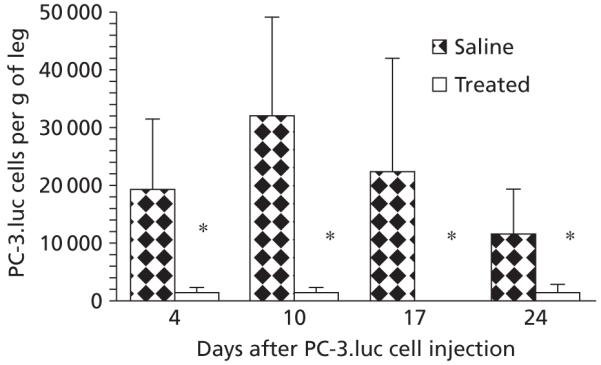
Inhibition of proliferation of PC-3.luc cells by Phor21-βCG. Mice (n = 8 per group) were inoculated with PC-3.luc into the thigh muscle and treated with 0.8 mg kg−1 of Phor21-βCG-ala (q.d.x3, i.v.) on days 1–3, 7–9, 14–16 or 21–23 after the inoculation. At necropsy on days 4, 10, 17 and 24 following the treatment, the numbers of PC-3.luc cells in the muscle were counted. Phor21-βCG(ala) produced a highly significant reduction in PC-3.luc cells in comparison with the controls (*P < 0.004, one-way analysis of variance).
Analytical method validation
The validated concentrations of Phor21-βCG(ala) ranged from 200 to 5000 ng mL−1 of all the biomatrices. The lower limit of quantitation was set at 200 ng mL−1. The intra-day accuracy was in the range 90.2–112.7% with the coefficient of variation 2.53–8.53%. The inter-day accuracy was in the range 89.3–107.5% with the coefficient of variation 2.09–12.0%. Figure 4 shows the mass spectra of Phor21-βCG(ala) separated from mouse plasma by the solid phase extraction. The following mass transitions were summed for analysis: 1003–129, 803–129 and 669–129.
Figure 4.
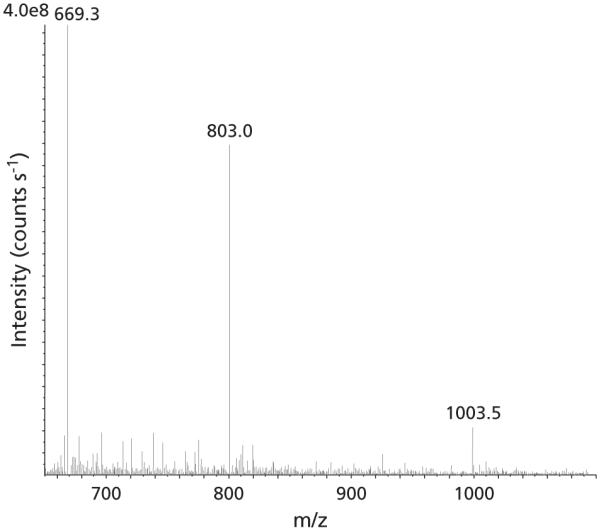
Mass spectra of Phor21-βCG(ala) separated from mouse plasma. The spectra showed multiply charged ions of the peptide at m/z 1003.5, (M+4H)+4; 803, (M+5H)+5; and 669.3, (M+6H)+6.
Phor21-βCG(ala) stability
When maintained at 37°C, Phor21-βCG(ala) (10 μg mL−1) was most stable in mouse and dog plasma followed by human plasma and least stable in rat plasma. After a 6-h incubation at 37°C, the extent of degradation of Phor21-βCG(ala) was 13, 15, 42 or 69% when incubated in dog, mouse, human or rat plasma, respectively (Figure 5). No degradation of Phor21-βCG(ala) was observed in mouse, rat, dog or human plasma when the samples were stored at 5°C for 2 h and at −20°C for 7 days (data not shown). After incubation of Phor21-βCG(ala) with either trypsin or chymotrypsin, several of the predicted fragments were observed in the incubation mixtures by mass analysis. For example, incubation of Phor21-βCG(ala) with trypsin resulted in peptide fragments KFAK, FAKFAK and SYAVALSAQAALAR, whereas incubation of Phor21-βCG (ala) with chymotrypsin produced peptide fragments KF, AKF and AKKF. These fragments are highly likely to be produced by hydrolysis of trypsin and chymotrypsin, which preferentially act at basic and hydrophobic amino acid residues of the peptide, respectively.
Figure 5.
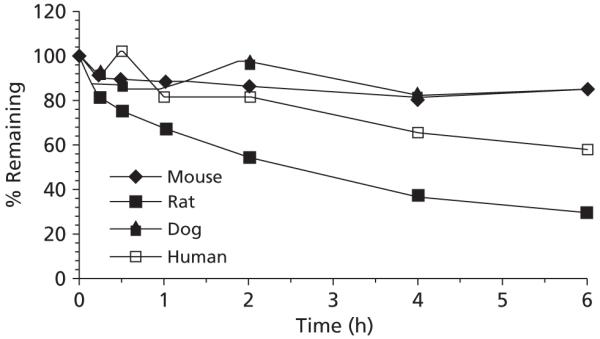
Stability of Phor21-βCG(ala) in mouse, rat, dog or human plasma at 37°C.
Pharmacokinetics of Phor21-βCG(ala) in mice and rats
For mice given the low dose (0.8 mg kg−1) of Phor21-βCG(ala), the data were best fit to a one-compartment model, suggesting that tissue concentrations of Phor21-βCG(ala) are proportional to its concentrations in blood. Data for mice given the high dose (8 mg kg−1) and data for rats were best fit to a two-compartment model. For mice given an intravenous dose of 0.8 and 8 mg kg−1, the mean plasma concentrations of Phor21-βGC(ala) observed at 2 min after dosing were 8.9 and 88.2 μg mL−1, respectively (Figure 6), demonstrating that the plasma concentrations of Phor21-βCG (ala) are proportional to the doses given. Thereafter, the concentration of Phor21-βCG(ala) in plasma declined and approached the limit of quantitation within 2 h (0.8 mg kg−1), or after 8 h (8 mg kg−1) following dosing (Figure 6). The Cmax extrapolated to time 0 was 18.8 ± 1.4 and 88.5 ± 3.5 μg mL−1 (or 4.7 ± 0.4 and 22.1 ± 0.9 μm) in mice with the calculated AUC0→∞ at 2.6 ± 0.3 and 50.8 ± 6.9 μg h mL−1 (or 0.64 ± 0.1 and 12.7 ± 1.7 μm h−1) corresponding to the two dose levels. The results suggest that the therapeutic levels (~μm) of Phor21-βCG(ala) tested in the in-vitro cytotoxic studies (Leuschner et al 2001, 2003a, b; Hansel et al 2007a) are achievable in the mice following i.v. administration. Plasma concentrations of Phor21-βCG(ala) declined slowly at clearance 157 (0.8 mg kg−1) or 313 (8 mg kg−1) mL kg−1 h−1 with terminal t½ 5.0 h (Table 1), indicating that the residual of Phor21-βCG(ala) in the body is long sustained. The mean volume of distribution at steady state (Vdss) for Phor21-βCG (ala) was in the range 170–287 mL kg−1, which is moderate in comparison with other drugs we studied (Jia et al 2003, 2005b, 2008a, b), indicating that Phor21-βCG(ala) was largely distributed within the blood compartment.
Figure 6.

Concentrations of Phor21-βCG(ala) in rodent plasma or RBCs over time following intravenous administration of 0.8 or 8 mg kg−1 to mice (A), or 6 mg kg−1 to the catheterized rats (B). Data are means ± s.e., n = 3 or 4 per time point.
Table 1.
Pharmacokinetic parameters for mice and rats dosed intravenously with Phor21-βCG(ala)
| Parameter | Mouse | Mouse | Rat |
|---|---|---|---|
| Dose (mg kg−1) | 0.8 | 8 | 6 |
| AUC0–∞ (μg · h mL−1) | 2.6 ± 0.3 | 50.8 ± 6.9 | 14.6 ± 2.5 |
| Cmax (μg mL−1) | 18.8 ± 1.4 | 88.5 ± 3.5 | 43.0 ± 4.0 |
| t½½α (h) | 0.46 ± 0.12 | 0.32 ± 0.02 | 0.13 ± 0.02 |
| t½½β (h) | ND | 5.0 ± 6.6 | 2.25 ± 1.4 |
| Cl (mL kg−1 h−1) | 313 ± 41 | 157 ± 22 | 412 ± 70 |
| Vdss (mL kg−1) | 170 ± 30 | 287 ± 32 | 666 ± 33 |
Data are means ± s.d., n = 3 or 4. Cmax, maximum plasma concentration extrapolated to time 0. t½α and t½β denote half-life of the distribution phase and the terminal elimination phase, respectively.
In rats, the mean plasma concentration of Phor21-βGC(ala) observed at 2 min after dosing (6 mg kg−1) was 37.3 μg mL−1. The Cmax extrapolated to time 0 was 43.0 ± 4.0 μg mL−1 (or 10.7 ± 1.0 μm) with the calculated AUC0→∞ at 14.6 ± 2.5 μg h mL−1 (or 3.6 ± 0.6 μm h−1), suggesting again that the therapeutic levels (~μm) of Phor21-βCG(ala) demonstrated in cytotoxicity studies (Leuschner et al 2001, 2003a; Hansel et al 2007a) are achievable following i.v. administration. Plasma concentrations of Phor21-βCG(ala) in rats declined relatively fast at clearance 412 ± 70 mL kg−1 h−1 with elimination t½ 2.25 h and Vdss 666 ± 337 mL kg−1 (Table 1).
To assess the extent of uptake or binding of Phor21-βCG(ala) by or to RBCs, the corresponding RBC samples were also analysed by LC/MS/MS. In general, the corresponding RBC concentration of Phor21-βCG(ala) in mice and rats was 8- to 12-fold lower than that in plasma, indicating that the majority of the peptide stays in plasma (Figure 6).
Discussion
In these studies, the cytotoxic effects of Phor21-βCG(ala) against androgen-independent PC-3 cells and carcinoma Caco-2 cells were confirmed. PC-3 cells showed sensitivity to Phor21-βCG(ala) 10-fold higher than Caco-2 (Figure 2). Previous investigations have demonstrated that IC50 values for lytic peptide βCG-conjugates and LH receptor binding capacities are highly correlated (R2 = 0.708) in a number of cell lines (Leuschner & Hansel 2005). Our results further established the specificity of Phor21-βCG(ala) for cells expressing LH/CG receptors.
Phor21-βCG(ala) treatment of mice bearing disseminated PC-3.luc cells resulted in a destruction of the tumour cells without overt drug toxicity, as indicated by the absence of toxic signs and treatment-related body-weight loss. Tumour necrosis and poor vascularization of tumours were observed in mice bearing PC-3 cells that were treated with Hecate-βCG or Phor14-βCG (Leuschner et al 2001; Hansel et al 2001). Although testicular damage, as demonstrated by the absence of primary and secondary spermatocytes and spermatids in the testicular tubules and spermatozoa in the epididymides, was observed in these mice bearing PC-3 cells, no histopathological lesions were noted in the liver, spleen, heart, kidney, adrenals, pancreas, lungs or pituitary. Similarly, the lytic peptide conjugates inhibit ovulation and corpus luteum formation in female mice (Leuschner & Hansel 2005).
Low levels of LH/CG receptors have been reported in extragonadal organs (brain, adrenal, hypothalamus, lymphocytes, blood vessels, skin) but no lesions have been seen in these organs after treatment. It was observed in the present studies that Phor21-βCG(ala) produced a highly significant (P < 0.004) reduction in the number of PC-3 cells in the legs of mice treated with Phor21-βCG(ala) on days 1–3, 7–9, 14–16 or 21–23 after tumour cell injection (Figure 3). At the time of the initiation of Phor21-βCG(ala) treatments, the PC-3 tumour cells were present as disseminated cells rather than vascularized tumours. The remarkable ability of Phor21-βCG to destroy disseminated PC-3 cells is in accord with previous studies in which all lytic peptide-βCG conjugates tested were shown to regress prostate and cancer cell xenografts and to destroy prostate and breast cancer metastatic cells in bones, lungs, lymph nodes and other tissues (Leuschner et al 2001, 2003; Hansel et al 2007a, b). In contrast to this study, these previous studies were conducted with nude mice bearing established tumours. The exact mechanism of action of Phor21-βCG(ala) and related lytic peptide conjugates has not been delineated; however, it has been shown that lytic peptides disrupt tumour cell membranes within minutes (Leuschner & Hansel 2004). It has been suggested that pore/ion channels are formed from disruption of lipids and plasma membrane proteins by the lytic peptides resulting in release of phospholipids from plasma membranes. These actions result in cell necrosis rather than apoptosis (Leuschner & Hansel 2005).
In-vitro plasma stability studies are important for predicting whether the in-vivo clearance of a compound is due to enzymatic degradation, and for determining proper methods for handling and processing of samples collected during pharmacokinetic, drug distribution and/or metabolism studies (Jia & Liu 2007). These studies also serve as a tool for identifying unstable drug candidates early in the drug development process (Liu & Jia 2007). The data obtained during the stability studies indicated that stability of Phor21-βCG(ala) in plasma was at least affected by thermodynamic process and protease hydrolysis. A possible species difference in the degradation of Phor21-βCG(ala) was also noted at 37°C (Figure 5), and it may be predictive of possible species differences in in-vivo efficacy and pharmacokinetics of Phor21-βCG(ala). The important class of serine proteases includes trypsin and chymotrypsin, which were used in our studies. All serine proteases have a serine residue that plays a critical role in the catalytic process, and preferentially cut a peptide chain just to the carboxyl side of specific kinds of amino acid. Trypsin cuts preferentially to the carboxylate side of basic amino acid residues, like lysine (Lys, or K) or arginine (Arg, or R), whereas chymotrypsin acts at hydrophobic residues, such as phenylalanine (Phe, or F) or leucine (Leu, or L) (Mathews & van Holde 1991). The trypsin-like activity in blood is well recognized. These enzymes enter the blood stream from either the pancreas or intestine (Proskuryakov & Dubinkin 1971). Incubation of Phor21-βCG(ala) with trypsin or chymotrypsin at 37°C resulted in mass profiles of trypsin- or chymotrypsin-hydrolysed peptide fragments (not shown), suggesting involvement of the proteases in metabolism of Phor21-βCG(ala).
The concentrations of Phor21-βCG(ala) in plasma at the early times after dosing were approximately 10-fold higher than the corresponding concentrations in RBCs, indicating that Phor21-βCG(ala) had a low affinity for RBCs. The mean plasma concentration of Phor21-βCG(ala) at 2 min after dosing was approximately 10-fold higher in mice given 8 mg kg−1 than in mice given 0.8 mg kg−1, suggesting that blood concentrations of Phor21-βCG(ala) are proportional to the doses given. It is important to establish relationships between systemic exposure measures to a drug and drug-induced effect or toxicity since the effect of the drug in the body is generally a function of its concentration at the site of action. Most commonly, drug plasma concentrations are measured as a more readily accessible surrogate to correlate between the AUC or Cmax of a drug in the plasma and the intensity of its pharmacodynamic effects or toxicity (Jia et al 2005a; 2008b). The measured mean AUC (12.7 μm h−1 in mice and 3.6 μm h−1 in rats) and Cmax (22.1 μm in mice and 10.7 μm in rats) data indicate that plasma levels of Phor21-βCG(ala) following i.v. administration can reach the therapeutic ranges tested in the in-vitro setting (Figure 2; Leuschner & Hansel 2005) and stay for a sufficient period. In fact, we have recently demonstrated that at intravenous doses as low as 8 and 80 μg kg−1 Phor21-βCG(ala) was potent enough to cause statistically significant tumour regression in xenograft nude mice (Hansel et al 2007a, b).
Programming the plasma concentration–time data confirmed the two-compartment model for Phor21-βCG(ala): the initial rapid decline of blood Phor21-βCG(ala) after administration at the distribution t½ within 0.5 h, and the terminal elimination t½ 5 h in mice and 2.3 h in rats (Table 1). The elimination t½ is a derived parameter that changes as a function of both Cl and Vdss. Of particular interest was the observation that Phor21-βCG(ala) possessed a moderate Vdss and relatively slow Cl in both mice and rats. The magnitude of the Vdss is a useful indicator for the amount of drug outside the blood compartment or in the peripheral tissues and tumours. The parameters elimination t½, Cl and Vdss determined for Phor21-βCG(ala) in mice and rats further support the notion that the lytic peptide can circulate long enough within the blood compartment and reach out to the peripheral tissues to scavenge the metastatic cells.
These studies concluded that Phor21-βCG(ala) is an effective inhibitor in-vitro of the growth of PC-3 cells and in-vivo of disseminated cells. The therapeutic possibilities for the use of Phor21-βCG(ala) and other targeted lytic peptides for the treatment of metastatic cancer offer a novel approach to cancer chemotherapy. In particular, a potential impact exists for the application of this treatment regimen in cases where early-stage disseminated tumour cells (or metastases) are to be treated. Because of its greater potency, ease of synthesis, acceptable biostability and pharmacokinetic profile, Phor21-βCG(ala) has been chosen for further toxicity studies and human trials.
Acknowledgments
Funding: The studies were supported by Gordon and Mary Cain Foundation, Grant #DAMD-17-03-1-0150 from the Department of Defense, and Contract No. N01-CM-07110 from the National Cancer Institute.
Contributor Information
Lee Jia, The National Cancer Institute, Rockville, MD, USA.
Patricia E. Noker, Southern Research Institute, Birmingham, AL, USA
Gary A. Piazza, Southern Research Institute, Birmingham, AL, USA
Carola Leuschner, Pennington Biomedical Research Center, Baton Rouge, LA, USA.
William Hansel, Pennington Biomedical Research Center, Baton Rouge, LA, USA.
Gregory S. Gorman, Southern Research Institute, Birmingham, AL, USA
Lori U. Coward, Southern Research Institute, Birmingham, AL, USA
Joseph Tomaszewski, The National Cancer Institute, Rockville, MD, USA.
References
- Bodek G, Rahman NA, Zaleska M, Soliymani R, Lankinen H, Hansel W, Huhtaniemi I, Ziecik AJ. A novel approach of targeted ablation of mammary carcinoma cells through luteinizing hormone receptors using Hecate-CGbetat conjugate. Breast Cancer Res. Treat. 2003;79:1–10. doi: 10.1023/a:1023351819956. [DOI] [PubMed] [Google Scholar]
- Gawronska B, Leuschner C, Enright F, Hansel W. Effects of a lytic peptide conjugated to beta-hCG on ovarian cancer studies in vitro and in vivo. Gynecol. Oncol. 2002;85:45–52. doi: 10.1006/gyno.2001.6558. [DOI] [PubMed] [Google Scholar]
- Hansel W. Targeting breast, prostate, ovarian, and testicular cancer through their hormone receptors. Biol. Reprod. 2005;73:850. doi: 10.1095/biolreprod.105.043471. [DOI] [PubMed] [Google Scholar]
- Hansel W, Leuschner C, Gawronska B, Enright F. Targeted destruction of prostate cancer cells and xenografts by lytic peptide-βLH conjugates. Reprod. Biol. 2001;1:20–32. [PubMed] [Google Scholar]
- Hansel W, Enright F, Leuschner C. Destruction of breast cancers and their metastases by lytic peptide conjugates in vitro and in vivo. Mol. Cell. Endocrinol. 2007a;260-262:183–189. doi: 10.1016/j.mce.2005.12.056. [DOI] [PubMed] [Google Scholar]
- Hansel W, Leuschner C, Enright F. Conjugates of lytic peptides and LHRH or βCG target and cause necrosis of prostate cancers and metastases. Mol. Cell. Endocrinol. 2007b;269:26–33. doi: 10.1016/j.mce.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics. CA Cancer J. Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- Jia L, Liu XD. The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr. Drug Metab. 2007;8:822–829. doi: 10.2174/138920007782798207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Wong H, Wang Y, Garza M, Weitman S. Carbendazim: disposition, cellular permeability, metabolite identification and pharmacokinetic comparison with its nanoparticle. J. Pharm. Sci. 2003;92:161–172. doi: 10.1002/jps.10272. [DOI] [PubMed] [Google Scholar]
- Jia L, Coward L, Gorman GS, Noker PE, Tomaszewski JE. Pharmacoproteomic effects of isoniazid, ethambutol, and N-Geranyl-N’-(2-adamantyl)ethane-1,2-diamine (SQ109) on Mycobacterium tuberculosis H37Rv. J. Pharmacol. Exp. Ther. 2005a;315:905–911. doi: 10.1124/jpet.105.087817. [DOI] [PubMed] [Google Scholar]
- Jia L, Tomaszewski J, Hanrahan C, Coward L, Noker PE, Gorman GS, Nikonenko B, Protopopova M. Pharmacodynamic and pharmacokinetic characteristics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005b;144:80–87. doi: 10.1038/sj.bjp.0705984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L, Coward LC, Kerstner-Wood CD, Gorman GS, Noker PE, Kitada S, Pellecchia MP, Reed JC. Comparison of pharmacokinetics and metabolic profiling among Gossypol, Apogossypol and Apogossypol Hexaacetate. Cancer Chemother. Pharmacol. 2008a;61:63–73. doi: 10.1007/s00280-007-0446-3. [DOI] [PubMed] [Google Scholar]
- Jia L, Schweikart K, Tomaszewski J, Page J, Noker PE, Buhrow SA, Reid JM, Ames M, Munn D. Toxicology and pharmacokinetics of 1-methyl-D-tryptophan: absence of toxicity due to saturating absorption. Food Chem. Toxicol. 2008b;46:203–211. doi: 10.1016/j.fct.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone SA, Gelmon K, Mayer LD, Hancock RE, Bally MB. In vitro characterization of the anticancer activity of membrane-active cationic peptides. I. Peptide-mediated cytotoxicity and peptide-enhanced cytotoxic activity of doxorubicin against wild-type and p-glycoprotein over-expressing tumor cell lines. Anticancer Drug Des. 2000;15:151–160. [PubMed] [Google Scholar]
- Leuschner C, Hansel W. Membrane disrupting lytic peptides for cancer treatments. Curr. Pharm. Des. 2004;10:2299–2310. doi: 10.2174/1381612043383971. [DOI] [PubMed] [Google Scholar]
- Leuschner C, Hansel W. Targeting breast and prostate cancers through their hormone receptors. Biol. Reprod. 2005;73:860–865. doi: 10.1095/biolreprod.105.043471. [DOI] [PubMed] [Google Scholar]
- Leuschner C, Enright FM, Melrose PA, Hansel W. Targeted destruction of androgen-sensitive and insensitive prostate cancer cells and xenografts through luteinizing hormone receptors. Prostate. 2001;46:116–125. doi: 10.1002/1097-0045(20010201)46:2<116::aid-pros1015>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Leuschner C, Enright F, Gawronska B, Hansel W. Membrane disrupting lytic peptide conjugates destroy hormone dependent and independent breast cancer cells in vitro and in vivo. Breast Cancer Res. Treat. 2003a;78:17–27. doi: 10.1023/a:1022169525521. [DOI] [PubMed] [Google Scholar]
- Leuschner C, Enright F, Gawronska B, Hansel W. Targeted destruction of prostate cancer xenografts through luteinizing hormone releasing hormone receptors. Prostate. 2003b;56:239–249. doi: 10.1002/pros.10259. [DOI] [PubMed] [Google Scholar]
- Liu XD, Jia L. The conduct of drug metabolism studies considered good practice (I): analytical systems and in vivo studies. Curr. Drug Metab. 2007;8:815–821. doi: 10.2174/138920007782798153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews CK, van Holde KE. Enzymes: biological catalysts. In: Mathews CK, van Holde KE, editors. Biochemistry. 2nd edn Benjamin/Cummings; California: 1991. pp. 339–380. [Google Scholar]
- Proskuryakov MT, Dubinkin DV. Nature of the trypsin-like activity of the blood. Biochem. Biophys. 1971;76:786–788. [Google Scholar]
- Rubio N, Villacampa MM, Blanco J. Traffic to lymph nodes of PC-3 tumor cells in nude mice visualized using the luciferase gene as a tumor cell marker. Lab. Invest. 1998;78:1315–1325. [PubMed] [Google Scholar]
- Rubio N, Villacampa N, Hilali NE, Blanco J. Metastatic burden in nude mice organs measured using prostate tumor PC-3 cells expressing the luciferase gene as a quantifiable tumor cell marker. Prostate. 2000;44:133–143. doi: 10.1002/1097-0045(20000701)44:2<133::aid-pros6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Zaleska M, Bodek G, Jana B, Hansel W, Ziecik AJ. Targeted destruction of normal and cancer cells through lutropin/choriogonadotropin receptors using Hecate-betaCG conjugate. Exp. Clin. Endocrinol. Diabetes. 2003;111:146–153. doi: 10.1055/s-2003-39787. [DOI] [PubMed] [Google Scholar]
- Zaleska M, Waclawik A, Bodek G, Zezula-Szpyra A, Li X, Janowski T, Hansel W, Rahman NA, Ziecik AJ. Growth repression in diethylstilbestrol/dimethylbenz[a]anthracene-induced rat mammary gland tumor using Hecate CGbeta conjugate. Exp. Biol. Med. 2004;229:335–344. doi: 10.1177/153537020422900408. [DOI] [PubMed] [Google Scholar]