

Abstract
The metal-dependent histone deacetylases (HDACs) catalyze hydrolysis of acetyl groups from acetyllysine side chains and are targets of cancer therapeutics. Two bound monovalent cations (MVCs) of unknown function have been previously observed in crystal structures of HDAC8; site 1 is near the active site, whereas site 2 is located >20 Å from the catalytic metal ion. Here we demonstrate that one bound MVC activates catalytic activity (K1/2 = 3.4 mm for K+), whereas the second, weaker-binding MVC (K1/2 = 26 mm for K+) decreases catalytic activity by 11-fold. The weaker binding MVC also enhances the affinity of the HDAC inhibitor suberoylanilide hydroxamic acid by 5-fold. The site 1 MVC is coordinated by the side chain of Asp-176 that also forms a hydrogen bond with His-142, one of two histidines important for catalytic activity. The D176A and H142A mutants each increase the K1/2 for potassium inhibition by ≥40-fold, demonstrating that the inhibitory cation binds to site 1. Furthermore, the MVC inhibition is mediated by His-142, suggesting that this residue is protonated for maximal HDAC8 activity. Therefore, His-142 functions either as an electrostatic catalyst or a general acid. The activating MVC binds in the distal site and causes a time-dependent increase in activity, suggesting that the site 2 MVC stabilizes an active conformation of the enzyme. Sodium binds more weakly to both sites and activates HDAC8 to a lesser extent than potassium. Therefore, it is likely that potassium is the predominant MVC bound to HDAC8 in vivo.
Keywords: Enzymes/Catalysis, Enzymes/Hydrolases, Enzymes/Mechanisms, Enzymes/Metallo, Histones/Deacetylase, Protein/Post-translational Modification, Inhibition, Monovalent Cation
Introduction
The post-translational acetylation of lysine residues regulates an array of biological processes, including transcription, protein degradation, cellular proliferation, and apoptosis (1). Lysine acetylation is catalyzed by histone acetyltransferases, which acetylate-specific lysine side chains, whereas histone deacetylases (HDACs)3 reverse this modification. The interest in HDACs has grown rapidly over recent years, as it was discovered that HDAC inhibitors have the potential to effectively treat cancer (1). Currently, one HDAC inhibitor has been approved by the Food and Drug Administration, with several more in clinical trials (2, 3). The majority of these inhibitors target the HDACs of classes I, II, and IV, which require a divalent metal ion such as Zn(II) or Fe(II) as an obligate cofactor (4). The class III HDACs are a separate family and are not related to the metal-dependent HDACs.
HDAC8 is a member of the class I HDAC family, which includes HDACs 1, 2, 3, and 8. HDAC8 is predominantly expressed in smooth muscle tissues, where it associates with the cytoskeletal protein smooth muscle α-actin (5, 6). Upon RNAi knockdown of HDAC8, the contractile ability of smooth muscle cells is decreased (6). In addition, abnormally low levels of HDAC8 are associated with severe colorectal motility disorders such as Hirschprung disease and idiopathic megacolon (7). HDAC8 may also play a role in the progression of specific cancers given that selective inhibition of HDAC8 induces apoptosis in tumor cells derived from T-cell lymphomas (8).
The catalytic mechanism originally proposed for HDAC8 and the other class I HDACs is based on the crystal structure of the histone deacetylase-like protein (HDLP) from Aquifex aeolicus (9). In this general acid-base catalytic pair mechanism, His-142 functions as a general base, deprotonating the metal-activated catalytic water molecule for attack on the substrate amide, and a second active site residue, His-143, serves as a general acid, protonating the leaving group.
Crystal structures reveal that HDAC8 binds two monovalent cations (MVCs), either Na+ or K+, in addition to the essential divalent catalytic metal ion (10–12). These two MVC sites in HDAC8 have been designated as site 1 and site 2 (10). The MVC of site 1 is 7 Å from the divalent catalytic metal ion and is coordinated by the side-chain oxygens of Asp-176 and Ser-199 and the backbone carbonyl oxygens of Asp-176, Asp-178, His-180, and Leu-200 (Fig. 1). Site 2 is 21 Å from the divalent catalytic metal ion, and this MVC is ligated by two water molecules and the backbone carbonyl oxygens of Phe-189, Thr-192, Val-195, and Tyr-225. Other class I and II human HDACs that have been crystallized also bind K+ in these same sites, as does the bacterial histone deacetylase-like amidohydrolase (13–15). Given this conservation of the MVC sites, it is likely that the functions revealed here for the MVCs in HDAC8 also apply to other class I and II HDACs.
FIGURE 1.
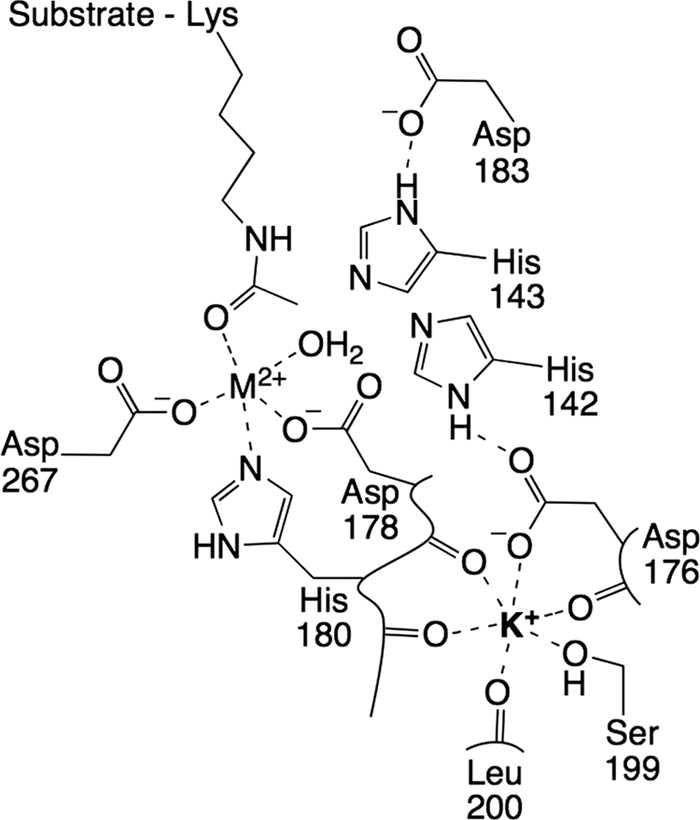
HDAC8 active site and monovalent site 1. The site 1 MVC is coordinated by the backbone carbonyl oxygens of Asp-176, Asp-178, His-180, and Leu-200 and by the side chains of Asp-176 and Ser-199. The carboxylate of Asp-176 also forms a hydrogen bond with His-142. With MVC 1 bound, as shown, the catalytic activity is less than maximal, likely due to the deprotonation of His-142.
Current knowledge of the functions of MVCs in HDAC8 is quite limited; the enzyme has greater thermal stability in KCl than in NaCl, refolds more rapidly when 10 mm KCl is present (16), and is slightly more active in 10 mm KCl than 1 mm KCl (10, 17). Suggested roles for the site 1 MVC include stabilizing a tight turn of the polypeptide backbone and altering the pKa of His-142 (10, 12). The purpose of the distant site 2 MVC is not readily apparent. The identity of the MVC that HDAC8 uses in vivo is also unclear, as crystal structures have been obtained with either Na+ or K+ bound, depending on the salt that was included during crystallization.
To understand the roles of the two MVCs visualized in the HDAC8 crystal structures (10–12), we have examined the effects of MVCs on the deacetylase activity of both wild-type HDAC8 (wt-HDAC8) and active site mutants. The results from these studies demonstrate that the MVC bound to site 1 inhibits catalytic activity, likely by altering the pKa of His-142, whereas the site 2 MVC enhances activity, likely by an allosteric effect. However, K+ binding to site 1 enhances the affinity of HDAC8 for SAHA. Under in vivo sodium and potassium concentrations, the activation site is saturated with potassium. Furthermore, the enzyme is partially inhibited by potassium under these conditions, rendering the enzyme activity sensitive to changes in the potassium concentration.
EXPERIMENTAL PROCEDURES
Materials
Unless specified, chemicals and supplies were purchased from Fisher. All chemicals were of the highest quality available. Chromatography resins were purchased from GE Healthcare.
Expression and Purification of HDAC8
Recombinant human HDAC8 was expressed and purified as previously described (4). After divalent metal ions were removed from the enzyme (4), apoHDAC8 (> 240 μm) was stored at −80 °C in 25 mm MOPS, pH 7.5, 100 mm NaCl, and 0.1 μm EDTA. HDAC8 mutants were constructed in the pHD2-TEV-His plasmid (4) using a QuikChange site-directed mutagenesis kit (Stratagene).
HDAC8 Activity Assay
The deacetylase activity of HDAC8 was measured using the commercially available Fluor de Lys H4-AcK16 and Fluor de Lys HDAC8 fluorescent substrates (Biomol) as previously described (4). Before assaying catalytic activity, apoHDAC8 was preincubated for 1 h on ice with stoichiometric Co(II) or Zn(II) and varying concentrations of KCl and/or NaCl in 25 mm Tris base, pH 8.0. The steady-state kinetic parameter kcat/Km was calculated from initial reaction rates measured under conditions of subsaturating substrate (50–200 μm) and enzyme (0.2–20 μm). The monovalent dependence of HDAC8 was primarily characterized using Co(II)-HDAC8. Although HDAC8 may use the Fe(II) cofactor in vivo under some conditions, both Co(II) and Fe(II) support higher levels of catalysis in HDAC8 than does Zn(II) (4), allowing for greater sensitivity in measuring the activity of HDAC8 mutants. Additionally, Co(II)-HDAC8 is stable in the presence of oxygen and behaves similarly to Fe(II)-HDAC8 in other respects. In all assays the NaCl contributed by the enzyme storage buffer is less than 3 mm.
The bell-shaped MVC dependence of wt-HDAC8 was fit by Equation 1, which is derived from the sequential two-site binding model shown in Scheme 1. Here the enzyme is catalytically active when either one or two monovalent ions are bound but with a different level of activity when two MVCs are bound (kobs2) than when one is bound (kobs1).
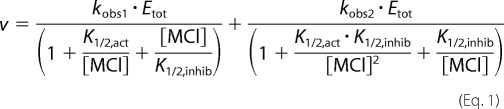 |
The concentration of either KCl or NaCl is represented by [MCl], Etot is the total enzyme concentration, and the apparent binding affinities for activation and inhibition are represented by K1/2,act and K1/2,inhib. When the values of the apparent affinities of the activating and inhibitory MVCs are not clearly resolved, as in the dependence of activity on NaCl, it is possible that the identities of sites 1 and 2 could be switched. For the D176N and D176A mutants as well as wt-Co(II)-HDAC8 with NaCl, the value of K1/2,inhib is too high to accurately fit a value to kobs,2. In these cases Equation 2 is used, which is derived from a model in which the binding of a second monovalent metal ion completely inhibits catalysis (kobs2 = 0).
 |
SCHEME 1.
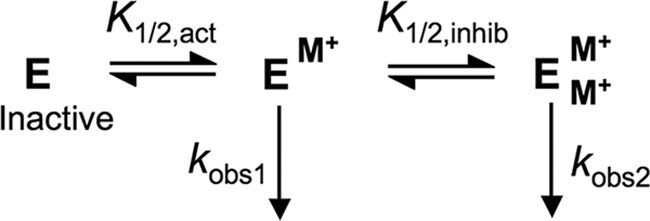
MVC binding of HDAC8. The first MVC activates HDAC8, whereas the second MVC partially inhibits activity (kobs1 > kobs2).
In the absence of inhibition by KCl, the data are fit to the single binding isotherm described by Equation 3.
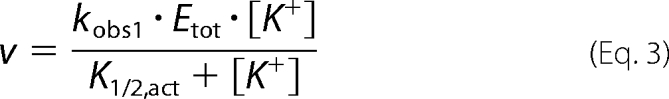 |
When only the inhibition phase is observed, the data are fit to the single binding isotherm described by Equation 4.
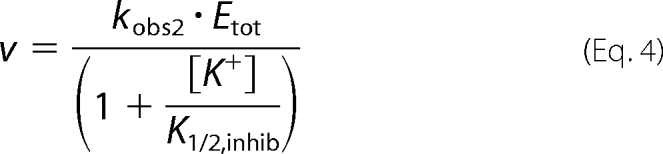 |
Time Dependence of KCl Activation
The rate of HDAC8 activation by KCl was determined by incubating Co(II)-HDAC8 in 5 mm KCl with 25 mm Tris, pH 8.0, at 25 °C for 0–20 min before measuring the catalytic activity. The initial rates were fit to a single exponential (Equation 5) as a function of preincubation time with a non-zero rate of deacetylation (v0) when t = 0.
The term A represents the amplitude, which is the difference between the rate measured at t = 0 and after the rate has reached a maximum. The rate constant for this increase is represented by kobs. The half-life for the KCl-mediated activation (t½) is related to kobs by the equation t½ = ln 2/kobs.
The rate at t = 0 min was measured by initiating the reaction with the addition of substrate and 5 mm KCl (final concentration). The last assay time point under these conditions was quenched 80 s after initiation. There was no significant deviation from linearity in the formation of product versus time.
Inhibition by SAHA
For measurement of inhibition by SAHA, reconstituted Co(II)-HDAC8 was incubated with 1 mm KCl on ice for 1 h in 25 mm Tris, pH 8.0. Before assaying activity at 25 °C, Co(II)-HDAC8 (0.2 μm) was incubated with varied concentrations of KCl (0.04–200 mm) and SAHA (0.2–5 μm) in 25 mm Tris, pH 8.0, on ice for 1 h and then at 25 °C for 4 min. The initial velocity for deacetylation was measured as described above. The inhibition constant for SAHA (Ki,obs) was obtained by fitting a binding isotherm that accounts for ligand depletion (Equation 6) to the initial velocities. The concentration of SAHA (Itot) was varied (0.2–5 μm) while using fixed concentrations of HDAC8 (Etot, 0.2 μm), KCl (0.04–200 mm), and subsaturating substrate (50 μm).
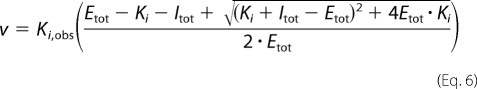 |
The dependence of the observed inhibition constant for SAHA (Ki,obs) on the KCl concentration was fit using an equation for a two-state model (Equation 7) where the SAHA affinity is coupled to KCl binding where Ki is the SAHA dissociation constant at saturating K+, and K2 and K4 are the dissociation constants for K+ from HDAC8 and the HDAC8·SAHA complex, respectively.
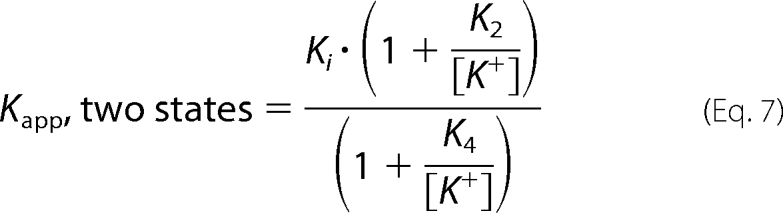 |
These data were also fit with an equation derived for the three-state model shown in Scheme 2 that accounts for K+ binding to both MVC sites using the values for the K+ dissociation constants, K1 and K2, determined from K1/2,act and K1/2,inhib (Equation 1, Table 1), respectively, from the K+ dependence of activity. In Equation 8, Ki3 and Ki2 reflect the SAHA dissociation constants from I·EK2+ and I·EK+, respectively, whereas K3 reflects the K+ dissociation constant from I·EK+ (see Scheme 2). The SAHA dissociation constant from I·E is calculated from the equilibrium box as Ki1 = K1 Ki2/K3.
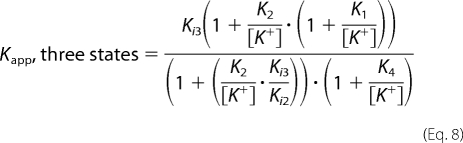 |
SCHEME 2.

Coupled binding of SAHA and K+ to HDAC8. The influence of potassium binding to MVC1 and MVC2 sites of wt-HDAC8 on the affinity of this enzyme for SAHA is described by this simplified three-state scheme where SAHA binds to the three major enzyme species, HDAC8 with no bound K+ (E), K+ bound to MVC site 2 (EK), and K+ bound to both MVC sites 1 and 2 (EK2). For simplicity, this scheme does not include SAHA binding to HDAC8 with K+ bound to MVC2, which constitutes only a minor fraction of the total enzyme. Fits to this model demonstrate that the affinity of SAHA decreases as K+ dissociates from the enzyme.
TABLE 1.
Apparent MVC affinities of wt-HDAC8 and mutants
HDAC8 activity was measured, and K1/2,act and K1/2,inhib were determined as described in the legends of Figs. 2 and 3.
| HDAC8 form | Monovalent salt | K1/2,act | K1/2,inhib |
|---|---|---|---|
| mm | mm | ||
| Co(II)-wta | KCl | 3.4 ± 0.3 | 26 ± 3 |
| Co(II)-wtb | NaCl | 90 ± 40 | 320 ± 140 |
| Zn(II)-wta | KCl | 14 ± 2 | 130 ± 30 |
| Co(II)-D176Nb | KCl | 41 ± 2 | 320 ± 30 |
| Co(II)-D176Ab | KCl | 9.3 ± 2.4 | 1200 ± 300 |
| Co(II)-H142Ac | KCl | 0.3 ± 0.1 | ≫1000 |
a MVC affinities are determined by fitting the initial rate data to Equation 1.
b Equation 2, in which activity is completely inhibited at high KCl, is fit to the initial rate data.
c Equation 3, in which there is no MVC inhibition, is fit to the initial rate data.
RESULTS
KCl and NaCl Modulate HDAC8 Activity
The roles of the two MVCs observed in the HDAC8 crystal structures were determined by measuring activity as a function of KCl concentration. The KCl dependence of Co(II)-HDAC8 is bell-shaped (Fig. 2), with maximal deacetylase activity observed at 10 mm KCl (Fig. 2) and partial inhibition at high KCl. These data are described well by Equation 1, which is derived from a two-site sequential binding model in which the enzyme is inactive until the first site is occupied, whereas the binding of a second cation decreases the rate of catalysis but does not lead to complete inhibition (Scheme 1). It is also possible that the MVCs could bind in random order rather than sequentially. K+ binding to the tighter affinity site (K1/2,act = 3.4 ± 0.3 mm) activated enzymatic activity 45-fold between 0.03 and 10 mm KCl. From a fit of Equation 1 to the data, the calculated value of kcat/Km for HDAC8 with a single K+ ion bound was 37,000 ± 2,000 m−1 s−1. The maximal observed rate constant was lower than this due to K+ binding to the inhibitory site at KCl concentrations less than that required to saturate the activating MVC site. The inhibitory K+ ion bound with weaker affinity (K1/2,inhib = 26 ± 3 mm), and Co(II)-HDAC8 was 11-fold less active with two K+ ions bound (kcat/Km = 3400 m−1 s−1) than with one K+ bound.
FIGURE 2.
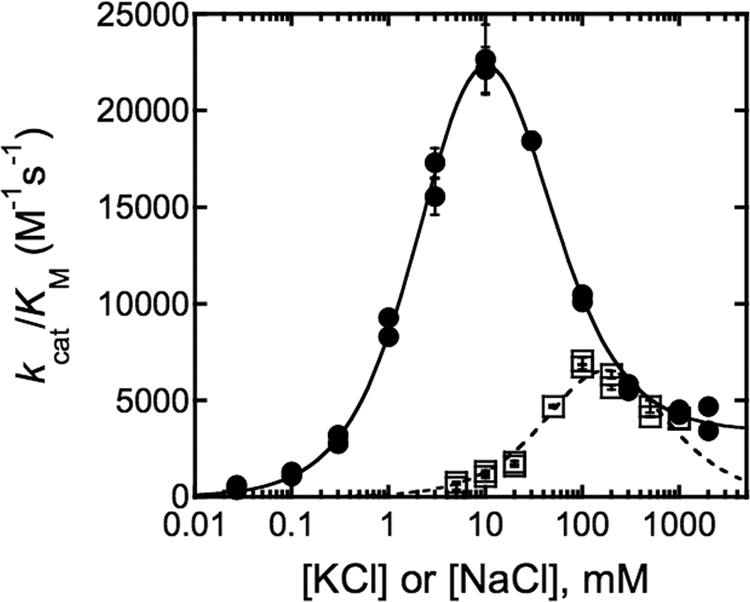
HDAC8 activation and inhibition by monovalent ions. Co(II)-wt-HDAC8 was incubated with KCl (●) or NaCl (□) for 1 h on ice before assaying activity with 0.4 μm enzyme and 50 μm Fluor de Lys HDAC8 substrate in 25 mm Tris pH 8.0, 25 °C. Initial velocities were determined based on changes in fluorescence, and Equations 1 and 2 are fit to the resulting rates at varying KCl and NaCl, respectively, yielding the K1/2,act and K1/2,inhib values shown in Table 1.
The second most abundant MVC inside the cell after K+ is Na+ (18); the dependence of HDAC8 activity on sodium concentration was also bell-shaped, demonstrating both activation and inhibition (Fig. 2). Differing from K+, Na+ activated HDAC8 to a smaller extent and bound both sites more weakly. The higher value of K1/2,inhib for Na+ prevented the measurement of deacetylase activity at saturating NaCl. Therefore, the Na+-dependent data were fit using Equation 2. Because the binding affinities of Na+ for both sites are within 4-fold of each other, it is difficult to accurately determine K1/2,act and K1/2,inhib for NaCl, and thus, these values have a large margin of error (Table 1).
The KCl dependence of Zn(II)-HDAC8 was also measured to ensure that the effects of MVCs on HDAC8 activity were not limited to Co(II)-HDAC8. The Zn(II)-bound form shows similar distinct activation and inhibition by KCl, with weaker apparent binding affinities for both sites than observed with Co(II)-HDAC8 (Table 1).
Inhibitory MVC Binds to Site 1 and Activating MVC Binds to Site 2
To understand how MVCs alter the catalytic activity of HDAC8, it is important to know where the activating and inhibitory MVCs bind. According to previously published crystal structures (10–12), the MVC that is distant from the divalent catalytic metal ion (site 2) is coordinated by peptide backbone oxygens and water molecules and is, therefore, not amenable to perturbation by site-directed mutagenesis. Site 1, which is near the active site, is composed of the side-chain oxygen atoms of Asp-176 and Ser-199 and four backbone carbonyl oxygens.
To determine whether site 1 binds the activating or the inhibitory MVC, Asp-176 was mutated to either alanine, which cannot coordinate to MVCs, or to asparagine, which is expected to bind MVCs more weakly than aspartate. Both the D176A and D176N mutants showed a significant decrease in affinity of the inhibitory MVC, with K1/2,inhib becoming progressively larger from wt-HDAC8 (26 mm) to D176N (320 mm) and D176A (1200 mm) (Fig. 3 and Table 1). Because of the weakening of K1/2,inhib in D176A and D176N, the activity of these mutants with K+ bound to both sites could not be accurately measured. The KCl dependence data for D176A and D176N were, therefore, fit by Equation 2 in which the activity is modeled as completely inhibited at high KCl. Although a moderate increase was also seen in the value of K1/2,act (3-fold) for the D176A mutant, the increase in the value of K1/2,inhib (46-fold) was much larger. The weakened affinity for the inhibitory MVC upon mutating Asp-176 strongly suggests that this residue forms part of the binding site for the inhibitory MVC.
FIGURE 3.
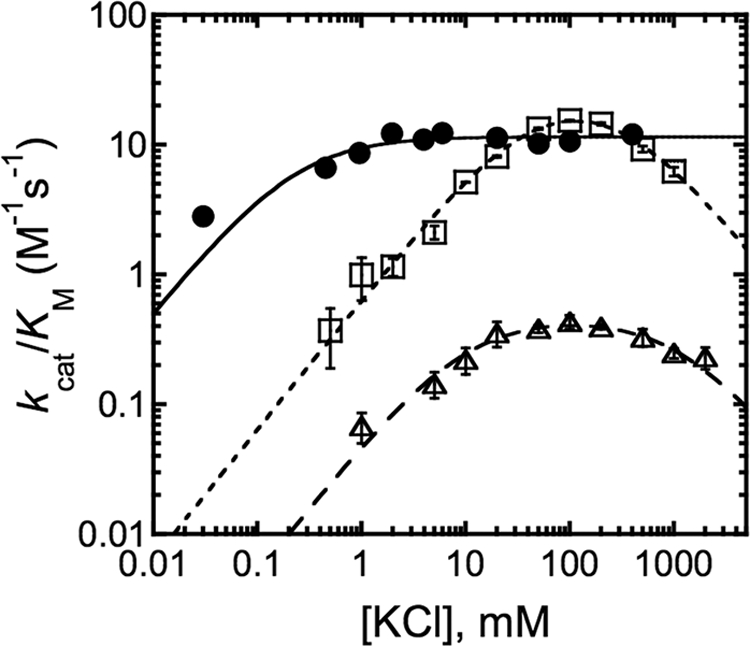
Dependence of the activity of HDAC8 mutants on KCl. Initial rates for HDAC8-catalyzed deacetylation of Fluor de Lys H4-Ack16 (wt, H142A, D176N) or HDAC8 (D176A) substrate were measured at 25 °C in 25 mm Tris pH 8.0 with 0.03–2000 mm KCl for the Co(II)-HDAC8 mutants H142A (●, 10 μm enzyme, 50 μm substrate), D176N (□, 10 μm enzyme, 200 μm substrate), and D176A (△, 20 μm enzyme, 150 μm substrate). Equation 2 is fit to the data for for D176N and D176A, and Equation 3 is fit to the data for H142A with the resulting apparent binding affinities listed in Table 1.
To confirm the assignment of the inhibitory MVC to site 1, the K+ dependence was examined for the H142A mutant. This residue was targeted because it is a second-shell K+ ligand via a hydrogen bond with Asp-176 (Fig. 1). Binding of K+ to site 1 is predicted to decrease the strength of the His-Asp hydrogen bond, thereby lowering the pKa of His-142. Strikingly, the H142A mutant showed a complete loss of inhibition by K+ (Fig. 3), with a 10-fold decrease in the apparent value of K1/2,act (Table 1). This loss of K+ inhibition in the H142A mutant provides further evidence that the inhibitory K+ binds in site 1 and demonstrates that His-142 plays a role in K+ inhibition.
Based on the assignment of site 1 as the inhibitory site, we propose that site 2 is the activating site. To further investigate the activation by MVCs, the activity of Co(II)-HDAC8 was measured as a function of preincubation time with 5 mm KCl. The activation by KCl was found to occur in two phases with the initial phase completed before the first time point was measured (Fig. 4). The amplitude of the first activation phase, measured by comparison to the activity of HDAC8 in the absence of KCl, was ∼50% of the final deacetylase activity. The slower activation phase is well described by a single exponential curve (Equation 5) with a t½ of 1.5 ± 0.1 min. Possible models to explain the biphasic activation by KCl are presented under “Discussion.”
FIGURE 4.
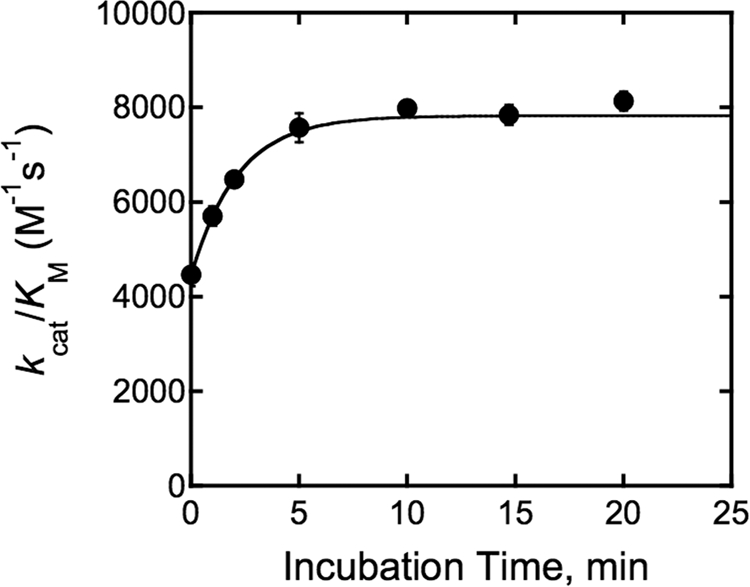
Time dependence of HDAC8 activation by KCl. Co(II)-wt-HDAC8 (0.44 μm) was incubated with 5 mm KCl in 25 mm Tris, pH 8.0, at 25 °C for the indicated length of time before assaying activity with 0.4 μm HDAC8 and 50 μm Fluor de Lys HDAC8 substrate. A single exponential curve (Equation 5) was fit to these data to determine the observed rate constant, kobs, for activation of Co(II)-HDAC8 by KCl.
KCl Alters Inhibition of HDAC8 by SAHA
Inhibition of Co(II)-HDAC8 by the hydroxamic acid inhibitor SAHA was also dependent on the KCl concentration. In contrast to the decreased HDAC8 activity observed in the presence of high potassium, SAHA exhibited higher potency as an inhibitor under saturating potassium conditions. As the KCl concentration decreased, the observed value of the Ki for SAHA increased until it reached a plateau near 0.4 μm at low KCl (Fig. 5). These data can be described most simply with a two-state model (Equation 7) where the SAHA affinity increases as K+ binds to HDAC8; at saturating K+ the value of Ki was 0.041 ± 0.03 μm, indicating that potassium binding to HDAC8 enhances the affinity of SAHA by 10-fold. Similarly, the K+ dissociation constant decreased ∼10-fold when SAHA was bound to HDAC8, from 29 ± 6 mm (consistent with the value of K1/2,inhib determined from steady-state kinetics, Table 1) to 3.2 ± 0.6 mm. Because this two-state scheme accurately modeled the observed data with a Ki comparable with the K+ affinity of the site 1 MVC in HDAC8, it is likely that potassium binding to the site 2 MVC has little effect on the SAHA affinity. To further analyze the coupling between SAHA and K+ binding to HDAC8, we modeled a three-state scheme that includes distinct changes in SAHA binding affinity upon K+ binding to both MVC sites 1 and 2 (Scheme 2; Equation 8). Fitting the data in Fig. 5 using Equation 8 (derived from Scheme 2) with the dissociation constants for K+ determined from the steady-state kinetic data yielded values for Ki,SAHA of 0.4, 0.2, and 0.040 μm for the E, EK, and EK2 species, respectively. Therefore, SAHA had the highest affinity for the potassium-inhibited form of HDAC8 (EK2) with the affinity decreasing up to 10-fold as potassium dissociates from the enzyme.
FIGURE 5.
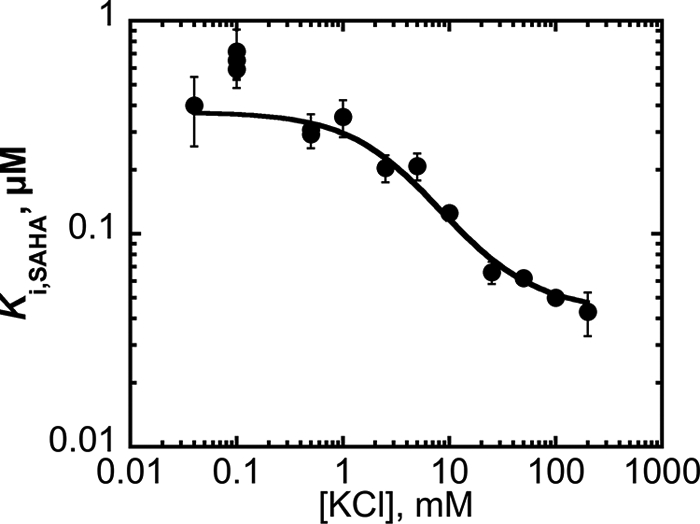
Dependence of the SAHA inhibition constant for HDAC8 on KCl. Co(II)-HDAC8 (0.2 μm) was incubated with SAHA at a given K+ concentration. Initial rates for deacetylation of Fluor de Lys HDAC8 (50 μm) were measured at 25 °C in 25 mm Tris, pH 8.0, containing 0.2–5 μm SAHA and 0.04–200 mm KCl. The SAHA inhibition constant at each K+ concentration was obtained from a fit of Equation 6 to the dependence of activity on the concentration of SAHA. Equations 7 and 8 are fit to these data to determine affinity of SAHA for E, EK, and EK2.
Effects of Physiological Levels of Monovalent Ions
In smooth muscle cells, where HDAC8 is predominantly found, the potassium ion concentration is ∼100 mm (19). When this concentration of KCl was present in vitro, the addition of NaCl did not lead to any further activation of Co(II)-HDAC8, and the value of K1/2,inhib for NaCl decreased 3-fold to 100 mm (Table 2). The loss of Na+-mediated activation in the presence of 100 mm KCl was expected, as the activating site is saturated by KCl (K1/2,act = 3.4 mm) before adding NaCl. In the converse experiment, the addition of physiological concentrations of NaCl (4 mm) did not measurably alter either activation or inhibition by K+ (Table 2). This provides further evidence that potassium functions as the in vivo MVC.
TABLE 2.
Apparent affinities of NaCl and KCl in the presence of physiological levels of the other MVC
HDAC8 activity was measured as described in the legends of Figs. 2 and 3.
| Monovalent ion varied | Monovalent ion held constant | K1/2,act | K1/2,inhib |
|---|---|---|---|
| mm | mm | ||
| KCl | 4 mm NaCl | 3.3 ± 0.2 | 28 ± 3 |
| NaCla | 100 mm KCl | –a | 100 ± 20 |
a Activation by NaCl was not observed in the presence of 100 mm KCl. Equation 4 is fit to the initial rates for deacetylation as a function of NaCl concentration.
DISCUSSION
MVCs Modulate HDAC8 Activity
Before the outset of this study, two bound monovalent cations of unknown function were identified in crystal structures of HDAC8 (10, 12). Here, we demonstrate that one of these two MVCs is essential for catalysis, as no measurable activity was observed in the absence of added KCl or NaCl. The binding of a second MVC resulted in partial inhibition of deacetylase activity (Fig. 2). HDAC8 shows moderate specificity for K+, which supports higher levels of activity than Na+, and is bound with tighter affinity. This preference for K+ is commonly seen in intracellular enzymes, given the greater abundance of K+ than Na+ in this environment (20). The cytosol of smooth muscle cells, where HDAC8 is predominantly observed, contains free MVC concentrations of ∼100 mm KCl and 4 mm NaCl (19, 21). Under these conditions, the activating site of both Co-HDAC8 and Zn-HDAC8 would be saturated and the inhibitory site partially occupied. Although the intracellular K+ concentration is tightly controlled, it varies during certain conditions, such as apoptosis, where intracellular K+ decreases to ∼35 mm (22). It is, thus, possible that HDAC8 activity may be regulated by changes in the fractional occupancy of the inhibitory site, which has a K1/2 value of 130 mm for Zn(II)-HDAC8 and 26 mm for Co(II)-HDAC8 (Fig. 2 and Table 1).
Although non-natural Fluor de Lys peptide substrates were used for these studies, the MVC affinities and the affects on catalytic activity will be the same for subsaturating concentrations of native protein substrates. This is because the MVC binding affinities measured here are for the free species of HDAC8 rather than for the HDAC8·substrate complex, as subsaturating substrate conditions were used (kcat/Km conditions). However, at saturating concentrations of substrate (kcat conditions), it is possible that the extent of activation and inhibition by MVCs may depend on the substrate.
Locations of Activating and Inhibitory MVCs
To delineate which MVC site is activating and which is inhibitory, mutants of the site 1 MVC ligand Asp-176 were characterized. The D176A mutant had a larger effect on K1/2,inhib than K1/2,act, and the inhibitory K+ affinity became progressively weaker as the residue at position 176 was changed from aspartate to asparagine to alanine (Table 1), whereas the effect of the mutations on the affinity of the activating site was variable, indicating that the inhibitory MVC is bound by site 1. The complete loss of MVC inhibition upon mutation of His-142, which is linked to site 1 by a hydrogen bond with Asp-176 (Fig. 1), reveals that His-142 is required for MVC inhibition. The mechanistic implications of inhibition resulting from site 1 occupancy are discussed below.
Given the strong evidence supporting site 1 as the location of the inhibitory MVC and the observation of only two MVC sites by crystallographic (10, 12) and kinetic studies (Fig. 2), site 2 is the most reasonable location for the activating MVC. As the MVC of site 2 is ∼21 Å from the active site, this MVC is considered to be an allosteric effector. A likely role for the site 2 MVC is to stabilize the active conformation of HDAC8. Most enzymes that are activated by allosteric MVCs retain some activity in the absence of added MVC (18); in contrast, HDAC8 is inactive when no MVC is bound (Fig. 2). This suggests that the active conformation of HDAC8 is strongly disfavored when site 2 is unoccupied.
Interestingly, the time-dependent activation of HDAC8 by 5 mm KCl is biphasic with an initial rapid increase in activity followed by a slow doubling of activity with a t½ of 1.5 min (Fig. 4). One model that can explain this time dependence is shown in Scheme 3 and is similar to the activation of thrombin by Na+ (23). Here, HDAC8 is in equilibrium between a minimum of two slowly interconverting conformations, E* and E, with E* unable to bind K+. When K+ is added, it is rapidly bound by E but not by E*. After this fast activation phase, E* slowly converts to E, which can then bind K+ and become catalytically active. One alternative mechanism is that this slow interconversion step occurs but only after potassium binding. These mechanisms cannot be distinguished from the current data. Similar slow conformational transitions have been reported for other proteins (24, 25), and conformational plasticity has been linked to allosteric activation by MVCs (26). Crystal structures of HDAC8 complexes with different inhibitors or substrates demonstrate that the protein displays significant structural plasticity (11, 12, 27). Because both MVC sites are occupied in all of the published crystal structures of HDAC8, the nature of this proposed conformational change upon MVC binding to site 2 is not known but would be expected to influence the positioning of active site residues.
SCHEME 3.
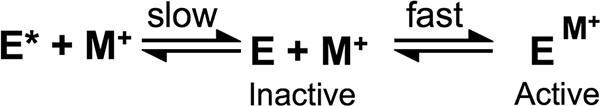
A conformational change may explain the slow activation of HDAC8 by KCl. In this model E and E* are two slowly interconverting conformations of HDAC8. Until M+ binds, the enzyme is inactive, and E* must convert to E before it can bind M+. Rapid binding of M+ to E activates catalysis, as shown in Scheme 1.
The activation and inhibition of HDAC8 by MVCs is not limited to Co(II)-HDAC8, as demonstrated by the qualitatively similar K+ dependence for Zn(II)- and Co(II)-substituted HDAC8. However, the identity of the divalent catalytic metal influences the affinities of the activating and inhibiting MVCs, as both decrease 4–5-fold when Zn(II) is substituted for Co(II) (Table 1). The differences in K1/2,inhib between Zn(II)- and Co(II)-HDAC8 may be due to the linkage of the inhibitory MVC site and the divalent metal ion by Asp-178 and His-180, as these residues contribute to the coordination spheres of both cations (Fig. 1). Furthermore, if the divalent metal ion has differential affinity for the active and inactive conformations, the affinities of the metal ion and the distant site 2 MVC could be coupled. This would result in the affinity of the site 2 MVC depending on the identity of the metal ion.
Maximal Activity When His-142 Is Protonated
The finding that catalysis is partially inhibited when an MVC binds to site 1 in wt-HDAC8 (Fig. 2), with this MVC inhibition eliminated by the H142A mutation (Fig. 3), indicates that the side chain of His-142 is required for MVC inhibition. One interpretation of these data is that the MVC binding site is completely disrupted by loss of a hydrogen bond between Asp-176 and His-142. However, this model is not consistent with the observation of potassium inhibition of the D176A and D176N mutants. A more likely model is that binding of the positively charged inhibitory K+ to site 1 in wt-HDAC8 diminishes catalytic activity by decreasing the fraction of protonated His-142, which is linked to the inhibitory MVC by Asp-176 (Fig. 6). The decrease in activity is ∼11-fold, consistent with a decrease in the pKa by 1 unit. Thus, these data argue that His-142 is protonated in the catalytically active form of HDAC8, in contrast to the previously proposed mechanism with His-142 functioning as the general base (9). Two possible catalytic functions for the protonated His-142 are 1) to function as a general acid to protonate the amine leaving group or 2) to serve as an electrostatic catalyst stabilizing the negative charge that develops during the formation of the tetrahedral intermediate, analogous to the function of His231 in thermolysin and Arg-127 in carboxypeptidase A (28, 29). The H142A mutant of HDAC8 lacks the positive charge of this histidine under all conditions and is, therefore, less active than wt-HDAC8 but not inhibited by K+. Although site 1 MVC has previously been suggested to control the basicity of His-142 (12), it was not known whether the proposed decrease in the pKa of His-142 would activate or inhibit catalysis. The MVC dependence data presented here indicate that the protonated form of His-142 is most active, providing insight into the mechanism of this important enzyme.
FIGURE 6.

Inhibitory K+ binding influences His-142 protonation. The protonated form of His-142 is expected to be thermodynamically more favored in the absence of the inhibitory K+ (upper panel) than when K+ is bound (lower panel).
Inhibition of enzymatic activity by a specifically bound MVC is much less common than activation by MVCs (18, 30). Two other enzymes that are also inhibited by MVCs are the Kex2 protease and homocitrate synthase; in both cases the inhibitory MVC is thought to prevent effective substrate binding (31, 32). Interestingly, the apparent mode of MVC inhibition in HDAC8, affecting the protonation state of a catalytic residue, may be somewhat unique.
MVCs Enhance SAHA Inhibition of HDAC8
In contrast to the inhibitory effect of high KCl on the catalytic activity, potassium binding to the MVC site near the catalytic metal ion (MVC1) enhances the affinity of SAHA by at least 5-fold (Fig. 5). As discussed previously, under physiological conditions (∼100 mm KCl and 4 mm NaCl (19, 21)) the MVC1 of both Co-HDAC8 and Zn-HDAC8 should be partially occupied by K+ (Table 1), enhancing the potency of SAHA. Structures of hydroxamate inhibitors bound to HDAC8 (10) demonstrate that the hydroxamate moiety chelates the active site zinc, with the hydroxamate NH and OH groups making hydrogen bond interactions with His-142, His-143, and Asp-178. Therefore, the increase in SAHA affinity caused by K+ binding to the site 1 MVC is most likely mediated via the hydrogen bond with His-142, which is linked to site 1 by a hydrogen bond with Asp-176. Because the increase in SAHA affinity is comparable with the decrease in catalytic activity caused by K+ binding to this site, it is possible that the molecular origin of both effects is deprotonation of His-142.
Conclusions
The roles of the two MVCs bound by metal-dependent HDACs have now been determined for HDAC8. This enzyme is activated by low concentrations of both KCl and NaCl, and elevated concentrations of these MVCs result in partial inhibition. The activating MVC binds to site 2, which is 21 Å from the catalytic site. These results are consistent with MVC2 being an allosteric effector, which stabilizes the active conformation of HDAC8. The inhibitory MVC binds near the active site and decreases activity likely by depressing the pKa of His-142, which is protonated for maximal activity. Potassium binding to MVC1 also enhances the affinity of SAHA, likely by a similar mechanism. The affinities of both MVCs are sensitive to the identity of the divalent catalytic metal ion, and it is possible that HDAC8 activity may be regulated in vivo by changes in the potassium concentration and/or changes in the divalent metal ion identity.
Acknowledgments
We thank members of the Fierke laboratory and Daniel Dowling (University of Pennsylvania) for helpful discussions and comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM40602 (to C. A. F.).
- HDAC
- histone deacetylase
- SAHA
- suberoylanilide hydroxamic acid
- MVC
- monovalent cation
- MOPS
- 3-(N-morpholino)-propanesulfonic acid
- wt
- wild type.
REFERENCES
- 1.Glozak M. A., Seto E. (2007) Oncogene 26, 5420–5432 [DOI] [PubMed] [Google Scholar]
- 2.Mann B. S., Johnson J. R., Cohen M. H., Justice R., Pazdur R. (2007) Oncologist 12, 1247–1252 [DOI] [PubMed] [Google Scholar]
- 3.Paris M., Porcelloni M., Binaschi M., Fattori D. (2008) J. Med. Chem. 51, 1505–1529 [DOI] [PubMed] [Google Scholar]
- 4.Gantt S. L., Gattis S. G., Fierke C. A. (2006) Biochemistry 45, 6170–6178 [DOI] [PubMed] [Google Scholar]
- 5.Waltregny D., De Leval L., Glénisson W., Ly Tran S., North B. J., Bellahcène A., Weidle U., Verdin E., Castronovo V. (2004) Am. J. Pathol. 165, 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waltregny D., Glénisson W., Tran S. L., North B. J., Verdin E., Colige A., Castronovo V. (2005) FASEB J. 19, 966–968 [DOI] [PubMed] [Google Scholar]
- 7.Wedel T., Van Eys G. J., Waltregny D., Glénisson W., Castronovo V., Vanderwinden J. M. (2006) Neurogastroenterol. Motil. 18, 526–538 [DOI] [PubMed] [Google Scholar]
- 8.Balasubramanian S., Ramos J., Luo W., Sirisawad M., Verner E., Buggy J. J. (2008) Leukemia 22, 1026–1034 [DOI] [PubMed] [Google Scholar]
- 9.Finnin M. S., Donigian J. R., Cohen A., Richon V. M., Rifkind R. A., Marks P. A., Breslow R., Pavletich N. P. (1999) Nature 401, 188–193 [DOI] [PubMed] [Google Scholar]
- 10.Vannini A., Volpari C., Filocamo G., Casavola E. C., Brunetti M., Renzoni D., Chakravarty P., Paolini C., De Francesco R., Gallinari P., Steinkühler C., Di Marco S. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 15064–15069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowling D. P., Gantt S. L., Gattis S. G., Fierke C. A., Christianson D. W. (2008) Biochemistry 47, 13554–13563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Somoza J. R., Skene R. J., Katz B. A., Mol C., Ho J. D., Jennings A. J., Luong C., Arvai A., Buggy J. J., Chi E., Tang J., Sang B. C., Verner E., Wynands R., Leahy E. M., Dougan D. R., Snell G., Navre M., Knuth M. W., Swanson R. V., McRee D. E., Tari L. W. (2004) Structure 12, 1325–1334 [DOI] [PubMed] [Google Scholar]
- 13.Nielsen T. K., Hildmann C., Dickmanns A., Schwienhorst A., Ficner R. (2005) J. Mol. Biol. 354, 107–120 [DOI] [PubMed] [Google Scholar]
- 14.Bottomley M. J., Lo Surdo P., Di Giovine P., Cirillo A., Scarpelli R., Ferrigno F., Jones P., Neddermann P., De Francesco R., Steinkühler C., Gallinari P., Carfí A. (2008) J. Biol. Chem. 283, 26694–26704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuetz A., Min J., Allali-Hassani A., Schapira M., Shuen M., Loppnau P., Mazitschek R., Kwiatkowski N. P., Lewis T. A., Maglathin R. L., McLean T. H., Bochkarev A., Plotnikov A. N., Vedadi M., Arrowsmith C. H. (2008) J. Biol. Chem. 283, 11355–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kern S., Riester D., Hildmann C., Schwienhorst A., Meyer-Almes F. J. (2007) FEBS J. 274, 3578–3588 [DOI] [PubMed] [Google Scholar]
- 17.Hu E., Chen Z.., Fredrickson T., Zhu Y., Kirkpatrick R., Zhang G. F., Johanson K., Sung C. M., Liu R., Winkler J. (2000) J. Biol. Chem. 275, 15254–15264 [DOI] [PubMed] [Google Scholar]
- 18.Page M. J., Di Cera E. (2006) Physiol. Rev. 86, 1049–1092 [DOI] [PubMed] [Google Scholar]
- 19.Kasner S. E., Ganz M. B. (1992) Am. J. Physiol. 262, F462–F467 [DOI] [PubMed] [Google Scholar]
- 20.Woehl E. U., Dunn M. F. (1995) Coord. Chem. Rev. 144, 147–197 [Google Scholar]
- 21.Borin M. L., Goldman W. F., Blaustein M. P. (1993) Am. J. Physiol. 264, C1513–C1524 [DOI] [PubMed] [Google Scholar]
- 22.Hughes F. M., Jr., Cidlowski J. A. (1999) Adv. Enzyme Regul. 39, 157–171 [DOI] [PubMed] [Google Scholar]
- 23.Bah A., Garvey L. C., Ge J., Di Cera E. (2006) J. Biol. Chem. 281, 40049–40056 [DOI] [PubMed] [Google Scholar]
- 24.Hayashi H., Szászi K., Coady-Osberg N., Orlowski J., Kinsella J. L., Grinstein S. (2002) J. Biol. Chem. 277, 11090–11096 [DOI] [PubMed] [Google Scholar]
- 25.Sanghamitra N. J., Mazumdar S. (2008) Biochemistry 47, 1309–1318 [DOI] [PubMed] [Google Scholar]
- 26.Gandhi P. S., Chen Z., Mathews F. S., Di Cera E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1832–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vannini A., Volpari C., Gallinari P., Jones P., Mattu M., Carfí A., De Francesco R., Steinkühler C., Di Marco S. ( 2007) EMBO Rep. 8, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lipscomb W. N., Sträter N. (1996) Chemical Reviews 96, 2375–2434 [DOI] [PubMed] [Google Scholar]
- 29.Hernick M., Fierke C. A. (2005) Arch. Biochem. Biophys. 433, 71–84 [DOI] [PubMed] [Google Scholar]
- 30.Di Cera E. (2006) J. Biol. Chem. 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 31.Holyoak T., Kettner C. A., Petsko G. A., Fuller R. S., Ringe D. (2004) Biochemistry 43, 2412–2421 [DOI] [PubMed] [Google Scholar]
- 32.Andi B., West A. H., Cook P. F. (2005) J. Biol. Chem. 280, 31624–31632 [DOI] [PubMed] [Google Scholar]