

Abstract
Background and purpose:
The effects of statins on diabetes mellitus (DM) are controversial, and their effects on pancreatic fibrosis are poorly defined. We investigated the effect of long- and short-term treatments with pravastatin on the development of DM and pancreatic fibrosis in DM-prone Otsuka–Long–Evans–Tokushima Fatty (OLETF) rats.
Experimental approach:
Male OLETF rats were divided into four groups at 12 weeks of age. The first group received a standard rat diet until the end of the experimental period at age 80 weeks. The second group was given a diet containing 0.05% pravastatin from 12 weeks of age, before the onset of DM and pancreatic fibrosis, and the third group was given the same pravastatin diet from 28 weeks of age, after the onset of DM and pancreatic fibrosis, until age 80 weeks. The fourth group received the same pravastatin diet only for 16 weeks, from 12 to 28 weeks of age, and switched to a standard diet. Progressions of DM and pancreatic fibrosis were evaluated.
Key results:
Long-term treatments with pravastatin, either from 12 or 28 weeks of age, decreased serum glucose concentration and fibrotic area, elevated superoxide dismutase activity and down-regulated transforming growth factor-β1 mRNA in the pancreas. In contrast, after a short-term treatment with pravastatin, these parameters markedly deteriorated after its cessation.
Conclusions and implications:
The results suggest that long-term treatment with pravastatin improves DM and pancreatic fibrosis via anti-oxidative and anti-fibrotic properties, whereas cessation of pravastatin abolishes these beneficial effects, and accelerates DM and pancreatic fibrosis.
Keywords: pravastatin, pancreatic fibrosis, diabetes mellitus, superoxide dismutase, transforming growth factor-β1, OLETF rat
Introduction
The Otsuka–Long–Evans–Tokushima Fatty (OLETF) rat is a diabetic strain established from the outbred colony of Long–Evans rat, and develops diabetes mellitus (DM) after 24 weeks of age (Kawano et al., 1992). During the progression of type 2-like DM, the OLETF rat eventually becomes hypo-insulinaemic and develops type 1-like DM after 70 weeks of age. Histologically, mild to moderate lymphocyte infiltration in the endocrine and exocrine pancreas is observed, and fibrosis becomes prominent especially around and in the islet at 6–20 weeks of age. After 70 weeks of age, the islets and exocrine glands are extremely atrophic and replaced by fatty and connective tissue (Kawano et al., 1992). Based on these histological alterations, OLETF rats are used as an animal model of pancreatic fibrosis as well as DM (Yoshikawa et al., 2002).
Pancreatic fibrosis is progressive and irreversible; therefore, chronic pancreatitis is considered to be one of the intractable pancreatic diseases. Recently, an experimental study in vitro has demonstrated that lovastatin inhibits the activation of pancreatic stellate cells (Jaster et al., 2003), which play a central role in pancreatic fibrosis (Apte et al., 1998). These results suggest that statins exert beneficial effects on pancreatic fibrosis, and improve the clinical course of chronic pancreatitis.
Statins are widely used in the first line management of hyperlipidaemia due to their known efficacy in improving plasma lipid profiles (Endo et al., 1976). However, recent studies have demonstrated that statins exert pleiotropic effects such as anti-oxidative (Moriyama et al., 2001), anti-inflammatory (Solheim et al., 2001; Li et al., 2004) and anti-fibrotic actions (Moriyama et al., 2001; Li et al., 2004) beyond cholesterol reduction. In addition, statins have been shown to exert beneficial effects on the progression of DM, not only by lowering plasma lipid levels (Freeman et al., 2001), but also by improving insulin sensitivity, insulin secretion (Paniagua et al., 2002) and leptin resistance (Yu et al., 2004), although not all the results reported are in agreement with these findings (Satoh et al., 2005; Takano et al., 2006). Because oxidative stress participates in the development of DM (Kaneto et al., 1999) and pancreatic fibrosis (Matsumura et al., 2001), we hypothesized that treatment with statin may exert beneficial effects on the progressions of DM and pancreatic fibrosis in the OLETF rats. In addition, we have previously demonstrated that the beneficial effects of acarbose on DM and pancreatic fibrosis persist for more than 40 weeks after its withdrawal in the OLETF rats (Yamamoto et al., 1999). Based on our previous findings and hypothesis, we examined the effects of pravastatin on the onset and progression of DM and pancreatic fibrosis in the OLETF rat during and after cessation of the treatment.
Methods
Ethical approval
The experimental protocol was approved by the animal welfare committee of our institute.
Animals and drugs
The OLETF rats were used as an animal model of type 2 DM and pancreatic fibrosis, and the age-matched Long–Evans–Tokushima–Otsuka (LETO) rats that are developed from the same colony of OLETF rats but do not show DM and pancreatic fibrosis were used as a normal control. Male OLETF and LETO rats aged 5 weeks were supplied by Otsuka Pharmaceutical Co. (Tokushima, Japan). These rats were maintained in a temperature- (23 ± 2°C) and humidity- (55 ± 5%) controlled room with a 12:12 h light–dark cycle (lights on at 0700 h), and received humane care according to the guidelines at our institution.
Pravastatin [monosodium(3R,5R)-3,5-dihydroxy-7-{(1S,2S,6S,8S,8aR)-6-hydroxy-2-methyl-8-[(2S)-2-methylbutanoyloxy]-1,2,6,7,8,8a-hexahydronaphthalen-1-yl} heptanoate] (Haruyama et al., 1986), a competitive inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA), was a generous gift from Sankyo Co. Ltd. (Tokyo, Japan). Because HMG-CoA is the key enzyme that regulates the synthesis of cholesterol from mevalonic acid, statins, by suppressing the conversion by HMG-CoA, exert their lipid-lowering effects (Endo et al., 1976).
Administration of pravastatin
Standard rat diet consisting of 61% carbohydrate, 26% protein and 13% fat (as a percentage of calories, 3.596 kcal·g−1 diet; Oriental Yeast, Tokyo, Japan) was powdered, and pravastatin was added and thoroughly mixed to a final concentration of 50 mg·100 g−1 food. The drug–diet powder mixture was reconstituted into pellets with a normal appearance. This pravastatin concentration was selected based on results from previous studies (Li et al., 2004; Yu et al., 2004). An OLETF rat of 600 g body wt. consumes almost 30 g food day−1. Therefore, the daily dosage of pravastatin taken by OLETF rat was approximately 15 mg per rat, which is equivalent to 25 mg·kg−1 body wt.
Experimental protocol
The OLETF rats were randomly divided into four groups at 12 weeks of age, each consisted of 9–10 rats. The first group of rats received a standard rat diet until the end of the experiment at age 80 weeks (O-C). The second group received a diet containing pravastatin from 12 weeks, before the onset of DM and pancreatic fibrosis, until the end of the experiment (O-P12-80). The third group received a pravastatin diet from 28 weeks, after the onset of DM and pancreatic fibrosis, until 80 weeks of age (O-P28-80). The fourth group of rats received the same pravastatin-containing diet for 16 weeks, from 12 to 28 weeks of age, and then switched to a standard rat diet until the end of the experiment (O-P12-28). The control group of rats, consisting of 10 LETO rats, received a standard rat diet throughout the experimental period (L-C). All groups of rats were allowed free access to food and water. The body weight and food intake were measured every 4 weeks. The food intake was determined for a 48 h period by weighing the full food cups, and then weighing the food cups again 48 h later, collecting for spillage for 8 days. The average food intake was estimated as the amount of food consumed per cage. Because the rats were fed as groups and housed two per cage, the value obtained for 48 h was then divided by four to obtain the approximate estimate of daily food consumption per rat. Blood samples were taken for the determination of fasting serum glucose concentration every 4 weeks, and for the determination of fasting serum total cholesterol (T-cho), triglycerides (TGs), free fatty acids (FFAs) and insulin concentrations every 8 weeks. At 12, 28, 36, 52, 60 and 72 weeks of age, the rats were deprived of food for 12 h before an intravenous glucose tolerance test (IVGTT) was performed under sodium pentobarbital anaesthesia (50 mg·kg−1 body wt., i.p.). A bolus dose of glucose 0.2 g·kg−1 body wt. was injected into the jugular vein, and blood samples were collected before and at 5 min after glucose loading to determine serum concentrations of glucose and insulin. Measurements of serum lipid concentration and IVGTT were conducted in eight rats in each group. At 80 weeks of age, the rats were deprived of food for 12 h before being killed by administration of an overdose of pentobarbital; the abdomen was opened to remove the pancreas and adipose depots. The pancreas was cleared of lymph nodes and fat, and weighed. A splenic portion of the pancreas was frozen at −80°C until assays. A duodenal portion of the pancreas was used for histological examination. White adipose depots were collected from the retroperitoneum, mesentery and epididymis, and then weighed.
Assays
For the measurement of pancreatic protein and DNA contents, the pancreas was homogenized in a 0.15 M sodium chloride solution using a motor-driven, Teflon-coated glass homogenizer. The homogenates were filtered through three layers of gauze, and then sonicated for 1 min. The aqueous phase was used for protein and DNA assay. Protein concentration in the pancreatic homogenate was measured using Folin phenol reagent with bovine serum albumin as a standard (Lowry et al., 1951). Pancreatic DNA was measured fluorometrically by the reaction between 3,5-diaminobenzoic acid and deoxyribose sugar using calf thymus DNA as a standard (Labarca and Paigen, 1980). The activity of a primary antioxidant enzyme, superoxide dismutase (SOD), in the pancreas was determined using water-soluble tetrazolium salt (SOD Assay Kit-WST; Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Insulin concentrations in serum and pancreatic homogenates were determined by RIA using a commercially available kit (ShionoRIA; Shionogi Pharmaceutical Co., Osaka, Japan) with crystalline rat insulin (Novo Industria, Copenhagen, Denmark) as a reference standard. The serum glucose concentration was determined by the glucose oxidase method (Bondar and Mead, 1974) using a commercially available kit (Glucose-E reagent; International Reagents Co., Kobe, Japan). Serum T-cho, TG and FFA concentrations were analysed enzymatically using commercially available kits (Wako Pure Chemical, Tokyo, Japan).
Evaluation of insulin secretory function and insulin resistance
Insulin secretory function was evaluated by insulinogenic index, a widely used index of early-phase insulin response, calculated using the formula: increment of plasma insulin (0–5 min)/increment of plasma glucose (0–5 min) (Kosaka et al., 1974). In addition, insulin resistance was estimated by the homeostasis model assessment of insulin resistance (HOMA-R) calculated from the following formula: fasting insulin (mU·mL−1) × fasting glucose (mM)/22.5 (Matthews et al., 1985).
Myeloperoxidase (MPO) estimation
Neutrophil sequestration in the pancreas was quantified by measuring tissue MPO activity (Bhatia et al., 2005). Tissue samples were thawed, homogenized in 20 mM phosphate buffer (pH 7.4) and centrifuged (10 000× g, 10 min, 4°C), and the resulting pellet was resuspended in 50 mM phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide (Sigma, St Louis, MO, USA). The suspension was subjected to four cycles of freezing and thawing, and was further disrupted by sonication. The sample was then centrifuged (10 000× g, 5 min, 4°C), and the supernatant was used for the MPO assay. The reaction mixture consisting of the supernatant, 1.6 mM tetramethylbenzidine (Sigma), 80 mM sodium phosphate buffer (pH 5.4) and 0.3 mM hydrogen peroxide was incubated at 37°C, and the reaction was terminated with 2 M H2SO4. Then, absorbance was measured at 450 nm and corrected for the weight of the tissue sample, and compared with the control in the OLETF rats (fold increase over control).
Quantitative real-time reverse transcription (RT)–polymerase chain reaction (PCR)
To investigate the mechanisms of the anti-inflammatory and anti-fibrotic actions of pravastatin, the expression levels of a transforming growth factor (TGF)-β1 and tumour necrosis factor (TNF)-α mRNA were determined by quantitative TaqMan PCR with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as a reference. For real-time RT–PCR, total RNA was extracted from the frozen pancreatic tissue by the acid guanidium thiocyanate/phenol/chloroform method (Korc et al., 1992), and 2 µg of total RNA was reverse transcribed using random hexamers and the TaqMan RT reagents (Applied Biosystems, Foster City, CA, USA). The PCR of TGF-β1, TNF-α and GAPDH was performed using the TaqMan probe, the TaqMan Universal PCR Master Mix and primer for TGF-β1 or TNF-α, as well as TaqMan rodent GAPDH control reagents (Applied Biosystems). The PCR products were amplified (50°C, 2 min; 95°C, 10 min; followed by 40 cycles of 95°C, 15 s, and 60°C, 1 min) and analysed on real-time PCR cycler, ABI PRISM 7000 sequence detection system (Applied Biosystems). For quantification, the fluorescence intensity was plotted against the PCR cycle number. The amplification cycle displaying the first significant increase of the fluorescence signal was defined as threshold cycle (CT). The CT value of each sample was compared with the CT values of the standardization series. The amounts of TGF-β1 and TNF-α transcripts were normalized to the amount of GAPDH transcripts in the same cDNA (expressed as fold change per GAPDH).
Histological examination
A duodenal portion of the pancreatic tissue was fixed overnight in 4% buffered neutral paraformaldehyde solution, embedded in paraffin and deparaffinized by standard procedure. Thin sections (5 µm) were stained with haematoxylin and eosin, and Azan–Mallory staining for light microscopic examination.
Quantitative analysis for cross-sectional area of islets and fibrosis
Quantitative evaluations of the cross-sectional area of the islets, and the proportion of fibrotic area in the islets and exocrine glands of the pancreas were performed by using an Axiophot microscope (Carl Zeiss, Eching, Germany) connected to an interactive image analysis system (IBAS; Carl Zeiss). Twenty non-overlapping islets were randomly selected in each experimental group (n= 5), and the cross-sectional area of the islets was determined by IBAS at ×100 magnification. For the determination of the proportion of fibrotic area in the islets and exocrine glands, 10 non-overlapping fields and 20 non-overlapping islets of Azan–Mallory staining were randomly selected in each experimental group (n= 5) at a ×100 magnification. The area of total pancreatic specimen or cross section of islet and that of blue-stained fibrosis was determined by IBAS. The proportion of fibrotic area in the islets and exocrine glands of the pancreas was calculated using the following equation: fibrosis area/total area.
Detection of apoptotic cells
Apoptotic cells were quantified immunohistochemically by a novel monoclonal antibody that recognizes exposed single-stranded regions in the DNA (ss-DNA) of apoptotic cells (Dako Corporation, Carpinteria, CA, USA). For immunohistochemistry, pancreatic tissue sections were immersed in phosphate-buffered saline (PBS) (pH 7.2) for 10 min, and then in PBS containing 3% H2O2 for 10 min to quench endogenous peroxidases. After further incubation in 0.25% casein solution for 10 min, the tissue sections were incubated with a specific primary antibody for ss-DNA diluted at 1:100 in PBS for 16 h at 4°C. The primary antibodies were visualized by the labelled streptavidin–biotin method using a commercially available kit (Dako Corporation), and all procedures were performed as recommended by the manufacturer. Twenty non-overlapping fields were randomly selected in each experimental group (n= 5) at ×200 magnification to determine acinar cell apoptotic index. At least 300 acinar cells were counted in each field, and the apoptotic index represented the number of positive cells per counted cells.
Statistical analysis
Results are expressed as the mean ± SEM. Statistical analysis was performed by analysis of variance followed by Fisher's protected least significant difference test using a commercial software StatView (Abacus Concepts/Brain Power, Berkeley, CA, USA). Differences with P < 0.05 were considered to be statistically significant.
Results
Daily food intake and body weight
The daily food intake of the OLETF rat was approximately 30 g, whereas that of the LETO rat was approximately 22 g. Pravastatin treatment had no influence on daily food intake in the OLETF rat (Figure 1A).
Figure 1.
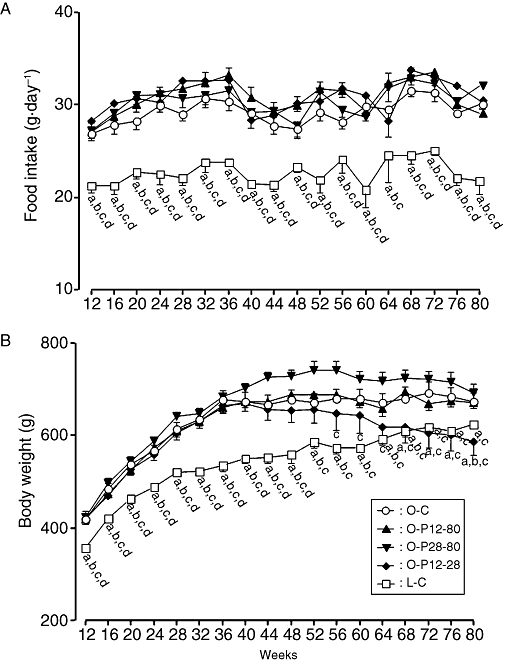
Serial changes in daily food intake (A) and body weight (B) in rats treated with or without pravastatin. The O-C group was given a standard rat diet during the entire experimental period. The O-P12-80 group was maintained on a diet containing pravastatin from 12 to 80 weeks of age. The O-P28-80 group were maintained on a diet containing pravastatin from 28 to 80 weeks of age. The O-P12-28 group received a diet containing pravastatin from 12 to 28 weeks of age, and then switched to standard rat diet until 80 weeks of age. The L-C group was given a standard rat diet during the entire experimental period. (a) Significant difference versus O-C group at corresponding age; (b) significant difference versus O-P12-80 group at corresponding age; (c) significant difference versus O-P28-80 group at corresponding age; (d) significant difference versus O-P12-28 group at corresponding age. Data are presented as mean ± SEM of seven to nine rats.
The body weight of the O-C and O-P12-80 groups increased until 36 weeks of age; thereafter, it remained at a steady state until the end of the observation period (Figure 1B). On the other hand, the body weight of the O-P28-80 group increased until age 52 weeks; thereafter, it decreased to the similar levels to that of O-C and O-P12-80 groups. In contrast, the body weight of the O-P12-28 group gradually decreased after 36 weeks of age, and was significantly lower than that of the O-C group after 68 weeks of age. The body weight of the L-C group showed a gradual increase, but was significantly lower than that of the O-C group at each corresponding time-point (Figure 1B).
Serial changes in fasting serum glucose and insulin concentrations
The fasting serum glucose concentration of the O-C group gradually increased until the end of the observation period, whereas those of the O-P12-80 and O-P28-80 groups gradually decreased to the initial level after commencement of pravastatin (Figure 2A). In contrast, the fasting serum glucose concentration of O-P12-28 gradually increased and was significantly higher than that of the O-C group from 12 weeks after cessation of pravastatin. The fasting serum glucose concentration of the L-C group remained at initial levels until age 72 weeks, but thereafter it gradually increased (Figure 2A).
Figure 2.
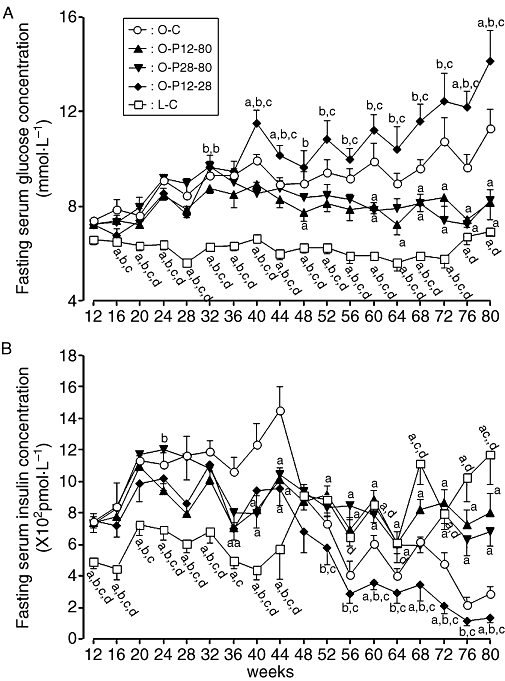
Serial changes in fasting serum glucose (A) and insulin (B) concentrations in rats treated with or without pravastatin. For key to groups, see Figure 1 legend. (a) Significant difference versus O-C group at corresponding age, and (b) significant difference versus O-P12-80 group at corresponding age; (c) significant difference versus O-P28-80 group at corresponding age; and (d): significant difference versus O-P12-28 group at corresponding age. Data are presented as mean ± SEM of seven to nine rats.
The fasting serum insulin concentration increased in the O-C group until 44 weeks of age; thereafter, it markedly decreased (Figure 2B). The fasting serum insulin concentrations of the O-P12-80 and O-P28-80 groups gradually decreased to the initial levels at age 12 weeks after commencement of pravastatin. In contrast, the fasting serum insulin concentration of the O-P12-28 group progressively decreased from 16 weeks after cessation of pravastatin. In the L-C group, the fasting serum insulin concentration gradually increased with age after 52 weeks.
Serial changes in insulin secretory function and insulin resistance
The insulin secretory function evaluated by the insulinogenic index peaked at 36 weeks of age in the O-C group, and thereafter it gradually decreased until the end of the experiment (Figure 3A). Administration of pravastatin before the onset of DM (O-P12-80 group) maintained the insulinogenic index at the initial level until the end of the experiment, and administration of pravastatin after the onset of DM (O-P28-80 group) lowered the elevated insulinogenic index to the initial level. In contrast, the insulinogenic index of the O-P12-28 group gradually, but progressively, decreased below the initial level after cessation of pravastatin, and was significantly lower than that of the O-C group at each corresponding time-point until age 72 weeks. In the L-C group, this index markedly increased after age 52 weeks.
Figure 3.
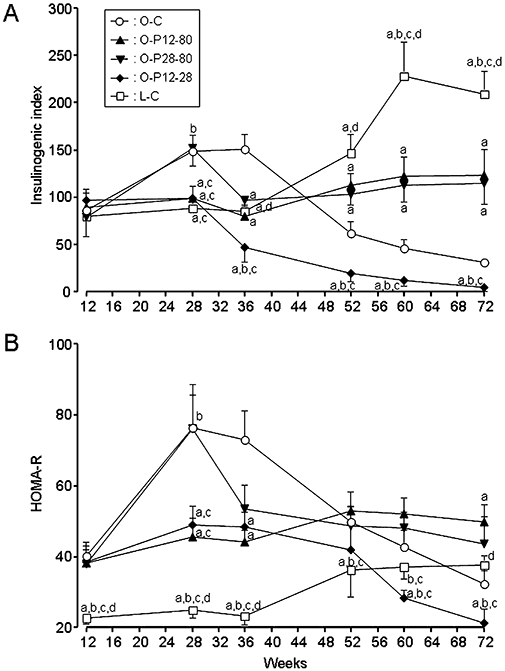
Serial changes in the insulinogenic index (A) and HOMA-R (B) in rats treated with or without pravastatin. For key to groups, see Figure 1 legend. (a) Significant difference versus O-C group at corresponding age; (b) significant difference versus O-P12-80 group at corresponding age; (c) significant difference versus O-P28-80 group at corresponding age; and (d) significant difference versus O-P12-28 group at corresponding age. Data are presented as mean ± SEM of seven to nine rats.
Chronological alteration of insulin resistance estimated by HOMA-R of the O-C group peaked at 28 weeks of age, and thereafter it gradually decreased, probably due to reduced insulin secretory function (Figure 3B). However, HOMA-R of the O-P12-80 group remained at the initial level until the end of the experiment. Commencement of pravastatin after the onset of DM (O-P28-80 group) reduced the elevated HOMA-R to the initial level. In contrast, cessation of pravastatin (O-P12-28 group) gradually, but progressively, decreased the HOMA-R to below the initial level, and it was significantly lower than that of the O-C group after 60 weeks of age. In the L-C group, HOMA-R was significantly lower than those in all four experimental groups of OLETF rats at 12, 28 and 36 weeks of age, but thereafter it gradually increased with age.
Serial changes in fasting serum T-cho, TG and FFA concentrations, and weight of white adipose deposits at 80 weeks of age
Fasting serum T-cho, TG and FFA concentrations of the OLETF rats increased with age and were similar irrespective of whether they were treated with pravastatin. The concentrations of these lipids in the L-C group gradually increased, but were significantly lower than those of the OLETF groups at almost all time-points (Figure 4A–C).
Figure 4.
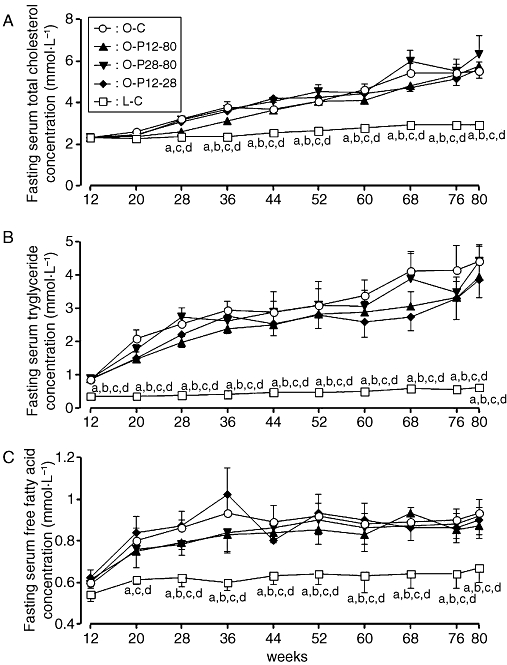
Serial changes of fasting total cholesterol (T-cho) (A), triglyceride (TG) (B) and free fatty acid (FFA) (C) concentrations in rats treated with or without pravastatin. For key to groups, see Figure 1 legend. (a) Significant difference versus O-C group at corresponding age; (b) significant difference versus O-P12-80 group at corresponding age; (c) significant difference versus O-P28-80 group at corresponding age; and (d) significant difference versus O-P12-28 group at corresponding age. Data are presented as mean ± SEM of six rats.
There were no significant differences in the weights of mesentery, epididymis and retoroperitoneum adipose depots among the O-C, O-P12-80 and O-P28-80 groups. In contrast, the weights of the adipose deposits in the O-P12-28 group were significantly lower than those in the O-C group, and were similar in levels to those in the L-C group (Table 1).
Table 1.
Effect of pravastatin on distribution of fat depots at 80 weeks of age
| O-C | O-P12-80 | O-P28-80 | O-P12-28 | L-C | |
|---|---|---|---|---|---|
| Mesentery (g) | 13.3 ± 1.4 | 14.9 ± 0.8 | 12.8 ± 1.7 | 7.9 ± 2.6 | 9.9 ± 0.9b |
| Epididymis (g) | 15.9 ± 1.0 | 17.4 ± 0.5 | 17.2 ± 2.1 | 8.5 ± 3.1a,b,c | 12.9 ± 0.5a,b,c |
| Retroperitoneum (g) | 45.6 ± 2.9 | 40.6 ± 3.5 | 52.2 ± 7.2 | 21.3 ± 6.5a,b,c | 14.1 ± 0.9a,b,c |
| Total (g) | 74.9 ± 3.6 | 72.9 ± 2.8 | 82.3 ± 10.4 | 37.6 ± 11.9a,b,c | 36.9 ± 1.6a,b,c |
Values were mean ± SEM of seven to nine rats: significant difference versus
O-C,
O-P12-80 and
O-P28-80.
The O-C and L-C groups were given standard rat diet the entire experimental period. The O-P12-80 group was maintained on a diet containing 0.05% pravastatin from 12 to 80 weeks of age. The O-P28-80 group was maintained on the same pravastatin diet from 28 to 80 weeks of age. The O-P12-28 group received the same pravastatin diet from 12 to 28 weeks of age, and then switched to standard rat chow until 80 weeks of age.
Pancreatic weight and its content of protein, DNA and insulin
At age 80 weeks, the pancreatic weight and its content of protein, DNA and insulin in the O-P12-80 and O-P28-80 groups were significantly higher than those of the O-C group, and were similar to those of the L-C group (Table 2). The pancreatic contents of these parameters in the O-P12-28 group were lower than those of the O-C group, and the difference was significant for the DNA and insulin content. When protein, DNA and insulin in the pancreas were expressed as total content g−1 pancreas, although these parameters in the O-P12-80 and O-P28-80 groups were higher than those in the O-C group, the difference was only significant for insulin, not for protein and DNA (Table 2). In addition, these parameters were lower in the O-P12-28 group than those of the O-C group, and a significant difference was only obtained for insulin (Table 2).
Table 2.
Effect of pravastatin on the pancreatic weight and contents of protein, DNA and insulin at 80 weeks of age
| O-C | O-P12-80 | O-P28-80 | O-P12-28 | L-C | ||
|---|---|---|---|---|---|---|
| Pancreatic weight (g) | 1.20 ± 0.07 | 1.55 ± 0.09a | 1.76 ± 0.10a | 0.94 ± 0.12b,c | 1.62 ± 0.06a,d | |
| (g·100 g BW) | 0.19 ± 0.01 | 0.24 ± 0.01a | 0.27 ± 0.02a | 0.16 ± 0.01b,c | 0.26 ± 0.01a,d | |
| Pancreatic protein, DNA and insulin contents | ||||||
| Protein | (mg/pancreas) | 54.5 ± 7.1 | 86.9 ± 10.3a | 92.9 ± 7.4a | 36.5 ± 5.1b,c | 106.1 ± 9.0a,d |
| (mg/g pancreas) | 46.5 ± 6.9 | 55.4 ± 4.3 | 52.8 ± 3.3 | 38.6 ± 5.5b,c | 65.2 ± 4.4a,d | |
| DNA | (mg/pancreas) | 1.5 ± 0.2 | 2.4 ± 0.3a | 2.7 ± 0.3a | 0.7 ± 0.1a,b,c | 3.3 ± 0.4a,d |
| (mg/g pancreas) | 1.3 ± 0.1 | 1.6 ± 0.1 | 1.6 ± 0.2 | 0.8 ± 0.2b,c | 2.0 ± 0.2a,d | |
| Insulin | (mmol/pancreas) | 15.8 ± 1.3 | 32.1 ± 4.1a | 46.1 ± 4.6a, b | 5.9 ± 1.2a,b,c | 46.8 ± 5.3a,d |
| (mmol·g−1 pancreas) | 13.2 ± 1.0 | 20.5 ± 1.7a | 26.4 ± 2.6a | 6.0 ± 0.9a,b,c | 28.5 ± 2.5a,d | |
Values were mean ± SEM of seven to nine rats: significant difference versus
O-C,
O-P12-80,
O-P28-80 and
O-P12-28.
For key to groups, see Table 1.
Activity of SOD and MPO, and levels of expression of TGF-β1 and TNF-α mRNA in the pancreas
Long-term treatment with pravastatin (O-P12-80 and O-P28-80 groups) significantly increased the SOD activity, and significantly decreased the MPO activity and the expression levels of TGF-β1 and TNF-α mRNA in the pancreas compared with the O-C group, and these parameters were similar to levels found in the L-C group (Table 3). In contrast, short-term treatment with pravastatin (O-P12-28 group) decreased the SOD activity, and increased the MPO activity and the levels of expression levels of TGF-β1 and TNF-α mRNA in the pancreas compared with those in the O-C group (Table 3).
Table 3.
Effect of pravastatin on activities of SOD and MPO, expression levels of TGF-β1 and TNF-α mRNA and ss-DNA index in the pancreas at 80 weeks of age
| O-C | O-P12-80 | O-P28-80 | O-P12-28 | L-C | |
|---|---|---|---|---|---|
| SOD activity (×103 U/pancreas) | 0.50 ± 0.13 | 1.52 ± 0.29a | 1.62 ± 0.25a | 0.21 ± 0.04a,b,c | 1.80 ± 0.13a,d |
| MPO activity (fold increase over O-C group) | 1.00 ± 0.18 | 0.48 ± 0.05a | 0.33 ± 0.04a | 1.71 ± 0.26a,b,c | 0.26 ± 0.03a,b,d |
| Expression levels of mRNA (/GAPDH) (fold increase over O-C group) | |||||
| TGF-β1 | 1.00 ± 0.18 | 0.57 ± 0.05a | 0.43 ± 0.14a | 1.40 ± 0.20b,c | 0.31 ± 0.09a,b,d |
| TNF-α | 1.00 ± 0.19 | 0.21 ± 0.14a | 0.15 ± 0.09a | 1.37 ± 0.20b,c | 0.01 ± 0.01a,d |
| ss-DNA index (%) | 0.55 ± 0.08 | 0.12 ± 0.05a | 0.05 ± 0.02a | 1.19 ± 0.23a,b,c | 0.03 ± 0.01a,d |
Values were mean ± SEM of seven to nine rats: significant difference versus
O-C,
O-P12-80,
O-P28-80 and
O-P12-28.
For key to groups, see Table 1.
Histological findings
At 12 weeks of age, the islet structure was normal, and fibrosis was rarely seen in the O-C group (Figure 5A). However, at 28 weeks of age, hyperplastic changes in the islets and fibrosis around the islets and pancreatic ducts were observed in the O-C group (Figure 5B). At 80 weeks of age, fibrosis and infiltration of inflammatory cells into the inter- and intralobular area in the pancreas occurred in the O-C group. In addition, most of the islets were enlarged and fibrotic, and some of them were atrophic (Figure 5C). However, these histological alterations were almost completely inhibited in the O-P12-80 and O-P28-80 groups (Figure 5D, E). In contrast, the exocrine glands and almost all the islets in the O-P12-28 group were extremely atrophic and fibrotic, and were noticeably infiltrated with inflammatory cells (Figure 5F). In the L-C group, some of the islets were enlarged, but histological alterations were rarely noted in the pancreas (Figure 5G).
Figure 5.
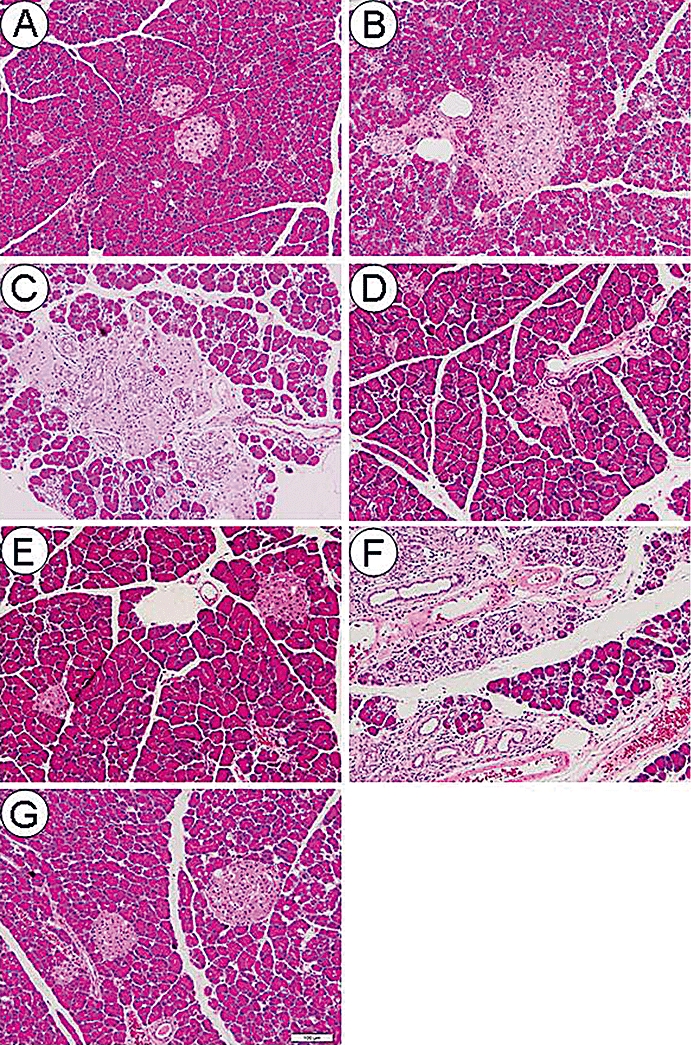
Representative light microscopic appearances of the pancreas stained with H&E (A–G). A duodenal portion of the pancreatic tissue was used for light microscopic examination. Original magnification, ×100. Scale bar indicates 100 µm. (A) Minimal histological changes were observed at 12 weeks of age in the O-C group. (B) Islets were enlarged, and fibrosis was distributed around islets and pancreatic ducts at 28 weeks of age in the O-C group. (C) Fibrosis and inflammatory cells markedly infiltrated into the inter- and intralobular areas in the pancreas of the O-C group at 80 weeks of age. In addition, most of the islets were enlarged and fibrotic, but some of them were atrophic. (D) and (E) Fibrosis and minimal infiltration of inflammatory cells were observed in the pancreas in the O-P12-80 (D) and O-P28-80 (E) groups at 80 weeks of age. (F) Exocrine glands and almost all islets were extremely atrophic with marked infiltration of inflammatory cells and fibrosis in the O-P12-28 group at 80 weeks of age. (G) Although some islets were enlarged, histological changes were rarely noted in the pancreas of the L-C group at 80 weeks of age.
Quantitative analysis using IBAS demonstrated that the fibrotic areas in the exocrine glands and islets were significantly reduced in the O-P12-80 and O-P28-80 groups compared with those in the O-C group (Table 4). The cross-sectional areas of the islets in the O-P12-80 and O-P28-80 groups were significantly smaller than that in the O-C group, and were similar to that in the L-C group. However, in the O-P12-28 group, the fibrotic areas in both the exocrine glands and islets were significantly larger, and the cross-sectional area of the islets was significantly smaller than those in the O-C group.
Table 4.
Effect of pravastatin on the proportion of fibrotic area in exocrine glands and islets, and size of islet at 80 weeks of age
| O-C | O-P12-80 | O-P28-80 | O-P12-28 | L-C | |
|---|---|---|---|---|---|
| Exocrine glands | |||||
| Proportion of fibrosis (%) | 7.9 ± 0.9 | 2.0 ± 0.3a | 0.7 ± 0.2a,b | 12.7 ± 1.2a,b,c | 0.4 ± 0.1a,b,c,d |
| Islets | |||||
| Cross-sectional area (µm2) | 25 063.9 ± 6417.1 | 6425.2 ± 1472.3a | 5261.1 ± 1073.3a | 1714.5 ± 358.8a,b,c | 6493.0 ± 1053.5a,d |
| Proportion of fibrosis (%) | 30.7 ± 4.4 | 6.2 ± 1.6a | 4.4 ± 1.0a | 47.5 ± 4.8a,b,c | 0.16 ± 0.1a,b,c,d |
Values were mean ± SEM of seven to nine rats. Significant difference versus
O-C,
O-P12-80,
O-P28-80 and
O-P12-28.
For key to groups, see Table 1.
Detection of apoptotic cells
Apoptotic cells detected with antibody for ss-DNA were mainly located in the acinar lesions of the pancreas. In the O-C group, a few apoptotic cells were observed in one intralobular area of the pancreas (Figure 6A). However, in the O-P12-80 (Figure 6B) and O-P28-80 (Figure 6C) groups, apoptotic cells were rarely noted in the pancreas. In the O-P12-28 group, apoptotic cells were markedly present especially in the atrophic lesion of the pancreas (Figure 6D). In contrast, apoptotic cells were very rarely seen in the pancreas of the L-C group (Figure 6E). The ss-DNA index of the O-P12-80 and O-P28-80 groups significantly decreased compared with that of the O-C group, and remained at a similar level to that of the L-C group (Table 3). In contrast, the ss-DNA index of the O-P12-28 group markedly increased compared with that of the O-C group (Table 3).
Figure 6.
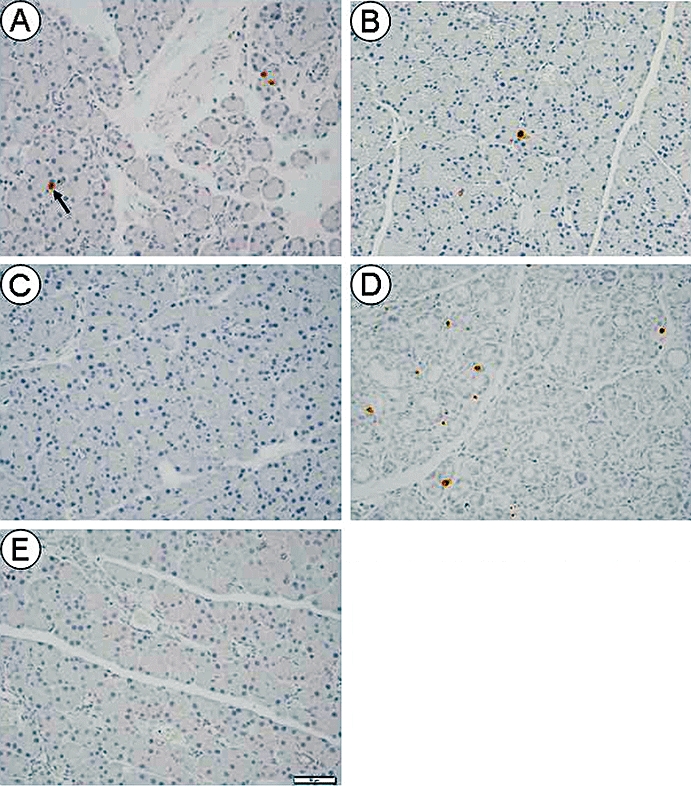
Immunohistochemistry for ss-DNA in the pancreas at 80 weeks of age. Original magnification, ×200. Scale bar indicates 50 µm. An arrow shows an apoptotic cell detected with the antibody for ss-DNA. (A) A few apoptotic cells were observed in one intralobular area of the pancreas in the O-C group. (B) and (C) A minimal number of apoptotic cells was observed in the pancreas of the O-P12-80 (B) and O-P28-80 (C) groups. (D) Apoptotic cells were markedly distributed especially in the atrophic portion of the pancreas of the O-P12-28 group. (E) Apoptotic cells were rarely detected in the pancreas of the L-C group.
Discussion and conclusions
The present study clearly demonstrated that long-term treatment with pravastatin until the end of experimental period exerts beneficial effects on the progression of DM and pancreatic fibrosis in the OLETF rat, whether pravastatin treatment was started before or after the onset of DM and pancreatic fibrosis. In contrast, a transient short-term treatment with pravastatin completely abolished these beneficial effects, and accelerated the DM and pancreatic fibrosis after its withdrawal.
A large-scale and randomized clinical trial has demonstrated that pravastatin therapy reduces by 30% the probability of becoming DM, probably by lowering the plasma lipid level (Freeman et al., 2001). However, because pravastatin treatment had no effects on serum lipid levels in the OLETF rats, the beneficial effects of pravastatin on DM are not via lowering serum lipid levels, but via a pleiotropic effect. Previous studies have also demonstrated that pravastatin attenuates inflammation and fibrosis in the kidney (Li et al., 2004) and the coronary arteries (Yu et al., 2004), and this effect is independent of its cholesterol-lowering actions.
It is well known that insulin resistance contributes to the pathogenesis of type 2 DM (Polonsky et al., 1996), and is closely associated with obesity, in particular, abdominal adiposity (Barzilai et al., 1998; Weyer et al., 2000). However, pravastatin treatment exerted beneficial effects on the progression of DM without reducing body weight gain or the amount of abdominal adipose deposits. These results suggest that pravastatin improves the control of diabetes by maintaining insulin sensitivity. In fact, HOMA-R in the OLETF rats continuously treated with pravastatin (O-P12-80 and O-P28-80 groups) remained nearly at the initial level after commencement of the treatment. Recent experimental studies have also demonstrated that statins improved insulin sensitivity in obese and insulin-resistant Zucker fatty rats (Wong et al., 2006), and obese and diabetic db/db mice (Takagi et al., 2008).
Consistent with the results from a previous study (Moriyama et al., 2001), we confirmed that pravastatin enhances the activity of SOD in the pancreas when it is given long-term treatment. Previous studies have shown that diabetic conditions induce oxidative stress (Kaneto et al., 1999) and reduce insulin sensitivity (Rudich et al., 1997). In addition, oxidative stress promotes the progression of fibrosis and inflammation in the rat pancreas (Matsumura et al., 2001; Su et al., 2002). In the current study, long-term treatment with pravastatin down-regulated the levels of expression of TNF-α and TGF-β1 mRNA, and attenuated atrophic and fibrotic changes in the pancreas. These results suggest that pravastatin prevents the decline in insulin sensitivity and insulin secretory capacity, and suppresses pancreatic fibrosis and inflammation in the OLETF rats by attenuating oxidative stress. In support of this view, antioxidant treatment can preserve in vivoβ-cell function (Kaneto et al., 1999), and suppress pancreatic fibrosis and inflammation (Matsumura et al., 2001; Su et al., 2002). In addition, statin treatment has been shown to reduce fibrosis and inflammation, along with an attenuation of oxidative stress in the kidney (Moriyama et al., 2001) and heart (Habibi et al., 2007). Recent experimental studies have also shown that rosuvastatin exerts a beneficial effect on damage to the glomerular filtration barrier (Whaley-Connell et al., 2008) and cerebrovascular dysfunction (Erdös et al., 2006) by inhibiting oxidative stress in insulin-resistant Zucker obese rats.
In the present study, pravastatin treatment exerted therapeutic effects on the progression of DM in the OLETF rats by maintaining insulin secretory function and insulin sensitivity. In addition, results from the current study suggest that pravastatin exerts therapeutic effects on established fibrosis in the pancreas; regardless of whether pravastatin treatment was started after the onset of fibrosis (O-P28-80 group), pravastatin down-regulated the expression levels of TGF-β1 and TNF-α, and decreased the proportion of fibrosis in the pancreas of OLETF rat to levels similar to those of LETO rats. Previous studies have shown that statins prevent fibrosis in several organs (Moriyama et al., 2001; Yu et al., 2004), but one experimental study showed that pravastatin exerts an anti-fibrotic action on established radiation-induced intestinal fibrosis by inhibiting the Rho/CCN2/extracellular matrix cascade in rats (Haydont et al., 2007).
The present study revealed that long-term treatment with pravastatin suppresses apoptosis in the pancreas, as the ss-DNA index in acinar cells was markedly reduced. In support of these observations, long-term treatment with pravastatin significantly increased pancreatic weight, and its DNA and protein contents compared with those in the control group of OLETF rat. Because the increase in protein and DNA content indicates growth and regeneration of the pancreas by cellular proliferation and/or hypertrophy (Pearson et al., 1977), these findings suggest that pravastatin maintained pancreatic tissue by regeneration and inhibition of apoptosis followed by atrophy. Because oxidative stress (Manna et al., 1998) and inflammatory mediators, such as TGF-β1 and TNF-α (Zhang et al., 2007), strongly induce apoptosis, it seems likely that pravastatin inhibits apoptosis via its anti-oxidative and anti-inflammatory actions. Previous studies demonstrated that statins induce apoptosis in myofibroblasts in the pancreas (Jaster et al., 2003) and liver (Aprigliano et al., 2008). In addition, myofibroblasts undergo increased apoptosis during wound healing (Desmoulière et al., 1995). These studies indicate that apoptotic elimination of myofibroblasts by pravastatin also played an important role in the improvement of pancreatic fibrosis in the OLETF rat. On the other hand, the total protein, DNA and insulin contents of the pancreas in the O-C group were significantly decreased compared with those in the O-P12-80 and O-P28-80 groups. However, when these parameters were expressed as total content g−1 pancreas (indicates concentration of these parameters in the pancreatic tissue), the insulin, but not the protein or the DNA, was significantly decreased in the O-C group compared with those in the O-P12-80 and O-P28-80 groups. These results suggest that pravastatin exerts pleiotropic effects more potently on the endocrine pancreas than on the exocrine pancreas in this rat model.
The most striking finding of the present study is that a short-term transient treatment of pravastatin accelerated the progression of DM accompanied by atrophied islets, and promoted pancreatic fibrosis. Because oxidative stress participates in the progression of insulin resistance (Rudich et al., 1997) and pancreatic fibrosis (Matsumura et al., 2001; Su et al., 2002), the marked decrease in pancreatic SOD activity, to below the control level in the pancreas, after a short-term treatment with pravastatin might induce pancreatic inflammation and fibrosis, and insulin secretory dysfunction. In support of our observations, there is a growing evidence that withdrawal of statin increases cardiovascular events (Heeschen et al., 2002) and mortality (Daskalopoulou et al., 2008). It is assumed that the underlying mechanism for the withdrawal effects of statins is caused by an impairment in vasoreactivity due to a decreased bioavailability of nitric oxide (NO), which is mediated by an increased activity of the small GTP-binding protein rho (Laufs et al., 2000) or increased oxidative stress (Vecchione and Brandes, 2002).
In conclusion, our results suggest that pravastatin exerts a preventive and therapeutic effect on the progression of DM and pancreatic fibrosis in the OLETF rat by pleiotropic effects, as it was found to have anti-oxidative, anti-inflammatory and anti-fibrotic actions. However, these beneficial effects were completely abolished after its withdrawal, and, instead, the progression of DM and pancreatic fibrosis was found to be accelerated. These results support the clinical use of pravastatin for patients with DM or chronic pancreatitis, but indicate that withdrawal of this treatment should be avoided.
Acknowledgments
We thank Naoko Kamichi, Megumi Teramoto and Hitomi Kamio for their excellent technical assistance. This work was supported in part by a Grant-in-Aid for Exploratory Research (no. 17659222) from Japan Society for the Promotion of Science.
Glossary
Abbreviations:
- DM
diabetes mellitus
- FFA
free fatty acid
- H&E
haematoxylin and eosin
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- HOMA-R
homeostasis model assessment of insulin resistance
- IBAS
interactive image analysis system
- IVGTT
intravenous glucose tolerance test
- LETO
Long–Evans–Tokushima–Otsuka
- MPO
myeloperoxidase
- OLETF
Otsuka–Long–Evans–Tokushima Fatty
- SOD
superoxide dismutase
- T-cho
total cholesterol
- TG
triglyceride
- TGF-β1
transforming growth factor-β1
- TNF-α
tumour necrosis factor-α
References
- Aprigliano I, Dudas J, Ramadori G, Saile B. Atorvastatin induces apoptosis by a caspase-9-dependent pathway: an in vitro study on activated rat hepatic stellate cells. Liver Int. 2008;28:546–557. doi: 10.1111/j.1478-3231.2008.01682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N, Banerjee S, Hawkins M, Chen W, Rossetti L. Caloric restriction reverses hepatic insulin resistance in aging rats by decreasing visceral fat. J Clin Invest. 1998;101:1353–1361. doi: 10.1172/JCI485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Ramnath RD, Chevali L, Guglielmotti A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1259–1265. doi: 10.1152/ajpgi.00435.2004. [DOI] [PubMed] [Google Scholar]
- Bondar RJ, Mead DC. Evaluation of glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides in the hexokinase method for determining glucose in serum. Clin Chem. 1974;20:586–590. [PubMed] [Google Scholar]
- Daskalopoulou SS, Delaney JA, Filion KB, Brophy JM, Mayo NE, Suissa S. Discontinuation of statin therapy following an acute myocardial infarction: a population-based study. Eur Heart J. 2008;29:2083–2091. doi: 10.1093/eurheartj/ehn346. [DOI] [PubMed] [Google Scholar]
- Desmoulière A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- Endo A, Kuroda M, Tanzawa K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976;72:323–326. doi: 10.1016/0014-5793(76)80996-9. [DOI] [PubMed] [Google Scholar]
- Erdös B, Snipes JA, Tulbert CD, Katakam P, Miller AW, Busija DW. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am J Physiol Heart Circ Physiol. 2006;290:H1264–1270. doi: 10.1152/ajpheart.00804.2005. [DOI] [PubMed] [Google Scholar]
- Freeman DJ, Norrie J, Sattar N, Neely RD, Cobbe SM, Ford I, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation. 2001;103:357–362. doi: 10.1161/01.cir.103.3.357. [DOI] [PubMed] [Google Scholar]
- Habibi J, Whaley-Connell A, Qazi MA, Hayden MR, Cooper SA, Tramontano A, et al. Rosuvastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, decreases cardiac oxidative stress and remodeling in Ren2 transgenic rats. Endocrinology. 2007;148:2181–2188. doi: 10.1210/en.2006-1355. [DOI] [PubMed] [Google Scholar]
- Haruyama H, Kuwano H, Kinoshita T, Terahara A, Nishigaki T, Tamura C. Structure elucidation of the bioactive metabolites of ML-236B (mevastatin) isolated from dog urine. Chem Pharm Bull (Tokyo) 1986;34:1459–1467. doi: 10.1248/cpb.34.1459. [DOI] [PubMed] [Google Scholar]
- Haydont V, Bourgier C, Pocard M, Lusinchi A, Aigueperse J, Mathé D, et al. Pravastatin inhibits the rho/CCN2/extracellular matrix cascade in human fibrosis explants and improves radiation-induced intestinal fibrosis in rats. Clin Cancer Res. 2007;13:5331–5340. doi: 10.1158/1078-0432.CCR-07-0625. [DOI] [PubMed] [Google Scholar]
- Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD, et al. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation. 2002;105:1446–1452. doi: 10.1161/01.cir.0000012530.68333.c8. [DOI] [PubMed] [Google Scholar]
- Jaster R, Brock P, Sparmann G, Emmrich J, Liebe S. Inhibition of pancreatic stellate cell activation by the hydroxymethylglutaryl coenzyme A reductase inhibitor lovastatin. Biochem Pharmacol. 2003;65:1295–1303. doi: 10.1016/s0006-2952(03)00075-3. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes. 1999;48:2398–2406. doi: 10.2337/diabetes.48.12.2398. [DOI] [PubMed] [Google Scholar]
- Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes. 1992;41:1422–1428. doi: 10.2337/diab.41.11.1422. [DOI] [PubMed] [Google Scholar]
- Korc M, Chandrasekar B, Yamanaka Y, Friess H, Bücher M, Beger HG. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest. 1992;90:1352–1360. doi: 10.1172/JCI116001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka K, Hagura R, Kuzuya T, Kuzuya N. Insulin secretory response of diabetics during the period of improvement of glucose tolerance to normal range. Diabetologia. 1974;10:775–782. doi: 10.1007/BF01219540. [DOI] [PubMed] [Google Scholar]
- Labarca C, Paigen K. A simple, rapid and sensitive DNA assay procedure. Anal Biochem. 1980;102:344–352. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- Laufs U, Endres M, Custodis F, Gertz K, Nickenig G, Liao JK, et al. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation. 2000;102:3104–3110. doi: 10.1161/01.cir.102.25.3104. [DOI] [PubMed] [Google Scholar]
- Li C, Yang CW, Park JH, Lim SW, Sun BK, Jung JY, et al. Pravastatin treatment attenuates interstitial inflammation and fibrosis in a rat model of chronic cyclosporine-induced nephropathy. Am J Physiol Renal Physiol. 2004;286:F46–57. doi: 10.1152/ajprenal.00428.2002. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Manna SK, Zhang HJ, Yan T, Oberley LW, Aggarwal BB. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappaB and activated protein-1. J Biol Chem. 1998;273:13245–13254. doi: 10.1074/jbc.273.21.13245. [DOI] [PubMed] [Google Scholar]
- Matsumura N, Ochi K, Ichimura M, Mizushima T, Harada H, Harada M. Study on free radicals and pancreatic fibrosis – pancreatic fibrosis induced by repeated injections of superoxide dismutase inhibitor. Pancreas. 2001;22:53–57. doi: 10.1097/00006676-200101000-00009. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Kawada N, Nagatoya K, Takeji M, Horio M, Ando A, et al. Fluvastatin suppresses oxidative stress and fibrosis in the interstitium of mouse kidneys with unilateral ureteral obstruction. Kidney Int. 2001;59:2095–2103. doi: 10.1046/j.1523-1755.2001.00724.x. [DOI] [PubMed] [Google Scholar]
- Paniagua JA, Lopez-Miranda J, Escribano A, Berral FJ, Marin C, Bravo D, et al. Cerivastatin improves insulin sensitivity and insulin secretion in early-state obese type 2 diabetes. Diabetes. 2002;51:2596–2603. doi: 10.2337/diabetes.51.8.2596. [DOI] [PubMed] [Google Scholar]
- Pearson KW, Scott D, Torrance B. Effects of partial surgical pancreatectomy in rats. I. Pancreatic regeneration. Gastroenterology. 1977;72:469–473. [PubMed] [Google Scholar]
- Polonsky KS, Sturis J, Bell GI. Non-insulin-dependent diabetes mellitus – a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med. 1996;334:777–783. doi: 10.1056/NEJM199603213341207. [DOI] [PubMed] [Google Scholar]
- Rudich A, Kozlovsky N, Potashnik R, Bashan N. Oxidant stress reduces insulin responsiveness in 3T3-L1 adipocytes. Am J Physiol. 1997;272:E935–940. doi: 10.1152/ajpendo.1997.272.5.E935. [DOI] [PubMed] [Google Scholar]
- Satoh K, Keimatsu N, Kanda M, Kasai T, Takaguri A, Sun F, et al. HMG-CoA reductase inhibitors do not improve glucose intolerance in spontaneously diabetic Goto–Kakizaki rats. Biol Pharm Bull. 2005;28:2092–2095. doi: 10.1248/bpb.28.2092. [DOI] [PubMed] [Google Scholar]
- Solheim S, Seljeflot I, Arnesen H, Eritsland J, Eikvar L. Reduced levels of TNF alpha in hypercholesterolemic individuals after treatment with pravastatin for 8 weeks. Atherosclerosis. 2001;157:411–415. doi: 10.1016/s0021-9150(00)00725-5. [DOI] [PubMed] [Google Scholar]
- Su SB, Motoo Y, Xie MJ, Mouri H, Asayama K, Sawabu N. Superoxide dismutase is induced during rat pancreatic acinar cell injury. Pancreas. 2002;24:146–152. doi: 10.1097/00006676-200203000-00005. [DOI] [PubMed] [Google Scholar]
- Takagi T, Matsuda M, Abe M, Kobayashi H, Fukuhara A, Komuro R, et al. Effect of pravastatin on the development of diabetes and adiponectin production. Atherosclerosis. 2008;196:114–121. doi: 10.1016/j.atherosclerosis.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Takano T, Yamakawa T, Takahashi M, Kimura M, Okamura A. Influences of statins on glucose tolerance in patients with type 2 diabetes mellitus. J Atheroscler Thromb. 2006;13:95–100. doi: 10.5551/jat.13.95. [DOI] [PubMed] [Google Scholar]
- Vecchione C, Brandes RP. Withdrawal of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors elicits oxidative stress and induces endothelial dysfunction in mice. Circ Res. 2002;91:173–179. doi: 10.1161/01.res.0000028004.76218.b8. [DOI] [PubMed] [Google Scholar]
- Weyer C, Hanson K, Bogardus C, Pratley RE. Long-term changes in insulin action and insulin secretion associated with gain, loss, regain and maintenance of body weight. Diabetologia. 2000;43:36–46. doi: 10.1007/s001250050005. [DOI] [PubMed] [Google Scholar]
- Whaley-Connell A, DeMarco VG, Lastra G, Manrique C, Nistala R, Cooper SA, et al. Insulin resistance, oxidative stress, and podocyte injury: role of rosuvastatin modulation of filtration barrier injury. Am J Nephrol. 2008;28:67–75. doi: 10.1159/000109394. [DOI] [PubMed] [Google Scholar]
- Wong V, Stavar L, Szeto L, Uffelman K, Wang CH, Fantus IG, et al. Atorvastatin induces insulin sensitization in Zucker lean and fatty rats. Atherosclerosis. 2006;184:348–355. doi: 10.1016/j.atherosclerosis.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Jia DM, Fukumitsu KI, Imoto I, Kihara Y, Hirohata Y, et al. Metabolic abnormalities in the genetically obese and diabetic Otsuka Long–Evans Tokushima Fatty rat can be prevented and reversed by alpha-glucosidase inhibitor. Metabolism. 1999;48:347–354. doi: 10.1016/s0026-0495(99)90084-7. [DOI] [PubMed] [Google Scholar]
- Yoshikawa H, Kihara Y, Taguchi M, Yamaguchi T, Nakamura H, Otsuki M. Role of TGF-beta1 in the development of pancreatic fibrosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol. 2002;282:G549–558. doi: 10.1152/ajpgi.00323.2001. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ohmori K, Chen Y, Sato C, Kiyomoto H, Shinomiya K, et al. Effects of pravastatin on progression of glucose intolerance and cardiovascular remodeling in a type II diabetes model. J Am Coll Cardiol. 2004;44:904–913. doi: 10.1016/j.jacc.2004.04.050. [DOI] [PubMed] [Google Scholar]
- Zhang XP, Lin Q, Zhou YF. Progress of study on the relationship between mediators of inflammation and apoptosis in acute pancreatitis. Dig Dis Sci. 2007;52:1199–1205. doi: 10.1007/s10620-006-9388-6. [DOI] [PubMed] [Google Scholar]