

Abstract
Background and purpose:
N-methyl-D-aspartate (NMDA) receptors represent an attractive drug target for the treatment of neurological and neurodegenerative disorders associated with glutamate-induced excitotoxicity. The aim of this study was to map the binding domain of high affinity 5-substituted benzimidazole derivatives [N-{2-[(4-benzylpiperidin-1-yl)methyl]benzimidazol-5-yl}methanesulphonamide (XK1) and N-[2-(4-phenoxybenzyl)benzimidazol-5-yl]methanesulphonamide (XK2)] on the GluN2B subunit of the NMDA receptor.
Experimental approach:
The pharmacological antagonistic profiles of XK1 and XK2 were assessed using in vitro rat primary cerebrocortical neurones and two-electrode voltage clamp on Xenopus oocytes expressing heterologous GluN1/GluN2B receptors. Direct ligand binding was determined using the recombinant amino-terminal domain (ATD) of GluN2B.
Key results:
XK1 and XK2 effectively protected against NMDA-induced excitotoxicity in rat primary cortical neurones. Low concentrations of XK1 (10 nM) and XK2 (1 nM) significantly reversed neuronal death. Both compounds failed to inhibit currents measured from oocytes heterologously expressing GluN1-1a subunit co-assembled with the ATD-deleted GluN2B subunit. XK1 and XK2 showed specific binding to recombinant protein of GluN2B ATD with low nanomolar affinities. Several residues in the recombinant ATD of GluN2B were identified to be critical for conferring XK1 and XK2 sensitivity. The inhibitory effects of XK1 and XK2 were pH-sensitive, being increased at acidic pH.
Conclusions and implications:
These results demonstrate that XK1 and XK2 are effective neuroprotective agents in vitro and indicate that 5-substituted benzimidazole derivatives inhibit GluN1/GluN2B receptors via direct binding to the ATD of the GluN2B subunit. These compounds represent valuable alternatives to the classical antagonist ifenprodil as pharmacological tools for studying GluN2B-containing NMDA receptors.
Keywords: NMDA receptors, benzimidazole, recombinant protein, amino-terminal domain, GluN2B subunit, circular dichroism, ifenprodil
Introduction
N-methyl-D-aspartate (NMDA) receptors belong to the family of ionotropic glutamate receptors. They regulate excitatory synaptic transmission, which mediates physiological processes in the central nervous system, for example, learning, memory and neuronal development (Dingledine et al., 1999). Overactivation of NMDA receptors by glutamate can lead to excitotoxicity caused by elevated levels of intracellular Ca2+ ions in neurones. Compounds that block this overactivation, such as ifenprodil and ketamine, have been shown to prevent stroke and alleviate pain in rodent models (Dingledine et al., 1999; Le and Lipton, 2001). Clinical trials for the treatment of stroke using non-selective NMDA receptor antagonists gave discouraging outcomes and were terminated due either to lack of efficacy or major side effects such as hallucinations, dysphoria and loss of motor coordination (Birmingham, 2002; Hocking and Cousins, 2003; Reisberg et al., 2003; Witt et al., 2004).
The NMDA receptor GluN1 (NR1) and GluN2A (NR2A) subunits are widely expressed throughout the mammalian central nervous system with the remaining GluN2B (NR2B), GluN2C (NR2C) and GluN2D (NR2D) subunits heterogeneously distributed. The GluN2B subunit is primarily localized in the dorsal horn of the spinal cord and in structures of the forebrain, including the hippocampus, cortex and striatum but not in the cerebellum (Monyer et al., 1994). The different levels of the GluN2B subunit in various regions may provide a way with which to separate efficacy (e.g. neuroprotection) from the undesirable side effects (e.g. hallucinations, reduced locomotive coordination) associated with non-specific NMDA receptor antagonists.
Each NMDA receptor subunit shares a characteristic modular structure with three transmembrane segments (M1, M3 and M4), a re-entrant loop (commonly known as M2) which forms the pore-lining region, an intracellular C-terminal domain and large extracellular domains (Dingledine et al., 1999; Erreger et al., 2004; Paoletti and Neyton, 2007; Figure 1A). The large extracellular domains of the mature GluN2B subunit include amino acids 27–557 (Accession number U11419) situated pre-M1 and amino acids 649–817 between M3 and M4. The endogenous agonist, glutamate, binds to two discontinuous stretches of amino acids known as the S1 and S2 domains. The non-agonist binding segment constitutes the proximal N-terminus amino acids 27–393 which is known as the amino-terminal domain (ATD) (Figure 1A).
Figure 1.
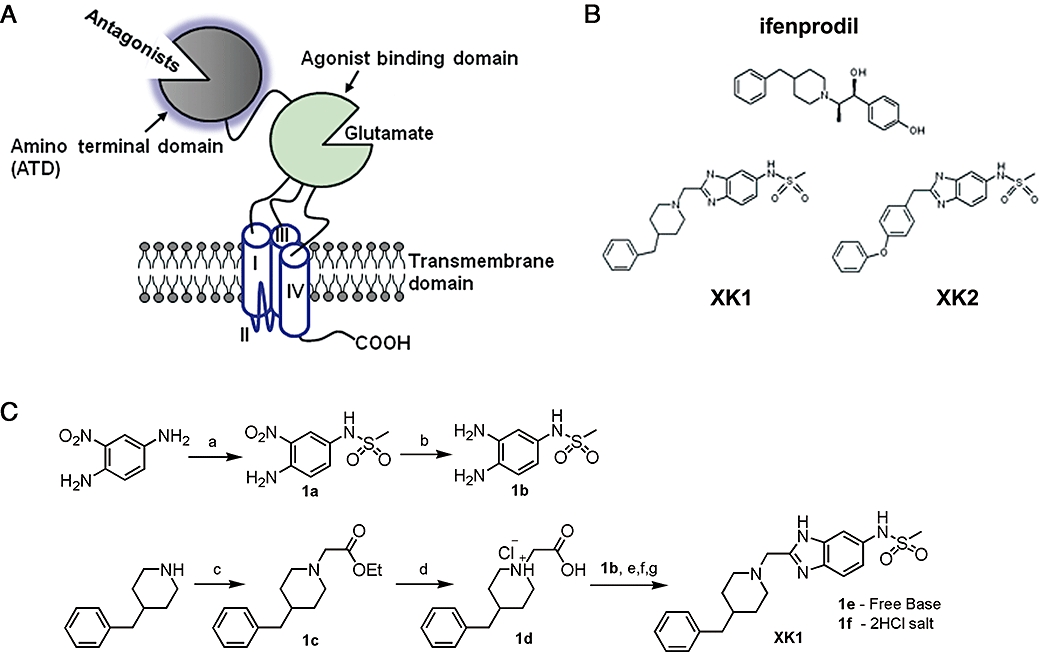
Functional organization of the GluN2B subunit and its selective antagonists. (A) GluN2B subunit consists of the antagonist-binding domain – amino-terminal domain (ATD) and the glutamate-binding domain (S1-S2) for various antagonists. These ligand binding domains are linked to three transmembrane domains (I, III and IV) with a re-entrant loop (II). The C-terminal tail (-COOH) resides in the cytoplasmic face of the plasma lipid bilayer. (B) Chemical structures of the classical reference GluN2B-selective antagonist – ifenprodil and two 5-substituted benzimidazole derivatives (XK1 and XK2). (C) Chemical synthesis of XK1. Reagents and conditions: (a) methanesulphonyl chloride, N,N-diisopropylethylamine, r.t., 4 h; (b) H2, 10% Pd/C, r.t., 2 h; (c) ethyl chloroacetate, triethylamine, anhydrous toluene, microwave-assisted condition, 170°C, 10 min; (d) 6M HCl, microwave-assisted condition, 150°C, 4 min; (e) Mukaiyama reagent (2-chloro-1-methyl-pyridinium iodide), 4-dimethylaminopyridine, triethylamine, anhydrous dimethylformamide, 50°C, 1h; (f) glacial acetic acid, 140°C, 15 min; (g) ether/HCl. Pd/C, palladium on carbon catalyst; r.t., room temperature condition.
The ATD presents as an allosteric site to either positively or negatively modulate NMDA receptor activity (reviewed by Mony et al., 2009a,b;). Phenylethanolamines, exemplified by ifenprodil and CP-101,606, are GluN2B-selective non-competitive antagonists acting at the GluN2B ATD (Williams, 1993; Chernard et al., 1995). Ifenprodil produces desensitization of GluN1/GluN2B receptors and its binding to GluN2B ATD leads to enhancement of tonic proton inhibition at physiological pH (Mott et al., 1998).
A variety of structurally diverse compounds such as phenylethanolamine-like and non-phenylethanolamine-like (e.g. aminoquinoline-, sulfoxide- and benzamidine-) analogues have been shown to be selective for GluN2B receptors (Williams, 1993; Kew et al., 1996; Chenard and Menniti, 1999; Chizh et al., 2001; Nikam and Meltzer, 2002; Pinard et al., 2002). Apart from phenylethanolamine-like analogues (exemplified by ifenprodil; Figure 1B; Borza and Domány, 2006), the site(s) of action of non-phenylethanolamine-like analogues (Layton et al., 2006) on the GluN2B subunit remain largely unknown. Recently, McCauley et al. (2004) reported two ultra high-affinity GluN2B-subunit binding benzimidazole derivatives, N-{2-[(4-benzylpiperidin-1-yl)methyl]benzimidazol-5-yl}methanesulphonamide (hereafter named XK1), and N-[2-(4-phenoxybenzyl)benzimidazol-5-yl]methanesulphonamide (hereafter named XK2) with Ki values of 0.99 nM and 0.68 nM respectively (Figure 1B; McCauley et al., 2004). In vivo, XK1 displayed excellent activity in the carrageenan-induced mechanical hyperalgesia assay in rats as well as good pharmacokinetic profiles in dogs (McCauley et al., 2004).
To date, the demonstration of the GluN2B-selective antagonism of XK1 and XK2 is limited to stably expressed heterologous GluN1/GluN2B receptors. Although XK1 and XK2 show some structural similarity to ifenprodil (e.g. benzylpiperidine side chain of XK1), they possess a bulkier and more isosteric replacement of the phenolic electrophilic methanesulphonamide side chain as compared with the phenolic group in ifenprodil. In this study, we (i) present the pharmacological characterization of the neuroprotective properties of XK1 and XK2 in a primary culture of rat cerebrocortical neurones; (ii) determine the binding of these benzimidazoles to the ATD of the GluN2B subunit; and (iii) show that the inhibition of GluN1/GluN2B receptors by XK1 and XK2 is pH-sensitive.
Methods
Animals
Pregnant adult Sprague-Dawley rats (n= 3) were supplied by the Centre for Animal Resources, National University of Singapore (NUS). For the preparation of cerebrocortical neurones, pregnant rats were killed by an overdose of CO2 followed by cervical dislocation. Primary cortical neurones were obtained from 18-day-old Sprague Dawley rat embryos (E18). All the protocols used for the animal studies were approved by the Institutional Animal Care and Use Committee of NUS.
Preparation of XK1 and XK2
XK1 (N-{2-[(4-benzylpiperidin-1-yl)methyl]benzimidazol-5-yl}methanesulphonamide) and XK2 (N-[2-(4-Phenoxybenzyl)benzimidazol-5-yl]methanesulphonamide) (Figure 1B) were prepared as described by McCauley et al. (2004), with several modifications (Figure 1C); 1a was obtained by selective sulphonylation of 2-nitro-p-phenylenediamine. Palladium on carbon catalyst reduction of the nitro group on 1a yielded 1b. Microwave-assisted synthesis was used to synthesize compounds 1c and 1d in place of the conventional reflux method (McCauley et al., 2004). Instead of the conventional EDC coupling, 2-(4-benylpiperidin-1-yl)acetic acid HCl (1d, 370.7 µmol) and triethylamine (1.483 mmol) was added to anhydrous dimethylformamide (10 mL), followed by Mukaiyama reagent (2-choloro-1-methyl-pyridium iodide) (448 µmol), dimethylaminopyridine (74.1 µmol) and N-(3,4-diaminophenyl)methanesulphonamide (1b, 370.7 µmol) and the mixture was stirred at 50°C for 1 h. The reaction was quenched with excess 0.1 M NaHCO3(aq), extracted with ethyl acetate, dried over anhydrous MgSO4 concentrated in vacuo to give a pale yellow oil followed by the addition of glacial acetic acid (10 mL). This mixture was refluxed at 140°C for 15 min. Toluene (30 mL) was added to the cooled crude mixture, concentration in vacuo and the mixture was purified by flash chromatography using ethyl acetate as eluent to obtain a colourless oil, 1e; 1e was then converted into XK1.HCl using ether/HCl, recrystallized using methanol/ethyl acetate (4:1) to give 1f, as white fluffy crystals (62% yield) and was pure as determined by 1H-NMR (500 MHz, MeOD4) with characteristics as follows: δ 7.79(d, J= 8.7 Hz, 1H), 7.74(d, J= 1.5 Hz, 1H), 7.42(dd, J= 1.9 Hz, J= 10.9 Hz, 1H), 7.28–7.14(m, 5H), 4.82(s, 2H), 3.67(d, J= 12.0 Hz, 2H), 3.22(t, J= 12.1 Hz, 2H), 2.99(s, 3H), 2.61(d, J= 6.4 Hz, 2H), 1.94–1.90(m, 3H), 1.71–1.59(m, 2H); melting point: 230–233°C, HRMS(ESI) m/z 399.1861 [(M+H)+, calculated for C21H27N4O2S, 399.1849]; Anal. (C21H26N4O2S·2HCl) C, H, N, Cl. Both compounds were converted to their respective hydrochloride salts (XK1.2HCl, 1f; XK2.HCl) to facilitate dispersion in aqueous buffers for biological evaluation. XK2 was prepared via the conventional EDC coupling method between commercially available 2-(phenoxyphenyl)acetic acid and intermediate 1b as described previously (McCauley et al., 2004)
Preparation of cerebrocortical neuronal culture
Primary cultures of cerebrocortical neurones were obtained from E18 Sprague Dawley rat embryos as described by Cheung et al. (1998) with modifications. Cortices were aseptically dissected from the brains. The meninges and the thoroid plexus were carefully removed under a dissecting microscope (Nikon SMZ645, Japan) placed in a Class I laminar flow hood. Cortical tissues were digested in 0.2 mg·mL–1 trypsin and 40 µg·mL–1 deoxyribonuclease in Hank's balanced salts solution (HBSS) supplemented with glucose (7.4 mM), sodium pyruvate (1 mM), HEPES (10 mM), NaHCO3 (4.2 mM), MgSO4 (1.2 mM) and bovine serum albumin (0.3% w/v) at 37°C for 5 min followed by mechanical trituration. Dissociated cells were harvested by centrifugation and resuspended in Neurobasal medium supplemented with 10% v/v dialysed heat-inactivated foetal bovine serum (FBS), 2.5% v/v B27, 2 mM GlutaMAX-1 and 1% v/v penicillin-streptomycin. Cells were seeded at densities of 0.5 × 106 cells/cm2 in 24-well plates (Nunc, Denmark) coated with 0.1 mg·mL–1 poly-D-lysine and cultured at 37°C in a humidified 5% CO2 incubator. Culture medium was replaced with the above Neurobasal medium and their supplements but in the absence of FBS a day later. Cytosine-β-D-arabino-furanoside (10 µM) was added on day-in-vitro (DIV) 4 and the cultures were used on DIV10-11 for immunostaining and NMDA-mediated neuronal excitotoxcity assay.
Biotinylation of neuronal surface proteins
Biotinylation of DIV10-11 primary cerebrocortical cultures were carried out using a Cell Surface Labeling kit according to the manufacturer's protocol. Neurones were placed on ice, washed twice with ice-cold PBS followed by incubation in PBS containing 0.25 mg·mL–1 sulpho-NHS-SS-biotin for 30 min at 4oC with gentle rocking. Biotinylation was stopped by adding quenching solution to the mixture. Biotinylated neurones were further rinsed 3x in Tris-buffered saline (TBS, 50 mM Tris, 115 mM NaCl, pH 7.5). Neurones were harvested and lysed with ice-cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150 mM NaCl, 1% (v/v) NP-40, 0.5% (w/v) deoxycholic acid containing protease inhibitors (2 µg·mL–1 aprotinin, 2 µg·mL–1 leupeptin, 1 mg·mL–1 pepstatin A and 574 µM phenylmethylsulphonyl fluoride). Protein concentration was determined using the bicinchoninic acid protein assay. Lysate (50 µg) was taken for total lysate fraction while a separate 500 µg of the lysate was incubated with Immobilized NeutrAvidin™ gel slurry for 1 h at room temperature on a mechanical arm rotator. After incubation, the sample was centrifuged at 16 000×g for 2 min at 4oC and supernatant was removed. The gel slurry was washed 5× using RIPA buffer and boiled in 4× modified Laemmli sample buffer (120 mM Tris-HCl pH 6.8, 200 mM dithiothreitol (DTT), 0.002% (w/v) bromophenol blue, 4% (w/v) sodium dodecyl sulphate (SDS), 20% (v/v) glycerol) for 5 min at 95oC. The gel slurry was centrifuged at 16 000×g for 2 min and the supernatant (the surface fraction) was removed and subjected to SDS-PAGE separation and transferred to polyvinylidene fluoride membrane for immunoblot analysis. Primary antibodies used were anti-GluN1 (1:2000) and anti-GluN2B (1:500). Secondary HRP-conjugated antibodies used were anti-rabbit (1:20 000) and anti-goat (1:15 000).
Immunocytochemistry
E18 cells were plated on glass coverslips coated with poly-D-lysine (0.1 mg·mL–1) and cultured as described above. DIV10-11 cultures were fixed in 4% w/v paraformaldehyde for 15 min at 4°C. The cells were washed in 1x phosphate buffered saline supplemented with 0.1% v/v Triton X-100 (PBS-Tx). This was followed by blocking in 3% w/v bovine serum albumin in PBS-Tx for 1 h at room temperature. Subsequently, the cells were incubated with primary antibodies for 24 h at 4°C and washed 3 × 5 min with PBS-Tx. The cells were then incubated with secondary antibodies in the dark for 1 h at room temperature. After washing, cells were treated with 100 µg·mL–1 of DNase-free RNase A for 30 min at 37°C, followed by incubation in 1 µg·mL–1 propidium iodide for 5 min at room temperature. The cells were washed 3× 5 min and mounted on glass slides using Gel Mount aqueous mounting medium.
Confocal microscopy
Fluorescent images were viewed and captured using a Zeiss LSM510 confocal microscope (Carl Zeiss, Germany) as previously described in Wee et al. (2008). Primary antibodies used for immunodetection were goat anti-GluN1 (1:100), rabbit anti-GluN2B (1:100), mouse anti-beta III tubulin TUJ1 (1:500) and rabbit anti-glial fibrillary acidic protein (GFAP) (1:2000). Secondary antibodies used were Alexa Fluor 488 donkey anti-rabbit (1:400), Alexa Fluor 594 donkey anti-goat (1:400) and Alexa Fluor 647 donkey anti-mouse (1:400).
NMDA-mediated neuronal excitotoxicity
Cerebrocortical neurones (DIV10-11) were treated with 500 µM NMDA in Neurobasal medium (free of serum and supplements) for 3 h at 37°C in 5% CO2 incubator. NMDA elicited on average 29% of neuronal death in this study. All drugs and control treatments were prepared in Neurobasal medium. MK-801 (10 µM), ifenprodil (0.11–10 µM), XK1 and XK2 were added simultaneously with or without NMDA-containing medium to the neurones and incubated for 3 h. The viability of cells was then analysed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
MTT cell viability assay
The quantitative colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay measures the conversion of the yellow tetrazolium salt to the coloured formazan product by mitochondrial enzymes in viable neurones as described in Cheung et al. (2004). Briefly, after drug treatment, MTT was added to the neurones and incubated for 30 min at 37°C in 5% CO2. The formazan crystals formed were then dissolved in dimethyl sulphoxide. Absorbance readings were taken using a 96-well plate reader at 570 nm (SpectraMAX Gemini EM, Molecular Devices, USA). Each cell survival assay was performed in triplicate and repeated at least three times on neurones harvested from three separate pregnant rats.
NMDA receptor subunit cDNAs
The NMDA receptor subunits previously referred to as ‘NR1’, ‘NR2A’, ‘NR2B’, ‘NR2C’ and ‘NR2D’ are now known as GluN1, GluN2A, GluN2B, GluN2C and GluN2D respectively (see Alexander et al., 2008; Collingridge et al., 2009). This revised nomenclature is adopted but when the expression plasmids are referred to for the first time, the new and old terminologies will be used for clarity. cDNA coding Gln29 to Arg393 of the GluN2B subunit was deleted using standard molecular method. Briefly, Bgl II cloning sites were inserted in between Ser28 and Ser31 (sense: 5′-AAAGCTCGTTCCagAtctAGCCCCCCCAGC-3′; antisense 5′-GCTGGGGGGGCTagaTctGGAACGAGCTTT-3′) and Met394 and Glu397 (sense 5′-AGTATTATGTGTGGagatctATGTGTCCTGAGAC-3′; antisense 5′-GTCTCAGGACACATagatctCCACACATAATACT-3′) sequentially on pcDNA1 GluN2B wild-type plasmid as template by QuikChange site-directed mutagenesis. The plasmid harbouring two new Bgl II restriction sites on pcDNA1 GluN2B was digested with Bgl II, gel purified followed by ligation using T4 ligase. The ligated product was transformed into XL2-Blue by ultracompetent E. coli and positive clones were verified by dideoxy sequencing to confirm the successful deletion of Gln29-Arg393. A two amino acid linker, Arg-Ser (Bgl II sequence), replaced the deleted stretch of amino acids Gln29-Arg393 in pcDNA1 GluN2B, which retained the putative endogenous rat GluN2B signal peptide (hereafter named GluN2BΔM394).
Expression of GluN1/GluN2B receptors in Xenopus oocytes
cRNA was synthesized from linearized template cDNA according to manufacturer's specifications (Ambion, USA). The quality of the synthesized cRNA was assessed by 0.8% agarose gel electrophoresis, and the quantity was estimated by spectroscopy and gel electrophoresis. Stage V and VI oocytes were surgically removed from the ovaries of Xenopus laevis anaesthetized with 3-amino-benzoic acid ethylester (1 g·L–1). Clusters of oocytes were incubated with 292 U·mL–1 Worthington type IV collagenase or 1.3 mg·mL–1 collagenase (17018–029) for 2 h in Ca2+-free solution (composition in mM: 115 NaCl, 2.5 KCl and 10 HEPES, pH 7.5) with slow agitation to remove the follicular cell layer. Oocytes were then washed extensively in the same solution supplemented with 1.8 mM CaCl2 and maintained in Barth's solution comprised of (in mM): 88 NaCl, 1 KCl, 24 NaHCO3, 10 HEPES, 0.82 MgSO4, 0.33 Ca(NO3)2, and 0.91 CaCl2 and supplemented with 100 mg·mL–1 gentamycin, 40 mg·mL–1 streptomycin and 50 mg·mL–1 penicillin. Oocytes were injected under the magnification of a Nikon SMZ645 dissecting scope (Nikon, Japan) within 24 h of isolation with 5–15 ng of cRNAs synthesized in vitro from linearized template cDNA in a 50 nL volume using oocyte microinjection pipette (Drummond Scientific Co., Broomall, PA, USA) mounted on a Marzhauser MM33 micromanipulator (SDR, Australia). The ratios of GluN1-1a to GluN2Bwt and GluN1-1a to GluN2BΔM394 injected cRNAs were 1:2 and 1:8 respectively. The injected oocytes were incubated in Barth's solution at 17°C for 3–7 days.
Electrophysiology
Two-electrode voltage clamp recordings on oocytes were as described previously (Traynelis et al., 1998; Low et al., 2000; 2003; Yuan et al., 2005). Oocytes were placed in a dual-track recording chamber with a single perfusion line that split to perfuse two oocytes. Two-electrode voltage-clamp recordings were made 2–3 days postinjection at room temperature (23°C) using Warner model OC-725C two-electrode voltage clamps (SDR Clinical Technology, Australia) as recommended by the manufacturer. The bath clamps communicated across Ag-AgCl2 pellets (Warner Instrument Corp., Hamden, CT, USA) placed each side of the recording chamber, both of which were assumed to be at a reference potential of 0 mV. The recording solution contained (in mM): NaCl 90, KCl 1, HEPES 10, BaCl2 0.5, pH adjusted to 7.3 with 5 N NaOH. Solution exchange was computer-controlled through 8-modular valve positioner (Digital MVP Valve, Reno, NV, USA) using the EasyOocyte software (a gift from Professor Stephen F. Traynelis, Emory University, Atlanta, GA, USA). Voltage and current electrodes were filled with 0.3–3.0 M KCl, and current responses were recorded at a holding potential of –30 to –50 mV; 100 µM glutamate, 100 µM glycine and 0.3–500 nM XK1 or XK2 were used in all oocyte experiments unless otherwise stated. Experimental manipulations were expressed as a % of pre-event and post-event control responses, and the data were pooled. Benzimidazole derivative inhibition data were fitted (least square criterion) to the equation:
 |
where n is the Hill slope, IC50 is the nominal concentration of benzimidazole that produces 50% inhibition, and minimum is a residual current response. For XK1, fitting algorithm returned a minimum of negative and hence was fixed at 0; for XK2, the algorithm returned a minimum of 6.7%
Expression, purification and refolding of 6xHis-ATD2B
Overexpressed recombinant protein (Arg27-Arg393) of the amino terminus of the GluN2B subunit (6xHis-ATD2B) was obtained as reported by our team recently (Wong et al., 2005; Ng et al., 2008). In brief, pET30b(+)EG ATD2B transformed BL21-CodonPlus-RIL E. coli was induced by 0.5 mM of isopropyl-β-D-1-thiogalactopyranoside (5 h at 37°C) The washed cells were lysed by French Pressure (ThermoSpectronics, USA). Inclusion bodies were centrifuged, washed and dissolved in a buffer containing 50 mM Tris HCl, 5 mM EDTA, 8 M guanidine HCl, 5 mM DTT, (pH 7.4). The supernatant isolated (crude 6xHis-ATD2B protein) was purified, concentrated to 0.2 mg·mL–1 and refolded in a buffer containing 10 mM NaCl, 0.4 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 0.5 M arginine HCl, 10 µM Ro25,6981, 10 mM glutamic acid, 1 mM DTT, 0.05% PEG w/v, pH 6.0. The refolded protein was concentrated to 1 mg·mL–1 for circular dichroism (CD) characterization and ligand binding assay.
Ligand binding determined by CD
The CD measurements were performed at 25°C using a Jasco J-715 spectropolarimeter equipped with a 1.0-mm path-length cuvette. The protein-ligand samples were scanned from 190 to 250 nm and accumulated 10 times at a resolution of 0.1 nm with a scanning speed of 50 nm·min–1 and sensitivity of 200 mdeg. All of the CD data were expressed as molar ellipticity. Increasing amounts of ifenprodil, XK1 or XK2 were added in small aliquots from stock solutions to 350 µL of protein solution and were allowed to bind for 35 min at room temperature. The CD spectra of the protein in the absence and presence of increasing concentrations of ligands (ifenprodil 0.02–5 µM; XK1 0.1–30 nM; XK2 0.1–10 nM) were recorded. The final change of assay volume was <3%. The shifts in ellipticity of the CD spectra at 215.5 nm (where the signal change on all three ligand bindings were maximal) were recorded. Dose-dependent titration curves of shifts in ellipticity versus drug concentration were plotted and the IC50 values were calculated. Duplicates were obtained per batch of induced/refolded 6xHis-ATD2B. Dose-titrations of ligand binding to 6xHis-ATD2B were determined in two separate batches of induced/refolded proteins and each batch repeated at least twice.
Data and statistical analyses
The amplitude of currents recorded from oocytes was measured using EasyOocyte. Raw current traces were generated using Clampfit 9.2 (Axon Instruments, Foster City, CA, USA) and Origin 7 (OriginLab Corporation, Northampton, MA, USA). CD experimental data were analysed by nonlinear curve fitting (GraphPad Prism 4, GraphPad Software Inc., La Jolla, CA, USA or ORIGIN 7, OriginLab Corp.). Data were analysed using one-way analysis of variance (anova) followed by post hoc Tukey test to determine significant differences. Values of P < 0.05 were considered to be statistically significant. Results are presented as means ± SEM. All experiments were performed at least three times unless stated otherwise.
Materials
Trypsin, deoxyribonuclease, FBS, HBSS, penicillin-streptomycin, poly-D-lysine, DNase-free RNase A, Gel Mount and ifenprodil were obtained from Sigma (St. Louis, MO, USA); Neurobasal medium and B27 from Invitrogen (Carlsbad, CA, USA); GlutaMAX-1 from Gibco (Carlsbad, CA, USA); Cell Surface Labeling kit from Pierce (Rockford, IL, USA). The primary antibodies anti-GluN1, anti-rabbit, anti-goat and goat anti-GluN1 were from Santa Cruz Biotechnology (sc1467, Santa Cruz, CA, USA); anti-GluN2B and rabbit anti-GluN2B from Alomone Labs AGC-003 (Jerusalem, Israel); mouse anti-beta III tubulin TUJ1 from Covance (Princeton, NJ, USA); rabbit GFAP from Dako (Copenhagen, Denmark). All secondary antibodies were purchased from Molecular Probes (Eugene, OR, USA). Propidium iodide was from Molecular Probes; NMDA and MK-801 from Tocris (Ellisville, MO, USA); T4 ligase from Promega (Madison, WI, USA); E. coli and BL21-CodonPlus-RIL E. coli from Strategene (La Jolla, CA, USA); Worthington type IV collagenase; was from Freehold (Freehold, NJ, USA) and collagenase from Life Technologies (Gaithersburg, MD, USA).
cDNAs encoding the GluN1 (NR1) subunit splice variant GluN1-1a (NR1-1a) (GenBank accession number U11418) as well as GluN2B (NR2B) (GenBank accession number U11419) subunits were generously provided by Professor S. F. Heinemann (Salk Institute, La Jolla, CA, USA). cDNAs coding for GluN2B mutants were generously provided by Professor S. F. Traynelis (Emory University).
Results
Chemistry
The 5-substituted benzimidazole derivatives, XK1 and XK2 were successfully synthesized from starting reagents as reported by McCauley et al. (2004) with modifications (Figure 1C). We adopted the microwave-assisted method, which generated comparable yields of XK1 and XK2 (above 90%). The times required to synthesize the intermediate compounds 1c and 1d were significantly reduced from 7 h to 10 min and 1 h to 4 min respectively.
Surface expression of GluN1 and GluN2B proteins on E18 DIV10-11 cerebrocortical neurones
Both GluN1 and GluN2B subunit proteins were readily detectable in total neuronal lysates as well as plasma membranes of cerebrocortical neurones at DIV10-11 from E18 rat embryos (Figure S1). Our GluN1 and GluN2B protein expression data are in good agreement with other studies (Monyer et al., 1994; Sheng et al., 1994; Portera-Cailliau et al., 1996; Li et al., 1998; Sans et al., 2000; Liu et al., 2004; Yuen et al., 2008).
Antagonistic effects of XK1 and XK2 on NMDA-induced neuronal excitotoxicity
Of the membrane integrity-based assays, the MTT assay is potentially more sensitive than the lactate dehydrogenase assay because the MTT assay shows cell abnormality at an earlier stage (Ying et al., 2000). Figure 2A shows that incubation of E18 cerebrocortical neurones at DIV10-11 with 500 µM NMDA in the presence of Ca2+ and glycine (both from media) induced neuronal death of ∼29% (P < 0.001) as determined by the MTT cell viability assay which concurred with other studies (Wamil and McLean, 1992; Hartnett et al., 1997; Kambe et al., 2008). This cell death was completely blocked by the NMDA receptor open channel blocker, MK-801 (10 µM) (P < 0.001; Figure 2A). The classical GluN2B-selective non-competitive antagonist, ifenprodil (10 µM), significantly blocked the NMDA induced neuronal death (P < 0.001, Figure 2A). Figure 2B and C show that both XK1 and XK2 inhibited NMDA-induced neuronal cell death in a dose-dependent manner. XK2 appeared to be more potent as a GluN2B-antagonist than XK1 (at 1 nM; P > 0.05 for XK1 and P < 0.05 for XK2). At higher concentrations, both XK1 and XK2 inhibited NMDA-induced neuronal death to a comparable degree as ifenprodil and MK-801 (compared with Figure 2A).
Figure 2.
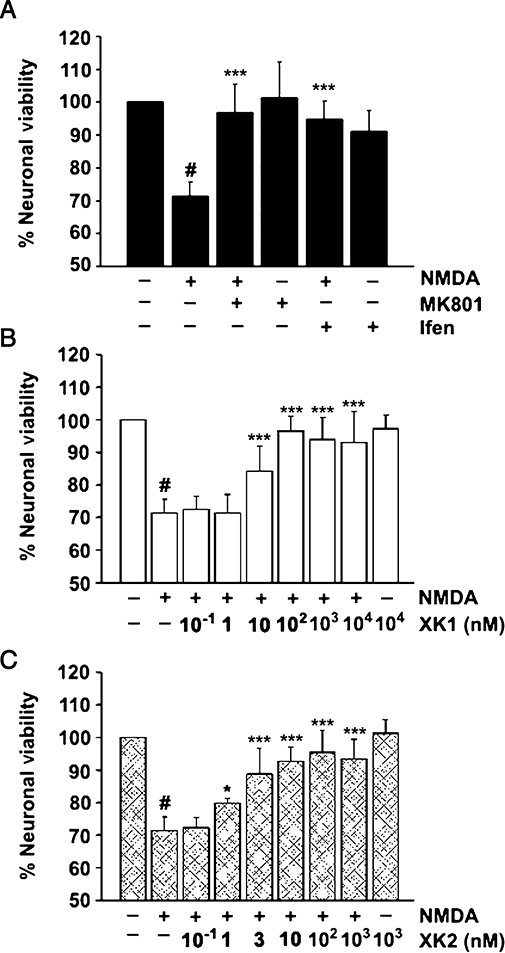
Neuroprotection of rat cerebrocortical neurones by NMDA receptor antagonists. (A) NMDA induced significant degree of neuronal death (#P < 0.001, compared with -NMDA, -MK801, -ifenprodil) which was reversed completely in the presence of the open channel blocker MK-801 (10 µM) and GluN2B-selective non-competitive antagonist ifenprodil (P < 0.001). Blockers alone did not affect neuronal viability (P > 0.05). Concentration-dependent antagonism of NMDA-induced neuronal cell death by XK1 (B) and XK2 (C). Data are the mean of at least three independent experiments on cerebrocortical neuronal cultures harvested from three separate pregnant rats (±SEM). Triplicates per concentration tested were performed on each independent experiment. XK2 showed relatively higher potency than XK1 in reversing neuronal death (compare at XK2 1 nM, *P < 0.05 and XK1 3 nM, ***P < 0.001). NMDA, N-methyl-D-aspartate.
Fitting the mean data with a single isotherm binding site model yielded an IC50 value of 2.4 ± 1.0 nM (n= 4) for XK2 which is ∼6-fold more potent as a neuroprotectant than XK1 (IC50 13.5 ± 5.8 nM, n= 4). The results showing the relatively higher potency of XK2 compared with XK1, determined here with the neuronal culture using an MTT assay, are summarized in Table 1. Both XK1 and XK2, when tested alone at the highest concentration (10 µM and 1 µM respectively), had no effect on neuronal death (Figure 2B and C).
Table 1.
In vitro binding data for ifenprodil, XK1 and XK2
| IC50 (nM) (cell line Ca2+influx) | IC50 (nM) (neuronal MTT assay) | KD (nM) (6xHis-A TD2B CD assay) | |
|---|---|---|---|
| Ifenprodil | 110 | N.D.* | 90.8 ± 21.3 (4) |
| XK1 | 1.9 | 13.5 ± 5.8 (4) | 1.2 ± 0.2 (4) |
| XK2 | 0.72 | 2.4 ± 1.0 (4) | 1.0 ± 0.2 (4) |
All values are the geometric mean of at least four measurements. The IC50 values (cell line Ca2+ influx) for ifenprodil were extracted from Kiss et al. (2005) while those for XK1 and XK2 were extracted from McCauley et al. (2004). IC50 (neuronal MTT assay) and KD (6xHis-ATD2B CD assay) values were determined in this study. Values in parentheses refer to number of repeats of experiments performed.
N.D., not determined in this study.
The ATD of the GluN2B subunit confers high sensitivity to XK1 and XK2
To test the hypothesis that the target-binding domain of the benzimidazole derivatives XK1 and XK2 resides within the ATD of GluN2B, the effects of XK1 and XK2 on NMDA receptors in which the ATD had been truncated on GluN2B (GluN2BΔM394; see Methods for details) were assessed. GluN1 and GluN2B wild-type (wt) or GluN1 and GluN2BΔM394 subunits were expressed in Xenopus oocytes, and NMDA currents were induced by saturating concentrations of L-glutamate (100 µM) and glycine (100 µM). As shown in Figure 3A (top and bottom panels), XK1 inhibited GluN1-1a/GluN2B with an IC50 of 37.6 ± 10.3 nM (n= 7 oocytes) while XK2 inhibited GluN1-1a/GluN2B with an IC50 of 15.7 ± 8.3 nM (n= 7 oocytes). Truncating the proximal N-terminus up to Arg393 of GluN2B (GluN2BΔM394) completely abolished the specific, high sensitivity, inhibition by both XK1 (IC50 > 1000 nM, n= 5 oocytes) and XK2 (IC50 > 1000 nM, n= 4 oocytes) (Figure 3B).
Figure 3.
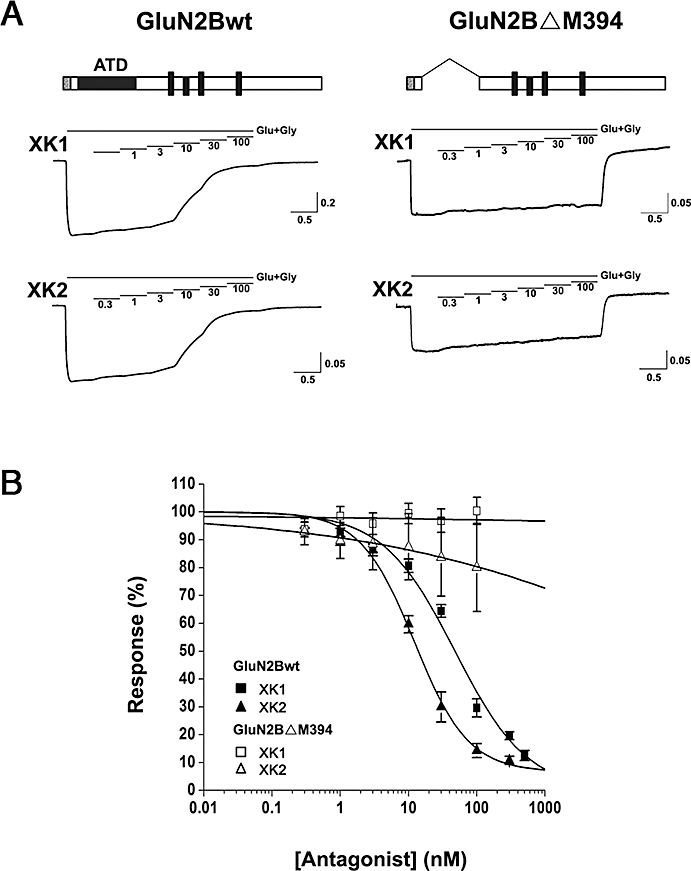
Inhibitory activities of XK1 and XK2 mediated via ATD of GluN2B. (A) Representative current traces recorded from Xenopus oocytes co-expressing rat GluN1-1a/GluN2Bwt (left traces) and GluN1-1a/GluN2BΔM394 (right traces). The horizontal lines show applications of solutions containing 0.3, 1, 3, 10, 30 or 100 nM XK1 or XK2 in the presence of maximal glutamate (Glu, 100 µM) and glycine (Gly, 100 µM). Schematic representation of the wt and ATD-truncated GluN2B subunits are depicted above the current traces; grey box denotes signal peptide, black rectangle denotes ATD and black sticks denote three transmembrane domains and a re-entrant loop. The holding potential was –50 mV. Calibration: current in nA and time in min. (B) Composite XK1 and XK2 inhibition curves are shown for oocytes expressing GluN1-1a/GluN2Bwt and GluN1-1a/GluN2BΔM394. Response (%) represents the percentage of current measured at each antagonist concentration to the maximum current elicited by glutamate (100 µM) and glycine (100 µM). Each point is the mean ± SEM value of four to seven oocytes. The mean IC50 values for XK1 and XK2 on GluN1-1a/GluN2Bwt are 37.6 nM and 15.7 nM respectively. The IC50 values for the high affinity binding of XK1 and XK2 on GluN1-1a/GluN2BΔM394 could not be determined within the antagonists' concentration range studied. ATD, amino-terminal domain.
XK1 and XK2 bind to ATD of GluN2B
The results obtained above suggest that the inhibitory activities exhibited by XK1 and XK2 on GluN2B-containing NMDA receptors are very likely to be mediated through their binding to the proximal N-terminus of GluN2B. To demonstrate direct binding of XK1 and XK2 to ATD of GluN2B, an additional and independent biochemical approach using the isolated recombinant protein of ATD of GluN2B was adopted. A 6xHis-ATD2B recombinant protein previously demonstrated to bind ifenprodil at a dissociation constant (KD) (Wong et al., 2005; Han et al., 2008; Ng et al., 2008) similar to that of [3H]-ifenprodil binding to rat brain cortex and mouse L(tk-) cell line (Schoemaker et al., 1990; McCauley et al., 2004; Kiss et al., 2005) was expressed, refolded and purified to carry out ligand binding assay using CD. Figure 4A shows that the recombinant 6xHis-ATD2B protein bound ifenprodil with a KD value of 90.8 ± 21.3 nM (n= 3), in agreement with that reported previously (Wong et al., 2005; Han et al., 2008; Ng et al., 2008). Increasing concentrations of XK1 or XK2 were incubated with 6xHis-ATD2B proteins and the concentration-dependent changes in CD spectra induced by each ligand were recorded (Figure 4B and C). XK1 showed a KD of 1.2 ± 0.2 nM (n= 4) while XK2 yielded 1.0 ± 0.2 nM (n= 5) (Table 1).
Figure 4.
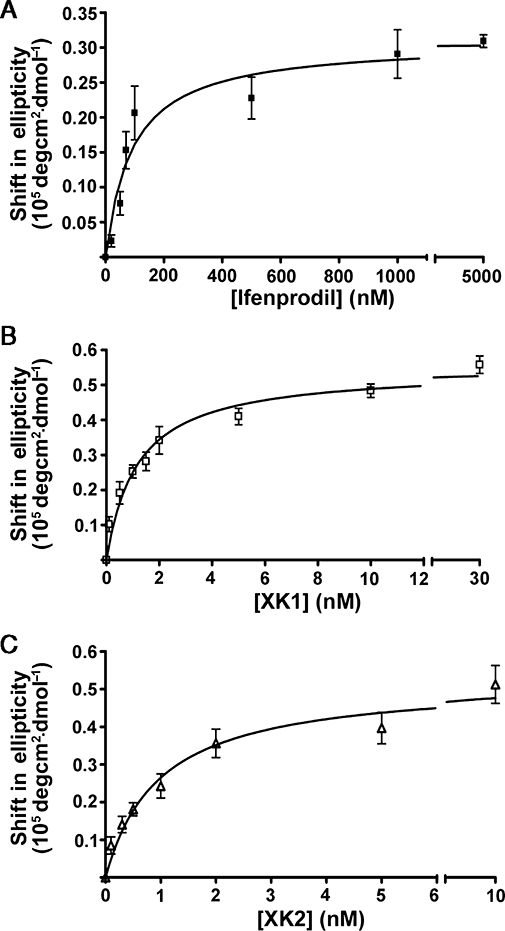
XK1 and XK2 bind to recombinant ATD protein of GluN2B. (A) Ifenprodil, (B) XK1 and (C) XK2 bound to 6xHis-ATD2B in a concentration-dependent manner yielding KD values 91 nM, 1.2 nM and 1.0 nM respectively. The x axes were broken between the last two highest antagonist concentrations to highlight the curve fitting of data at the lower concentrations as well as saturated bindings. ATD, amino-terminal domain.
Molecular determinants of XK1 and XK2 sensitivity within the ATD of GluN2B
Site-directed mutagenesis experiments have delineated numerous critical amino acids within ATD of GluN2B subunit that influence the high-affinity inhibition of GluN1/GluN2B receptors induced by ifenprodil (Perin-Dureau et al., 2002; Mony et al., 2009a). Selective mutations at positions that strongly (>60-fold shift in ifenprodil IC50– D101A, I150A, F176A and Y231A), moderately (5>IC50 fold shift>60 – D104A) and weakly (IC50 < 5-fold shift – Y282A) affect ifenprodil sensitivity were screened by measuring inhibition of agonist-induced current at two concentrations of XK1 and XK2: 30 nM, a concentration approximately the IC50, and 300 nM, a nearly saturating concentration for recombinant GluN1-1a/GluN2Bwt receptors. The sensitivities of D101A, I150A and F176A to XK1 and XK2 were significantly decreased at both concentrations tested (P < 0.01, Table 2). Interestingly, substitution of Asp104 with alanine affected the sensitivity to XK1 much more than that to XK2. Y231A mutation reduced the sensitivity to XK2 at both 30 nM and 300 nM but only to XK1 at 300 nM. The Y282A mutation showed reduced sensitivities to XK1 and XK2 only at 300 nM (Table 2).
Table 2.
Screening of point mutations in the ATD of GluN2B for effects on XK1 and XK2 sensitivity
| GluN2B mutant |
Mean relative current (%) |
|||||
|---|---|---|---|---|---|---|
|
XK1 |
XK2 |
|||||
| 30 nM | 300 nM | n | 30 nM | 300 nM | n | |
| wt | 64 ± 1* | 20 ± 2 | 8 | 43 ± 2 | 11 ± 1 | 7 |
| D101A | 95 ± 5** | 70 ± 2** | 4 | 93 ± 6** | 46 ± 7** | 6 |
| D104A | 92 ± 7** | 50 ± 9** | 5 | 64 ± 12* | 15 ± 3 | 5 |
| I150A | 98 ± 18** | 100 ± 26** | 4 | 88 ± 9** | 79 ± 13** | 4 |
| F176A | 95 ± 7** | 73 ± 11** | 4 | 108 ± 5** | 79 ± 4** | 5 |
| Y231A | 81 ± 10 | 108 ± 5** | 3 | 75 ± 12** | 50 ± 12** | 4 |
| Y282A | 79 ± 10 | 51 ± 14** | 4 | 58 ± 10 | 30 ± 12** | 4 |
Significance of the differences in the mean relative currents were calculated at 30 nM and 300 nM of XK1 and XK2 using anova post hoc Tukey test. For both XK1 and XK2, the mutations D101A, I150A and F176A yielded significant differences (**P < 0.01) when compared with wt at both concentrations tested. The D104A mutation had a relatively weak effect on inhibition by XK2 (*P < 0.05 at 30 nM; P > 0.05 at 300 nM). The Y231A mutation showed reduced inhibition by XK1 at 300 nM and XK2 at both concentrations (**P < 0.01). The Y282A mutation resulted in a significant reduction in inhibition sensitivities to both ligands at only 300 nM (**P < 0.01). All mutations tested yielded significant differences for ifenprodil (P < 0.001, data not shown).
ATD, amino-terminal domain.
Extracellular pH influences inhibition by XK1 and XK2
In recombinant GluN1/GluN2B receptors expressed in oocytes, ifenprodil inhibition was found to be pH-sensitive with a smaller inhibition at alkaline pH. (Pahk and Williams, 1997; Mott et al., 1998). Decreasing the pH from 8.0 to 6.8 significantly increased the degree of inhibition by 30 nM XK1 (P < 0.01) and XK2 (P < 0.001) (Figure 5A and B). Lowering the pH from 7.3 to 6.8 increased XK2 potency, but had no significant effect on the inhibitory potency of XK1.
Figure 5.
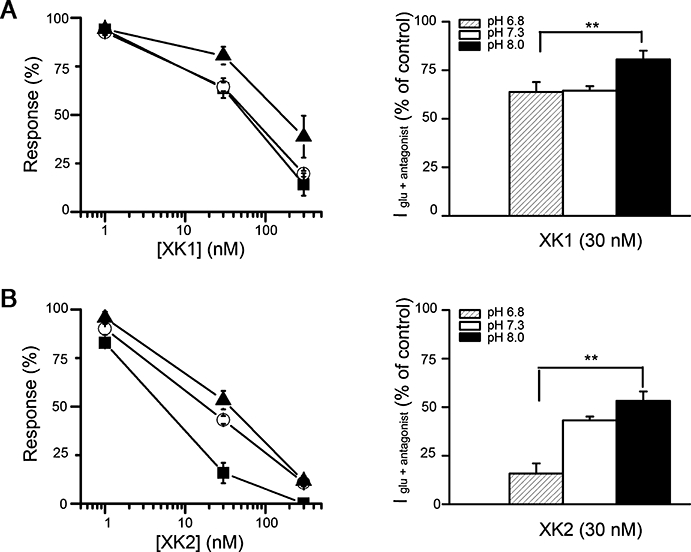
The XK1 and XK2 inhibition of recombinant GluN1/GluN2Bwt receptors is pH sensitive. (A) (Left panel) The % inhibition produced by 1 nM, 30 nM and 300 nM XK1 and (B) XK2 were measured at pH 6.8 (solid squares), pH 7.3 (open circles) and pH 8.0 (solid triangles) from oocytes injected with GluN1-1a/GluN2Bwt. (Right panel) Histograms comparing the potency of 30 nM XK1 or XK2 at three pHs tested. **Indicates a significant increase in inhibition by XK1 and XK2 at acidic pH (P < 0.01). Values are mean ± SEM from n= 4–16 for each condition tested and are expressed as a percentage of the control response to glutamate (100 µM) and glycine (100 µM) at each pH.
Discussion
The present study was carried out to investigate the domain at which 5-substituted benzimidazole derivatives, exemplified by XK1 and XK2, exert their negative modulatory effects on the GluN1/GluN2B receptor. We report here, for the first time, that XK1 and XK2 bind to the ATD of GluN2B to exert their inhibitory properties. The ATD of the GluN2B subunit has been shown previously to be the domain where ifenprodil binds to exert its negative allosteric modulation of GluN1/GluN2B receptor (Kew et al., 1996; Perin-Dureau et al., 2002; Wong et al., 2005; Rosahl et al., 2006; Han et al., 2008; Ng et al., 2008; Mony et al., 2009b; reviewed by Williams, 2001).
In mice, inhibition of hippocampal neuronal activity by ifenprodil exhibited a biphasic concentration-response curve with the high-affinity component IC50 318 nM and the low-affinity component IC50 at 95–139 µM (Rosahl et al., 2006). Rosahl et al. suggested that the high affinity site resides within the proximal N-terminus 282 amino acids while the low affinity site presumably represents the non-selective inhibition of NMDA receptor subtypes at very high concentrations of ifenprodil. The ifenprodil concentration used in the present study was 10 µM, approximately 10-fold lower than the low-affinity component (IC50 95 µM; Rosahl et al., 2006) and hence it is assumed to be insufficient to exert a non-selective, inhibitory effect on other NMDA receptors.
XK1 and XK2 were very potent at preventing NMDA-induced cerebrocortical neuronal death; this effect is most likely mediated through GluN2B-containing NMDA receptors. The embryonic cerebrocortical neurones used in this study are appropriate native cell types that have physiological relevance. The GluN2B mRNA level of day 16–18 embryonic Sprague-Dawley rat cortical neurones has been shown to peak at in vitro culture day 10–21 (Cheng et al., 1999). This time course of the increase in GluN2B mRNA most closely followed the increase in glutamate (100 µM and glycine 10 µM)-stimulated changes in magfura-2 signal and neuronal injury (Cheng et al., 1999). In addition, increased glutamate-induced currents over time in cultures of E17-19 DIV7-15 rat cortical and hippocampal neurones has been demonstrated by the patch electrode voltage-clamp technique (Ozawa et al., 1988; Murphy and Baraban, 1990). The concomitant increases in GluN2B mRNA, glutamate-induced intracellular [Ca2+] and currents, and neuronal injury suggest that NMDA receptors (especially GluN2B) are involved in the development of susceptibility to excitotoxicity in of rat cerebrocortical neurones in vitro. Similar glutamate-induced neurotoxicity was ascribed to GluN1/GluN2B receptors in primary cultures of E15-16 murine cerebrocortical neurones DIV 7-11 (Mizuta et al., 1998; Gu et al., 2009). Incubation of rat E18 DIV10-11 cerebrocortical neurones in increasing concentrations of XK1 or XK2 could prevent neuronal cell death. Such concentration-dependent inhibition by XK1 and XK2 of neuronal cell death (mediated through the fraction of NMDA receptors that contained the GluN2B subunit) yielded low nanomolar IC50 values. XK2 displayed ∼6-fold relatively higher potency than XK1 which is in good agreement with that reported by McCauley et al. (2004) using stably transfected L(tk-) cells of human GluN1/GluN2B receptors (∼3-fold). Next, the inhibitory effects of XK1 and XK2 were studied using voltage-clamp recordings of GluN1/GluN2B wild-type receptors expressed in Xenopus oocytes. The cationic inward current elicited by glutamate and glycine (saturating concentrations) activation of GluN1/GluN2B receptors could be reduced by XK1 and XK2 in an antagonist concentration-dependent manner at physiological pH.
To determine which part of the extracellular domains of the GluN2B subunit is responsible for the inhibitory activity of both XK1 and XK2, the ATD truncated GluN2B subunit was constructed and co-expressed in Xenopus oocytes with the GluN1-1a subunit. The amino terminal 1-28 residues of the rat GluN2B cDNA, which contained the rat GluN2B putative signal peptide, were left intact. This ensured proper protein trafficking and targeting. Oocytes expressing GluN1-1a/GluN2Bwt receptors yielded progressively attenuated responses in the presence of increasing concentrations of XK1 and XK2. The fitted IC50 values of XK1 (37.6 nM) and XK2 (15.7 nM) were consistent with the higher potency of XK2 (McCauley et al., 2004). In contrast, oocytes expressing GluN1-1a/GluN2BΔM394 receptors were sensitive to saturating concentrations of glutamate and glycine but completely insensitive to XK1 and XK2. Due to the low nanomolar IC50 values of XK1 and XK2, it is likely that they share a similar interaction domain with that of ifenprodil on GluN2B ATD. Indeed, Rosahl et al. (2006) did elegant experiments to demonstrate that ifenprodil-sensitivity and -binding reside within the ATD of the GluN2B subunit. Using a gene ‘knock-in’ technique, amino acids 35–282 of GluN2B were replaced by the corresponding domain of GluN2A. This generated GluN2B ‘knock-in’ mice where the hippocampal neurones displayed a greatly reduced sensitivity to ifenprodil.
After the N-terminal ∼363 amino acids of GluN2B had been implicated as the putative high-affinity binding site responsible for the efficacy of XK1 and XK2, a recombinant protein of GluN2B ATD was used to demonstrate direct binding of XK1 and XK2 to this domain. Ligand-induced changes in protein structure were inferred from changes in the CD spectra, and the concentration-dependence of these changes was used to determine binding constants for XK1 and XK2. In line with the above functional findings, XK1 and XK2 showed specific binding to the recombinant 6xHis-ATD2B protein, just like ifenprodil (Wong et al., 2005; Han et al., 2008; Ng et al., 2008). It is noted that, in this study, there is a 2- to 10-fold difference between the neuronal IC50 and 6xHis-ATD2B KD values for both XK1 and XK2. Still, the relative potency of XK2 to XK1 remained consistent. The IC50 values determined from the functional assays demonstrated that the reduction in cation flux through the aqueous channel pore induced by the antagonists is concentration-dependent. This functional measure thus takes into account the effects of allosteric interactions between domains, long-range intra-protein interactions, in addition to effects on the ligand binding site. It is likely that the differences between KD and functional IC50 values observed here are partially attributed to the study of ATD2B in isolation.
Ifenprodil is representative of a class of NMDA receptor antagonists (phenylethanolamines) which (i) possesses high selectivity for GluN2B-containing receptors (reviewed by Williams, 2001); (ii) inhibits GluN2B-containing receptors non-competitively and voltage-independently (Carter et al., 1988; Legendre and Westbrook, 1991; Williams, 1993; Perin-Dureau et al., 2002); (iii) binds to ATDs of GluN2B (Perin-Dureau et al., 2002; Wong et al., 2005; Rosahl et al., 2006; Han et al., 2008; Ng et al., 2008) and GluN1 (Masuko et al., 1999; Han et al., 2008); (iv) has its potency strongly dependent on the extraceullar pH but weakly on GluN1 exon 5 (Pahk and Williams, 1997; Mott et al., 1998); and finally (v) displays use-dependence such that binding of ifenprodil increases binding of glutamate and vice versa (Kew et al., 1996; Zheng et al., 2001). Interestingly, benzimidazole derivatives (exemplified by XK1 and XK2), like ifenprodil, share a similar sensitivity to extracellular pH and critical molecular determinants within ATD of GluN2B subunit. The structure of XK1 bears resemblance and differences to ifenprodil in several ways. Firstly, both XK1 and ifenprodil share the same benzylpiperidine side chain. In contrast, XK1 bears a sulphonamide functional group as an isosteric replacement of the phenolic group in ifenprodil to serve as a hydrogen bond donor (McCauley et al., 2004). The aromatic ring, the positively charged nitrogen of benzylpiperidine side chain and the hydrogen bond donor group represented by the phenolic group of ifenprodil were regarded as key pharmacophoric features for this class of ligands (Borza and Domány, 2006). Recent computational studies on the binding mode of ifenprodil and Ro25,6981 indicated that an additional hydrogen bond acceptor moiety in proximity to the positively charged piperidine ring may aide in the binding (Gitto et al., 2008). It is interesting to note that the benzimidazole ring nitrogen in XK1 and XK2 can potentially function as a hydrogen bond acceptor group. An interesting observation from our mutagenesis studies revealed that replacing Asp104 with alanine differentially affected the inhibition by XK1 much more strongly than XK2. One plausible explanation could be that the central basic nitrogen of XK1, which is absent in XK2, may interact with the negatively charged Asp104 residue.
The similarity shown in this study between benzimidazole derivatives (XK1 and XK2) and ifenprodil in terms of their binding site on GluN2B and pH-sensitivity suggests that these two classes of GluN2B-selective NMDA receptor antagonists could share a common mechanism of modulation at the structural level. The knowledge on where the new class of benzimidazoles bind to the GluN2B subunit has practical implications for rational drug design that can be used as valuable research tools and may ultimately lead to novel treatment of the wide variety of neurological disorders such as stroke and pain that have been suggested to involve GluN2B-containing NMDA receptors.
Acknowledgments
We are grateful to Dr Stephen F. Heinemann (Salk Institute) for sharing cDNAs of GluN1-1a and GluN2B (GenBank Accession Nos. U11418 and U11419 respectively); Dr Stephen F. Traynelis (Emory University) for pCIneo GluN1-1a, GluN2B mutant cDNAs and EasyOocyte software; Dr M.L. Go (NUS) for supervision on part of synthesis work; Dr. G. Dawe (NUS) for kindly sharing Xenopus oocytes and Dr Philip K. Moore (King's College London) for critical reading of this manuscript. We thank the NUH-NUS Medical Publications Support Unit, Singapore, for assistance in the preparation of the article. Support for this research was provided by Biomedical Research Council (R-184-000-119-305) and NUS Academic Research Funds (R-184-000-114-112 and Final Year Project) to C-M.L. K-S.N., H-W.L., K-H.K. and F-M.N. are recipients of graduate research scholarships from the NUS.
Glossary
Abbreviations:
- CD
circular dichroism
- DIV
day-in-vitro
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Characterization of primary E18 DIV10-11 cerebrocortical neuronal cultures using immunofluorescence microscopy and immunoblotting. (a) Double immunolabelling with neuronal and astrocyte markers. Significantly more βIII tubulin-positive (blue) neurones than GFAP-positive (green) astrocytes showed that the culturing protocol reported here produced pure neuronal cultures. (b) Triple immunolabelling of the neurones with anti-βIII tubulin (blue), GluN1 (red) and GluN2B (green). Merged immunofluorescence confocal images showed nearly all neurones co-expressed GluN1 and GluN2B proteins (arrowhead; magenta). The single neurone that did not stain positively for either GluN1 or GluN2B is indicated by an arrow. (c) Representative Western blots of GluN1 and GluN2B proteins in E18 DIV10-11 neurones: total neuronal lysate (50μg) versus surface. All of the biotinylated plasma membrane surface proteins eluted from the NeutrAvidin™ beads previously incubated with 500 μg of total neuronal lysates were loaded (right lane). A crude estimation, from the band intensity, suggested there were approximately 10–15% of GluN1 and GluN2B total proteins were localized to the plasma membranes of E18 DIV10-11 neurones. Scale bar = 20 μm.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham K. Future of neuroprotective drugs in doubt. Nat Med. 2002;8:5. doi: 10.1038/nm0102-5a. [DOI] [PubMed] [Google Scholar]
- Borza I, Domány G. NR2B selective NMDA antagonists: the evolution of the ifenprodil-type pharmacophore. Curr Top Med Chem. 2006;6:687–695. doi: 10.2174/156802606776894456. [DOI] [PubMed] [Google Scholar]
- Carter C, Benavides J, Legendre P, Vincent JD, Noel F, Thuret F, et al. Ifenprodil and SL 82.0715 as cerebral anti-ischemic agents. II. Evidence for N-methyl-D-aspartate receptor antagonist properties. J Pharmacol Exp Ther. 1988;247:1222–1232. [PubMed] [Google Scholar]
- Cheung NS, Pascoe CJ, Giardina SF, John CA, Beart PM. Micromolar L-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurons. Neuropharmacology. 1998;37:1419–1429. doi: 10.1016/s0028-3908(98)00123-3. [DOI] [PubMed] [Google Scholar]
- Cheung NS, Koh CH, Bay BH, Qi RZ, Choy MS, Li Q-T, et al. Chronic exposure to U18666A induces apoptosis in cultured murine cortical neurons. Biochem Biophys Res Communs. 2004;315:408–417. doi: 10.1016/j.bbrc.2004.01.066. [DOI] [PubMed] [Google Scholar]
- Chernard BL, Bordner J, Butler TW, Chambers LK, Collins MA, De Costa DL, et al. 1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: a potent new neuroprotectant which blocks N-methyl-D-aspartate responses. J Med Chem. 1995;38:3138–3145. doi: 10.1021/jm00016a017. [DOI] [PubMed] [Google Scholar]
- Chenard BL, Menniti FS. Antagonists selective for NMDA receptors containing the NR2B subunit. Curr Pharm Des. 1999;5:381–404. [PubMed] [Google Scholar]
- Cheng C, Fass DM, Reynolds IJ. Emergence of excitotoxicity in cultured forebrain neurons coincides with larger glutamate-stimulated [Ca2+]i increases and NMDA receptor mRNA levels. Brain Res. 1999;849:97–108. doi: 10.1016/s0006-8993(99)01995-2. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Headley PM, Tzschentke TM. NMDA receptor antagonists as analgesics: focus on the NR2B subtype. Trends Pharamacol Sci. 2001;22:636–642. doi: 10.1016/s0165-6147(00)01863-0. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Erreger K, Chen PE, Wyllie DJ, Traynelis SF. Glutamate receptor gating. Crit Rev Neurobiol. 2004;16:187–224. doi: 10.1615/critrevneurobiol.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- Gitto R, De Luca L, Ferro S, Occhiuto F, Samperi S, De Sarro G, et al. Computational studies to discover a new NR2B/NMDA receptor antagonist and evaluation of pharmacological profile. ChemMedChem. 2008;3:1539–1548. doi: 10.1002/cmdc.200800124. [DOI] [PubMed] [Google Scholar]
- Gu B, Nakamichi N, Zhang W-S, Nakamura Y, Kambe Y, Fukumori R, et al. Possible protection by notoginsenoside R1 against glutamate toxicity mediated by N-methyl-D-aspartate receptors composed of an NR1/NR2B subunit assembly. J Neurosci Res. 2009;87:2145–2156. doi: 10.1002/jnr.22021. [DOI] [PubMed] [Google Scholar]
- Han X, Tomitori H, Mizuno S, Higashi K, Füll C, Fukiwake T, et al. Binding of spermine and ifenprodil to a purified, soluble regulatory domain of the N-methyl-D-aspartate receptor. J Neurochem. 2008;107:1566–1577. doi: 10.1111/j.1471-4159.2008.05729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartnett KA, Stout AK, Ragdev S, Rosenberg PA, Reynolds IJ, Aizenman E. NMDA receptor-mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro. J Neurochem. 1997;68:1836–1845. doi: 10.1046/j.1471-4159.1997.68051836.x. [DOI] [PubMed] [Google Scholar]
- Hocking G, Cousins MJ. Ketamine in chronic pain management: an evidence-based review. Anesth Analg. 2003;97:1730–1739. doi: 10.1213/01.ANE.0000086618.28845.9B. [DOI] [PubMed] [Google Scholar]
- Kambe Y, Nakamichi N, Geogiev DD, Nakamura N, Taniura H, Yoneda Y. Insensitivity to glutamate neurotoxicity mediated by NMDA receptors in association with delayed mitochondrial membrane potential disruption in cultured rat cortical neurons. J Neurochem. 2008;105:1886–1900. doi: 10.1111/j.1471-4159.2008.05270.x. [DOI] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol (Lond) 1996;497:761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L, Cheng G, Bednar B, Bednar RA, Bennett PB, Kane SA, et al. In vitro characterization of novel NR2B selective NMDA receptor antagonists. Neurochem Int. 2005;46:453–464. doi: 10.1016/j.neuint.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Layton ME, Kelly MJ, 3rd, Rodzinak KJ. Recent advances in the development of NR2B subtype-selective NMDA receptor antagonists. Curr Top Med Chem. 2006;6:697–709. doi: 10.2174/156802606776894447. [DOI] [PubMed] [Google Scholar]
- Le DA, Lipton SA. Potential and current use of N-methyl-D-aspartate (NMDA) receptor antagonists in diseases of aging. Drugs Aging. 2001;18:717–724. doi: 10.2165/00002512-200118100-00001. [DOI] [PubMed] [Google Scholar]
- Legendre P, Westbrook GL. Ifenprodil blocks N-methyl-D-aspartate receptors by a two-component mechanism. Mol Pharmacol. 1991;40:289–298. [PubMed] [Google Scholar]
- Li JH, Wang YH, Wolfe BB, Krueger KE, Corsi L, Stocca G, et al. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur J Neurosci. 1998;10:1704–1715. doi: 10.1046/j.1460-9568.1998.00169.x. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci. 2004;24:8885–8895. doi: 10.1523/JNEUROSCI.2476-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low C-M, Zheng F, Lyuboslavsky P, Traynelis SF. Molecular determinants of coordinated proton and zinc inhibition of N-methyl-D-aspartate NR1/NR2A receptors. Proc Natl Acad Sci USA. 2000;97:11062–11067. doi: 10.1073/pnas.180307497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low C-M, Lyuboslavsky P, French A, Le P, Wyatte K, Thiel WH, et al. Structural determinants of proton-sensitive N-methyl-D-aspartate receptor gating. Mol Pharmacol. 2003;63:1212–1222. doi: 10.1124/mol.63.6.1212. [DOI] [PubMed] [Google Scholar]
- McCauley JA, Theberge CR, Romano JJ, Billings SB, Anderson KD, Claremon DA, et al. NR2B-selective N-methyl-D-aspartate antagonists: synthesis and evaluation of 5-substituted benzimidazoles. J Med Chem. 2004;47:2089–2096. doi: 10.1021/jm030483s. [DOI] [PubMed] [Google Scholar]
- Masuko T, Kashiwagi K, Kuno T, Nguyen ND, Pahk AJ, Fukuchi J, et al. A regulatory domain (R1–R2) in the amino terminus of the N-methyl-D-aspartate receptor: effects of spermine, protons, and ifenprodil, and structural similarity to bacterial leucine/isoleucine/valine binding protein. Mol Pharmacol. 1999;55:957–969. doi: 10.1124/mol.55.6.957. [DOI] [PubMed] [Google Scholar]
- Mizuta I, Katayama M, Watanabe M, Ishii K. Developmental expression of NMDA receptor subunits and the emergence of glutamate neurotoxicity in primary cultures of murine cerebral cortical neurons. Cell Mol Life Sci. 1998;54:721–725. doi: 10.1007/s000180050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mony L, Krzaczkowski L, Leonetti M, Goff AL, Alarcon K, Neyton J, et al. Structural basis of NR2B-selective antagonist recognition by N-methyl-D-aspartate receptors. Mol Pharmacol. 2009a;75:60–74. doi: 10.1124/mol.108.050971. [DOI] [PubMed] [Google Scholar]
- Mony L, Kew JNC, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanism and therapeutic potential. Br J Pharmacol. 2009b;157:1301–1317. doi: 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, et al. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Baraban JM. Glutamate toxicity in immature cortical neurons precedes development of glutamate receptor currents. Dev Brain Res. 1990;57:146–150. doi: 10.1016/0165-3806(90)90195-5. [DOI] [PubMed] [Google Scholar]
- Ng F-M, Geballe M, Snyder JP, Traynelis SF, Low C-M. Structural insights into phenylethanolamines high-affinity binding site in NR2B from binding and molecular modeling students. Mol Brain. 2008;1:16. doi: 10.1186/1756-6606-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikam SS, Meltzer LT. NR2B selective NMDA receptor antagonists. Curr Pharm Des. 2002;8:845–855. doi: 10.2174/1381612024607072. [DOI] [PubMed] [Google Scholar]
- Ozawa S, Nakamura T, Yuzaki M. Cation permeability change caused by L-glutamate in cultured rat hippocampal neurons. Brain Res. 1988;443:85–94. doi: 10.1016/0006-8993(88)91601-0. [DOI] [PubMed] [Google Scholar]
- Pahk AJ, Williams K. Influence of extracellular pH on inhibition by ifenprodil at N-methyl-D-aspartate receptors in Xenopus oocytes. Neurosci Lett. 1997;225:29–32. doi: 10.1016/s0304-3940(97)00176-6. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:9–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Perin-Dureau F, Rachline J, Neyton J, Paoletti P. Mapping the binding site of the neuroprotectant ifenprodil on NMDA receptors. J Neurosci. 2002;22:5955–5965. doi: 10.1523/JNEUROSCI.22-14-05955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinard E, Alanine A, Bourson A, Büttelmann B, Heitz M-P, Mutel V, et al. 4-Aminoquinolines as a novel class of NR1/NR2B subtype selective NMDA receptor antagonists. Bioorg Med Chem Lett. 2002;12:2615–2619. doi: 10.1016/s0960-894x(02)00470-5. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. N-methyl-D-aspartate receptor proteins NR2A and NR2B are differentially distributed in the developing rat central nervous system as revealed by subunit-specific antibodies. J Neurochem. 1996;66:692–700. doi: 10.1046/j.1471-4159.1996.66020692.x. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Wingrove PB, Hunt V, Fradley RL, Lawrence JM, Heavens RP, et al. A genetically modified mouse model probing the selective action of ifenprodil at the N-methyl-D-aspartate type 2B receptor. Mol Cell Neurosci. 2006;33:47–56. doi: 10.1016/j.mcn.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoemaker H, Allen J, Langer SZ. Binding of [3H]ifenprodil, a novel NMDA antagonist, to a polyamine-sensitive site in the rat cerebral cortex. Eur J Pharmacol. 1990;176:249–250. doi: 10.1016/0014-2999(90)90539-i. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamil AW, McLean MJ. Use-, concentration- and voltage-dependent limitation by MK-801 of action potential firing frequency in mouse central neurons in cell culture. J Pharmacol Exp Ther. 1992;260:376–383. [PubMed] [Google Scholar]
- Wee KS-L, Zhang Y, Khanna S, Low C-M. Immunolocalization of NMDAreceptor subunit NR3B in selected structures in the rat forebrain, cerebellum, and lumbar spinal cord. J Comp Neurol. 2008;509:118–135. doi: 10.1002/cne.21747. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: Selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Williams K. Ifenprodil, a novel NMDA receptor antagonist: site and mechanism of action. Curr Drug Targets. 2001;2:285–298. doi: 10.2174/1389450013348489. [DOI] [PubMed] [Google Scholar]
- Witt A, Macdonald N, Kirkpatrick P. Memantine hydrochloride. Nat Rev Drug Discov. 2004;3:109–110. doi: 10.1038/nrd1311. [DOI] [PubMed] [Google Scholar]
- Wong ES-P, Ng F-M, Yu C-Y, Lim P, Lim L-H, Traynelis SF, et al. Expression and characterization of soluble amino-terminal domain of NR2B subunit of N-methyl-D-aspartate receptor. Protein Sci. 2005;14:2275–2283. doi: 10.1110/ps.051509905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying HS, Gottron FJ, Choi DW. Assessment of cell viability in primary neuronal cultures. Curr Protoc Neurosci. 2000. Chapter 7, Unit 7.18. [DOI] [PubMed]
- Yuan H, Erreger K, Dravid SM, Traynelis SF. Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. J Biol Chem. 2005;280:29708–29716. doi: 10.1074/jbc.M414215200. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Ren Y, Yan Z. Postsynaptic density-95 (PSD-95) and calcineurin control the sensitivity of N-methyl-D-aspartate receptors to calpain cleavage in cortical neurons. Mol Pharmacol. 2008;74:360–370. doi: 10.1124/mol.108.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Erreger K, Low C-M, Banke T, Lee CJ, Conn PJ, et al. Allosteric interaction between the amino terminal domain and the ligand binding domain of NR2A. Nat Neurosci. 2001;4:894–901. doi: 10.1038/nn0901-894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.