

Abstract
Background and purpose:
Melanin-concentrating hormone (MCH) is an orexigenic neuropeptide expressed in the lateral hypothalamus that is involved in feeding and body weight regulation. Intracerebroventricular infusion of a peptidic MCH1 receptor antagonist ameliorated obesity in murine models. Recently, small molecule MCH1 receptor antagonists have been developed and characterized for the treatment of obesity. However, little is known of the mechanism of the anti-obesity effects of MCH1 receptor antagonists.
Experimental approach:
To examine the mechanisms of action of the anti-obesity effect of MCH1 receptor antagonists more precisely, we conducted a pair-feeding study in mice with diet-induced obesity (DIO), chronically treated with an orally active and highly selective MCH1 receptor antagonist and examined changes in mRNA expression levels in liver, brown and white adipose tissues. We also assessed the acute effects of the MCH1 receptor antagonist in energy expenditure under thermoneutral conditions.
Key results:
Treatment with the MCH1 receptor antagonist at 30 mg·kg−1 for 1 month moderately suppressed feeding and significantly reduced body weight by 24%. In contrast, pair-feeding resulted in a smaller weight reduction of 10%. Treatment with the MCH1 receptor antagonist resulted in a higher body temperature compared with the pair-fed group. TaqMan and calorimetry data suggested that the MCH1 receptor antagonist also stimulated thermogenesis.
Conclusions and implications:
Our results indicate that an MCH1 receptor antagonist caused anti-obesity effects im mice by acting on both energy intake and energy expenditure.
Keywords: melanin-concentrating hormone, antagonist, pair-feeding, anti-obesity mechanism
Introduction
Melanin-concentrating hormone (MCH) is a cyclic 19 amino acid peptide expressed predominantly in the lateral hypothalamus and zona incerta of the brain (Nahon, 2006). Several lines of evidence have shown that MCH plays an important role in the control of feeding and energy metabolism. Hypothalamic expression of prepro-MCH (Pmch) mRNA is higher in genetically obese ob/ob mice as well as in fasted mice (Qu et al., 1996; Mizuno et al., 1998). Intracerebroventricular (ICV) administration of MCH stimulates food intake (Qu et al., 1996; Rossi et al., 1997), whereas chronic ICV infusion of MCH produces hyperphagia, increased adiposity and hyperinsulinaemia (Gomori et al., 2003;Ito et al., 2003). Pmch over-expressing mice show increased susceptibility to obesity (Ludwig et al., 2001), whereas genetic ablation of Pmch results in a lean phenotype with hypophagia and increased metabolic rate (Shimada et al., 1998).
The effects of MCH are mediated through G protein-coupled receptors in the CNS. Two distinct MCH receptors, the MCH1 receptor and the MCH2 receptor, have been identified (Fried et al., 2002; Tan et al., 2002; nomenclature follows Alexander et al., 2008). Because rodents possess only MCH1 receptors, whereas MCH2 receptors are also present in other species including humans (Fried et al., 2002; Tan et al., 2002), the pharmacological effects of MCH in rodents are solely mediated via MCH1 receptors. In agreement with this, Mch1 receptor knockout (KO) mice are resistant to diet-induced obesity (DIO; Marsh et al., 2002; Kokkotou et al., 2005). We also have reported that chronic ICV infusion with a peptidic MCH1 receptor antagonist, which did not show any effects in Mch1 receptor KO mice, remarkably improved DIO and ovariectomy-induced obesity in mice (Mashiko et al., 2005; Gomori et al., 2007; Ito et al., 2008). The peptidic MCH1 receptor antagonist caused fat-selective weight reduction and ameliorated hyperglycaemia, hyperinsulinaemia, hypercholesterolaemia and fatty liver (Mashiko et al., 2005; Gomori et al., 2007; Ito et al., 2008). There are few studies of MCH2 receptor signalling are minimal, due to the species-specific expression pattern of MCH2 receptors.
Recently a number of small molecule MCH1 receptor antagonists have been developed for the treatment of obesity, and these antagonists produced anti-obesity effects in obese rodent models (Luthin, 2007; Mendez-Andino and Wos, 2007). However, limited studies have addressed the mechanism of the anti-obesity effect of MCH1 receptor antagonists. Huang et al. conducted a pair-feeding study in rats. They showed that an MCH1 receptor antagonist-treated group lost significantly more weight than a pair-fed group (Huang et al., 2005), although no further evaluation regarding mechanism of action and no information about MCH1 receptor specificity of this compound was described. On the other hand, Kowalski et al. reported that body weight and fat mass were not different between mice treated with the MCH1 receptor antagonist, SCH-A, and a pair-fed group (Kowalski et al., 2006), suggesting that the effect of SCH-A on body weight was principally due to feeding suppression. However, the latter group also reported that acute dosing of SCH-A prevented the compensatory decrease in energy expenditure induced by hypophagia in pair-fed animals. Thus, observations regarding the anti-obesity mechanism(s) of MCH1 receptor antagonists are somewhat contradictory and the precise mechanisms of action of MCH1 receptor antagonists remain unclear.
Recently we identified a highly selective and potent MCH1 receptor antagonist that has high brain penetrability and oral bioavailability. To examine the manner in which MCH1 receptor antagonists cause anti-obesity effects more precisely, we conducted a 1-month pair-feeding study in DIO mice orally treated with this MCH1 receptor antagonist. We show here that the MCH1 receptor antagonist causes its anti-obesity effects by acting on both energy intake and expenditure.
Methods
Binding and functional assays
CHO-K1-derived cell lines (CCL-61; American Type Culture Collection, Manassas, VA, USA) that stably express human MCH1 receptor from pEF/myc/cyto (Invitrogen, Carlsbad, CA, USA) and human MCH2 receptor from pEF1/V5-HisB (Invitrogen) were used. For the binding assays, preparation of cell membranes and binding reactions were done as described previously (Mashiko et al., 2005). The MCH1 receptor and MCH2 receptor binding assays were conducted with [125I]MCH (PerkinElmer, Wellesley, MA, USA) and [Phe13,[125I]Tyr19]-MCH (PerkinElmer) respectively. For the MCH1 receptor functional assay, fluorescence output from MCH1 receptor/CHO-K1 cells loaded with Fluo-4-AM (Molecular Probes, Inc., Eugene, OR, USA) was measured using FLIPR (Molecular Devices, Sunnyvale, CA, USA) as described previously (Mashiko et al., 2005). The maximum fluorescence change induced by 5 nM MCH was determined in the presence of several concentrations of the test substance. The half maximal inhibitory concentration (IC50) and equilibrium dissociation constant (Ki) values for both the binding and functional assays were calculated using Prism Version 4.00 software (GraphPad Software, Inc., San Diego, CA, USA).
Animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee. Male Sprague-Dawley (SD) rats (Charles River Japan, Kanagawa, Japan), C57BL/6J mice (CLEA Japan, Tokyo, Japan) and male Mch1 receptor wild-type (WT) and littermate Mch1 receptor KO mice (F2-F4 50% C57BL/6J × 50% 129SvEv, in-house bred, derived from F1-F3 heterozygous crosses) were used. The generation of Mch1 receptor KO mice was described previously (Marsh et al., 2002). Animals were housed individually in plastic cages under controlled temperature, humidity (23 ± 2°C, 55 ± 15%) and a 12-h light–dark cycle (lights on 0700–1900). Animals had ad libitum access to a regular diet (CE-2, CLEA Japan; 59.3% energy as carbohydrate, 29.2% as protein and 11.5% as fat; 3.42 kcal·g−1) and tap water unless otherwise stated.
Experimental designs
Pharmacokinetics
SD rats were fasted overnight. The MCH1 receptor antagonist was administered (1 mg·kg−1, i.v. or 3 mg·kg−1, p.o.) and blood samples were collected at 15 and 30 min, 1, 2, 4, 6 and 24 h after dosing. For brain penetrability, lean C57BL/6J mice were given the MCH1 receptor antagonist (30 mg·kg−1) orally and plasma and brain samples were taken at 2 h after dosing. For plasma exposure, lean C57BL/6J mice were given the MCH1 receptor antagonist (30 mg·kg−1) orally and plasma and brain samples were taken at 2 and 15 h after dosing. Drug exposure levels were measured by high performance liquid chromatography.
Food intake in WT and Mch1r KO mice
Mch1 receptor KO and WT mice were fed a moderately high-fat (MHF) diet (Oriental BioService Kanto, Ibaraki, Japan; 52.4% energy as carbohydrate, 15.0% as protein and 32.6% as fat, 4.4 kcal·g−1) for about 6 months before the experiment. The mice of each genotype were divided into two groups, and each group was given p.o. either vehicle (0.5% methylcellulose in water) or the MCH1 receptor antagonist at a dose of 30 mg·kg−1, once a day for 3 days (n= 9–10). Drug administration and measurements of food intake and body weight were carried out 1–2 h before the beginning of the dark period.
Chronic administration of a MCH1 receptor antagonist and in DIO C57BL/6J mice
C57BL/6J mice were fed a MHF diet for about 10 months before the experiment. The mice were then divided into three groups, and each group was given p.o. either vehicle (0.5% methylcellulose in water) or the MCH1 receptor antagonist at a dose of 30 mg·kg−1 once-daily for 35 days (n= 10–13). Drug was administered after measurement of daily food intake and body weight, 1–2 h before the beginning of the dark period. In the pair-fed group, mice were fed the same amount of MHF diet as that consumed by the MCH1 receptor antagonist-treated group using a synchronized pellet pair-feeding apparatus (Nakagawa et al., 2000), Osaka Micro Systems, Settsu, Japan). The apparatus is composed of a controller, counter printer and cage units. Mice were supplied with pellet type MHF diet (∼13 mg per pellet). A pellet feeder and a pellet detector are attached to each cage, and the number of pellets consumed was recorded per unit time. MCH1 receptor antagonist- and vehicle-treated control mice were allowed to eat ad libitum, whereas the supply of pellets to the pair-fed mice was limited to the mean number of pellets consumed by the MCH1 receptor antagonist-treated mice.
Five days before and 6, 13 and 34 days after the start of drug administration, rectal temperature was measured in the morning with a digital thermometer (BAT-12, Physitemp Instruments, Clifton, NJ, USA) equipped with a rectal probe (IT-14, Physitemp Instruments) inserted to a depth of 1.5 cm. The mice were gently wrapped with a paper towel and lightly held during insertion of the probe. To reduce methodological stress, mice were acclimated to the procedure before the experiment.
At the end of the experiment, mice were fasted for 2 h (0700–0900), and blood samples were collected from the intra-orbital plexus for measurement of plasma glucose, insulin and leptin levels. Body composition was determined using a nuclear magnetic resonance analyzer (Minispec, Bruker Optics, Billerica, MA, USA). Subsequently, mice were killed by collecting whole blood from the abdominal vein under isoflurane anaesthesia. Mesenteric white adipose tissue (WAT) and liver were weighed. Mesenteric WAT, liver and brown adipose tissue (BAT) were sampled for measurement of mRNA expression, measurement of triglyceride (TG) contents and fatty acid (FA) oxidation.
Measurement of plasma parameters and TG contents
Plasma glucose, insulin and leptin levels were measured by commercial kits (Determiner GL-E, Kyowa Medex, Tokyo, Japan; leptin or insulin ELISA kit, Morinaga, Kanagawa, Japan). Plasma TG and total cholesterol levels were measured using commercial kits (Determiner L TGII, L TCII). To measure the TG contents, total lipids in liver and muscle were extracted using the procedure of Folch et al. (Folch et al., 1957). After drying, the extracts were dissolved in isopropanol and the TG content in the samples was measured enzymatically using a commercial kit (Determiner L TGII).
FA oxidation
Small fragments of BAT (5–10 mg wet weight) were incubated in 1.5 mL of Krebs-Ringer bicarbonate, pH 7.4, containing 4% fatty acid-free bovine serum albumin and 2 µCi [1-14C]palmitic acid (PerkinElmer) in vials. The vials were gassed for 15 s with 95% O2–5% CO2, capped and incubated for 40 min in a shaking water bath at 30°C. At the end of the incubation period, the reaction was terminated by the addition of 0.5 mL of 9 M H2SO4, and the vials were incubated further for 1 h at 37°C. Gaseous 14CO2 produced during incubation was trapped in filter paper soaked in 25% phenylethylamine. This filter paper was contained in a trapping vial, which was connected to the incubation vial. The radioactivity of the filter paper was determined using a liquid scintillation counter. The conversion of [14C]palmitic acid to 14CO2 was expressed as palmitate converted per gram of wet tissue in 40 min.
TaqMan analysis
Total RNA was isolated from the liver, mesenteric WAT and BAT samples using ISOGEN reagent (Nippongene, Toyama, Japan). PCR reactions and analysis were performed according to the manufacturer's protocol. An ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA) was used to determine levels of mRNA for sterol-regulatory element-binding protein-1c (SREBP1c), fatty acid synthase (FAS) and liver (L) type carnitine palmitoyltransferase 1 (CPT1) in the liver, uncoupling protein-1 (UCP1), -2 (UCP2), -3 (UCP3), β3-adrenoceptor, SREBP1c, FAS and GLUT4 in the mesenteric WAT, and UCP1 and muscle (M) type CPT1 in the BAT. Details of the TaqMan analysis, including sequences and fluorogenic probes, were as described previously (Ito et al., 2003; Mashiko et al., 2008). β-Actin was used as an internal standard for liver, WAT and BAT transcripts (Niiya et al., 2001).
Acute effect on metabolic rate under thermoneutrality
DIO C57BL/6J mice were placed into chambers of an indirect open circuit calorimeter (Oxymax, Columbus Instruments, OH) without food overnight at thermoneutrality (29°C). On the next day, 3 h into the light cycle, the mice were orally dosed with either the MCH1 receptor antagonist at 30 mg·kg−1 or the vehicle (n= 8). Metabolic rate was measured for 8 h. Baseline was determined from the 3 h period prior to dosing. Spontaneous motor activity was simultaneously measured by photobeam arrays attached to the calorimetry cages.
Statistical analysis
Data are expressed as means or means ± standard deviation (pharmacokinetics), or means ± standard error (other results). Significant differences were analyzed by repeated-measures one-way analysis of variance (anova) (body weight changes), or one-way anova (other results) and Bonferroni test.
Materials
The MCH1 receptor antagonist, 4-(benzyloxy)-1-[4-(2-pyrrolidin-1-ylethoxy)phenyl]pyridin-2(1H)-one (WO 2005085200) was synthesized by Banyu Pharmaceutical Co., Ltd (Ibaraki, Japan). Other chemicals were of analytical grade.
Results
Profiles of the MCH1R antagonist
The binding affinities of MCH and the MCH1 receptor antagonist were evaluated in CHO-K1 cells that stably expressed human MCH1 and MCH2 receptors (Table 1). The MCH1 receptor antagonist exhibited high affinity for mouse Mch1 receptor, with a Ki value of 3.8 nM and >2000-fold selectivity over MCH2 receptor. The MCH1 receptor antagonist showed antagonistic activity in a functional assay, with an IC50 value of 26.5 nM. Significant affinities was not observed for >160 other receptors and enzymes at 10 µM (data not shown).
Table 1.
In vitro profile of the MCH1 receptor antagonist on human and murine MCH receptors
|
Binding assaya |
[Ca2+]i mobilizationb |
|||
|---|---|---|---|---|
| Ki (nM) | IC50 (nM) | |||
| hMCH1 | mMch1 | hMCH2 | hMCH1 | |
| MCH1 antagonist | 2.8 ± 0.4 | 3.8 ± 0.6 | >9700 ± 100 | 26.5 ± 0.9 |
| MCH | 0.03 ± 0.01 | 0.05 ± 0.01 | 1.6 ± 0.6 | |
Ki values for MCH1 and MCH2 receptors were determined using [125I] MCH and [Phe13, [125I] Tyr19]-MCH respectively. The values are means ± standard error (n= 3).
IC50, half maximal inhibitory concentration; MCH, melanin-concentrating hormone.
In fasted SD rats, the MCH1 receptor antagonist was orally bioavailable and showed a good pharmacokinetic profile suitable for once daily dosing (Table 2). In lean C57BL/6J mice, the MCH1 receptor antagonist showed high brain penetrability with brain/plasma ratio of 2.0 (plasma and brain levels were 6.1 ± 0.4 µM and 12.5 ± 1.7 nmol/g 2 h after oral administration at 30 mg·kg−1). The duration of plasma exposure was longer in DIO than in lean C57BL/6J mice (6.1 ± 0.4 and 0.2 ± 0.1 µM in lean C57BL/6J, and 6.6 ± 0.7 and 1.2 ± 0.3 µM in DIO C57BL/6J mice, respectively, at 2 and 15 h after oral dosing at 30 mg·kg−1).
Table 2.
Pharmacokinetic profiles of the MCH1 receptor antagonist in SD rats
| AUC0-µ (µM·h) | CLp (mL·min−1·kg−1) | Vd (L·kg−1) | T1/2 (h) | Cmax (µM) | BA (%) | |
|---|---|---|---|---|---|---|
| i.v.a | 0.98 | 46 | 8.7 | 2.5 | ||
| p.o.b | 1.46 | 3.4 | 0.27 | 50 |
Data are means of n= 3.
1 mg·kg−1, i.v.
3 mg·kg−1, given orally.
AUC, area under the curve; BA, bioavailability; CLp, plasma clearance; Cmax, peak concentration; Vd, volume of distribution.
Food intake in WT and Mch1 receptor KO mice
Three-day treatment of DIO mice (initial body weights were 44.3 ± 0.9 and 41.2 ± 1.1 g in WT and Mch1 receptor KO mice, respectively) with the MCH1 receptor antagonist (30 mg·kg−1 p.o.) significantly reduced cumulative food intake (Figure 1A,B) and body weight (Figure 1C,D) only in WT mice, indicating that the effect of this compound was dependent on the presence of MCH1/Mch1 receptors.
Figure 1.
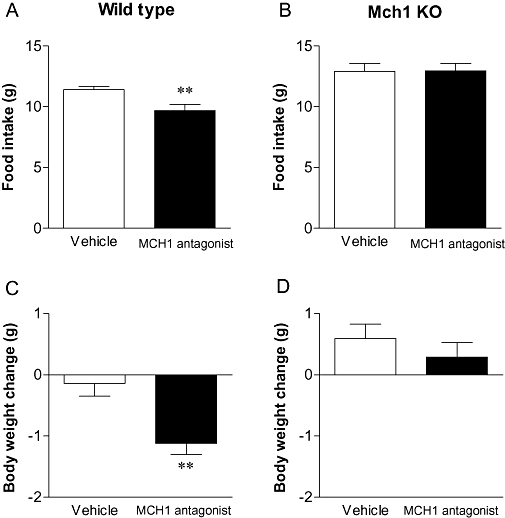
Food intake (upper) and body weight change (lower) of moderately high-fat (MHF) diet-fed wild type (A, C) and Mch1 receptor knockout mice (B, D) orally treated with the MCH1 receptor antagonist at 30 mg·kg−1 for 3 days. Data are mean ± standard error (n= 9–10). **P < 0.01 versus vehicle-treated group. MCH, melanin-concentrating hormone.
Pair-feeding in DIO C57BL/6J mice
DIO C57BL/6J mice had a stable body weight at the start of the experiment (initial body weights were 49.3 ± 0.8, 49.1 ± 0.9 and 48.2 ± 0.9 g in vehicle-treated, the MCH1 receptor antagonist and a pair-fed group respectively). Chronic administration of the MCH1 receptor antagonist suppressed food intake and continuously reduced body weight in DIO mice. The body weight difference versus the vehicle-treated group on day 36 was 25% (Figure 2A). In contrast, the pair-fed group, which received the same amount of food as the MCH1 receptor antagonist-treated group (Figure 2B,C), only transiently showed similar weight loss to that of the MCH1 receptor antagonist-treated group during the initial days of the study. The weight reduction on day 36 for the pair fed group was only 10% (Figure 2A).
Figure 2.
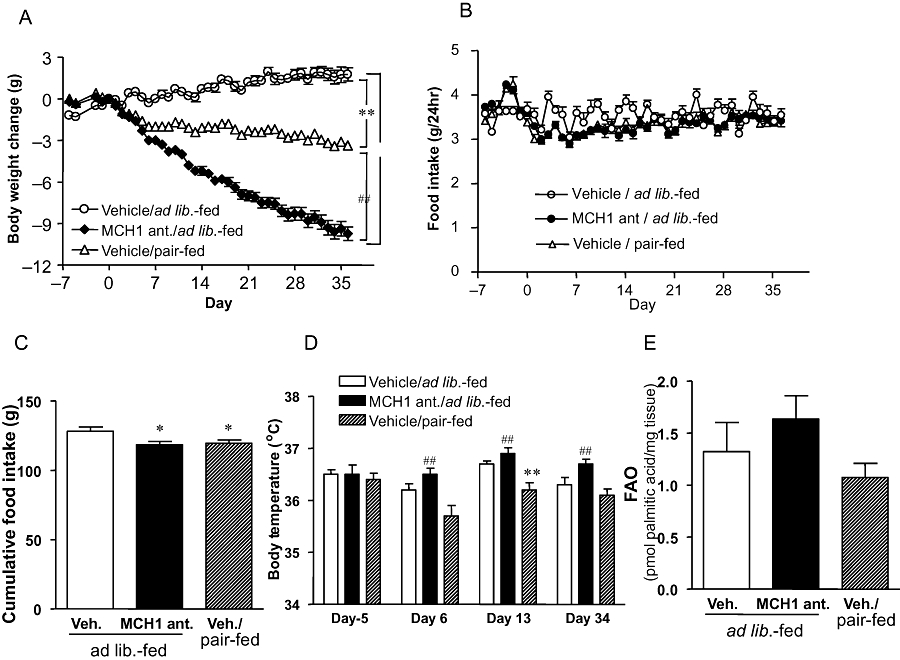
Body weight change (A), daily food intake (B), cumulative food intake (C), rectal temperature (D) and brown adipose tissue fatty acid oxidation (FAO; E) in mice treated with the MCH1 receptor antagonist (MCH1 ant) or pair-fed mice. Data are mean ± standard error (n= 10–13). *P < 0.05, **P < 0.01 versus vehicle-treated/ad lib.-fed group, ##P < 0.01 versus vehicle/pair-fed group. MCH, melanin-concentrating hormone.
During the month of drug administration, the pair-fed group showed relatively lower rectal temperature, whereas the mice given the MCH1 receptor antagonist had a significantly higher body temperature than the pair-fed group (Figure 2D).
After the 1 month drug treatment, ex vivo FA oxidation activity in isolated BAT tended to be higher in the MCH1 receptor antagonist-treated group compared with the pair-fed group, although the difference was not statistically significant (Figure 2E). Mesenteric fat weight and total body fat mass were significantly reduced in the MCH1 receptor antagonist-treated group (Table 3). The change in lean mass in this group was very slight, indicating that the MCH1 receptor antagonist mainly reduced body fat. Although pair-feeding also significantly reduced fat mass, the reduction was about half that induced by the MCH1 receptor antagonist. The MCH1 receptor antagonist and pair-feeding groups showed similar reduction in liver weight and liver TG contents (Table 3).
Table 3.
Tissue weights, TG contents and plasma parameters in MCH1 receptor antagonist-treated or pair-fed DIO mice
| Vehicle/ad libitum-fed | MCH1 ant. | Vehicle/pair-fed | |
|---|---|---|---|
| Mesenteric fat (g) | 1.27 ± 0.09 | 0.39 ± 0.04**,# | 0.75 ± 0.04** |
| Body fat mass (g) | 21.4 ± 0.9 | 10.5 ± 0.8**,# | 15.8 ± 0.8** |
| Body lean mass (g) | 27.8 ± 0.5 | 26.4 ± 0.3* | 26.8 ± 0.3 |
| Liver weight (g) | 2.1 ± 0.1 | 1.6 ± 0.1** | 1.5 ± 0.1** |
| Liver TG (mg·g−1 tissue) | 74.2 ± 8.0 | 29.5 ± 3.2** | 37.2 ± 3.0** |
| Glucose (mg·L−1) | 2102 ± 64 | 1707 ± 74** | 1750 ± 55** |
| Insulin (ng·mL−1) | 8.9 ± 1.0 | 3.0 ± 0.3** | 4.0 ± 0.4** |
| Leptin (ng·mL−1) | 62.4 ± 3.6 | 17.1 ± 1.9**,# | 34.5 ± 2.2** |
| TG (mg·L−1) | 170 ± 10 | 170 ± 20 | 150 ± 10 |
| Total cholesterol (mg·L−1) | 1740 ± 400 | 1160 ± 80** | 1280 ± 60** |
| FFAs (µEq·L−1) | 561 ± 40 | 459 ± 20** | 501 ± 21 |
Data are means ± standard error (n= 10–13).
P < 0.05;
P < 0.01 versus vehicle/ad libitum-fed group;
P < 0.01 versus vehicle/pair-fed group.
DIO, diet-induced obesity; FFAs, free fatty acids; TG, triglycerides.
The MCH1 receptor antagonist significantly reduced plasma leptin and free fatty acids (FFAs) more than the pair-fed group. Plasma glucose, insulin and total cholesterol levels were similarly reduced by the MCH1 receptor antagonist and pair-feeding (Table 3).
We measured mRNA expression levels of several key genes related to lipid metabolism and energy expenditure in BAT, mesenteric WAT and liver (Table 4). In BAT, expression levels of UCP1 increased in both the MCH1 receptor antagonist-treated and the pair-fed groups by 1.6 to 1.7-fold. CPT1M mRNA was significantly increased in the MCH1 receptor antagonist-treated mice. In WAT, the MCH1 receptor antagonist significantly increased mRNA of UCP3, β3AR, SREBP1c, FAS and GLUT4, whereas pair-feeding did not affect expression levels of these genes. In the liver, both the MCH1 receptor antagonist and pair-feeding similarly reduced mRNA expression levels of SREBP1c and FAS, whereas CPT1L mRNA was not affected.
Table 4.
Expression levels of mRNA in brown adipose tissue (BAT), white adipose tissue (WAT) and liver from MCH1 receptor antagonist-treated or pair-fed DIO mice
| Tissue/mRNA | Vehicle/ad lib.-fed | MCH1 antagonist | Vehicle/pair-fed |
|---|---|---|---|
| BAT | |||
| UCP1 | 0.79 ± 0.11 | 1.31 ± 0.15** | 1.26 ± 0.12* |
| UCP2 | 0.84 ± 0.04 | 0.78 ± 0.06 | 0.84 ± 0.06 |
| UCP3 | 0.81 ± 0.16 | 1.50 ± 0.28* | 1.42 ± 0.14* |
| CPT1M | 0.66 ± 0.14 | 1.26 ± 0.14** | 1.02 ± 0.08 |
| WAT | |||
| UCP1 | 0.68 ± 0.31 | 1.53 ± 0.69 | 1.19 ± 0.72 |
| UCP2 | 0.55 ± 0.05 | 0.72 ± 0.08 | 0.66 ± 0.03 |
| UCP3 | 1.13 ± 0.30 | 2.31 ± 0.29** | 1.53 ± 0.17 |
| β3AR | 0.72 ± 0.12 | 1.21 ± 0.13** | 0.97 ± 0.09 |
| SREBP1c | 1.01 ± 0.17 | 1.73 ± 0.19**,# | 0.98 ± 0.09 |
| FAS | 0.90 ± 0.19 | 1.96 ± 0.26**,# | 1.21 ± 0.11 |
| GLUT4 | 2.45 ± 0.18 | 3.61 ± 0.31** | 2.79 ± 0.19 |
| Liver | |||
| SREBP1c | 1.03 ± 0.05 | 0.82 ± 0.07* | 0.71 ± 0.06** |
| FAS | 1.32 ± 0.21 | 0.90 ± 0.10* | 0.84 ± 0.12* |
| CPT1L | 1.39 ± 0.09 | 1.32 ± 0.11 | 1.42 ± 0.16 |
Data are means ± standard error (n= 10–13).
P < 0.05;
P < 0.01 versus vehicle/ad lib-fed group;
P < 0.01 versus vehicle/pair-fed group.
β3AR, β3-adrenoceptor; CPT1L, liver carnitine palmitoyltransferase 1; CPT1M, muscle carnitine palmitoyltransferase 1; FAS, fatty acid synthase; GLUT4, glucose transporter 4; SREBP1, sterol-regulatory element-binding protein-1; UCP, uncoupling protein.
Acute effect on metabolic rate under thermoneutrality
Acute dosing of the MCH1 receptor antagonist stimulated heat production in DIO mice (Figure 3A). There was a 17% increase in average metabolic rate 1–8 h after dosing (Figure 3B). During the experiment, the MCH1 receptor antagonist did not significantly affect spontaneous motor activity (vehicle: 43.5 ± 3.6, MCH1 antagonist: 50.2 ± 3.4 counts/h) or respiratory exchange ratio (data not shown).
Figure 3.
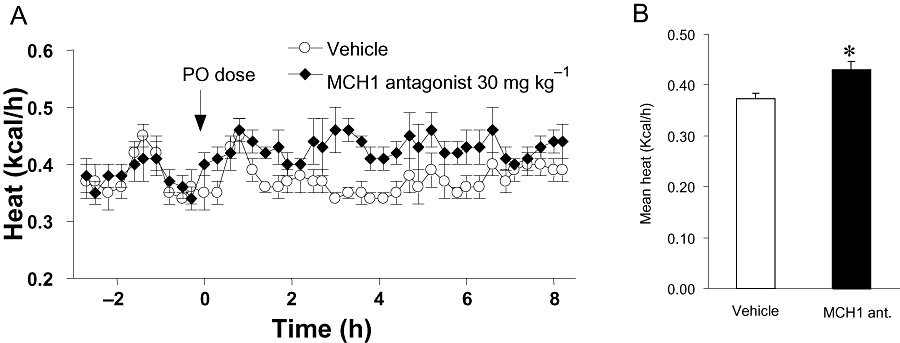
Metabolic rate (kcal/h) of diet-induced obesity mice under thermoneutral conditions after acute dosing with the MCH1 receptor antagonist at 30 mg·kg−1 or the vehicle. (A) Time course changes. (B) Average heat output (kcal/h) over 1–8 h after oral dosing. Data are mean ± standard error (n= 8). *P < 0.05 versus vehicle-treated group. MCH, melanin-concentrating hormone.
Discussion and conclusions
Chronic oral treatment with the MCH1 receptor antagonist reduced food intake and body weight in DIO mice. The MCH1 receptor antagonist showed good pharmacokinetic profiles, oral bioavailability and high brain penetrability. We showed that the MCH1 receptor antagonist did not affect food intake and body weight change after 3 days of dosing in Mch1 receptor KO mice, indicating that the effects of this compound on energy homeostasis are selectively mediated by MCH1 receptor blockade. No behavioural abnormalities were observed during the treatment period. Together with the in vitro binding specificity, these results indicate that the compound used in this study is highly specific for MCH1 receptors. Our recent report indicated that long-lasting and high receptor occupancy (∼90% for 24 h) is required to exert weight reduction by MCH1 receptor antagonists (Ito et al., unpublished). In the current study, we confirmed that, at the dose used in our experiments, the plasma levels of the MCH1 receptor antagonist were 0.6 ± 0.1 µM at 24 h after a single dose, which is enough for ∼90% receptor occupancy (Ito et al., unpublished, and data not shown).
The anti-obesity effects of the MCH1 receptor antagonist after chronic dosing caused a reduction of fat mass with a negligible effect on lean mass. Liver weight, liver TG content, plasma glucose, insulin, leptin and total cholesterol levels were also significantly decreased. These results are in agreement with our previous observations that peptide and non-peptide MCH1 receptor antagonists ameliorated obesity and obesity-related abnormal changes in DIO and ovariectomy-induced obese mice (Mashiko et al., 2003; Kowalski et al., 2006; Gomori et al., 2007; Ito et al., 2008).
In contrast to the remarkable 25% weight reduction in the MCH1 receptor antagonist-treated mice, the pair-fed group lost only 10% of their body weight. Body fat mass, muscle TG contents and plasma leptin levels at the end of the experiment were also significantly higher in the pair-fed group than in the antagonist-treated group. These results are consistent with the previous report by Huang et al. which showed that rats treated with an MCH1 receptor antagonist lost significantly more weight than pair-fed rats (Huang et al., 2005), and indicate that the anti-obesity effects of the MCH1 receptor antagonist are due not only to suppression of feeding, but also to an increase in energy metabolism. In agreement with this, the MCH1 receptor antagonist-treated mice had a significantly higher rectal temperature than the pair-fed group throughout the experiment. The relatively low rectal temperature in the pair-fed mice may be due to a compensatory reduction of energy expenditure, in response to reduced food intake (Gavrilova et al., 1999; Severinsen and Munch, 1999). We also showed that acute dosing with the MCH1 receptor antagonist stimulated heat production in DIO mice under thermoneutrality. During the experiment, the MCH1 receptor antagonist did not affect spontaneous motor activity, so the elevation of metabolic rate was not due to activity changes.
The observations that the MCH1 receptor antagonist affected thermogenic function are also consistent with previous reports. Zheng et al. (2005) reported that administration of MCH into the fourth ventricle of rats induced suppression of thermogenesis and body temperature. Acute treatment with the MCH1 receptor antagonist, SCH-A, prevented the compensatory decrease in energy expenditure induced by hypophagia in pair-fed animals (Kowalski et al., 2006). Mch1 receptor KO mice showed higher energy expenditure compared with WT mice (Chen et al., 2002; Marsh et al., 2002). The body weight of Mch1 receptor KO mice was less than that of WT mice under high fat diet accompanied by hyperphagia. Deletion of both MCH and leptin (MCH–/–: ob/ob) in mice, inhibited the sharp reduction by exposure to cold, compared with ob/ob mice (Segal-Lieberman et al., 2003)and ablation of MCH neurons increased energy expenditure (Alon and Friedman, 2006). Together with these reports, our results indicate that MCH1 receptor antagonists stimulate energy expenditure.
One possible target tissue involved in the thermogenic effects of MCH1 receptor antagonist is BAT. Significant increases in expression of CPT1M and UCP1, genes involved in fatty acid catabolism and oxidation, were observed in BAT of the group treated with the MCH1 receptor antagonist. FA oxidation tended to be higher in the BAT of the MCH1 receptor antagonist-treated mice compared with pair-fed animals, although the difference was not significant. These data suggest that the MCH system may regulate energy expenditure via BAT, at least in part. In agreement with these findings, there are several reports that the MCH system affects BAT functions. Thus, MCH neurons which project to the brain stem connect to the sympathetic innervation of BAT (Oldfield et al., 2002; Zheng et al., 2005). Antisense against MCH increased BAT mass and UCP1 expression in cold-exposed rats (Pereira-da-Silva et al., 2003). Although a physiological role for BAT was thought to be unimportant in adult humans because of the low amounts of BAT, recent studies showed that active and hypermetabolic BAT exist in several regions of adult humans (Nedergaard et al., 2007), and that white adipocytes can transdifferentiate into brown adipocytes and vice versa (Cinti, 2008; Kajimura et al., 2008). These findings re-introduce BAT as a therapeutic target for obesity and associated disorders.
In the current study, UCP1 expression levels and rectal temperature did not change together. UCP1 expression was similarly increased by the MCH1 receptor antagonist and pair-feeding, despite the higher rectal temperature in the antagonist-treated group. The reason for this dissociation is not clear. Other mechanism(s) than UCP1 are also involved in controlling body temperature, as diet-induced thermogenesis occurs similarly in UCP1-deficient and WT mice fed an obesogenic diet (Anunciado-Koza et al., 2008).
Another possible target tissue may be WAT. In WAT, treatment with the MCH1 receptor antagonist, but not pair-feeding, significantly increased expression levels of β3 adrenoceptors and UCP3. The β3 adrenoceptor plays a key role in lipolysis in WAT via sympathetic stimulation and is known to correlate with the development of obesity (Collins et al., 1999). The increased β3 adrenoceptor expression following the MCH1 receptor antagonist-treatment may have caused mobilization of TG from WAT, leading to a reduction of body fat mass. The UCP3 expression level was also increased in WAT in the MCH1 receptor antagonist-treated group. This suggests that the MCH1 receptor antagonist also stimulated energy expenditure in WAT by inducing BAT-like functions (Ravussin and Kozak, 2009), although the role of UCP3 in whole body thermogenesis is still controversial and might be limited under specific conditions (Echtay, 2007).
It was surprising that mRNA of genes involved in glucose uptake and lipogenesis, such as GLUT4, SREBP1c and FAS, were also significantly increased in the WAT of the MCH1 receptor antagonist-treated group. These changes are apparently inconsistent with the reduction of body fat content. The MCH1 receptor antagonist might stimulate lipid utilization by simultaneously activating fuel supply and expenditure. Alternatively, these might be compensatory changes induced by rapid reduction of body fat. Further studies are necessary to clarify the physiological meaning of these changes.
In the liver, mRNA levels of SREBP1c and FAS, genes involved in lipogenesis, as well as liver weight and TG content, were significantly decreased in both groups. Because these changes were observed in both MCH1 receptor antagonist-treated and pair-fed groups, they may be mainly due to feeding suppression, or a consequence of reduced insulin levels, which were observed in both antagonist-treated and pair-fed groups. We previously reported that a peptidic MCH1 receptor antagonist decreased liver weight and liver TG content in non-alcoholic steatohepatitis (NASH) models, induced by methionine- and choline-deficient (MCD) diet or by exposure for 1 year to 60% high-fat diet (Ito et al., 2008). In the MCD diet model, the MCH1 receptor antagonist did not affect plasma insulin levels or body weight, suggesting that MCH1 receptor antagonists could ameliorate fatty liver via a mechanism(s) independent from insulin. In the current study, because the extent of fatty liver was not as severe, feeding suppression and/or the reduction of plasma insulin may have led to improved liver parameters.
Although we show here effects of the MCH1 receptor antagonist on some peripheral tissues, the main mechanism and sites of actions within the brain is still unclear. As MCH1 receptors are expressed mainly in brain, the antagonist would act centrally and the effect would be conveyed from brain to peripheral tissues such as BAT, liver, WAT and so on, via the autonomic nervous system. On the other hand, it was reported that MCH1 receptors are also expressed in peripheral tissues such as WAT, pancreas and small intestine with small amount (Saito and Nagasaki, 2008). MCH stimulated leptin secretion by isolated rat adipocytes (Bradley et al., 2000). Thus, there is a possibility that a part of the effects of systemically administered MCH1 receptor antagonist may be direct influence on peripheral tissues. A denervation study might be useful to test this possibility.
As described previously, earlier reports regarding the anti-obesity mechanism(s) of MCH1 receptor antagonists are not totally compatible with the current results. In contrast to our results, the body weight and fat mass did not differ between mice treated with the MCH1 receptor antagonist SCH-A and a pair-fed group (Kowalski et al., 2006). These discrepancies may reflect the length of the treatment period. SCH-A was administered for only 5 days, whereas in our experiment, differences in body weight between the MCH1 receptor antagonist treatment and pair-feeding became obvious after about 1 week. Also, SCH-A did not increase energy expenditure above the vehicle treatment level, in contrast to our findings. Unlike potent thermogenic drugs such as β3-adrenoceptor agonists that directly activate BAT functions, the thermogenic effects of MCH1 receptor antagonists are relatively mild and are detected under specific conditions like thermoneutrality.
In conclusion, the MCH1 receptor antagonist induced anti-obesity effects by acting on both energy intake and energy expenditure. Treatment with the MCH1 receptor antagonist resulted in a significantly larger reduction of body weight and body fat than pair feeding. Together with previous observations, the current results indicate that MCH1 receptor signaling plays an important role in the central regulation of food intake and body weight. Hence, blockade of MCH1 receptors may be a useful treatment for obesity and metabolic disorders by suppressing caloric intake and stimulating energy expenditure.
Acknowledgments
We thank Satoshi Mashiko, Junko Ito, Rei Sakoyama, Naoko Fujino and Naomi Morita (Banyu Pharmaceutical Co. Ltd.) for the technical support. We also thank Norikazu Ohtake (Banyu Pharmaceutical Co. Ltd.) for preparing the compound.
Glossary
Abbreviations:
- BAT
brown adipose tissue
- CPT1
carnitine palmitoyltransferase 1
- DIO
diet-induced obesity
- FA
fatty acid
- FAS
fatty acid synthase
- ICV
intracerebroventricular
- MCD
methionine- and choline-deficient
- MCH
melanin-concentrating hormone
- MCH1 receptor
melanin-concentrating hormone 1-receptor
- MCH2 receptor
melanin-concentrating hormone 2-receptor
- MHF
moderately high-fat
- NASH
non-alcoholic steatohepatitis
- Pmch
prepro-MCH
- SREBP1c
sterol-regulatory element-binding protein-1c
- TG
triglyceride
- UCP
uncoupling protein
- WAT
white adipose tissue
Conflict of interest
M Ito, A Ishihara, A Gomori, H Matsushita, M Ito, Y Haga, H Iwaasa, S Tokita, N Takenaga, N Sato, M Moriya and A Kanatani are employed by and have equity interests in Banyu Pharmaceutical Co., Ltd. JM Metzger, DJ MacNeil and DJ Marsh are employed by and have equity interests in Merck Research Laboratories.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon T, Friedman JM. Late-onset leanness in mice with targeted ablation of melanin concentrating hormone neurons. J Neurosci. 2006;11:389–397. doi: 10.1523/JNEUROSCI.1203-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anunciado-Koza R, Ukropec J, Koza RA, Kozak LP. Inactivation of UCP1 and the glycerol phosphate cycle synergistically increases energy expenditure to resist diet-induced obesity. J Biol Chem. 2008;283:27688–27697. doi: 10.1074/jbc.M804268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R, Kokkotou EG, Maratos-Flier E, Cheatham B. Melanin-concentrating hormone regulates leptin synthesis and secretion in rat adipocytes. Diabetes. 2000;49:1073–1077. doi: 10.2337/diabetes.49.7.1073. [DOI] [PubMed] [Google Scholar]
- Chen YY, Hu CZ, Hsu CK, Zhang Q, Bi C, Asnicar M, et al. Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology. 2002;143:2469–2477. doi: 10.1210/endo.143.7.8903. [DOI] [PubMed] [Google Scholar]
- Cinti S. Reversible transdifferentiation in the adipose organ. Int J Pediatr Obes. 2008;3(Suppl 2):21–26. doi: 10.1080/17477160802404665. [DOI] [PubMed] [Google Scholar]
- Collins S, Daniel KW, Rohlfs EM. Depressed expression of adipocyte beta-adrenergic receptors is a common feature of congenital and diet-induced obesity in rodents. Int J Obes Relat Metab Disord. 1999;23:669–677. doi: 10.1038/sj.ijo.0800894. [DOI] [PubMed] [Google Scholar]
- Echtay KS. Mitochondrial uncoupling proteins – what is their physiological role? Free Radic Biol Med. 2007;43:1351–1371. doi: 10.1016/j.freeradbiomed.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Fried S, O'Neill K, Hawes BE. Cloning and characterization of rhesus monkey MCH-R1 and MCH-R2. Peptides. 2002;23:1401–1408. doi: 10.1016/s0196-9781(02)00077-3. [DOI] [PubMed] [Google Scholar]
- Gavrilova O, Leon LR, Marcus-Samuels B, Mason MM, Castle AL, Refetoff S, et al. Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc Natl Acad Sci. 1999;96:14623–14628. doi: 10.1073/pnas.96.25.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomori A, Ishihara A, Ito M, Mashiko S, Matsushita H, Yumoto M, et al. Chronic intracerebroventricular infusion of MCH causes obesity in mice. Melanin-concentrating hormone. Am J Physiol Endocrinol Metab. 2003;284:E583–E588. doi: 10.1152/ajpendo.00350.2002. [DOI] [PubMed] [Google Scholar]
- Gomori A, Ishihara A, Ito M, Matsushita H, Ito M, Mashiko S, et al. Blockade of MCH1 receptor signalling ameliorates obesity and related hepatic steatosis in ovariectomized mice. Br J Pharmacol. 2007;151:900–908. doi: 10.1038/sj.bjp.0707292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CQ, Baker T, Schwarz D, Fan J, Heise CE, Zhang M, et al. 1-(4-Amino-phenyl)-pyrrolidin-3-yl-amine and 6-(3-amino-pyrrolidin-1-yl)-pyridin-3-yl-amine derivatives as melanin-concentrating hormone receptor-1 antagonists. Bioorg Med Chem Lett. 2005;15:3701–3706. doi: 10.1016/j.bmcl.2005.05.130. [DOI] [PubMed] [Google Scholar]
- Ito M, Gomori A, Ishihara A, Oda Z, Mashiko S, Matsushita H, et al. Characterization of MCH-mediated obesity in mice. Am J Physiol Endocrinol Metab. 2003;284:E940–E945. doi: 10.1152/ajpendo.00529.2002. [DOI] [PubMed] [Google Scholar]
- Ito M, Gomori A, Suzuki J, Tsujioka S, Sasaki M, Matsuda M, et al. Antagonism of central melanin-concentrating hormone 1 receptor alleviates steatohepatitis in mice. J Endocrinol. 2008;198:309–315. doi: 10.1677/JOE-08-0087. [DOI] [PubMed] [Google Scholar]
- Ito M, Ishihara A, Gomori A, Egashira S, Matsushita H, Mashiko S, et al. Melanin-concentrating hormone 1-receptor (MCH1R) antagonist suppresses body weight gain correlated with high receptor occupancy levels in diet-induced obesity mice. Eur J Pharmacol. in press. [DOI] [PubMed]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, et al. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008;22:1397–1409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkotou E, Jeon JY, Wang XM, Marino FE, Carlson M, Trombly DJ, et al. Mice with MCH ablation resist diet-induced obesity through strain-specific mechanisms. Am J Pysiol Regul Integr Comp Physiol. 2005;289:R117–R124. doi: 10.1152/ajpregu.00861.2004. [DOI] [PubMed] [Google Scholar]
- Kowalski TJ, Spar BD, Weig B, Farley C, Cook J, Ghibaudi L, et al. Effects of a selective melanin-concentrating hormone 1 receptor antagonist on food intake and energy homeostasis in diet-induced obese mice. Eur J Pharmacol. 2006;535:182–191. doi: 10.1016/j.ejphar.2006.01.062. [DOI] [PubMed] [Google Scholar]
- Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, et al. Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest. 2001;107:379–386. doi: 10.1172/JCI10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthin DR. Anti-obesity effects of small molecule melanin-concentrating hormone receptor 1 (MCHR1) antagonists. Life Sci. 2007;81:423–440. doi: 10.1016/j.lfs.2007.05.029. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci. 2002;99:3240–3245. doi: 10.1073/pnas.052706899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiko S, Ishihara A, Iwaasa H, Sano H, Oda Z, Ito J, et al. Characterization of neuropeptide Y (NPY) Y5 receptor-mediated obesity in mice: chronic intracerebroventricular infusion of D-Trp(34)NPY. Endocrinology. 2003;144:1793–1801. doi: 10.1210/en.2002-0119. [DOI] [PubMed] [Google Scholar]
- Mashiko S, Ishihara A, Gomori A, Moriya R, Ito M, Iwaasa H, et al. Antiobesity effect of a melanin-concentrating hormone 1 receptor antagonist in diet-induced obese mice. Endocrinology. 2005;146:3080–3086. doi: 10.1210/en.2004-1150. [DOI] [PubMed] [Google Scholar]
- Mashiko S, Ishihara A, Iwaasa H, Moriya R, Kitazawa H, Mitobe Y, et al. Effects of a novel Y5 antagonist in obese mice: combination with food restriction or sibutramine. Obesity (Silver Spring) 2008;16:1510–1515. doi: 10.1038/oby.2008.223. [DOI] [PubMed] [Google Scholar]
- Mendez-Andino JL, Wos JA. MCH-R1 antagonists: what is keeping most research programs away from the clinic? Drug Discov Today. 2007;12:972–979. doi: 10.1016/j.drudis.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–297. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Tsuchida A, Itakura Y, Nonomura T, Ono M, Hirota F, et al. Brain-derived neurotrophic factor regulates glucose metabolism by modulating energy balance in diabetic mice. Diabetes. 2000;49:436–444. doi: 10.2337/diabetes.49.3.436. [DOI] [PubMed] [Google Scholar]
- Nahon JL. The melanocortins and melanin-concentrating hormone in the central regulation of feeding behavior and energy homeostasis. C R Biol. 2006;329:623–638. doi: 10.1016/j.crvi.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293:E444–E452. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- Niiya T, Osawa H, Onuma H, Suzuki Y, Taira M, Yamada K, et al. Activation of mouse phosphodiesterase 3B gene promoter by adipocyte differentiation in 3T3-L1 cells. FEBS Lett. 2001;505:136–140. doi: 10.1016/s0014-5793(01)02807-1. [DOI] [PubMed] [Google Scholar]
- Oldfield BJ, Giles ME, Watson A, Anderson C, Colvill LM, Mckinley MJ. The neurochemical characterisation of hypothalamic pathways projecting polysynaptically to brown adipose tissue in the rat. Neuroscience. 2002;110:515–526. doi: 10.1016/s0306-4522(01)00555-3. [DOI] [PubMed] [Google Scholar]
- Pereira-Da-Silva M, Torsoni M, Nourani HV, Augusto VD, Souza C, Gasparetti AL, et al. Hypothalamic melanin-concentrating hormone is induced by cold exposure and participates in the control of energy expenditure in rats. Endocrinology. 2003;144:4831–4840. doi: 10.1210/en.2003-0243. [DOI] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, et al. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature. 1996;380:243–247. doi: 10.1038/380243a0. [DOI] [PubMed] [Google Scholar]
- Ravussin E, Kozak LP. Have we entered the brown adipose tissue renaissance? Obes Rev. 2009;10:265–268. doi: 10.1111/j.1467-789X.2008.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Choi SJ, O'Shea D, Miyoshi T, Ghatei MA, Bloom SR. Melanin-concentrating hormone acutely stimulates feeding, but chronic administration has no effect on body weight. Endocrinology. 1997;138:351–355. doi: 10.1210/endo.138.1.4887. [DOI] [PubMed] [Google Scholar]
- Saito Y, Nagasaki H. The melanin-concentrating hormone system and its physiological functions. Results Probl Cell Differ. 2008;46:159–179. doi: 10.1007/400_2007_052. [DOI] [PubMed] [Google Scholar]
- Segal-Lieberman G, Bradley RL, Kokkotou E, Carison M, Trombly DJ, Wang XM, et al. Melanin-concentrating hormone is a critical mediator of the leptin-deficient phenotype. Proc Natl Acad Sci USA. 2003;100:10085–10090. doi: 10.1073/pnas.1633636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinsen T, Munch IC. Body core temperature during food restriction in rats. Acta Physiol Scand. 1999;165:299–305. doi: 10.1046/j.1365-201x.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- Tan CP, Sano H, Iwaasa H, Pan J, Sailer AW, Hreniuk DL, et al. Melanin-concentrating hormone receptor subtypes 1 and 2: species-specific gene expression. Genomics. 2002;79:785–792. doi: 10.1006/geno.2002.6771. [DOI] [PubMed] [Google Scholar]
- Zheng H, Patterson LM, Morrison C, Banfield BW, Randall JA, Browning KN, et al. Melanin concentrating hormone innervation of caudal brainstem areas involved in gastrointestinal functions and energy balance. Neuroscience. 2005;135:611–625. doi: 10.1016/j.neuroscience.2005.06.055. [DOI] [PubMed] [Google Scholar]