

Abstract
Background and purpose:
Stress on the endoplasmic reticulum (ER) can trigger rescuer responses such as the unfolded protein response (UPR). However, pharmacological modulators of these ER-regulated stress responses are not well understood. In the present study, we found that amiloride, a potassium-sparing diuretic, has unique properties relating to such stress.
Experimental approach:
We treated mouse primary cultured glial cells with amiloride, in the absence and presence of the ER stress-inducing reagents tunicamycin (Tm) or dithiothreitol, and measured UPR and ER stress-induced cell death. IRE1α phosphorylation, eIF2α phosphorylation, X-box binding protein 1 (XBP1) splicing, glucose regulated protein 78 (GRP78) and CCAAT/enhancer-binding protein homologous protein (CHOP) expression by reverse transcription-polymerase chain reaction and Western blotting were used to assess UPR and lactate dehydrogenase activity was determined to measure ER stress-induced cell death.
Key results:
Amiloride completely inhibited ER stress-induced activation of IRE1α, an ER-localized stress sensor protein, splicing of XBP1, and subsequent expression of GRP78 at the mRNA and protein levels. ER stress induces the phosphorylation of eIF2α, leading to the expression of CHOP or an attenuation of translation in cells. Surprisingly, treatment with amiloride alone markedly promoted the phosphorylation but actually inhibited ER stress-induced CHOP expression. Finally, we found that amiloride (200 µM) synergistically enhanced ER stress-induced cell death, which was mediated through caspases. On the other hand, a low dose of amiloride (20 µM) significantly prevented Tm-induced cell death.
Conclusions and implications:
These results suggest that amiloride can modulate UPR. They also suggest amiloride to be an important pharmacological agent and provide basic information for understanding and preventing ER stress-related diseases.
Keywords: ER stress, amiloride, eIF2α, CHOP, XBP1, GRP78, apoptosis, caspase
Introduction
Proteins must be correctly folded in the endoplasmic reticulum (ER) before they can be carried to intercellular organelles or the cell surface. However, stresses which affect ER function can lead to the accumulation of misfolded or unfolded proteins. There is evidence to suggest that ER stress is involved in several diseases, including neurodegenerative disorders, cancer, obesity and diabetes.
When unfolded or misfolded proteins accumulate in the ER, cells activate rescuer responses, such as the unfolded protein response (UPR). UPR: includes (i) eIF2α-mediated translational attenuation; (ii) the expression of ER chaperones [i.e. glucose regulated protein 78 (GRP78)]; and (iii) the degradation of unfolded proteins (i.e. ER-associated protein degradation) (Ron and Walter, 2007). Moreover, when ER functions are severely impaired, (iv) apoptotic pathways are activated. This apoptosis is mediated by the activation of caspases (Nakagawa et al., 2000; Hitomi et al., 2004) or CCAAT/enhancer-binding protein homologous protein (CHOP) (Zinszner et al., 1998). However, the intracellular mechanisms and pharmacological modulators of these ER-regulated stress responses are not well understood. Therefore, it is of great interest to identify novel drugs, which can modulate UPR. Indeed, we (Hosoi et al., 2008a) and others (Zhang et al., 2008; Ozcan et al., 2009) have recently found chemical chaperons, which can modulate ER stress, to be effective in treating ER stress-induced leptin resistance associated with obesity.
We previously reported that vanadate inhibited the ER stress-induced expression of GRP78 and CHOP, suggesting that vanadate-responsive proteins are involved in regulating UPR (Hosoi et al., 2008b). Vanadate has various pharmacological effects. It has been reported to inhibit protein-tyrosine phosphatases (Swarup et al., 1982), Na+/K+ ATPase (Cantley et al., 1977) and alkaline phosphatases (Lopez et al., 1976; Seargeant and Stinson, 1979). Thus, it is possible that other drugs that can modulate these pharmacological effects, would affect ER stress. In the light of these findings, we focused on the possible pharmacological effect of amiloride in regulating ER stress for the following reason: amiloride has similar (but not the same) pharmacological effects to vanadate. Thus, if amiloride has a similar regulatory effect to that of vanadate on ER stress, a common amiloride/vanadate-responsive protein(s) would be a novel target for controlling ER stress and we thought it would be of interest to evaluate these possibilities. In addition, amiloride is a drug already used clinically and could, therefore, be used to treat ER stress-related disease if it is found to be effective at inhibiting ER stress.
Upon the accumulation of unfolded or misfolded protein in the ER, an ER resident stress-sensor protein called inositol-requiring enzyme-1 (IRE1) is activated (Cox et al., 1993; Tirasophon et al., 1998). The activated IRE1 induces mRNA splicing of X-box binding protein 1 (XBP-1) by cleaving off the intron of XBP-1 (Yoshida et al., 2000; Calfon et al., 2002). The translated XBP-1 (spliced XBP-1) then functions as a transcription factor specifically recognizing ER-stress chaperons such as GRP78 (Yoshida et al., 2001).
We investigated whether amiloride affects the ER stress-induced pathways, including IRE1-XBP1-GRP78 pathway, translational attenuation and eIF2α-CHOP pathway.
Methods
Cell culture and induction of ER stress
Primary cultured glial cells were prepared from the whole brains of neonatal C57BL/6 mice as described previously (Hosoi et al., 2009). The cells were allowed to grow to confluency (10 days) in DMEM with 10% FCS, 100 U·mL−1 of penicillin G, 100 µg·mL−1 of streptomycin and 0.25 µg·mL−1 of amphotericin B (Nacalai Tesque, Kyoto, Japan). All the cultured cells were kept at 37°C in 5% CO2/95% air. Subsequently, mixed glial cells were shaken at 120 rpm for 18 h, cultured again for 6–8 days in dishes, and then used in experiments.
ER stress can be induced by agents that interfere with protein glycosylation (e.g. tunicamycin, Tm) or reduce the disulphide bonds of proteins (e.g. dithiothreitol, DTT). We treated cells with Tm (0.01 µg·mL−1) or DTT (3 mM) for 5 h and analysed IRE1α activation. IRE1α activation during ER stress correlates with phosphorylation, which reduces the mobility of IRE1α on SDS-polyacrylamide gel (Bertolotti et al., 2000).
Western blot analysis
Western blotting was performed as described previously (Hosoi et al., 2007). Cells were washed with ice-cold PBS and lysed in a buffer containing (in mM) HEPES-NaOH 10 (pH 7.5), NaCl 150, EGTA 1, Na3VO4 1, NaF 10, phenylmethylsulphonyl fluoride (PMSF) 1, aprotinin 10 µg·mL−1, leupeptin 10 µg·mL−1 and 1% NP-40 for 20 min. The lysate was centrifuged at 20 630×g for 20 min at 4°C, and the supernatant was collected. The samples were boiled with laemmli buffer for 3 min, fractionated by sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred at 4°C to nitrocellulose membranes. In case of IRE1α, we did electrophoresis for longer periods (3–4 h at 130 V) to obtain a difference in the small shift of phosphorylated IRE1α. The membranes were incubated with anti-KDEL (1:1000), CHOP (1:500), anti-IRE1α (1:1000), anti-XBP-1 (1:1000), anti-phospho-eIF2α (1:1000) and anti-GAPDH (1:1000) antibodies, followed by an anti-horseradish peroxidase-linked antibody. Peroxidase was detected using an enhanced chemiluminescence system.
Semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated using TRI reagent. RT-PCR was performed as described previously (Hosoi et al., 2009). Specifically, cDNA was synthesized from the total RNA by reverse transcription using 100 U of Superscript III Reverse Transcriptase and the Oligo (dt)12-18 primer in 20 µL of reaction mixture containing Superscript buffer, 1 mM dNTP mix, 10 mM DTT and 40 U of RNase inhibitor. The total RNA and Oligo (dt)12-18 primer were incubated at 70°C for 10 min prior to RT. After incubation for 1.5 h at 46°C, the RT reaction was terminated by denaturing the reverse transcriptase for 15 min at 70°C. For PCR amplification, 1.2 µL of cDNA was added to 12 µL of a reaction mix containing 0.2 µM of each primer, 0.2 µM of dNTP mix, 0.6 U of Taq polymerase and reaction buffer. PCR was performed in a DNA Thermal Cycler (GeneAmp® PCR System 9700, Life Technologies, Carlsbad, CA, USA). The following primers were used: XBP-1 upstream, 5′-cct tgt ggt tga gaa cca gg-3′; XBP-1 downstream, 5′-cta gag gct tgg tgt ata c-3′ (451 bp and 425 bp for unspliced and spliced band of XBP-1, respectively); GRP78 upstream, 5′-ctg ggt aca ttt gat ctg act gg-3′; GRP78 downstream, 5′-gca tcc tgg tgg ctt tcc agc cat tc-3′ (398 bp); CHOP upstream, 5′-ccc tgc ctt tca cct tgg-3′; CHOP downstream, 5′-ccg ctc gtt ctc ctg ctc-3′ (371 bp); GAPDH upstream, 5′-aaa ccc atc acc atc ttc cag-3′; and GAPDH downstream, 5′-agg ggc cat cca cag tct tct-3′ (361 bp). The PCR products (10 µL) were resolved by electrophoresis in an 8% polyacrylamide gel in TBE buffer. The gels were stained with ethidium bromide and then photographed under ultraviolet light. Each cycle of PCR used in the present study produced a linear relationship between the amount of input cDNA and the resulting PCR product.
Translation assay
Semi-confluent cells were treated with drugs and washed with PBS twice. The cells were then incubated with methionine/cysteine-free medium including 35S-labelled methionine/cysteine (10 µCi·mL−1) and L-glutamine (4 mM), with or without drugs for 30 min. After being washed with PBS, the cells were lysed in a buffer containing (in mM): HEPES-NaOH (pH 7.5) 10, NaCl 150, EGTA 1, Na3VO4 1, NaF 10, PMSF 1, aprotinin 10 µg·mL−1, leupeptin 10 µg·mL−1 and 1% NP-40 for 20 min. The lysate was centrifuged at 20 630 ×g for 20 min at 4°C, and the supernatant was collected. The samples were boiled with laemmli buffer for 3 min and fractionated by SDS-PAGE. Radiolabelled proteins were then visualized by autoradiography.
Lactate dehydrogenase leakage assays
The viability of cells was estimated by the lactate dehydrogenase (LDH) leakage method using a cytotoxicity detection kit according to the manufacturer's protocol. LDH activity was measured as the optimal density at 492 nm.
Statistics
Results are expressed as the mean ± SE. Statistical analyses were performed using Student's t-test, the paired t-test, or Dunnett's test.
Materials and reagents
Tm and DTT were obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Amiloride hydrochloride was obtained from Calbiochem (Darmstadt, Germany). TRI Reagent and methionine/cysteine-free medium were obtained from Sigma-Aldrich (St. Louis, MO, USA); 35S-labelled methionine/cysteine was from Perkin Elmer (Waltham, MA, USA). Superscript III Reverse Transcriptase, the Oligo (dt)12-18 primer and the Superscript buffer were obtained from Invitrogen (Life Technologies). The antibodies: anti-KDEL was from StressGen (Ann Arbor, MI, USA); anti-XBP-1 and CHOP were from Santa Cruz (Santa Cruz, CA, USA); anti-IRE1α and anti-phospho-eIF2α were from Cell Signaling (Boston, MA, USA); and anti-GAPDH was from Chemicon (Millipore, Billerica, MA, USA). The cytotoxicity detection kit was obtained from Roche Molecular Biochemicals (Indianapolis, IN, USA).
The drug/molecular target nomenclatures used are in accordance with BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Results
Amiloride inhibited the ER stress-induced IRE1-XBP1-GRP78 pathway
As assessed by Western blotting, Tm or DTT alone increased IRE1 phosphorylation (Figure 1). DTT treatment caused a larger shift in IRE1α mobility than did Tm treatment, though precisely why is unclear. We next investigated whether amiloride affects these responses. The activation of IRE1α was examined in cells pretreated with amiloride (200–500 µM) for 1 h and then treated with Tm (0.01 µg·mL−1) or DTT (3 mM) for 5 h. Interestingly, the Tm-induced phosphorylation of IRE1α was attenuated by amiloride, whereas the DTT-induced phosphorylation was not. In both cases, however, expression levels of total-IRE1α were inhibited by amiloride whether it was phosphorylated or not (Figure 1). This suggests that amiloride inhibits IRE1α by preventing its phosphorylation and/or expression levels.
Figure 1.
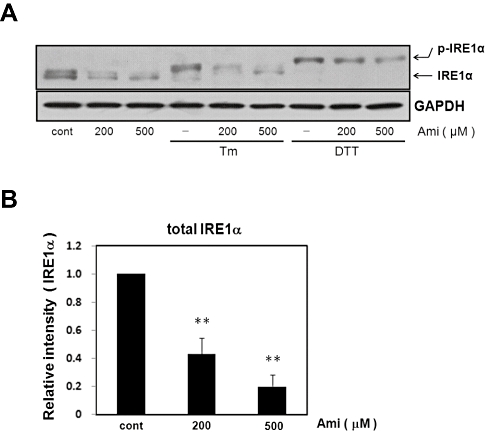
Effect of amiloride (Ami) on the expression of IRE1α and endoplasmic reticulum stress-induced IRE1α phosphorylation. (A) Cells were treated with Ami (200–500 µM) for 1 h and then stimulated with tunicamycin (Tm: 0.01 µg·mL−1) or dithiothreitol (DTT: 3 mM) for 5 h. Western blotting was performed using anti-IRE1α antibody. The band was detected at about 130 kDa∼ for normal and shifted band for IRE1α. Amiloride inhibited Tm-induced IRE1α phosphorylation. Amiloride inhibited the expression of IRE1α. (B) Densitometric analysis of total IRE1α expression using image analyzing software. **P < 0.01 versus control (n= 5). Cont, control.
We next examined the XBP-1 splicing of mRNA for XBP-1, a down-stream regulator of IRE1α activated by ER stress. As assessed by RT-PCR, treatment of cells with Tm or DTT increased XBP-1 mRNA splicing, whereas pretreatment with amiloride inhibited it (Figure 2A). Interestingly, we observed a novel band of XBP-1 mRNA in the amiloride-treated cells (Figure 2A). However, the precise characteristics of this band are unknown and further analysis is needed. On the other hand, amiloride did not affect the levels of spliced + unspliced + unknown forms of XBP-1 (total transcripts of XBP-1) mRNAs (Figure 2A), indicating that amiloride would not affect XBP-1 transcription. Spliced XBP-1 mRNA can be translated and act as a highly active transcription factor (Yoshida et al., 2001). Thus, we subsequently measured protein levels of the spliced form of XBP-1. As shown in Figure 2B, treatment of cells with ER stress-inducing reagents for 18 h increased the amount of spliced XBP-1 protein and the effect was inhibited by amiloride pretreatment.
Figure 2.
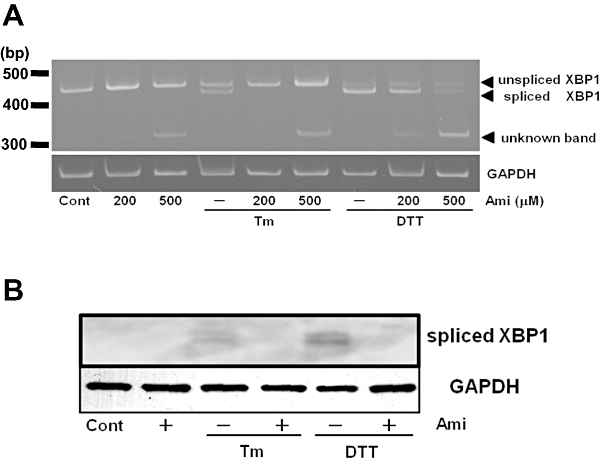
Amiloride (Ami) inhibited endoplasmic reticulum stress-induced X-box binding protein 1 (XBP1) splicing. (A) Cells were treated with Ami (200–500 µM) for 1 h and then stimulated with tunicamycin (Tm: 0.01 µg·mL−1) or dithiothreitol (DTT: 3 mM) for 6 h. Reverse transcription-polymerase chain reaction was performed using specific primers for XBP1 and GAPDH. (B) Cells were treated with amiloride (Ami: 200 µM) for 1 h and then stimulated with tunicamycin (Tm: 0.01 µg·mL−1) or dithiothreitol (DTT: 3 mM) for 18 h. Western blotting was performed using anti-XBP1 (54 kDa) and anti-GAPDH (37 kDa) antibodies. Cont, control.
Overall, these results indicate that amiloride can inhibit ER stress-induced XBP-1 mRNA splicing and the subsequent production of a spliced form of XBP-1 protein.
To further evaluate the inhibitory effect of amiloride on ER stress, we measured levels of GRP78, a molecular chaperon the expression of which is induced by the IRE1-XBP-1 pathway. Amiloride alone did not affect the mRNA or protein level of GRP78 (Figure 3). However, it markedly inhibited the ER stress-induced expression of GRP78 at the mRNA and protein levels (Figure 3). Together, these findings suggest that amiloride inhibits the ER stress-induced IRE1-XBP1-GRP78 pathway.
Figure 3.
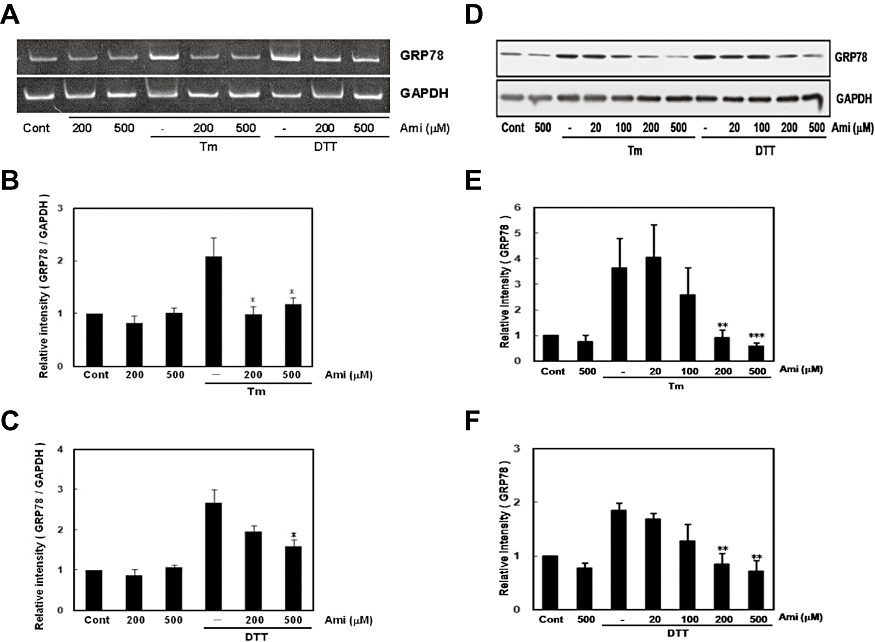
Amiloride (Ami) inhibited endoplasmic reticulum (ER) stress-induced glucose regulated protein 78 (GRP78) expression. (A) Cells were treated with Ami (20–500 µM) for 1 h and then stimulated with tunicamycin (Tm: 0.01 µg·mL−1) or dithiothreitol (DTT: 3 mM) for 6 h. Reverse transcription-polymerase chain reaction analysis was performed using specific primers for GRP78 and GAPDH. (B) and (C) Densitometric analysis of GRP78 mRNA using image analysing software. Ami dose-dependently inhibited ER stress-induced GRP78 expression. *P < 0.05, versus Tm or DTT (n= 5). (D) Cells were treated with Ami (20–500 µM) for 1 h and then stimulated with Tm (0.01 µg·mL−1) or DTT (3 mM) for 18 h. Western blotting was performed using anti-KDEL (78 kDa) or anti-GAPDH (37 kDa) antibodies. (E) and (F) Densitometric analysis of GRP78 protein using image analysing software. **P < 0.01, ***P < 0.001 versus Tm or DTT (n= 5). Cont, control.
Amiloride increased eIF2α phosphorylation and inhibited translation
UPR involves the activation of PERK, an ER-resident kinase that phosphorylates eIF2α, thereby blocking translational initiation (Shi et al., 1998; Harding et al., 1999). To investigate whether amiloride affects this part of UPR, we treated cells with amiloride with or without an ER stress-inducing reagent (Tm or DTT) and measured the level of phosphorylated eIF2α. To our surprise, the treatment with amiloride alone drastically increased eIF2α phosphorylation, and co-treatment with amiloride and the ER stress-inducing reagent (Tm or DTT) also increased eIF2α phosphorylation in primary cultured glial cells (Figure 4A). Amiloride (200 µM)-induced eIF2α phosphorylation was observed from 0.5 h, peaked at 2–5 h and declined thereafter (Figure 4D, E). Furthermore, the effect of amiloride on eIF2α phosphorylation was dose-dependent (Figure 4B, C). In addition, amiloride increased eIF2α phosphorylation in other types of cells including mouse DBT astrocytoma and human SH-SY5Y neuroblastoma cell lines (data not shown).
Figure 4.
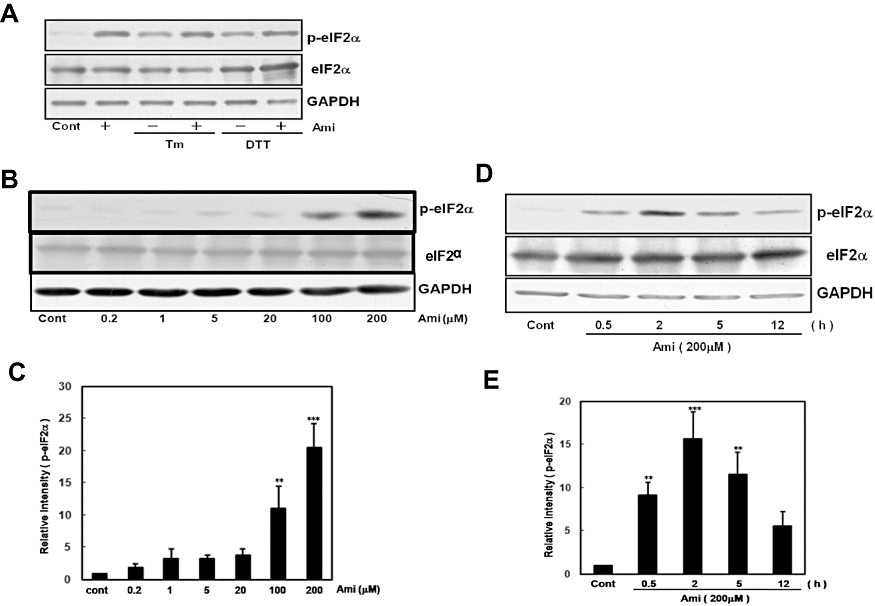
Amiloride (Ami) increased eIF2α phosphorylation. (A) Mouse primary cultured glial cells were treated with Ami (200 µM) for 1 h and then stimulated with tunicamycin (Tm: 0.01 µg·mL−1) or dithiothreitol (DTT: 3 mM) for 5 h. (B) Cells were treated with Ami (0.2–200 µM) for 5 h. (D) Cells were treated with Ami (200 µM) for the periods indicated. Western blotting was performed using anti-phospho-eIF2α (42 kDa) and anti-GAPDH (37 kDa) antibodies. (C) and (E) Densitometric analysis of the phosphorylation of eIF2α using image analysing software. **P < 0.01, ***P < 0.001 versus control (n= 4 and 5 respectively). Cont, control.
To investigate its functionality, we next assessed whether amiloride can inhibit translation by using 35S-labelled methionine/cysteine. Tm-induced ER stress inhibited translation (Figure 5). Furthermore, amiloride dose-dependently inhibited translation (Figure 5). These findings strongly suggest that amiloride can induce eIF2α phosphorylation, which decreases eIF2 activity and subsequently leads to attenuation of the translation.
Figure 5.
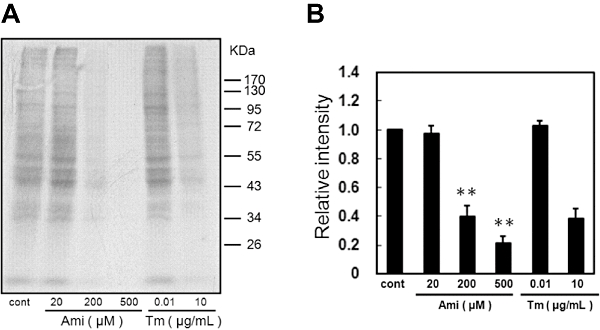
Amiloride (Ami) inhibited translation. (A) Rates of protein synthesis were measured based on the incorporation of [35S]-methionine/cystine into proteins during a 30 min pulse of labelling followed by a 90 min exposure to the indicated concentrations of Ami. (B) Densitometric analysis of translation rates using image analysing software. **P < 0.01 versus control (n= 3). Tm, tunicamycin. Cont, control.
Amiloride inhibited ER stress-induced CHOP expression
A decrease in eIF2 activity will exceptionally promote translation of activating transcription factor 4 and activate the CHOP promoter, which results in the production of CHOP protein (Fawcett et al., 1999; Harding et al., 2000; Scheuner et al., 2001; Jiang et al., 2004; Vattem and Wek, 2004). As we found that amiloride can induce eIF2α phosphorylation, we next examined whether amiloride would be able to induce CHOP expression. ER stress increased eIF2α phosphorylation and the subsequent expression of CHOP (Figure 6A). Thus, we next treated cells with amiloride for 0.5–12 h and analysed CHOP expression. However, to our surprise, although amiloride strongly increased eIF2α phosphorylation, we did not observe an increase in CHOP expression at any time point investigated (Figure 6A). To explore the possible pharmacological role of amiloride in regulating UPR, responsible for this paradoxical relationship, we next investigated the possibility that amiloride inhibits rather than induces ER stress-induced CHOP expression. We treated cells with amiloride and measured ER stress-induced CHOP expression. The results show that amiloride dose-dependently inhibited the expression of CHOP at the mRNA and protein levels (Figure 6B–G). These findings illustrate that amiloride-induced eIF2α phosphorylation was not linked to CHOP expression, but rather inhibited ER stress-induced CHOP expression at the transcriptional level.
Figure 6.
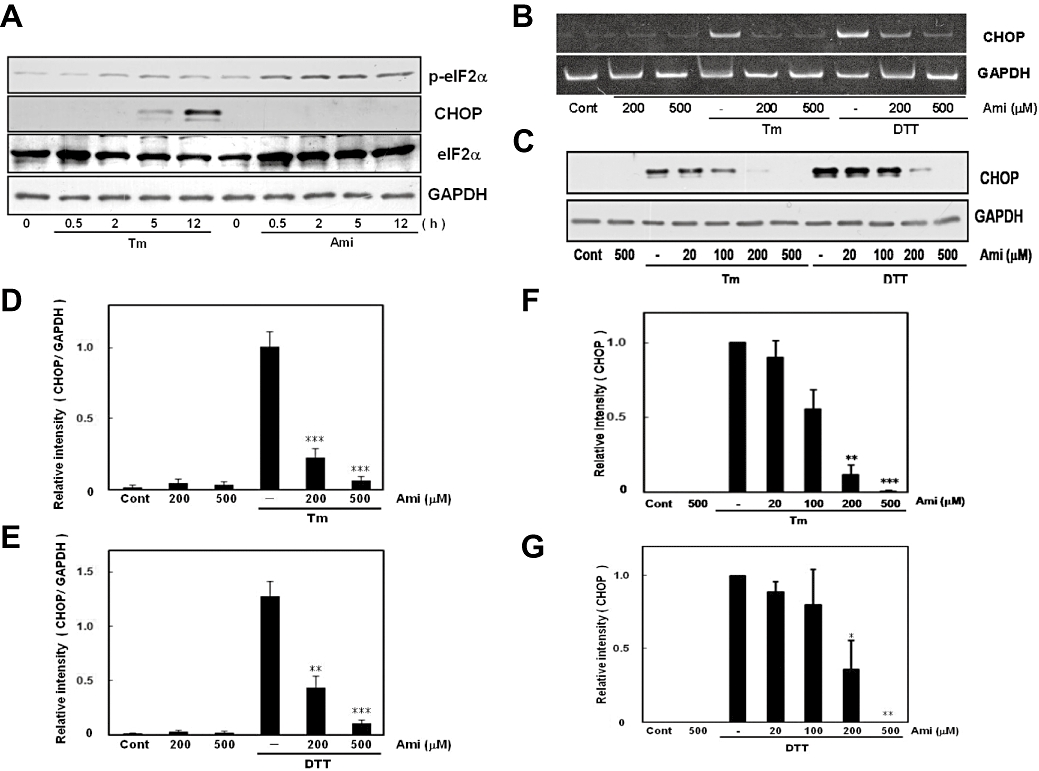
Amiloride (Ami) did not induce CCAAT/enhancer-binding protein homologous protein (CHOP) expression. (A) Cells were treated with tunicamycin (Tm: 0.01 µg·mL−1) or Ami (200 µM) for the periods indicated. Although Ami increased eIF2α phosphorylation, it did not induce CHOP expression. Western blotting was performed using anti-phospho-eIF2α (42 kDa), anti-eIF2α (40 kDa), anti-CHOP (29 kDa) and anti-GAPDH (37 kDa) antibodies. (B) Cells were treated with Ami (20–500 µM) for 1 h and then stimulated with Tm (0.01 µg·mL−1) or dithiothreitol (DTT: 3 mM) for 6 h. Reverse transcription-polymerase chain reaction was performed using specific primers for CHOP and GAPDH. (C) Cells were treated with Ami (20–500 µM) for 1 h and then stimulated with Tm (0.01 µg·mL−1) or DTT (3 mM) for 18 h. Western blotting was performed using anti-CHOP (29 kDa) and anti-GAPDH (37 kDa) antibodies. (D–G) Densitometric analysis of CHOP mRNA and protein using image analysing software. Ami dose-dependently inhibited ER stress-induced CHOP expression. *P < 0.05, **P < 0.01, ***P < 0.001 versus Tm or DTT (n= 6 for RT-PCR, n= 5 for Western blotting). Cont, control.
Dual effects of amiloride on ER stress-induced cell death
As amiloride has a unique regulatory role in ER stress-activated UPR, we next investigated its effect on ER stress-induced cell death. As assessed by the LDH leakage assay, amiloride alone at 200 µM slightly increased cell death (Figure 7A and B). Tm (0.1 µg·mL−1)- or DTT (3 mM)-induced ER stress also increased cell death (Figure 7A and B). Thus, we next treated cells with amiloride and then estimated ER stress-induced cell death. Co-treatment with amiloride 200 µM and ER stress synergistically increased cell death (Figure 7A, B). Z-VAD-fmk, an inhibitor of the proteases caspase and calpain (Waterhouse et al., 1998), reversed this increased death rate induced by amiloride in ER stressed cells (Figure 8).
Figure 7.
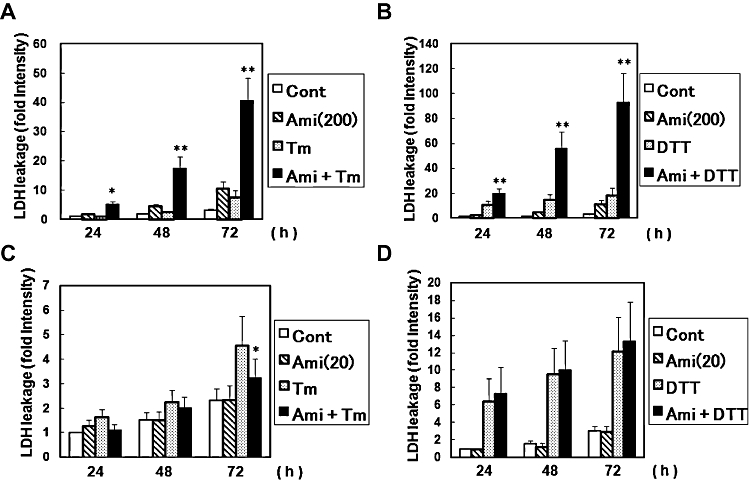
Effect of amiloride (Ami) on endoplasmic reticulum stress-induced cell death. (A) and (B) Cells were treated with Ami (200 µM) for 1 h and then stimulated with tunicamycin (Tm: 0.1 µg·mL−1) or dithiothreitol (DTT: 3 mM) for the periods indicated. Lactate dehydrogenase (LDH) activity was measured as an indicator of cytotoxicity. (C) and (D) Cells were treated with Ami (20 µM) for 1 h and then stimulated with Tm (0.1 µg mL−1) or DTT (3 mM) for the indicated times. *P < 0.05, **P < 0.01 Tm or DTT versus Ami + Tm or Ami + DTT (n= 3–8). Cont, control.
Figure 8.
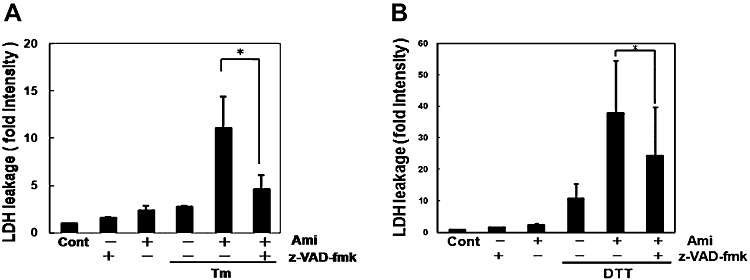
A caspase inhibitor prevented cell death caused by the combination of amiloride (Ami) and endoplasmic reticulum (ER) stress. (A) and (B) Cells were pretreated with the caspase inhibitor z-VAD-fmk (100 µM) for 30 min and treated with Ami (200 µM) for 1 h, and then stimulated with (A) tunicamycin (Tm: 0.1 µg·mL−1) or (B) dithiothreitol (DTT: 3 mM) for 72 h. Lactate dehydrogenase (LDH) activity was measured as an indicator of cytotoxicity. *P < 0.05 compared with Ami + ER stress (n= 3). Cont, control.
Next, we investigated the effect of a low dose of amiloride (20 µM) on ER stress-induced cell death. Amiloride (20 µM) alone did not affect cell viability (Figure 7C, D). However, we found that amiloride (20 µM) significantly inhibited the ER stress-induced cell death evoked by Tm (0.1 µg·mL−1) (Figure 7C), but, in contrast, had no effect on DTT (3 mM)-induced cell death in the present experimental conditions (Figure 7D).
Discussion
Increasing evidence suggests that ER stress is involved in neurodegenerative diseases, obesity, diabetes and cancer (Kaufman, 2002; Hosoi et al., 2008a). Drugs, which can modulate ER stress, may therefore be effective in treating these disorders. However, pharmacological modulators of ER stress are not well understood. In the present study, we found that amiloride has a novel/unique pharmacological action in regulating ER stress.
Amiloride drastically increased eIF2α phosphorylation: a mechanistic insight
In the present study, we found that amiloride induced the phosphorylation of eIF2α and a subsequent attenuation of translation. Protein kinases are responsible for the phosphorylation. Four types of kinases have been reported: the haeme-regulated inhibitor, HRI; the amino acid control kinase, GCN2; the double stranded RNA activated protein kinase, PKR; and the ER-localized protein kinase, PERK (Samuel, 1993; Kaufman, 1999). PERK has been shown to be activated by ER stress (Harding et al., 1999). However, in a preliminary study, we did not detect any phosphorylated PERK in amiloride-treated cells (data not shown). Thus, it is possible that other and/or novel kinases are involved in amiloride-induced eIF2α phosphorylation. Interestingly, similar to amiloride, salubrinal has been reported to be a novel inducer of eIF2α phosphorylation (Boyce et al., 2005). Salubrinal inhibits eIF2α dephosphorylation, thereby protecting cells from ER stress. Thus, it is also possible that amiloride can modulate eIF2α dephosphorylation.
Amiloride has been used clinically as a potassium-sparing diuretic that acts by inhibiting the epithelial sodium channel of renal tubes. In addition to this pharmacological effect, amiloride can inhibit Na+/H+ (Aickin and Thomas, 1977). Inhibition of Na+/H+ has been shown to produce acidification of gliomas but only minimal acidification in primary astrocytes (McLean et al., 2000). Thus, our present data obtained in primary astrocytes is unlikely to be due to inhibition of Na+/H+-mediated acidification. On the other hand, amiloride can also inhibit Na+/Ca2+ antiporters (Schellenberg et al., 1985), Na+/K+-ATPase (Soltoff and Mandel, 1983), acid-sensing ion channels (Waldmann et al., 1997), T-type Ca2+ channels (Tang et al., 1988) and urokinase-type plasminogen activator (Vassalli and Belin, 1987), and these amiloride-sensitive protein(s) are involved in the regulation of UPR. However, the mechanisms linking these known effects of amiloride and ER stress are at present unknown. Thus, it is of interest to elucidate these mechanisms and identify the molecules involved. Indeed, in a preliminary study, we observed that ouabain, a Na+/K+-ATPase inhibitor, increased eIF2α phosphorylation (unpublished observation).
Pharmacological properties of amiloride for treating ER stress-related diseases
PERK-deficient mice have a dysfunctional exocrine pancreas, resulting in diabetes, suggesting that PERK-mediated phosphorylation of eIF2α has a cytoprotective role (Harding et al., 2001; Lu et al., 2004). In addition, eIF2α phosphorylation promotes β-cell survival (Scheuner et al., 2001). These observations raise the possibility that a chemical modulator, which increases eIF2α phosphorylation, would be useful for treating ER stress-related disease. In the present study, amiloride increased eIF2α phosphorylation, whereas it did not induce expression of the pro-apoptotic transcription factor CHOP. Thus, amiloride, by selectively increasing eIF2α phosphorylation, would be effective against ER stress-related disease. Indeed, we found that a low dose of amiloride inhibited ER stress-induced cell death evoked by Tm, but, in contrast, had no effect on DTT-induced cell death in the present experimental conditions.
The reasons for this difference (Tm vs. DTT) are not known at present, but as Tm inhibits protein N-glycosylation, amiloride may specifically inhibit cells defective in N-type glycosylation-mediated-cell death (ER stress). Indeed, ER degradation-enhancing α-mannosidase-like protein has been reported to degrade misfolded glycoproteins (Hosokawa et al., 2001). Thus, misfolded glycoprotein-degradation systems do occur in response to ER stress. Overall, the results indicate that a high dose of amiloride synergistically enhances ER stress-mediated cell death, whereas the effect of a low dose is dependent on the type of compound used to evoke ER stress.
Interestingly, amiloride and its derivatives have been reported to be effective in treating type 2 diabetes (Gunawardana et al., 2008), Huntington's disease (Wong et al., 2008), Parkinson's disease (Arias et al., 2008) and brain ischaemia (Xiong et al., 2004). It is possible that the beneficial effects of amiloride on these diseases are in part achieved by inhibiting ER stress, as observed in the present study. On the other hand, a high dose of amiloride synergistically enhanced ER stress-induced cell death. The reason for this is a matter of debate. However, the high dose of amiloride caused a complete inhibition of ER stress-induced IRE1/XBP1/GRP78 signalling. As the activation of UPR, including the expression of GRP78, plays a protective role against ER stress, the amiloride-induced cell death would be due to the impaired activation of UPR. Moreover, the reason for the synergistic cytotoxicity of the high dose of amiloride is likely to be due to higher/sustained levels of eIF2α phosphorylation. Indeed, it has been recently reported that sustained PERK signalling impairs cell proliferation and promotes apoptosis (Lin et al., 2009). From our results combined with previous findings (Lin et al., 2009), it is speculated that the transient phosphorylation of eIF2α (attenuation of the translation) inhibits the ER stress-induced accumulation of unfolded or misfolded proteins, although, at the late phase of ER stress when unfolded proteins are cleared, cells would escape from apoptosis by inhibiting eIF2α phosphorylation (escape from excessive translational block, which results in cell death). Overall, it is likely that the magnitude and duration of eIF2α phosphorylation determine cell viability. Therefore, a low dose of amiloride can protect against ER stress by weakly increasing eIF2α phosphorylation, whereas with a high dose of amiloride, higher/sustained induction of eIF2α phosphorylation would result in a synergistic increase in cell death.
Conclusion
In the present study, we found that amiloride has a unique role in regulating ER stress. As amiloride has been used as a diuretic since 1967, the current findings are important from a pharmacological as well as clinical point of view. Moreover, the present results provide a basis for identifying new classes of drug, which target ER stress.
Acknowledgments
The authors thank Drs Yasuyuki Nomura, Yasunobu Okuma and Michiko Yoshii for helpful advice. This research was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture, Japan, and by the Astellas Foundation for Research on Metabolic Disorders. Animal studies were supported by the Institute of Laboratory Animal Science (Hiroshima University).
Glossary
Abbreviations:
- CHOP
CCAAT/enhancer-binding protein homologous protein
- eIF2α
eukaryotic initiation factor 2 α
- ER stress
endoplasmic reticulum stress
- GRP78
glucose regulated protein 78
- IRE1
inositol-requiring enzyme-1
- PERK
PKR-like ER kinase
- UPR
unfolded protein response
- XBP-1
X-box binding protein 1
Conflict of interest
The authors state no conflict of interest.
References
- Aickin CC, Thomas RC. An investigation of the ionic mechanism of intracellular pH regulation in mouse soleus muscle fibres. J Physiol. 1977;273:295–316. doi: 10.1113/jphysiol.1977.sp012095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias RL, Sung ML, Vasylyev D, Zhang MY, Albinson K, Kubek K, et al. Amiloride is neuroprotective in an MPTP model of Parkinson's disease. Neurobiol Dis. 2008;31:334–341. doi: 10.1016/j.nbd.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Jr, Josephson L, Warner R, Yanagisawa M, Lechene C, Guidotti G. Vanadate is a potent (Na,K)-ATPase inhibitor found in ATP derived from muscle. J Biol Chem. 1977;252:7421–7423. [PubMed] [Google Scholar]
- Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Gunawardana SC, Head WS, Piston DW. Dimethyl amiloride improves glucose homeostasis in mouse models of type 2 diabetes. Am J Physiol Endocrinol Metab. 2008;294:E1097–E1108. doi: 10.1152/ajpendo.00748.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek RC, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi T, Hyoda K, Okuma Y, Nomura Y, Ozawa K. Akt up- and down-regulation in response to endoplasmic reticulum stress. Brain Res. 2007;1152:27–31. doi: 10.1016/j.brainres.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M, et al. Endoplasmic reticulum stress induces leptin resistance. Mol Pharmacol. 2008a;74:1610–1619. doi: 10.1124/mol.108.050070. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Saito A, Kume A, Okuma Y, Nomura Y, Ozawa K. Vanadate inhibits endoplasmic reticulum stress responses. Eur J Pharmacol. 2008b;594:44–48. doi: 10.1016/j.ejphar.2008.07.034. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Sasaki M, Baba S, Ozawa K. Effect of pranoprofen on endoplasmic reticulum stress in the primary cultured glial cells. Neurochem Int. 2009;54:1–6. doi: 10.1016/j.neuint.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, et al. A novel ER α-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Zhang Y, Ron D, Walter P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE. 2009;4:e4170. doi: 10.1371/journal.pone.0004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez V, Stevens T, Lindquist RN. Vanadium ion inhibition of alkaline phosphatase-catalyzed phosphate ester hydrolysis. Arch Biochem Biophys. 1976;175:31–38. doi: 10.1016/0003-9861(76)90482-3. [DOI] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, et al. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean LA, Roscoe J, Jorgensen NK, Gorin FA, Cala PM. Malignant gliomas display altered pH regulation by NHE1 compared with nontransformed astrocytes. Am J Physiol Cell Physiol. 2000;278:C676–C688. doi: 10.1152/ajpcell.2000.278.4.C676. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Samuel CE. The eIF-2α protein kinases, regulators of translation in eukaryotes from yeasts to humans. J Biol Chem. 1993;268:7603–7606. [PubMed] [Google Scholar]
- Schellenberg GD, Anderson L, Swanson PD. Inhibition of Na+-Ca2+ exchange in rat brain by amiloride. Mol Pharmacol. 1985;24:251–258. [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Seargeant LE, Stinson RA. Inhibition of human alkaline phosphatases by vanadate. Biochem J. 1979;181:247–250. doi: 10.1042/bj1810247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltoff SP, Mandel LJ. Amiloride directly inhibits the Na,K-ATPase activity of rabbit kidney proximal tubules. Science. 1983;220:957–958. doi: 10.1126/science.6302840. [DOI] [PubMed] [Google Scholar]
- Swarup G, Cohen S, Garbers DL. Inhibition of membrane phosphotyrosyl-protein phosphatase activity by vanadate. Biochem Biophys Res Commun. 1982;107:1104–1109. doi: 10.1016/0006-291x(82)90635-0. [DOI] [PubMed] [Google Scholar]
- Tang CM, Presser F, Morad M. Amiloride selectively blocks the low threshold (T) calcium channel. Science. 1988;240:213–215. doi: 10.1126/science.2451291. [DOI] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli JD, Belin D. Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett. 1987;214:187–191. doi: 10.1016/0014-5793(87)80039-x. [DOI] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Waterhouse NJ, Finucane DM, Green DR, Elce JS, Kumar S, Alnemri ES, et al. Calpain activation is upstream of caspases in radiation-induced apoptosis. Cell Death Differ. 1998;5:1051–1061. doi: 10.1038/sj.cdd.4400425. [DOI] [PubMed] [Google Scholar]
- Wong HK, Bauer PO, Kurosawa M, Goswami A, Washizu C, Machida Y, et al. Blocking acid-sensing ion channel 1 alleviates Huntington's disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum Mol Genet. 2008;17:3223–3235. doi: 10.1093/hmg/ddn218. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]