

Abstract
Cytokinesis is the process by which a cell physically divides in two at the conclusion of a cell cycle. In animal and fungal cells, this process is mediated by a conserved set of proteins including actin, type II myosin, IQGAP proteins, F-BAR proteins, and the septins. To facilitate biochemical and ultrastructural analysis of cytokinesis, we have isolated and partially purified the Saccharomyces cerevisiae cytokinetic apparatus. The isolated apparatus contains all components of the actomyosin ring for which we tested—actin, myosin heavy and light chain, and IQGAP—as well as septins and the cytokinetic F-BAR protein, Hof1p. We also present evidence indicating that the actomyosin rings associated with isolated cytokinetic apparati may be contractile in vitro, and show preliminary electron microscopic imaging of the cytokinetic apparatus. This first successful isolation of the cytokinetic apparatus from a genetically tractable organism promises to make possible a deeper understanding of cytokinesis.
Keywords: Cytokinesis, cytokinetic apparatus, isolation and partial purification, actomyosin ring, Saccharomyces cerevisiae
Introduction
Understanding the structure and function of supra-macromolecular assemblies—from heterochromatin to the nuclear pore complex—is a crucial frontier in biology. Overcoming the technical challenges to investigating these large assemblies is likely to require several decades of intense effort. Here we focus on an extremely large supra-macromolecular assembly, the cytokinetic apparatus.
Cytokinesis is the process by which the cell physically cleaves in two. In fungal and animal cells, an actomyosin contractile ring mediates cytokinesis by constricting the plasma membrane at the plane of division (Balasubramanian et al. 2004; Glotzer 2005; Rappaport 1996; Wolfe and Gould 2005). While many conserved proteins involved in cytokinesis have been identified (Balasubramanian et al. 2004; Bielak-Zmijewska et al. 2008; Machesky 1998; Wolfe and Gould 2005)—including actin, type II myosins, F-BAR proteins, IQGAP proteins, and the septins—the mechanisms of ring activation, contraction and disassembly remain mysterious. Illuminating these mechanisms will require a definitive list of factors sufficient for ring contraction, as well as experimental tools enabling more detailed biochemical examination of the process. Achieving these goals will likely require a system for investigating ring contraction in vitro.
Also critical to understanding cytokinesis is detailed knowledge of actomyosin ring ultrastructure. Despite the more than thirty years that have passed since the first observations of the actomyosin ring, detailed understanding of its organization is still lacking. Just as determination of the structure of the sarcomere helped illuminate the process of muscle contraction (Huxley 1969), in depth knowledge of actomyosin ring ultrastructure should provide invaluable insight into the mechanism of ring contraction and disassembly. A groundbreaking first step in this understanding occurred recently with the detailed description of the actin filament framework of actomyosin rings from Schizosaccharomyces pombe (Kamasaki et al. 2007). The difficult next step will be to determine how the many other ring components fit into this framework and how ring architecture intersects with other critical cytokinetic factors, such as the septin proteins.
Isolation and purification of the cytokinetic apparatus—that is to say, intact actomyosin rings and associated cytokinetic factors such as septin proteins—is important for addressing these crucial issues (Fujimoto and Mabuchi 1997; Mabuchi et al. 1988; Yonemura et al. 1991). Isolation of the cytokinetic apparatus from an organism with sophisticated genetics has not yet been achieved. The ability to do so from the highly experimentally tractable organism, Saccharomyces cerevisiae, with its excellent genetics and short generation time, would be particularly valuable to the field of cytokinesis. The difficulties of working with an organelle of the size of the cytokinetic apparatus (∼1 μm in S. cerevisiae), as well as the challenges of yeast biochemistry—highly active vacuolar proteases and the lysis-resistant cell wall—have to date, however, presented a formidable challenge to such an approach.
Material and Methods
Yeast Strains
BYY116 (Mat a mob1-77 MYO1-GFP∷KANMX pep4Δ∷TRP1 ura3-52 trp1Δ1 his3Δ200 leu2-3,112); BYY124 (Mat a mob1-77 MYO1-GFP∷KANMX pep4Δ∷TRP1 MLC2-HPM (his9-precission2-myc9)∷HIS3 ura3-52 trp1Δ1 his3Δ200 leu2-3,112); BYY138 (Mat a CDC15L99G pep4Δ∷TRP1 ADE2); BYY143 (Mat α mob1-77 MYO1-GFP∷KANMX pep4Δ∷TRP1 HXT1-mRFP∷HIS3 ura3-52 trp1Δ1 his3Δ200 leu2-3,112 [YEp13-SIC1]); BYY154(Mat α mob1-77 MYO1-GFP∷KANMX IQG1-mRFP∷HIS3 ura3-52 trp1Δ1 his3Δ200 leu2-3,112); BYY155 (Mat α mob1-77 IQG1-mRFP∷HIS3 ura3-52 trp1Δ1 his3Δ200 leu2-3,112); BYY158(Mat α mob1-77 MYO1-GFP∷KANMX pep4Δ∷TRP1 HOF1-HA∷HIS3 ura3-52 trp1Δ1 his3Δ200 leu2-3,112); BYY162((Mat α mob1-77 HOF1-HA∷HIS3 ura3-52 trp1Δ1 his3Δ200 leu2-3,112). All yeast strains (except BYY138) were S288C background and were derived from FLY725 and its parent FLY97 (Luca et al. 2001). BYY138 was W303 background. Deletions and COOH-terminal RFP, HPM and HA tags were constructed by PCR integrations as previously described (Graumann J et al. 2004, Longtine et al. 1998, Toshima JY, et al. 2006). Mlc2p-HPM and Iqg1p-mRFP functionality were validated by: 1) cells carrying the tagged derivatives as the sole copy of the gene are viable; 2) constructs show localization for their respective proteins as previously described in publications; 3) Myo1-GFP ring contraction was verified to occur in strains carrying these constructs as the sole copy of the respective genes. The functionality of the Hof1p-HA construct has been previously described (Vallen et al. 2000).
Purification of Recombinant O. xanthineolytica β-1,3-Glucanase
Recombinant O. xanthineolytica β-1,3-Glucanase obtained by periplasmic shock of E. coli strain RSB805 was a kind gift of Randy Schekmans's laboratory. Briefly, cells induced at OD∼0.5 with 4 mM IPTG for 5 hours were washed (25 mM Tris, pH 7.4) and resuspended in 1/50 volume 25 mM Tris, pH 7.4, 2 mM EDTA. Cells were diluted 2-fold in 40% sucrose, 25 mM Tris, pH 7.4, mixed for 20 minutes and pelleted. Pellet was resuspended in 1/50 volume cold 0.5 mM MgSO4 and mixed 20 minute on ice. The β-1,3-Glucanase containing supernatant was collected by centifugation.
Isolation and Enrichment of the Cytokinetic Apparatus
mob1-77 yeast cells were grown in YPD media at the permissive temperature (∼20°C) to OD∼0.4 and then shifted to 37°C for 4 hours. (Intact ring yields were considerably improved by gradually increasing culture volume over several days while maintaining cultures between OD=0.05 and OD=0.5 as much as possible.) Large quantities (∼12 liters) of cells were pelleted at 4,000xg for 20 minutes in a rotor pre-warmed to 37°C. Smaller quantities (∼1 liter) of cells were harvested using Millipore Express PLUS bottle filters. Pellets were resuspended in ∼1/7 volume of sterile water and frozen by dripping into liquid N2. Care was taken to minimize time at the permissive temperature prior to freezing.
To isolate the cytokinetic apparatus, 0.3 g of frozen cells were rapidly brought to 37°C by addition of 1 ml pre-warmed Sorbitol Buffer (100 mM potassium phosphate, pH 7.0/1.33 M sorbitol/ 40 mM βME). Cells were pre-incubated in a 37°C water bath for 5 minutes before 125 μl (∼200 μg) of recombinant O. xanthineolytica β-1,3-glucanase was added and cells spheroplasted for 15 minutes. Cells were then pelleted at 1,000xg for 2 minutes, washed 2× with 1 ml Sorbitol Buffer and osmotically lysed by resuspending pellet in 2 ml NY buffer (50 mM Hepes KOH, pH 7.5/10 mM MgOAc, pH 7.5/ 60 mM potassium acetate, pH 7.5/1 mM EDTA/ 10% glycerol/ 1 mM DTT) supplemented with 1.8× (18 μl/1 ml) Calbiochem protease inhibitor cocktail set IV. Nonidet P-40 (NP-40; Calbiochem) was added to 0.5% and cells incubated on ice ≥ 10 minutes. Lysate was cleared of unbroken cells by 300xg spin for 5 minutes. (While it is difficult to determine, due to large quantities of unlysed cells, we do not believe significant amounts of rings were pelleted in the 300xg pellet. Two pieces of evidence support this conclusion: 1) repeated 300xg pelleting does not decrease ring titers; and 2) washing 300xg pellets does not release additional Myo1p.)
Protein concentration in purifications was measured by Bradford assay. For subcellular fractionation (Figure 2A), clarified lysates were centrifuged at 13,000xg for 10 minutes and samples of supernatant and resuspended pellet were collected. The 13,000xg supernatants were further centrifuged at 100,000xg for 1 hour and samples of the supernatant and pellet were collected. Samples were supplemented with SDS sample dye, boiled and quantitative immunoblotting was performed using the Li-Cor Odyssey system. Myo1p was probed with polyclonal rabbit α-GFP antibody (1000:1; Torrey Pines.) To count the number of rings in 13,000xg and 100,000xg pellet fractions, ring lysates were diluted into 5 ml of NY buffer supplemented with 0.5% NP-40; .1× (1 μl/ml) Calbiochem protease inhibitor cocktail set IV; and 3 mg/ml glucose. These solutions were spun at 13,000xg for 10 minutes onto glass coverslips using previously described modified Corex tubes (Mitchison and Kirschner 1984). To prevent drying of slides upon removal from buffer, coverslips affixed to metal washers with double-side tape (to create a buffer chamber) were used. After the 13,000xg spin coverslips were removed and solutions spun onto fresh coverslips for the equivalent of 100,000xg for 1 hour (27,750xg for 3 hours and 36 minutes). After centrifugations, catalase and glucose oxidase were added to slides to prevent photobleaching (to 20 μg/ml and 100 μg/ml respectively); additional Calbiochem protease inhibitor cocktail set IV was also added (to 1× concentration). In microscopic counting of rings (and thoroughout this paper) an Olympus IX-71 microscope with a 100× NA1.4 objective was used. A long pass GFP filter cube (GFPLP) was used in ring counting to allow differentiation of green GFP signal from yellow/orange autofluorescence.
Figure 2.
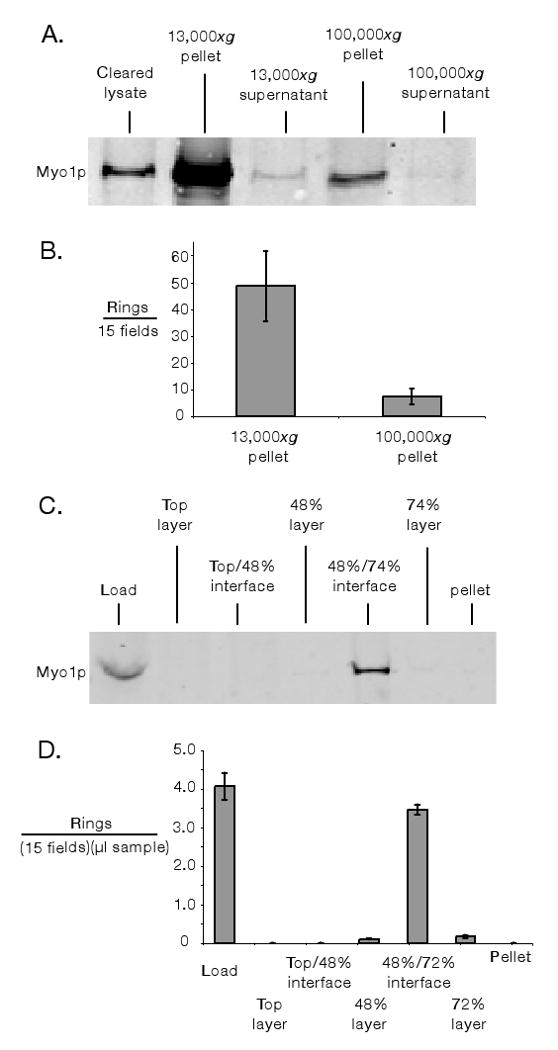
Concentration and enrichment of the budding yeast cytokinetic apparatus. A & B) Differential subcellular fractionation of lysate (cleared of unlysed cells by 300xg spin). In A) 10 μg of each fraction was assayed for Myo1-GFP by quantitative immunoblotting. In B) the number of intact Myo1-GFP rings pelleted in 13,000xg and 100,000xg fractions was measured by spinning 15 μg clarified lysate onto slides and counting rings using fluorescence microscopy. C & D) Discontinuous sucrose density gradient fractionation. In C) 25 μl of each fraction was assayed for Myo1p by quantitative immunoblotting. In D) fractions were assayed by pelleting Myo1-GFP rings onto slides and counting microscopically. In both B & D) only morphologically distinct rings (crisp fluorescent outline, closed figures where the outline does not cross itself) were counted. “Fields” refers to ocular fields using a 100× objective (Area= 38,013 μm2). Error bars represent standard error of the mean of three measurements of ring numbers.
For discontinuous sucrose gradient fractionation, isolation was as described above, except 0.7 g of frozen cells were osmotically lysed in a smaller volume, 1166 μl, of buffer NY with 1.8× Calbiochem inhibitors. 750 μl cleared lysate was layered over a 5.7 ml 48%/5.7 ml 74% sucrose density step gradient (layers contained buffer NY, 1× Calbiochem inhibitors, 0.5% NP-40) and centrifuged for 2 hours and 30 minutes at 150,000xg. Fractions of the top layer, interfaces, sucrose layers and pellet were collected. Myo1p fractionation was analysed by quantitative immunoblotting; ring fractionation was analyzed by microscopic counting of rings pelleted for 10 minutes at 13,000xg onto metal washer coverslips (see above).
Best yields of rings were obtained from freshly frozen cells; over a number of months yields seemed to decline. Isolated rings were stable in solution for hours even in very dilute form. Limited freeze/thaw (<2) of isolated rings did not lower ring number, or interfere with association of the known cytokinetic components tested.
Characterization of the Cytokinetic Apparati
To test for association of known cytokinetic components with Myo1-GFP rings, cells were lysed and fractionated on sucrose gradients as above. Aliquots of the 48%/74% interfaces were concentrated by 10 minute 13,000xg pelleting onto coverslips, as above. (Here, however, coverslip buffer chambers were formed by five layers of Scotch tape with a circular window punched in the center.) After spin, excess buffer was removed from coverslips with a Kimwipe.
To test for association of Iqg1p-RFP, 50 μl of buffer NYB (NY with 1× Calbiochem inhibitors, 0.5% NP-40, 3 mg/ml glucose, 20 μg/ml catalase, and 100 μg/ml glucose oxidase) was added to slides. Rings were located on microscope and GFPLP, TRITC (exposure=500 ms) and bright field images were obtained. (When possible rings were identified using the GFPLP cube; the TRITC cube could also be used but RFP's greater susceptibility to photobleaching led to weaker images.)
To test for association of actin, 30 μl of buffer NYBB (NYB with 1mg/ml BSA) was added to slides. Slides were washed 5× with 100 μl buffer NYBB, rings were identified and GFPLP, TRITC (exposure=500 ms) and bright field images were obtained. 0.2 U of Rhodamine Phalloidin (Calbiochem) was then added in 70 μl buffer NYBB. Slides were incubated for >1 hour on microsope. They were then were washed 10× 100 μl NYBB. GFPLP (500 ms), TRITC (50 ms) and bright field images were obtained.
For Mlc2p, Cdc11p antibody staining, 20 μl NYBB was added and slides washed 5× with 100 μl NYBB. Rings were identified and GFPLP, TRITC (exposure 500 ms) and bright fields images were obtained. Primary antibody was added in 20 μl NYBB (Mlc2p—500:1 (from 3 mg/ml) affinity purified mouse monoclonal 9E10 α-myc;; Cdc11p—200:1 (from 0.5 mg/ml) α-Cdc11p ((Carroll et al. 1998)). Slides were incubated for 1 hour at room temperature in primary stain. They were then washed 10× with 100 μl NYBB, 20 μl of solution was removed, and rhodamine coupled secondary antibody was added in 20 μl NYBB (Mlc2p—1000:1 donkey α-mouse; Cdc11p—100:1 donkey α-rabbit). Secondary staining was for 1 hour at room temperature. Slides were then washed 10× with 100 μl NYBB and images obtained on microscope. (GFPLP exposures—500ms. TRITC exposures—Mlc2p 50ms; Cdc11p 10ms). Hof1p antibody staining was similar (1°: 20 μl added of 500:1 (from 1 mg/ml) α-HA.11 rabbit polyclonal (Covance cat# PRB-101P; 2°: 20 μl added of 100:1 donkey α-rabbit Rhodamine; GFPLP=500ms; TRITC=500ms). However, there were the following exceptions. 1) Hof1p association with rings was sensitive to high temperatures, so staining was performed at 4°C and imaging at ∼18°C. 2) As the Hof1p signal is comparatively low, during washes all of the 1° & 2° antibodies were removed and slides washed 20× after 1° and 30× after 2°.
Testing Functionality of the Isolated Cytokinetic Apparati
Clarified lysate (300xg supernatant) containing isolated cytokinetic apparati was prepared as described in Isolation and Enrichment of the Cytokinetic Apparatus except: .15 g frozen BYY143 cells were lysed in 400 μl Buffer NY (1.8× Calbiochem protease inhibitor cocktail set IV and 0.5% NP-40). To test functionality of actomyosin rings, 2 μl of ATP/ATP Regenerating system (120 mM ATP, 400 mM creatine phosphate, 4 mg/ml creatine kinase in Buffer NY without DTT or salt) was added to 20 μl of clarified lysate. 20 μl of this mixture was removed and added to 20 μl of cytokinetic yeast extract (see below). In control reactions 2 μl of 2.5 U/μl Apyrase (Sigma, Grade VI) was added in place of ATP/ATP Regenerating system.
We tested for function by assessing Myo1-GFP ring titers after incubation times in excess of ring contraction/disappearance time in vivo. Reactions were incubated in a room temperature water bath for 40 minutes. Entire reactions were then concentrated by 10 minute 13,000xg pelleting onto metal washer coverslips, and the number of Myo-GFP rings titered as described above. In parallel, 20 μl of untreated cytokinetic apparatus lysate was spun onto a metal washer slide and titered for Myo1-GFP rings.
We also tested for function by assessing changes in the size distribution of Myo1-GFP rings treated with ATP and extract. Reactions (Figure 4C) were incubated for 4 minutes in a room temperature water bath before addition of 2 μl 2.5 U/μl Apyrase, and then spinning onto metal washer coverslips. Control reactions (Figure 4B), in which the ATP/ATP-regenerating system was replaced with 2 μl 2.5 U/μl Apyrase (see above) were incubated for 20 minutes in a room temperature water bath before spinning onto metal washer coverslips. To assess size distribution, fields were first checked by eye to confirm that “rings” photographed by CCD were not autofluorescent debris (were green rather than yellow/orange.) ImageJ software was used to threshold, skeletalize and measure the inner perimeter of rings. To minimize bias, all green figures that appeared to have empty space in the center were converted to skeletalized “rings”. Figures whose calculated inner diameter was less than the resolution of the light microscope (250 nm) were regarded as too morphologically indistinct to be considered.
Figure 4.

Evidence for functionality of isolated cytokinetic apparatus. A) Number of Myo1-GFP rings was measured for isolated rings alone, rings incubated with ATP and cytokinetic extracts; and rings incubated with cytokinetic extract and a highly active ATPase, apyrase, for ATP depletion. Consistent with in vitro ring contraction, ring titers declined in an energy-dependent fashion. Incubation time was in excess of that required for full contraction of rings in vivo. Rings were titered by pelleting onto slides and counted by fluorescence microscopy. “Fields” here refers to ocular fields using a 100× objective. Only morphologically distinct rings were counted. B) Size distribution of isolated Myo1-GFP rings in the absence of activation by ATP and cytokinetic extract (i.e. 0 minutes contraction time) was measured for rings incubated with cytokinetic extract & apyrase. C) Size distribution of isolated rings after incubation with ATP and cytokinetic extract for approximately half the time necessary for ring contraction in vivo. In B & C) ring sizes were determined by photographing pelleted rings and measuring them using ImageJ software. “Fields” here refers to CCD camera fields using a 100× objective. In both B & C) 3 sets of 15 fields were measured. Error bars represent the standard error of the mean.
To prepare cytokinetic extracts, a variety of cell synchronization techniques were explored to synchronize the cells at the time of ring contraction prior to lysate preparation. Ultimately, we found that cells released from the mitotic arrest caused by chemical inhibition of an ATP analog-sensitive allele of the mitotic exit kinase Cdc15p preceded quite synchronously into cytokinesis. Specifically we found that during a ∼15 minute interval after release (∼35-50 minutes), actomyosin rings were contracting in approximately 71% of cells and that at the peak of contraction (∼39 minutes after release), about 40% of the cells were engaged in actomyosin ring contraction (data not shown). To prepare such synchronized yeast, 1 L of BYY138 was grown to OD∼ 0.25 at 30°C (culture volumes were gradually increased over 2 days while maintaining cells in log phase as much as possible). The ATP analog 1NaPP1 was added to a final concentration of 5 μM and the culture grown for another 1.5 hours at 30°C. Cells were harvested using Millipore Express PLUS bottle filters. 1/5 cell volume of 2% glucose; 250 mM HEPES/KOH 7.5; 5 mM MgOAc; 5 mM EDTA; 50% Glycerol; 5× Calbiochem IV protease inhibitor cocktail; 5 mM DTT was added to cells and before freezing in liquid N2.
To prepare extract, frozen cells pellet was supplemented with 1/10 cell volume of frozen 10 mM Benzamidine, 20 μg/μl Aprotinin and 100 μg/μl Leupeptin. This mixture was then ground in a mortar and pestle bathed in liquid N2. This lysed cell powder was thawed, clarified by 5000xg, 30-second centrifugation, and re-frozen in liquid N2.
Correlative Electron Microscopy of Cytokinetic Apparati
Cytokinetic apparati were purified by sucrose gradient as above, but with the following modification. In the Figure 5A lysis buffer and the sucrose gradient layers 1 mM MgCl2/75 mM KCl was substituted for 10 mM MgOAc, pH 7.5/ 60 mM potassium acetate, pH 7.5. In Figure 5B, the lysis and sucrose gradient buffer were as described above; however cytokinetic apparati were incubated overnight with 3.5 μM Phalloidin and 0.8 μM Myosin S1. In Figure 5A, 10 μl of 48%/74% layer interface was spun onto a scotch tape window slide (see Characterization of the Cytokinetic Apparati) on which had been placed a formvar coated copper index grid. The position of Myo1-GFP rings on the index grid was visualized by epifluorescence. The copper grid was fixed with 2% formaldehyde for 15 minutes, washed with ddH20, stained with 1% Tungsten Phosphate for 1 minute and then washed again with ddH20. The grid was then dried and visualized in the electron microscope. In Figure 5B, the procedure was the same except 0.5 % Glutaraldehyde was used to fix the sample, and 1% Uranyl Acetate was used to stain the sample.
Results
We set out to isolate the intact S. cerevisiae cytokinetic apparatus. To allow simple light microscopic assessment of the release and enrichment of the cytokinetic apparatus from cells, S. cerevisiae type II myosin, Myo1p, was fused to GFP. A significant challenge to the isolation of the cytokinetic apparatus is its short life time—at least in fully assembled form—in the cell cycle. (Although S. cerevisiae type II myosin begins to localize to the plane of division early in the cell cycle, actomyosin rings do not form until late in mitosis (Bi et al. 1998; Epp and Chant 1997; Lippincott and Li 1998b; Wolfe and Gould 2005). Hence, we sought to enrich for yeast cells that contain a fully assembled cytokinetic apparatus. Previously, the conserved NDR kinase Mob1p/Dbf2p, the most downstream member of the mitotic exit network, was shown to be involved in triggering actomyosin ring contraction (Luca et al. 2001). At the non-permissive temperature, actomyosin rings assemble but fail to contract in mob1 mutants (Luca et al. 2001). Hence, we used yeast strains carrying a temperature sensitive mob1 allele and enriched for those carrying fully assembled cytokinetic apparatus by incubating cells at the non-permissive temperature. To prevent vacuolar proteases from damaging the cytokinetic apparatus upon lysis, we deleted the gene PEP4, which encodes a master vacuolar proteolytic regulator (Zubenko et al. 1983), from the isolation strain and performed lysis in the presence of a broad spectrum of protease inhibitors.
We explored a wide variety of lysis conditions for our attempts to isolate the cytokinetic apparatus. Strikingly, we found that intact rings of Myo1-GFP could indeed be liberated from S. cerevisiae cells (Figure 1). No such rings could be detected upon lysis of cells that did not express Myo1-GFP (data not shown). Yields were highest from cells briefly spheroplasted using recombinant purified Oerskovia xanthineolytica β-1,3-glucanase and lysed osmotically by dilution into lysis buffer (see Materials and Methods). We were unable to obtain rings from cells spheroplasted for either short or long time periods with the more highly active, commercially available, Zymolyase 100T (Seikagaku)(data not shown). Limited numbers of rings could be obtained when yeast were sheared open by bead beating. However, yields were low and autofluorescence in the lysate increased sharply, making detection of rings difficult (data not shown). Solubilizing membrane lipids using detergents such as NP-40 and octylglucoside (up to 0.5% and 1%, respectively) did not interfere with ring isolation and seemed to reduce autofluorescence (data not shown). As it was initially unclear in which state rings would be most stable, we attempted to isolate rings both from cells arrested in late anaphase due to the mitotic exit arrest phenotype of mob1, or from cells wherein rings persisted into subsequent cell cycles when mob1 mitotic arrest was overcome by high copy expression of Sic1p. We found that rings could be isolated under both conditions (data not shown).
Figure 1.
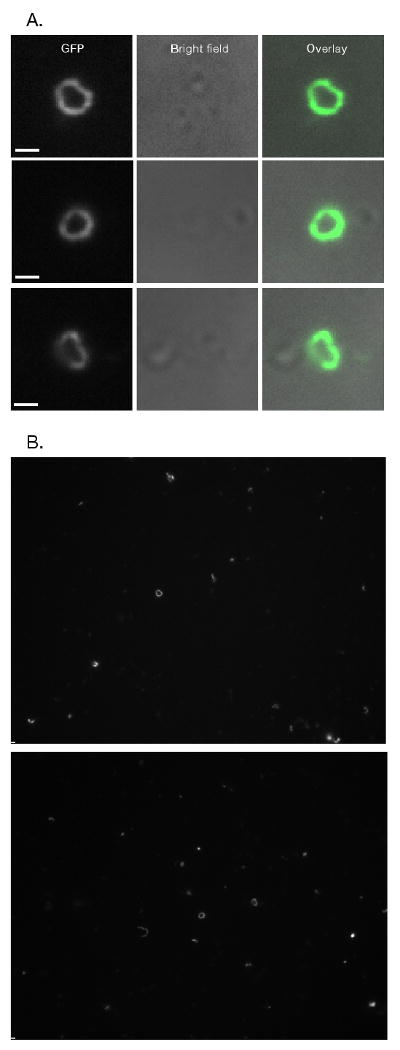
Release of intact Myo1-GFP rings from S. cerevisiae cells. A & B) Isolated Myo1-GFP rings from S. cerevisiae. Rings were concentrated for visualization by 13,000xg pelleting (See Figure 2). A) Comparisions of long pass GFP (GFPLP) fluorescent images and bright field images of close-up views of rings. Bright field images show that rings are not associated with cells. B) whole camera field views of rings. Bars ∼ 1 μm (1032 nm).
Differential centrifugation followed by subcellular fractionation of lysates was performed to enrich for and concentrate rings; fractionation was assayed by quantitative immunoblotting for Myo1-GFP (Figure 2A and Table I). Myo1-GFP was enriched in the 13,000xg pellet (20-fold enrichment), mildly depleted in the 100,000xg pellet (1.1 fold depletion) and strongly depleted from the supernatants (13,000xg supernatant—6.8 fold depletion; 100,000xg—14 fold depletion). The bulk of Myo1-GFP was concentrated in the 13,000xg pellet (75%). We tested if the pelleted Myo1-GFP detected by immunoblotting represented intact ring fractionation by direct counting the rings in the 13,000xg and 100,000xg pellets by fluorescence microscopy (Figure 2B). First attempts to visualize rings in these fractions indicated that when rings were pelleted in tubes they aggregated and resisted homogeneous resuspension. We found it expedient to instead directly pellet rings onto coverslips. This seemed to have the additional benefit of spreading out co-purifying autofluorescent cell debris, reducing out-of-focus fluorescence. Counting rings by fluorescence microscopy supported the conclusion that Myo1-GFP fractionation detected by immunoblotting was representative of fractionation of intact rings (Figure 2B). Typically, we were able to obtain ∼4 intact rings per 100× ocular field when 50 μg of clarified lysate was spun onto a slide. In addition to intact rings, many partial rings and ring fragments were visible in each field.
Table 1. Partial purifications of cytokinetic apparatus.
Fold enrichment of cytokinetic apparatus relative to cleared lysate (300xg supernatant) for two different purifications was measured by quantitative immunoblotting for Myo1p-GFP. For the sucrose gradient purification, enrichment was also quantified by comparing the number of pelletable Myo1-GFP rings in the purified fraction relative to pelletable rings in cleared lysates. (It is not possible to determine by microscopic count the enrichment of the 13,000×g pellet relative to clarified lysate due to our inability to accurately quantitate rings in solution.) Values are those of the representative purifications shown in Figure 2. Each purification was independently performed and quantitated at least twice with similar results.
| Myo1p units | Ring units | ||||
|---|---|---|---|---|---|
| Fraction | Purification | % yield | Fold enrichment | % yield | Fold enrichment |
| Pellet 13,000xg | Differential centrifugation | 75 | 20 | - | - |
| 48/74% interface | Sucrose density gradient | 91 | 34 | 59 | 22 |
We also purified the cytokinetic apparatus by density gradient fractionation. We found that Myo1-GFP rings could be enriched (34-fold) at the 48%/74% interface of a discontinuous sucrose density gradient (Figure 2 C and D and Table I). This purification gives moderately greater enrichment on a protein basis than differential centrifugation (assayed by immunoblotting; Table I), though it does not eliminate autofluorescence contamination. Concentrating dilute 48%/74% interfaces by 13,000xg pelleting did not further enrich the organelle preparation on a protein basis (data not shown).
We used fluorescence microscopy to determine if known components of the cytokinetic apparatus in addition to Myo1-GFP were present in the rings. All known components of the actomyosin ring for which we tested were associated with the isolated rings. S. cerevisiae IQGAP protein, Iqg1p, was found to be associated with rings by purification from a strain in which Iqg1 was tagged with RFP (Figure 3A). Actin was shown to be associated with rings by rhodamine phalloidin staining (Figure 3B). Rings also contain myosin regulatory light chain, Mlc2p, as assessed by antibody staining of rings purified from cells in which Mlc2p was tagged with the myc epitope (Figure 3C; control staining of untagged rings is shown in Supplement Figure S1A). Moreover, we also discovered that the important cytokinetic F-BAR protein, Hof1p, as well as a marker for the septin filaments, Cdc11p, were associated with rings. Hof1 binding was assessed by antibody staining of the cytokinetic apparatus from cells expressing Hof1-HA (Figure 3D; control staining of untagged rings is shown in Supplemental Figure S1B). Septin association was tested using a previously described (Carroll et al. 1998) affinity-purified antibody specific to Cdc11p (Figure 3E). In all cases, we eliminated the possibility that the signal of the component in question was due to fluorescent bleed through from, or photoconversion of, the co-localizing Myo1-GFP (Supplemental Figure S2).
Figure 3.
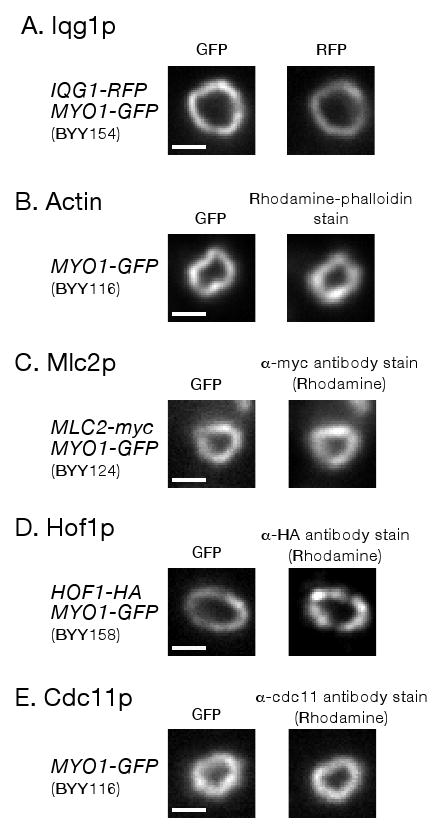
Known components of the cytokinetic apparatus are associated with Myo1-GFP rings purified by sucrose gradient. Rings were purified from cells containing markers indicated to the left (yeast strain number is in parenthesis). A) Iqg1p association was tested by RFP fluorescence imaging. B) Actin association was tested by rhodamine-phalloidin staining. C) Mlc2p-myc association was tested by α-myc antibody staining. D) Hof1p-HA association was tested by α-HA antibody staining. In some cases Hof1p staining throughout rings was less uniform compared to other components. E) Cdc11p (septin) association was tested with affinity purified α-Cdc11p antibody staining. Percentage of rings where component in question was associated: Iqg1p 96% (24/25); Actin 92% (23/25); Mlc2p 100%(15/15); Hof1p 93% (14/15); Cdc11p 100% (15/15). Only morphologically distinct rings (crisp fluorescent outline, closed figures where the outline does not cross itself) were tested for association. Bars ∼1 μm (1032 nm).
The association of all cytokinetic components which we tested for with the isolated cytokinetic apparati strongly implied that they might be functional in vitro (i.e. actomyosin rings might be contractile in vitro). As which mitotic regulators directly trigger contraction of assembled actomyosin ring is not known, we developed a method to add all known and unknown contraction regulators to rings by adding yeast extracts prepared from cells synchronized at cytokinesis. Because we were unsuccessful in establishing a method to observe Myo1-GFP rings free in solution, we assayed for contraction by incubating isolated cytokinetic apparati with ATP and yeast extract for sufficient time for full contraction and disappearance of actomyosin rings in vivo, and then pelleted the apparati and looked for a measurable drop in Myo1-GFP ring titers (Figure 4A). Both extract and cytokinetic apparati were prepared from protease minus (pep4Δ) cells and in the presence of a broad spectrum of protease inhibitors to prevent proteolysis from decreasing rings titers. Consistent with ring contraction in vitro, incubation of cytokinetic apparati with ATP and cytokinetic extracts led to a precipitous decline in Myo1-GFP ring numbers (Figure 4A). This decrease is ATP dependent, as expected for the energy intensive process of actomyosin ring contraction, as little decline was seen in reactions lacking exogenous ATP and containing an active ATPase to deplete any endogenous ATP (Figure 4A). To provide further evidence for in vitro contraction of actomyosin rings, we determined how the size of Myo1-GFP rings changed upon incubation with ATP and extract for intermediate amounts of time sufficient for only partial actomyosin ring contraction in vivo. Likely as a result of the cell size heterogeneity we have observed in our multiply mutant isolation strain (mob1, pep4Δ; data not shown), isolated Myo1-GFP rings show a distribution of sizes (Figure 4B). When rings were treated with ATP and extract for about half of the in vivo contraction time (4 minutes), the distribution shifted toward smaller Myo1-GFP rings (Figure 4C). This data, together with our observations that the isolated cytokinetic apparati include many known components, suggests that the cytokinetic apparati may remain functional when isolated from cells.
We also visualized isolated cytokinetic apparati by correlative electron microscopy. Cytokinetic apparati were pelleted onto formvar electron microscopy finder grids, and Myo1-GFP rings identified using a fluorescence microscope (Supplemental Figure 3A). These rings were then visualized in the EM by negative staining (Supplemental Figure 3B). Filaments are weakly visualizable in the cytokinetic apparati (Supplemental Figure 3C), but amorphous electron dense material obscured other details in the images. We expect that additional extraction of the cytokinetic apparati will be necessary for optimal imaging.
Discussion
As an important step toward studying this large supra-macromolecular assembly in vitro, we have isolated and partially purified the cytokinetic apparatus from S. cerevisiae. This achievement represents the first time that it has been possible to isolate the cytokinetic apparatus from a genetically tractable organism. We showed that the isolated cytokinetic apparatus contains all known components of S. cerevisiae actomyosin rings for which we tested—actin, myosin heavy and light chains, IQGAP—as well as an important cyokinetic component whose association with actomyosin rings is controversial, Hof1p (Lippincott and Li 1998a; Vallen et al. 2000). We also showed that the septin filament protein, Cdc11p, is associated with the isolated cytokinetic apparatus. Moreover, we present suggestive evidence that actomyosin rings associated with isolated cytokinetic apparati may be contractile in vitro. Finally, we presented a preliminary electron microscopic analysis of the isolated cytokinetic apparati.
Pioneering permeabilized cell models and a whole-cell cortex model for actomyosin ring contraction have been described (Cande 1980; Hoffmann-Berling 1954a; Hoffmann-Berling 1954b; Kinoshita and Hoffmann-Berling 1964; Walker et al. 1994). However, progress toward a pure system for studies of ring contraction requires isolation of the cytokinetic apparatus away from other cellular material. The first groundbreaking step towards this goal came with the hand dissection of functional cytokinetic apparati from eggs of the newt Cynops pyrrhogaster ((Mabuchi et al. 1988)). However, difficulties in obtaining large quantities of material by individually dissecting cytokinetic furrows, as well as the challenges of working with this non-canonical organism, have precluded continued biochemical (or structural) progress using this system. Impressively, mass isolation of the cytokinetic apparati from several species of sea urchin, however, has proven possible (Fujimoto and Mabuchi 1997; Yonemura et al. 1991). Here again, however, the difficulties associated with performing isolations from an organism that must be obtained from the wild appears to have impeded further progress.
S. cerevisae cells are readily cultured, easy to manipulate genetically, and have a short generation time, presenting a significant advantage for obtaining sufficient amounts of wild-type or mutant material for cytokinesis studies. The ease of molecular-genetic manipulation in yeast provides further advantages. For example, demonstration that known cytokinetic components in addition to actin are associated with partially purified sea urchin furrows has so far not proven possible (Fujimoto and Mabuchi 1997; Yonemura et al. 1991). (Though in one case, partially purified ring fractions were shown to contain type II myosin by immunoblotting (Yonemura et al. 1991).) In our studies, with the advantage of molecular-genetic tagging, we were able to demonstrate the association of six known cytokinetic components with partially purified S. cerevisiae apparati. Moreover, as Yonemura et al. (9) propose, reactivating contraction of mass isolated actomyosin rings, which has yet to be achieved for cytokinetic apparati purified away from cell cortexes, may require adding back cytoplasmic contraction regulators and/or weakly held subunits of the actomyosin ring. The availability of numerous genetic manipulations allowing cell cycle synchronization in S. cerevisiae may facilitate preparation of cytokinetic extracts to activate ring contraction. Furthermore, the opportunity in S. cerevisiae to use genetically encoded modern affinity purification tags (e.g. TAP tags) to further purify the cytokinetic apparatus and cytoplasmic contraction regulators may facilitate progress toward a pure system that recapitulates in vivo behavior. We note that many of the cases in which a cell-free system for a complex biological process has progressed to a pure system have involved genetic organisms for which very large quantities of cells for biochemical purifications are easily obtained (Kaguni and Kornberg 1984; Matsuoka et al. 1998).
Consistent with the conclusion that the isolated cytokinetic apparati retain functional properties, we observed that the numbers of rings in solution decrease markedly when incubated with ATP and cytokinetic extracts for sufficient time for full ring contraction in vivo. Our use of protease-minus yeast and a broad-spectrum of protease inhibitors made it unlikely that this decline is due to non-specific ring proteolysis. This phenomenon is energy (ATP) dependent as expected for the energy intensive process of contraction. Furthermore, also consistent with the possibility that we succeeded in reconstituting ring contraction in vitro, incubation of the rings with ATP and extract for intermediate lengths of time caused a pronounced shift of the size the distribution toward smaller rings. Unfortunately, when extracts were added to Myo1-GFP rings adhered (by pelleting) to glass coverslips and then visualized by total internal reflection microscopy, Myo1-GFP rings did not contract, likely because of ring/glass adherence (data not shown). Thus, we have not yet succeeded in observing ring contraction in real time. Significantly, when the rings were adhered to glass they did not disappear, suggesting that they are not disassembling due to proteolysis. Ultimately, definitive demonstration of ring contraction will require direct visualization of the process. In any case, it has long been clear that ring disassembly and contraction are tightly coupled (Schroeder 1972). As previously proposed, this coupling may exist because the ring uses disassembly to help drive the contraction process (Cande 1980). (Such a mechanism may explain how it is that rings contract when type II myosins are inactivated (Kanada et al. 2005; Lord et al. 2005; Neujahr et al. 1997)) Our work may provide a system with which to explore the still mysterious process of ring disassembly.
The ability to isolate the cytokinetic apparatus from S. cerevisiae should also facilitate a better ultrastructural understanding of how the components of the actomyosin ring fit into its actin framework. An excellent description of this actin framework has been recently obtained by electron microscopic study of permeabilized Schizosaccharomyces pombe cells (Kamasaki et al. 2007). However, progressing to the next step of understanding how other components fit into this framework is likely to be difficult. Immunogold labeling of permeabilized whole cells prior to plastic embedding for sectioning is unreliable due to the inherently high incidence of non-specific background signal. Conversely, when staining is done after embedding and sectioning, gold-labeled antibodies only have access to epitopes located at the surface of the sections (estimated at between 1-10% of the total amount of epitope within a section)(Griffins and 1996). With such limited access, localizing low abundance subunits of the cytokinetic apparatus may be virtually impossible, and exhaustive localization of even relatively abundant proteins may be problematic. Immunogold EM studies of the isolated budding yeast cytokinetic apparatus could circumvent these difficulties. First, non-specifically bound antibodies can be removed by extensive washing (e.g., see supplementary Figure 1A). Second, antibodies will have access to the entire cytokinetic apparatus prior to embedding, allowing three-dimensional labeling that can be visualized by electron tomography. Moreover, the availability of epitope tagging in S. cerevisiae (as opposed to organisms such as newt or sea urchin) should facilitate progress by allowing standardized use of epitope tag antibodies known to be effective in EM so all subunits can be localized. As the antibody requirements for immunogold EM are notoriously idiosyncratic, with most antibodies used for western blotting and immunofluorescence failing to be effective, the ability to use epitope tags should considerably facilitate progress.
Determining whether our method can be applied to the fission yeast, S. pombe, is of considerable interest. The more advanced state of cytokinesis studies in S. pombe (due to the earlier recognition of actomyosin rings and extensive genetic and image-based analysis in this organism) should make such an effort especially worthwhile. Potentially, however, the more dynamic nature of the actomyosin ring during contraction in S. pombe (Pelham and Chang 2002) relative to the less dynamic, self-contained ring (with components not exchanging with cytoplasm) observed in S. cerevisiae (Dobbelaere and Barral 2004) may make isolation more difficult. Indeed, we were inspired to attempt cytokinetic apparatus isolation from S. cerevisiae by observations suggesting a less dynamic nature for the S. cerevisiae actomyosin ring.
Conclusions
Historically, enabling biochemical interrogation of a well-developed genetic system (and vice versa) has contributed to biological understanding in respects not initially envisioned, as well as in the ways initially foreseen. The system described here promises to allow biochemical and ultrastructural analysis of the contractile mechanism to complement genetic and cell biological studies. As such, we believe our cytokinetic apparatus isolation has the potential to significantly contribute to understanding of the vitally important process of cytokinesis.
Supplementary Material
Acknowledgments
The authors would like to thanks H. Stimpson, V. Okreglak, and J. Woodruff for critical reading of the manuscript; F. Lucas for generously providing yeast strains FLY725 and FLY97; R. Cooke for generously providing Myosin S1; D. Kellogg for generously providing affinity-purified Cdc11p antibody; and Jennifer L. Paulson and Kevan Shokat for sharing their CDC15L99G yeast strain before publication. They would further like to thank all members of the Drubin and Barnes laboratories for useful discussions and experimental advice. This work was supported by a Ruth Kirschstein NIH postdoctoral fellowship to B.A.Y and NIH RO1 grant GM42759 to D.G.D.
References
- Balasubramanian MK, Bi E, Glotzer M. Comparative analysis of cytokinesis in budding yeast, fission yeast and animal cells. Curr Biol. 2004;14(18):R806–18. doi: 10.1016/j.cub.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Bi E, Maddox P, Lew DJ, Salmon ED, McMillan JN, Yeh E, Pringle JR. Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J Cell Biol. 1998;142(5):1301–12. doi: 10.1083/jcb.142.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielak-Zmijewska A, Kolano A, Szczepanska K, Maleszewski M, Borsuk E. Cdc42 protein acts upstream of IQGAP1 and regulates cytokinesis in mouse oocytes and embryos. Dev Biol. 2008;322(1):21–32. doi: 10.1016/j.ydbio.2008.06.039. [DOI] [PubMed] [Google Scholar]
- Cande WZ. A permeabilized cell model for studying cytokinesis using mammalian tissue culture cells. J Cell Biol. 1980;87(2 Pt 1):326–35. doi: 10.1083/jcb.87.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Altman R, Schieltz D, Yates JR, Kellogg D. The septins are required for the mitosis-specific activation of the Gin4 kinase. J Cell Biol. 1998;143(3):709–17. doi: 10.1083/jcb.143.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelaere J, Barral Y. Spatial coordination of cytokinetic events by compartmentalization of the cell cortex. Science. 2004;305(5682):393–6. doi: 10.1126/science.1099892. [DOI] [PubMed] [Google Scholar]
- Epp JA, Chant J. An IQGAP-related protein controls actin-ring formation and cytokinesis in yeast. Curr Biol. 1997;7(12):921–9. doi: 10.1016/s0960-9822(06)00411-8. [DOI] [PubMed] [Google Scholar]
- Fujimoto H, Mabuchi I. Isolation of cleavage furrows from eggs of regular sea urchins and identification of furrow-specific proteins. J Biochem (Tokyo) 1997;122(3):518–24. doi: 10.1093/oxfordjournals.jbchem.a021783. [DOI] [PubMed] [Google Scholar]
- Glotzer M. The molecular requirements for cytokinesis. Science. 2005;307(5716):1735–9. doi: 10.1126/science.1096896. [DOI] [PubMed] [Google Scholar]
- Graumann J, Dunipace LA, Seol JH, McDonald WH, Yates JR, 3rd, Wold BJ, Deshaies RJ. Applicability of tandem affinity purification MudPIT to pathway proteomics in yeast. Mol Cell Proteomics. 2004;3(3):226–237. doi: 10.1074/mcp.M300099-MCP200. [DOI] [PubMed] [Google Scholar]
- Griffins G, B B, Lucocq J. Fine Structure Immunocytochemistry. Berlin; New York: Springer; 1996. p. 459. [Google Scholar]
- Hoffmann-Berling H. Adenosinetriphosphate as the energy substance for cell movement. Biochim Biophys Acta. 1954a;14(2):182–94. doi: 10.1016/0006-3002(54)90157-2. [DOI] [PubMed] [Google Scholar]
- Hoffmann-Berling H. Glycerin water extracted telophase cells as a model of cytokinesis. Biochim Biophys Acta. 1954b;15(3):332–9. doi: 10.1016/0006-3002(54)90034-7. [DOI] [PubMed] [Google Scholar]
- Huxley HE. The mechanism of muscular contraction. Science. 1969;164(886):1356–65. doi: 10.1126/science.164.3886.1356. [DOI] [PubMed] [Google Scholar]
- Kaguni JM, Kornberg A. Replication initiated at the origin (oriC) of the E. coli chromosome reconstituted with purified enzymes. Cell. 1984;38(1):183–90. doi: 10.1016/0092-8674(84)90539-7. [DOI] [PubMed] [Google Scholar]
- Kamasaki T, Osumi M, Mabuchi I. Three-dimensional arrangement of F-actin in the contractile ring of fission yeast. J Cell Biol. 2007;178(5):765–71. doi: 10.1083/jcb.200612018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanada M, Nagasaki A, Uyeda TQ. Adhesion-dependent and contractile ring-independent equatorial furrowing during cytokinesis in mammalian cells. Mol Biol Cell. 2005;16(8):3865–72. doi: 10.1091/mbc.E05-03-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita S, Hoffmann-Berling H. Local Contraction as a Result of Cytoplasmic Cleavage of Fibroblast Cells. Biochim Biophys Acta. 1964;79:98–101. [PubMed] [Google Scholar]
- Lippincott J, Li R. Dual function of Cyk2, a cdc15/PSTPIP family protein, in regulating actomyosin ring dynamics and septin distribution. J Cell Biol. 1998a;143(7):1947–60. doi: 10.1083/jcb.143.7.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott J, Li R. Sequential assembly of myosin II, an IQGAP-like protein, and filamentous actin to a ring structure involved in budding yeast cytokinesis. J Cell Biol. 1998b;140(2):355–66. doi: 10.1083/jcb.140.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14(10):953–61. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lord M, Laves E, Pollard TD. Cytokinesis depends on the motor domains of myosin-II in fission yeast but not in budding yeast. Mol Biol Cell. 2005;16(11):5346–55. doi: 10.1091/mbc.E05-07-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca FC, Mody M, Kurischko C, Roof DM, Giddings TH, Winey M. Saccharomyces cerevisiae Mob1p is required for cytokinesis and mitotic exit. Mol Cell Biol. 2001;21(20):6972–83. doi: 10.1128/MCB.21.20.6972-6983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi I, Tsukita S, Sawai T. Cleavage furrow isolated from newt eggs: contraction, organization of the actin filaments, and protein components of the furrow. Proc Natl Acad Sci U S A. 1988;85(16):5966–70. doi: 10.1073/pnas.85.16.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM. Cytokinesis: IQGAPs find a function. Curr Biol. 1998;8(6):R202–5. doi: 10.1016/s0960-9822(98)70125-3. [DOI] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93(2):263–75. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Microtubule assembly nucleated by isolated centrosomes. Nature. 1984;312(5991):232–7. doi: 10.1038/312232a0. [DOI] [PubMed] [Google Scholar]
- Neujahr R, Heizer C, Gerisch G. Myosin II-independent processes in mitotic cells of Dictyostelium discoideum: redistribution of the nuclei, re-arrangement of the actin system and formation of the cleavage furrow. J Cell Sci. 1997;110(Pt 2):123–37. doi: 10.1242/jcs.110.2.123. [DOI] [PubMed] [Google Scholar]
- Pelham RJ, Chang F. Actin dynamics in the contractile ring during cytokinesis in fission yeast. Nature. 2002;419(6902):82–6. doi: 10.1038/nature00999. [DOI] [PubMed] [Google Scholar]
- Rappaport R. In: Cytokinesis in animal cells. Barlow PW, Bard JBL, Green PB, editors. Cambridge, U.K.: Cambridge University Press; 1996. [Google Scholar]
- Schroeder TE. The contractile ring. II. Determining its brief existence, volumetric changes, and vital role in cleaving Arbacia eggs. J Cell Biol. 1972;53(2):419–34. doi: 10.1083/jcb.53.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima JY, Toshima J, Kaksonen M, Martin AC, King DS, Drubin DG. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc Natl Acad Sci U S A. 2006;103(15):5793–5798. doi: 10.1073/pnas.0601042103. Translated from eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallen EA, Caviston J, Bi E. Roles of Hof1p, Bni1p, Bnr1p, and Myo1p in cytokinesis in Saccharomyces cerevisiae. Mol Biol Cell. 2000;11(2):593–611. doi: 10.1091/mbc.11.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker GR, Kane R, Burgess DR. Isolation and characterization of a sea urchin zygote cortex that supports in vitro contraction and reactivation of furrowing. J Cell Sci. 1994;107(Pt 8):2239–48. doi: 10.1242/jcs.107.8.2239. [DOI] [PubMed] [Google Scholar]
- Wolfe BA, Gould KL. Split decisions: coordinating cytokinesis in yeast. Trends Cell Biol. 2005;15(1):10–8. doi: 10.1016/j.tcb.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Mabuchi I, Tsukita S. Mass isolation of cleavage furrow from dividing sea urchin eggs. Journal of Cell Science. 1991;100:73–84. [Google Scholar]
- Zubenko GS, Park FJ, Jones EW. Mutations in PEP4 locus of Saccharomyces cerevisiae block final step in maturation of two vacuolar hydrolases. Proc Natl Acad Sci U S A. 1983;80(2):510–4. doi: 10.1073/pnas.80.2.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.