

Abstract
Thiolactomycin (TLM), a natural product thiolactone antibiotic produced by species of Nocardia and Streptomyces, is an inhibitor of the β-ketoacyl-acyl carrier protein synthase (KAS) enzymes in the bacterial fatty acid synthase pathway. Using enzyme kinetics and direct binding studies, TLM has been shown to bind preferentially to the acyl-enzyme intermediates of the KASI and KASII enzymes from Mycobacterium tuberculosis and Escherichia coli. These studies, which utilized acyl-enzyme mimics in which the active site cysteine was replaced by a glutamine, also revealed that TLM is a slow onset inhibitor of the KASI enzymes KasA and ecFabB but not of the KASII enzymes KasB and ecFabF. The differential affinity of TLM for the acyl-KAS enzymes is proposed to result from structural change involving the movement of helices α5 and α6 that prepare the enzyme to bind malonyl-AcpM or TLM and that is initiated by formation of hydrogen bonds between the acyl-enzyme thioester and the oxyanion hole. The finding that TLM is a slow onset inhibitor of ecFabB supports the proposal that the long residence time of TLM on the ecFabB homologues in Serratia marcescens and Klebsiella pneumonia is an important factor for the in vivo antibacterial activity of TLM against these two organisms despite the fact that the in vitro MIC values are only 100–200 μg/ml. The mechanistic data on the interaction of TLM with KasA will provide an important foundation for the rational development of high affinity KasA inhibitors based on the thiolactone skeleton.
Keywords: Cell/Wall/Bacteria, Enzymes/Inhibitors, Enzymes/Lipid, Lipid/Fatty Acid, Methods/Fluorescence, Organisms/Bacteria, Residence Time, Slow Onset Inhibition, Tuberculosis
Introduction
New antibacterial agents are needed to treat drug-resistant bacterial infections caused by pathogens such as Mycobacterium tuberculosis (MTB)2 and Staphylococcus aureus (1, 2). The emergence of resistance is driven by drug pressure and a lack of compliance to antibiotic therapy, exacerbated in the case of tuberculosis by a long and complex treatment regime that involves multiple antibiotics over a 6–9-month course of therapy. The World Health Organization estimates that 0.5 million new cases of multidrug-resistant tuberculosis occur each year (2009 WHO Report), whereas the emergence of extensively drug resistant strains of MTB that are resistant to both first and second line drugs pose a severe threat to human health (4). There is, thus, a requirement not only for novel compounds that circumvent existing drug resistance mechanisms but also that shorten the course of therapy for diseases such as tuberculosis.
Cell wall biosynthesis is a widely used target for chemotherapeutic intervention in a variety of bacterial infections (5). Although many antibiotics, such as the β-lactams and vancomycin, target peptidoglycan biosynthesis, there is growing evidence that fatty acid biosynthesis is a promising target for drug discovery (6, 7). This is particularly true for mycobacteria, where the frontline tuberculosis drug isoniazid compromises cell wall integrity by inhibiting the biosynthesis of mycolic acids, very long chain lipids that provide protection and allow the bacteria to persist in the human macrophage (8). Mycolic acids are synthesized from long chain (C50+) fatty acids that are in turn synthesized by the dissociated fatty acid (FAS-II) pathway, and isoniazid inhibits the synthesis of mycolic acids through an effect on one or more cellular targets including the FAS-II enoyl-AcpM reductase InhA (Fig. 1) (9–13). Although the enoyl-ACP reductase is the most heavily targeted FAS-II component for the development of novel antibacterial agents (7), the isolation of natural products such as thiolactomycin, cerulenin, and platensimycin (Fig. 1) that inhibit the FAS-II β-ketoacyl-ACP synthase (KAS) enzymes have demonstrated that the condensation step in fatty acid biosynthesis is also a very promising target for drug discovery (14–18).
FIGURE 1.

The FAS-II pathway in M. tuberculosis and the structures of several natural product KAS inhibitors. In M. tuberculosis, the FAS-II pathway elongates the C18+ acyl-CoAs from the FAS-I pathway to C54-C56 fatty acids. The pathway is primed by the CoA-dependent β-ketoacyl-AcpM synthase FabH, which condenses the acyl-CoA with malonyl-AcpM to generate a β-ketoacyl-AcpM. The β-ketoacyl-AcpM is converted into a saturated enoyl-AcpM by the sequential actions of a β-ketoacyl-AcpM reductase (MabA), a dehydrase, and a trans-2-enoyl-AcpM reductase (InhA). Subsequent rounds of elongation are initiated by the KasA or KasB β-ketoacyl-AcpM synthases. KasA is thought to be responsible for the early rounds of fatty acid elongation. Also shown is the malonyl-CoA AcpM transacylase (FabD) responsible for the synthesis of malonyl-AcpM. β-Ketoacyl-AcpM synthase inhibitors include the natural products thiolactomycin, cerulenin, and platensimycin.
The bacterial FAS-II pathway contains one KAS enzyme that is responsible for priming the initial round of fatty acid extension (KASIII) and one or two additional KAS enzymes (KASI and KASII) that function in subsequent rounds of fatty acid synthesis. In MTB both KASI and KASII enzymes are present and carry the designations KasA and KasB, whereas the corresponding enzymes in the very well characterized pathway from Escherichia coli are known as FabB (ecFabB) and FabF (ecFabF), respectively (19, 20). The KAS enzymes are members of the thiolase superfamily and catalyze the Claisen condensation reaction between malonyl-ACP and the growing fatty acid (acyl-CoA for KASIII and acyl-ACP for KASI/II) (Fig. 2) using a conserved active site triad that includes the nucleophilic cysteine and two histidines (KASI/II) or a histidine and an asparagine (KASIII) (21, 22).
FIGURE 2.

Ping-pong catalytic mechanism for KasA. Acyl-enzyme formation occurs after nucleophilic attack of the active site cysteine (Cys-171 in KasA) on the carbonyl carbon of acyl-AcpM. This reaction is facilitated by the oxyanion hole formed by the amide groups of Cys-171 and Phe-404. Dissociation of AcpM and binding of the second substrate, malonyl-AcpM, is followed by decarboxylation and carbanion formation. Condensation and carbon-carbon bond formation occurs through a nucleophilic attack by the malonyl-AcpM carbanion on the acyl-KasA thioester carbonyl group to form the β-keto acyl-AcpM product and free enzyme. Decarboxylation of malonyl-AcpM and subsequent condensation with the acyl group are facilitated by two conserved histidines (His-311 and His-345 in KasA). In the mechanism shown a conserved phenylalanine is proposed to destabilize the malonate anion, thereby promoting decarboxylation, in line with previous proposals for the mechanism of KASIII enzymes as well as thiolase homologues such as chalcone synthase (21). We note that formation of the acetyl carbanion has also been proposed to occur by attack of water on the malonate group and elimination of bicarbonate (51); however, in the case of KasA, a conserved phenylalanine (Phe-237) is appropriately positioned to destabilize the malonate anion, and no structured water molecule can be observed in the x-ray structures of wild-type and mutant KasA (45).
Thiolactomycin (TLM), a natural product thiolactone isolated from Nocardia sp., is a reversible KAS enzyme inhibitor (14, 16, 23, 24) with activity against both Gram-positive and Gram-negative bacteria (25, 26) as well as MTB (MIC 62.5 μm) (27, 28). Although TLM has also been reported to inhibit the human FAS-I enzyme (29), the low toxicity and relatively low affinity of TLM for FAS-I (IC50 100 μm) make it an attractive lead compound for antimicrobial drug discovery (30). TLM-resistant E. coli strains contain mutations in the fabB gene (31), and overproduction of ecFabB confers TLM resistance (32), suggesting that ecFabB is the major cellular target. Importantly, KasA, the ecFabB homologue in MTB, is known to be essential (33), and thus, there is significant interest in using TLM as a novel lead for developing selective and potent KasA inhibitors (27).
To provide a foundation for the development of TLM analogues that target KasA, we have performed a detailed kinetic analysis on the interaction of TLM with wild-type KasA as well as the C171Q KasA mutant that mimics the acyl-enzyme intermediate (17). In addition, because genetic evidence exists that ecFabB is the target for TLM in E. coli, we have also quantitated the interaction of TLM with this enzyme as well as with the corresponding KASII enzymes from MTB (KasB) and E. coli (ecFabF) in an attempt to rationalize previous published data on KAS enzyme inhibition by TLM (16, 17, 34). These studies have revealed that TLM binds preferentially to the acyl-enzyme form of both the KASI and KASII enzymes in each organism and that TLM is also a slow onset inhibitor of the KasA and ecFabB acyl-enzymes but not of the corresponding KASII enzyme-substrate intermediates. These studies are significant because they reveal that the relevant structure for TLM-lead optimization is the complex of TLM with the acyl-enzyme and not of TLM with the free enzyme. In addition, the observation that TLM is a slow onset inhibitor of the KasA and ecFabB acyl-enzymes is also of high importance given the growing realization that inhibitor residence time is a critical factor for in vivo drug activity (35–37), prolonging the activity of drugs even when the free drug concentration is low. The acyl-KasA·TLM EI* complex has a half-life of 14 min, and based on an analysis of the structural data, we propose a model for the slow onset inhibition of KasA that will be useful for the development of long residence time KasA inhibitors.
EXPERIMENTAL PROCEDURES
Materials
CoA substrates were purchased from Sigma, and PCR and mutagenesis primers were purchased from Integrated DNA Technologies. Precast SDS-PAGE gels, columns, and resins were purchased from Bio-Rad. pET expression vectors were purchased from Novagen, and restriction enzymes and buffers were purchased from New England Biolabs. Enantiomerically pure TLM was synthesized as previously described (38) or was generously provided by Cynthia Dowd and Clifton Barry (NIAID, National Institute of Allergy and Infectious Diseases). Buffers, growth media, and other reagents were purchased from Fisher.
KasA Expression and Purification
The M. tuberculosis kasA gene was cloned from genomic DNA into the mycobacterial expression vector pFPCA (39) after PCR amplification with the primers 5′-GGAAGATCTGGAGGATAACAAAGATGGG- 3′ (forward) and 5′-CCGGAATTCTCAGTAACGCCCGAAGGCA-3′ (reverse). Plasmid was isolated from E. coli XL1-Blue cells and transformed into Mycobacterium smegmatis strain mc2155 competent cells by electroporation. Colonies selected on 7H10 solid media containing 30 μg/ml kanamycin, 200 μg/ml ampicillin, and 15 μg/ml cycloheximide were cultivated in 7H9 liquid media supplemented with glycerol and grown to an optical density (A600) of 0.6–0.8, after which protein expression was induced with 0.2% acetamide. Cells were harvested by centrifugation and lysed by sonication. The KasA protein was subsequently purified using standard metal ion affinity chromatography followed by chromatography on G-25 resin using 50 mm sodium phosphate buffer, pH 8.5, 0.3 m NaCl as the eluent.
ecFabB, ecFabF, and KasB Expression and Purification
The E. coli fabB gene was cloned from genomic DNA into the pET28a expression vector using NheI and XhoI restriction sites after PCR amplification with the primers 5′-CTAGCTAGCATGAAACGTGCAGTGATTACTGGC-3′ and 5′-CCGCTCGACTTATTAATCTTTCAGCTTGCGCATTACC-3′. Similarly, the E. coli fabF gene was also cloned into the pET28a expression vector using the NheI and XhoI restriction sites after PCR amplification with the primers 5′-CTAGCTAGCGTGTCTAAGCGTCGTGTAGTT-3′ and 5′-CCGCTCGAGTTATTAGATCTTTTTAAAGATCAAAGA-3′. The KasB pET28a expression vector was generously provided by Merrill Schaeffer. All three KAS enzymes were expressed as N-terminal hexahistidine-tagged constructs in E. coli pLysS cells and purified using standard nickel affinity chromatography as described previously (16, 19).
Mutagenesis
The KasA and ecFabF Cys to Gln mutant constructs were prepared using the QuikChange mutagenesis method using the primers KasA C171Q (forward) (5′- ACCCCGGTGTCGGCCCAGTCGTCGGGCTCGGAA-3′), KasA C171Q (reverse) (5′-TTCCGAGCCCGACGACTGGGCCGACACCGGGGT-3′), ecFabF C164Q (forward) (5′- TCTATCGCGACTGCCCAGACTTCCGGCGTGCAC-3′), and ecFabF C164Q (reverse) (5′-GTGCACGCCGGAAGTCTGGGCAGTCGCGATAGA-3′). These mutant proteins were expressed and purified as described for the wild-type enzymes.
AcpM Expression and Substrate Synthesis
The mycobacterial acyl carrier protein (AcpM) was cloned, expressed, and purified as previously described using the pET24b(+) expression plasmid (40). Briefly, plasmid transformants were selected on LB agar containing 30 μg/ml kanamycin, and single colonies were cultivated to an A600 of 0.8–1.0 and then induced with 0.7 mm isopropyl 1-thio-β-d-galactopyranoside. Isopropyl alcohol precipitation and anion exchange column chromatography yielded three forms of AcpM: apo, holo, and acyl. After anion exchange chromatography, fractions containing the three AcpM species were dialyzed into 20 mm Tris-HCl, pH 8.0, buffer and subjected to ion exchange chromatography using a Mono Q 10/100 GL column equilibrated with 20 mm Tris-HCl, pH 8.0 buffer. Elution of the various AcpM species was achieved using a linear gradient of NaCl with apo-AcpM eluting at 320 mm NaCl.
The E. coli holo-ACP synthase was cloned, expressed, and purified as described previously (41). Briefly, transformants containing pET-ACP synthase were selected on LB agar containing 200 μg/ml ampicillin, and selected colonies were grown to an A600 of 0.8–1.0 on a 1-liter scale in YT media (16 g of Tryptone, 10 g of yeast, 5 g (85 mm) of NaCl, 2.4 g (20 mm) of sodium phosphate in 1 liter). Protein expression was then induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 30 °C for 3 h, after which cells were harvested by centrifugation, resuspended in 50 mm Tris-HCl, pH 8.0, buffer and lysed by sonication. Cell free extracts were gently mixed with 1 g of DE-52 resin to form a slurry that was kept at 4 °C for 15 min and then centrifuged to remove the DE-52. This step was repeated, and then the supernatant was adjusted to pH 6.5 with a saturated MES solution. The crude protein mixture was loaded onto an 8-ml SP-Sepharose column (8 × 1 cm) that had been equilibrated with 50 mm Tris-HCl, pH 6.5, buffer. Pure ACP synthase protein was eluted with a linear gradient of NaCl and concentrated using a YM-10 Centricon concentrator. Protein was stored at −20 or −80 °C with 10–50% glycerol. All samples retained holo-ACP synthase transfer activity for >2 years.
Homogeneous apo-AcpM was incubated in ACP synthase reaction buffer (50 mm Tris-HCl, pH 8.5, 25 mm MgCl2, 5 mm MnCl2, 1 mm dithiothreitol) with a 2-fold molar excess of either malonyl-CoA or palmitoyl-CoA (Sigma) and 6 μm ACP synthase for 90 min at 30 °C. The reaction mixture was exchanged into 20 mm Tris-HCl, pH 8.0, and purified by ion exchange chromatography as described above.
Enzyme Assays
KasA activity was monitored in 50 mm Tris-HCl, 1 mm dithiothreitol at pH 8.5 at 25 °C using a coupled assay with MabA, the NADPH-dependent FAS-II β-ketoacyl-ACP reductase (19). MabA was expressed in E. coli BL21-DE3 cells and purified by metal ion affinity chromatography as described previously (42). Reactions were initiated by the addition of either malonyl-AcpM (15 μm) or palmitoyl-AcpM (15 μm) to reaction mixtures containing 250 μm NADPH, 6 μm MabA, 15 μm of the other substrate, and 90 nm KasA. For the preincubation experiments, TLM was added to the reaction mixture 15 min before the addition of the second substrate after control experiments in which the IC50 values for TLM inhibition were determined as a function of preincubation time. Data-fitting was performed using Grafit 4.0 (Erithacus Software Ltd). The activity and inhibition of ecFabF was assayed in a similar fashion, except that CoA-based substrates were used. The kinetic parameters for ecFabF (500 nm) were determined in 50 mm potassium phosphate buffer, pH 6.5, containing 50 mm NaCl, 1 mm dithiothreitol by varying the concentration of lauroyl-CoA at a fixed concentration of malonyl-CoA (2 mm) or by varying the concentration of malonyl-CoA at a fixed concentration of lauroyl-CoA (100 μm). The IC50 for the inhibition of ecFabF with TLM was determined using this assay system by varying the concentration of TLM at fixed concentrations of lauroyl-CoA (100 μm) and malonyl-CoA (550 μm). All kinetic data were analyzed using Grafit 4.0.
Direct Binding Fluorescence Titrations
TLM binding was quantitated by monitoring the intrinsic tryptophan fluorescence of each enzyme using a Quanta Master fluorimeter (Photon Technology International). Excitation and emission wavelengths were 280 and 337 nm (KasA/ecFabB/ecFabF) or 280 and 331 nm (KasB), respectively, with an excitation slit width of 4.0 nm and an emission slit width of 8.0 nm. TLM solutions in buffer (50 mm Tris-HCl, pH 8.5, 1 mm dithiothreitol) were titrated into enzyme in the same buffer. The concentration of enzyme in the direct binding measurements was 500 nm in all cases except for acyl-ecFabF and C164Q-ecFabF, where 250 nm enzyme was used. All solutions were filtered and equilibrated to 25 °C. Titration curves were corrected for inner filter effects, and Kd values were calculated using the Scatchard equation (Grafit 4.0). To quantitate the slow onset binding of TLM to acyl-KasA, acyl-ecFabB, and the C171Q KasA mutant, the fluorescence emission was monitored as a function of time for 35 min. Fluorescence intensities were fit to a double exponential equation (Equation 1) to account for a slight time-dependent decrease in the background fluorescence (kback).
The values of kobs obtained from the exponential fitting were dependent on the concentration of TLM and were subsequently fit to Equation 2 to extract values for Ki, the dissociation constant of the initial enzyme-inhibitor complex (EI), as well as k1 and k2, the forward and reverse rate constants for conversion of EI to the final enzyme-inhibitor complex (EI*) (Scheme 1).
SCHEME 1.
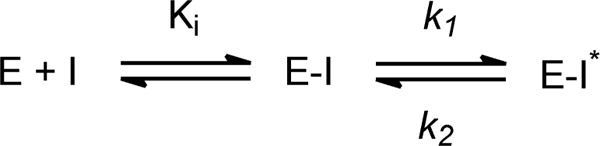
General scheme for the two-step formation of an enzyme-inhibitor complex. In slow onset inhibition, formation of the initial E-I complex is rapid compared with formation of E-I*.
Subsequently, Ki*, the overall dissociation constant of the final EI* complex, was calculated using Equation 3.
RESULTS
Expression, Purification, and Kinetic Characterization of the KAS Enzymes
KasB, ecFabF, and ecFabB were all obtained as soluble proteins after overexpression in E. coli. In contrast, expression of KasA in E. coli failed to yield soluble protein (19), and consequently this enzyme was expressed in M. smegmatis. Because KasA is inactive with CoA-based substrates, the natural malonyl-AcpM and palmitoyl-AcpM substrates were synthesized from apo-AcpM using the E. coli holo-ACP synthase (19). Kinetic parameters were determined by monitoring the NADPH-dependent reduction of the β-ketoacyl-AcpM KasA product in a coupled assay with MabA, the subsequent enzyme in the FAS-II pathway (19). The Km value for palmitoyl-AcpM of 2.6 ± 0.6 μm determined at a fixed concentration of malonyl-AcpM (54 μm) is similar to the Km value determined previously (3.2 μm), whereas the kcat value of 32 ± 2 min−1 is 6-fold larger than that determined by Schaeffer et al. (19) using the ACP from E. coli, in agreement with the observation that KasA displays higher activity with the natural mycobacterial acyl carrier protein compared with the E. coli homologue. Kinetic parameters have so far not been reported for ecFabF, and therefore these were determined using CoA-based substrates, giving kcat and Km values of 1.26 ± 0.08 min−1 and 25 ± 3 μm, respectively, when lauroyl-CoA was varied at a fixed concentration of malonyl-CoA (2 mm) and 1.25 ± 0.04 min−1 and 600 ± 70 μm, respectively, when malonyl-CoA was varied at a fixed concentration of lauroyl-CoA (100 μm) (Table 1).
TABLE 1.
Kinetic parameters for KasA and ecFabF
| Enzyme | Kma | Kmb | kcata | kcat/Kmc |
|---|---|---|---|---|
| μm | μm | min−1 | min−1/μm | |
| KasA | 2.6 ± 0.6 | NDd | 32 ± 2.0 | 12.3 |
| KasAe | 3.2 ± 0.3 | 13.5 ± 2.6 | 5.3 ± 0.1 | 1.7 |
| ecFabF | 25 ± 3 | 600 ± 70 | 1.25 ± 0.04 | 0.05 |
a Calculated by varying the acyl substrate (palmitoyl for KasA, lauroyl for ecFabF).
b Calculated by varying the malonyl substrate.
c With respect to the acyl substrate.
d Not determined.
e Previously reported using refolded KasA expressed in E. coli and ecACP substrates (19).
KasA Inhibition by TLM
We initially determined the IC50 for KasA inhibition by TLM using a kinetic assay in which KasA was the last reagent added to the reaction mixture. This experiment provided an IC50 value of greater than 200 μm (Table 2), in contrast to the values of 14 μm (27) and 20 μm (19) reported previously. Subsequently IC50 values were determined by first preincubating TLM and KasA followed by initiation of the reaction with one or other of the two substrates (Table 2). The IC50 value determined when palmitoyl-AcpM was used to initiate the preincubated reaction mixture containing KasA, TLM, and malonyl-AcpM was 240 μm. However, when the reaction was initiated with malonyl-AcpM after preincubation the KasA-TLM complex with palmitoyl-AcpM, the observed IC50 value decreased to 19 μm. These data strongly suggest that TLM binds with a higher affinity to the acyl-enzyme form of KasA. While these studies were being conducted, a similar result was reported with platensimycin, a potent and selective natural product inhibitor of the acyl-enzyme intermediate form of ecFabF (KASII E. coli) (17). The effect of palmitoyl-AcpM on the IC50 value for KasA inhibition was only observed when TLM was preincubated with the enzyme. Because the palmitoyl-KasA acyl-enzyme is expected to form rapidly, this observation additionally suggests that there is a slow onset component to the interaction of TLM with acyl-KasA.
TABLE 2.
Kinetic and thermodynamic parameters for the interaction of TLM with KasA and KasB
| Enzyme | IC50 | Kda | Kib | k1b | k2b | Ki*c |
|---|---|---|---|---|---|---|
| μm | μm | μm | s−1 | s−1 | μm | |
| Apo-KasA | 242 ± 60d | 226 ± 9 | NDe | ND | ND | ND |
| Acyl-KasA | 19.0 ± 2.3f | ND | 163 ± 28 | 0.19 ± 0.02 | 0.0004 ± 0.0009 | 0.3 ± 0.8 |
| C171Q KasA | ND | ND | 126 ± 16 | 0.046 ± .002 | 0.0008 ± 0.0005 | 2.2 ± 1.6 |
| Apo-KasB | ND | ≥400 | ND | Rapid | Rapid | Rapid |
| Acyl-KasB | ND | 53 + 5 | ND | Rapid | Rapid | Rapid |
a Kd values were determined by fluorescence titration and fitting the data to the Scatchard equation.
b Parameters were determined by fitting the kobs data for the slow binding of TLM using Equation 2.
c Ki* was calculated using Equation 3.
d IC50 value was determined by preincubating TLM with KasA and malonyl-AcpM and initiating the reaction by the addition of palmitoyl-AcpM.
e ND, not determined.
f IC50 value was determined by preincubating TLM with KasA and palmitoyl-AcpM and initiating the reaction by the addition of malonyl-AcpM.
Direct Binding Experiments
The interaction of TLM with KasA was further examined using a direct binding experiment in which the intrinsic tryptophan fluorescence of KasA was used to measure the affinity and slow rate of binding of TLM to KasA. Interaction of TLM with free KasA results in a rapid decrease in fluorescence. After correction of the data for the inner filter effect, the fluorescence titrations yielded a Kd value of 226 μm for the interaction of TLM with the free enzyme (Fig. 3a, Table 2). This Kd value is similar to the IC50 value obtained when TLM was not preincubated with the reaction mixture (see above and Table 2). Based on the decrease in IC50 value observed when TLM was preincubated with palmitoyl-ACP and KasA, we set out to quantitate the interaction of TLM with acyl-KasA. We synthesized the acyl-KasA intermediate by incubation of palmitoyl-CoA with KasA for 10 min as previously described (17). When the titration was repeated with the acylated form of KasA, the addition of TLM to the enzyme solution resulted in a much slower decrease in the fluorescence signal than the instantaneous change observed for the free enzyme. The rate of decay of the fluorescence signal was dependent on the concentration of TLM, and the data were fit to a double exponential to correct for a slight background decrease in intensity and to obtain kobs, the rate constant for TLM binding (43, 44). The kobs values obtained at different TLM concentrations were then fit to Equation 2, which describes the interaction of TLM with KasA using a two-step binding model (Scheme 1). This analysis yielded estimates for Ki* of 0.3 ± 0.8 μm for the formation of the final enzyme inhibitor complex EI* (Table 2). The poor estimate of Ki* is primarily due to significant errors in the observed values for k2. Based on the report that the C163Q mutant of ecFabF mimicked the structural change accompanying acyl-ecFabF formation (17), we repeated the fluorescence titration using the C171Q KasA mutant. As observed for palmitoyl-KasA, binding of TLM to C171Q KasA resulted in a time-dependent decrease in fluorescence (Fig. 3b), which after data analysis (Fig. 3c) provided a Ki* value of 2.2 ± 1.6 μm. These data are summarized in Table 2.
FIGURE 3.

Interaction of the KASI enzymes with TLM. a, change in fluorescence when 1 μm apo-KasA is titrated by TLM is shown. The solid line is the best fit of the data to the Scatchard equation. b, fluorescence decay curves obtained after the addition of TLM to 1 μm C171Q KasA are shown. Data are shown for TLM concentrations 10, 15, 20, and 50 μm, and the solid lines are the best fit of the data to Equation 1. c, plot of kobs as a function of TLM concentration obtained from the data shown in b, with the solid line showing the best fit to Equation 2. d, change in fluorescence when 1 μm apo-ecFabB is titrated by TLM is shown. The solid line is the best fit of the data to the Scatchard equation. e, fluorescence decay curves obtained after the addition of TLM to 1 μm acyl-ecFabB are shown. Data are shown for TLM concentrations 3, 5, 10, 20, and 40 μm, and the solid lines are the best fit of the data to Equation 1. f, shown is a plot of kobs as a function of TLM concentration obtained from the data shown in e, with the solid line showing the best fit to Equation 2. In each case fluorescence emission was monitored at 337 nm.
TLM Binding to Other KAS Enzymes
The discovery that TLM bound more tightly to acyl-KasA prompted us to analyze the interaction of this inhibitor with other KAS enzymes. We consequently extended our studies to KasB and the KASII enzyme from M. tuberculosis as well as ecFabB and ecFabF, the KASI and KASII enzymes from E. coli. In the case of ecFabF, we found the enzyme to be active with CoA-based substrates (see above). This assay system was used to measure the IC50 value of TLM with ecFabF. Unlike KasA, ecFabF showed no change in IC50 upon preincubation, and our value of 5.4 ± 0.5 μm (Table 3) is consistent with the previously published value of 6 μm (16). These data suggest that TLM is not a slow binding inhibitor of ecFabF. We further quantified TLM binding using the intrinsic tryptophan fluorescence of ecFabF for the free enzyme, acyl-enzyme, and C164Q mutant (Fig. 4). As observed for KasA, the measured Kd values were ∼10-fold lower with the acyl-enzyme or C164Q mutant compared with the free enzyme (6.5 or 7.5 μm compared with 61 μm; Table 3). However, in each case the change in fluorescence upon the addition of TLM was rapid, consistent with the notion that TLM is not a slow onset inhibitor of ecFabF.
TABLE 3.
Kinetic and thermodynamic parameters for the interaction of TLM with ecFabB and ecFabF
| Enzyme | IC50 | Kda | Kib | k1b | k2b | Ki*c |
|---|---|---|---|---|---|---|
| μm | μm | μm | s−1 | s−1 | μm | |
| Apo-ecFabB | 12d | 31 ± 5 | NDe | Rapid | Rapid | Rapid |
| Acyl-ecFabB | ND | ND | 41 ± 10 | 0.32 ± 0.04 | 0.012 ± 0.005 | 1.4 ± 1.0 |
| Apo-ecFabF | 5.4 ± 0.5f | 61 ± 7 | ND | Rapid | Rapid | Rapid |
| Acyl-ecFabF | ND | 6.5 ± 2.9 | ND | Rapid | Rapid | Rapid |
| C164Q-ecFabF | ND | 7.5 ± 0.3 | ND | Rapid | Rapid | Rapid |
a Kd values were determined by fluorescence titration and fitting the data to the Scatchard equation.
b Parameters were determined by fitting the kobs data for the slow binding of TLM using Equation 2.
c Ki* was calculated using Equation 3.
d E. coli (strain ANS1) MIC = 6 μm (lower than all values except Ki*) (27).
e ND, not determined.
f IC50 value was determined by using CoA substrates. Preincubation has no effect.
FIGURE 4.

Interaction of the KASII enzymes with TLM. a, change in fluorescence is shown when 250 nm apo-FabF (closed circles) or 250 nm acyl-FabF (open circles) are titrated by TLM. In each case the black line is the best fit of the data to the Scatchard equation. b, change in fluorescence is shown when 1 μm apo-KasB (closed circles) or 1 μm acyl-KasB (open circles) are titrated by TLM. In each case the black line is the best fit of the data to the Scatchard equation.
Fluorescence spectroscopy was also used to monitor the direct binding of TLM to KasB and ecFabB. Similar to KasA, ecFabB showed an instantaneous change in fluorescence upon the addition of TLM to the free enzyme, resulting in a calculated Kd value of 31 ± 5 μm (Fig. 3d, Table 3). Again, similar to KasA, a slow decrease in fluorescence that was dependent on TLM concentration was observed for binding of TLM to the acyl-enzyme synthesized by incubating ecFabB for 10 min with lauroyl-CoA (Fig. 3e). Kinetic analysis demonstrated that TLM is a slow onset inhibitor of acyl-ecFabB, with a Ki* value for the final inhibited complex of 1.4 ± 1.0 μm (Fig. 3f, Table 3).
Finally, the binding of TLM to KasB was quantified in a similar fashion. Similar to ecFabF, TLM showed preferential binding to the acyl-enzyme over the free enzyme with observed Kd values of 53 and ≥400 μm, respectively (Fig. 4, Table 2). However TLM was not a slow onset inhibitor of acyl-KasB. Taken together, these data demonstrate that TLM is a slow onset inhibitor of the KASI enzymes KasA and ecFabB but not of the KASII enzymes KasB and ecFabF. In each case TLM binds preferentially to the acyl-enzyme intermediate of each enzyme, and slow onset inhibition is only observed upon binding to the acyl-enzyme intermediates of the KASI enzymes (Scheme 2).
SCHEME 2.

Scheme for the interaction of TLM with KasA.
DISCUSSION
To establish a foundation for rational inhibitor design, we have undertaken a detailed characterization of KasA and its inhibition by the lead molecule TLM. We have shown through kinetic analysis and direct binding experiments that TLM shows preferential binding to the acyl-enzyme intermediate of KasA and to the Cys to Gln mutant that mimics this intermediate. In addition, we have also shown that TLM binds preferentially to the KasB, ecFabB, and ecFabF acyl-enzyme intermediates. The preference of TLM for the acyl-KAS enzymes is consistent with the proposal that TLM is a competitive inhibitor of malonyl-ACP (15, 16) and that TLM occupies the malonyl binding site (16, 45). This observation is also in line with previous studies on the interaction of platensimycin with ecFabF (17) and explains why the reported Kd value for TLM binding to free ecFabF of 60 μm is 10-fold larger than the IC50 value of 6 μm for enzyme inhibition (16). Additionally, our analysis has also revealed that TLM is a slow onset inhibitor of the KASI acyl-enzymes (KasA and ecFabB) but only a rapid reversible inhibitor of the KASII enzymes KasB and ecFabF. For the KASI enzymes, the increase in affinity of TLM for the acyl-enzyme is only manifest after time has been allowed for formation of EI*. Thus, for KasA and ecFabB, the Ki value for formation of the initial encounter complex EI between TLM and the acyl-enzyme intermediate is similar to the value of Kd for binding of TLM to the free enzyme. Formation of EI is then followed by a slow step, leading to formation of the final EI* complex where Ki* is 10–100-fold smaller than Ki. Importantly then, in cases where slow onset inhibition is observed, measured IC50 values for enzyme inhibition will depend on how the inhibition assays are conducted and, specifically, whether time is given for formation of EI* in the reaction mixture. Consequently, the slow onset inhibition of the acyl-KASI enzymes by TLM could account for some of the variation in previously reported IC50 values (19, 27, 46). In addition, the possibility that TLM might also be a slow onset inhibitor of the type I FAS (FAS-I) could also account for contradictory reports on the ability of TLM to inhibit the human and rat FAS-I enzymes (29, 47).
To direct the synthesis of TLM analogues with improved antibacterial activity, it is clearly important to understand the structural basis for the increased affinity of TLM for the KAS acyl-enzymes. Comparison of the structures of TLM bound to wild-type KasA and the C171Q acyl-enzyme mimic indicates that acyl-enzyme formation involves a 60° rotation and a 3.3 Å shift of Phe-404 (45). The change in position of Phe-404 is accompanied by movement of a flexible loop (residues 402–406), which increases the solvent-accessible volume of the active site. Increased access to the active site together with an edge to face interaction between Phe-404 and the thiolactone ring of the inhibitor are both thought to contribute to the increased affinity of TLM for acyl-KasA (45).
Although the structural data provide an explanation for the differential affinity of TLM for apo- and acyl-KasA, the data do not directly shed light on why TLM is a slow onset inhibitor of KasA. In particular, the structure of C171Q KasA in the presence and absence of TLM are very similar, raising the possibility that the structure of TLM bound to C171Q KasA is actually a structure of the initial encounter complex EI rather than EI*. Alternatively, it may be that the structural changes that accompany formation of EI* from EI are not readily visualized by x-ray crystallography, and we are currently using molecular dynamics simulations to explore the possibility that the transition from EI to EI* involves a change in the dynamics of helices α5 and α6 that are intimately involved in ligand binding. In this regard we can trace a series of interactions that propagate from the active site through the protein core to α5 and α6. Formation of hydrogen bonds between the acyl-enzyme thioester carbonyl, mimicked in this case by the Gln-171 amide carbonyl, and the backbone amides of Phe-404 and residue 171, which form the oxyanion hole, brings Phe-404 closer to Val-278 on an adjacent loop and results in a strengthening of an interaction between Met-277 and Met-146 in the neighboring monomer (Fig. 5). Increased contact with Met-146 leads to a conformational change that includes Met-146—Pro-147-Asn-148 and ultimately a shift in helices α5 and α6 that prepare the enzyme to bind malonyl-AcpM or, in this case, TLM, which binds in the malonyl binding site. This structural analysis predicts that the helices are more flexible in the free enzyme, which is in line with the model proposed by Reynolds and co-workers (48) for substrate binding to mtFabH, the KASIII β-ketoacyl-ACP synthase from MTB, and also with our previous analysis of the KasA·TLM structure (45). In KasA, we believe that this network of interactions provides a mechanism for sensing the binding of TLM to the acyl-enzyme that then results in a further decrease in the dynamics of helix motion, which would be the slow step in the transition from EI to EI*. Interestingly, the two methionines that we speculate are important for interactions across the KasA dimer interface are conserved in ecFabB but not in ecFabF (supplemental Fig. S1). In addition, although Met-146 and Met-277 are both present in KasB, analysis of the KasB structure (PDB 2gp6) (49) indicates that the side chains of these residues are rotated away from one another and do not interact.
FIGURE 5.

Interactions linking the KasA active site and helices α5 and α6. a, shown is the structure of wild-type and C171Q KasA bound to TLM. KasA is a homodimer and the wild-type subunits are colored cyan and magenta, whereas the C171Q subunits are colored yellow and silver. TLM is colored black. Arrows show the movement of helices α5 and α6 on adjacent subunits upon formation of the acyl-enzyme, mimicked in this case by mutation of the catalytic cysteine to a glutamine. Key residues that link the active site with helices α5 and α6 are shown as sticks. b, a detailed view shows the side chains, which alter their position upon transition from the free enzyme to the acyl-enzyme. Hydrogen-bond formation between the carbonyl oxygen of Gln-171 and the oxyanion hole NH donors Phe-204 and Gln-171 cause Phe-404 and loop 1 to move closer to loop 2 containing Met-277 and Val-278. The Met-277 side chain shifts closer to Met-146 at the base of helices α5 and α6 in the opposite subunit. This in turn decreases the distance between the Phe-147 residues from each subunit from 7.57 to 4.57 Å linking changes in one subunit with the corresponding structural changes in the second subunit. The figure was made using PyMOL (3) and the PDB entries 2WGD (wild type without TLM), 2WGE (wild type with TLM), 2WGF (C171Q without TLM), and 2WGG (C171Q with TLM).
Although this model for the inhibition of KasA by TLM remains to be thoroughly evaluated by molecular dynamics simulations and site-directed mutagenesis, the observation that TLM is a slow onset inhibitor of the KASI enzymes has important implications for the in vivo activity of this compound. In particular, it has previously been shown that TLM is able to provide protection from both urinary tract and oral infections caused by Serratia marcescens and Klebsiella pneumonia (30) despite the fact that the MIC values of TLM against these organisms are only 100–200 μg/ml (14). Although we have not quantitated the interaction of TLM with the KAS enzymes from S. marcescens and K. pneumonia, it is plausible that TLM is also a slow onset inhibitor of the FabB enzymes in these organisms (smFabB and kpFabB), which are 89 and 96% identical, respectively, to ecFabB (supplemental Fig. S1). Many successful therapeutics, such as the NSAID CeleCoxib, are thought to owe their clinical efficacy to long residence times on their therapeutic targets (35, 36), and recently we demonstrated a correlation between residence time and in vivo activity for a series of diaryl ether FabI inhibitors (37). Consequently, we propose that the long residence time of TLM on smFabB and kpFabB could explain the antibacterial activity of TLM in rodents challenged by these organisms despite the modest in vitro antibacterial activity of this antibiotic.
Although further experiments are in progress to determine the mechanism of KAS inhibition by TLM in other organisms, we believe the observation that TLM is a slow onset inhibitor of KasA provides critical support for the utility of TLM as a lead compound for developing potent and selective KasA inhibitors. Finally, although the slow onset inhibition of smFabB and kpFabB presents a plausible explanation for the in vivo antibacterial activity of TLM against S. marcescens and K. pneumonia, it may also be that the inherent resistance of the TLM producer species Nocardia and Streptomyces to TLM results from the reduced affinity and/or absence of slow onset inhibition of the antibiotic for the enzyme target(s) in these organisms, as recently shown in an analogous fashion for resistance of Pantoea agglomerans to the antibiotic andrimid (50).
Supplementary Material
Acknowledgment
We thank Dr. Scott Franzblau at the Institute for Tuberculosis Research, University of Illinois for the gift of the pFPCA plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grant R01AI044639 (to P. J. T.). This work was also supported by Deutsche Forschungsgemeinschaft Grants SFB 630 and Forschungszentrum FZ82 (to C. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- MTB
- Mycobacterium tuberculosis
- ACP
- acyl carrier protein
- KAS
- β-ketoacyl-ACP synthase
- FAS
- fatty acid synthase
- TLM
- thiolactomycin
- MES
- 4-morpholineethanesulfonic acid
- EI
- enzyme-inhibitor complex
- MIC
- minimal inhibitory concentration.
REFERENCES
- 1.Bloom B. R., Murray C. J. (1992) Science 257, 1055–1064 [DOI] [PubMed] [Google Scholar]
- 2.de Lencastre H., Oliveira D., Tomasz A. (2007) Curr. Opin. Microbiol. 10, 428–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 4.Shenoi S., Friedland G. (2009) Annu. Rev. Med. 60, 307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh C. T. (2003) Antibiotics: Actions, Origins, Resistance, pp. 23–49, American Society for Microbiology, Washington, DC [Google Scholar]
- 6.Heath R. J., Rock C. O. (2004) Curr. Opin. Investig. Drugs 5, 146–153 [PMC free article] [PubMed] [Google Scholar]
- 7.Lu H., Tonge P. J. (2008) Acc. Chem. Res. 41, 11–20 [DOI] [PubMed] [Google Scholar]
- 8.Liu J., Barry C. E., 3rd, Besra G. S., Nikaido H. (1996) J. Biol. Chem. 271, 29545–29551 [DOI] [PubMed] [Google Scholar]
- 9.Banerjee A., Dubnau E., Quemard A., Balasubramanian V., Um K. S., Wilson T., Collins D., de Lisle G., Jacobs W. R., Jr. (1994) Science 263, 227–230 [DOI] [PubMed] [Google Scholar]
- 10.Quémard A., Sacchettini J. C., Dessen A., Vilcheze C., Bittman R., Jacobs W. R., Jr., Blanchard J. S. (1995) Biochemistry 34, 8235–8241 [DOI] [PubMed] [Google Scholar]
- 11.Rozwarski D. A., Grant G. A., Barton D. H., Jacobs W. R., Jr., Sacchettini J. C. (1998) Science 279, 98–102 [DOI] [PubMed] [Google Scholar]
- 12.Slayden R. A., Lee R. E., Barry C. E., 3rd (2000) Mol. Microbiol. 38, 514–525 [DOI] [PubMed] [Google Scholar]
- 13.Argyrou A., Jin L., Siconilfi-Baez L., Angeletti R. H., Blanchard J. S. (2006) Biochemistry 45, 13947–13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oishi H., Noto T., Sasaki H., Suzuki K., Hayashi T., Okazaki H., Ando K., Sawada M. (1982) J. Antibiot. 35, 391–395 [DOI] [PubMed] [Google Scholar]
- 15.Nishida I., Kawaguchi A., Yamada M. (1986) J. Biochem. 99, 1447–1454 [DOI] [PubMed] [Google Scholar]
- 16.Price A. C., Choi K. H., Heath R. J., Li Z., White S. W., Rock C. O. (2001) J. Biol. Chem. 276, 6551–6559 [DOI] [PubMed] [Google Scholar]
- 17.Wang J., Soisson S. M., Young K., Shoop W., Kodali S., Galgoci A., Painter R., Parthasarathy G., Tang Y. S., Cummings R., Ha S., Dorso K., Motyl M., Jayasuriya H., Ondeyka J., Herath K., Zhang C., Hernandez L., Allocco J., Basilio A., Tormo J. R., Genilloud O., Vicente F., Pelaez F., Colwell L., Lee S. H., Michael B., Felcetto T., Gill C., Silver L. L., Hermes J. D., Bartizal K., Barrett J., Schmatz D., Becker J. W., Cully D., Singh S. B. (2006) Nature 441, 358–361 [DOI] [PubMed] [Google Scholar]
- 18.Wright H. T., Reynolds K. A. (2007) Curr. Opin. Microbiol. 10, 447–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaeffer M. L., Agnihotri G., Volker C., Kallender H., Brennan P. J., Lonsdale J. T. (2001) J. Biol. Chem. 276, 47029–47037 [DOI] [PubMed] [Google Scholar]
- 20.Edwards P., Nelsen J. S., Metz J. G., Dehesh K. (1997) FEBS Lett. 402, 62–66 [DOI] [PubMed] [Google Scholar]
- 21.Heath R. J., Rock C. O. (2002) Nat. Prod. Rep. 19, 581–596 [DOI] [PubMed] [Google Scholar]
- 22.Olsen J. G., Kadziola A., von Wettstein-Knowles P., Siggaard-Andersen M., Larsen S. (2001) Structure 9, 233–243 [DOI] [PubMed] [Google Scholar]
- 23.Sato T., Suzuki K., Kadota S., Abe K., Takamura S., Iwanami M. (1989) J. Antibiot. 42, 890–896 [DOI] [PubMed] [Google Scholar]
- 24.Brown M. S., Akopiants K., Resceck D. M., McArthur H. A., McCormick E., Reynolds K. A. (2003) J. Am. Chem. Soc. 125, 10166–10167 [DOI] [PubMed] [Google Scholar]
- 25.Hamada S., Fujiwara T., Shimauchi H., Ogawa T., Nishihara T., Koga T., Nehashi T., Matsuno T. (1990) Oral Microbiol. Immunol. 5, 340–345 [DOI] [PubMed] [Google Scholar]
- 26.Noto T., Miyakawa S., Oishi H., Endo H., Okazaki H. (1982) J. Antibiot. 35, 401–410 [DOI] [PubMed] [Google Scholar]
- 27.Kim P., Zhang Y. M., Shenoy G., Nguyen Q. A., Boshoff H. I., Manjunatha U. H., Goodwin M. B., Lonsdale J., Price A. C., Miller D. J., Duncan K., White S. W., Rock C. O., Barry C. E., 3rd, Dowd C. S. (2006) J. Med. Chem. 49, 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slayden R. A., Lee R. E., Armour J. W., Cooper A. M., Orme I. M., Brennan P. J., Besra G. S. (1996) Antimicrob. Agents Chemother. 40, 2813–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McFadden J. M., Medghalchi S. M., Thupari J. N., Pinn M. L., Vadlamudi A., Miller K. I., Kuhajda F. P., Townsend C. A. (2005) J. Med. Chem. 48, 946–961 [DOI] [PubMed] [Google Scholar]
- 30.Miyakawa S., Suzuki K., Noto T., Harada Y., Okazaki H. (1982) J. Antibiot. 35, 411–419 [DOI] [PubMed] [Google Scholar]
- 31.Jackowski S., Zhang Y. M., Price A. C., White S. W., Rock C. O. (2002) Antimicrob. Agents Chemother. 46, 1246–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsay J. T., Rock C. O., Jackowski S. (1992) J. Bacteriol. 174, 508–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhatt A., Kremer L., Dai A. Z., Sacchettini J. C., Jacobs W. R., Jr. (2005) J. Bacteriol. 187, 7596–7606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones A. L., Gane A. M., Herbert D., Willey D. L., Rutter A. J., Kille P., Dancer J. E., Harwood J. L. (2003) Planta 216, 752–761 [DOI] [PubMed] [Google Scholar]
- 35.Copeland R. A., Pompliano D. L., Meek T. D. (2006) Nat. Rev. Drug Discov. 5, 730–739 [DOI] [PubMed] [Google Scholar]
- 36.Tummino P. J., Copeland R. A. (2008) Biochemistry 47, 5481–5492 [DOI] [PubMed] [Google Scholar]
- 37.Lu H., England K., am Ende C., Truglio J. J., Luckner S., Reddy B. G., Marlenee N. L., Knudson S. E., Knudson D. L., Bowen R. A., Kisker C., Slayden R. A., Tonge P. J. (2009) ACS Chem. Biol. 4, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McFadden J. M., Frehywot G. L., Townsend C. A. (2002) Org. Lett. 4, 3859–3862 [DOI] [PubMed] [Google Scholar]
- 39.Changsen C., Franzblau S. G., Palittapongarnpim P. (2003) Antimicrob. Agents Chemother. 47, 3682–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaeffer M. L., Agnihotri G., Kallender H., Brennan P. J., Lonsdale J. T. (2001) Biochim. Biophys. Acta 1532, 67–78 [DOI] [PubMed] [Google Scholar]
- 41.Lambalot R. H., Walsh C. T. (1995) J. Biol. Chem. 270, 24658–24661 [DOI] [PubMed] [Google Scholar]
- 42.Cohen-Gonsaud M., Ducasse S., Hoh F., Zerbib D., Labesse G., Quemard A. (2002) J. Mol. Biol. 320, 249–261 [DOI] [PubMed] [Google Scholar]
- 43.Tsai Y. C., Johnson K. A. (2006) Biochemistry 45, 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rawat R., Whitty A., Tonge P. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13881–13886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luckner S. R., Machutta C. A., Tonge P. J., Kisker C. (2009) Structure 17, 1004–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaeffer M. L., Carson J. D., Kallender H., Lonsdale J. T. (2004) Tuberculosis 84, 353–360 [DOI] [PubMed] [Google Scholar]
- 47.Hayashi T., Yamamoto O., Sasaki H., Kawaguchi A., Okazaki H. (1983) Biochem. Biophys. Res. Commun. 115, 1108–1113 [DOI] [PubMed] [Google Scholar]
- 48.Sachdeva S., Musayev F. N., Alhamadsheh M. M., Scarsdale J. N., Wright H. T., Reynolds K. A. (2008) Chem. Biol. 15, 402–412 [DOI] [PubMed] [Google Scholar]
- 49.Sridharan S., Wang L., Brown A. K., Dover L. G., Kremer L., Besra G. S., Sacchettini J. C. (2007) J. Mol. Biol. 366, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X., Fortin P. D., Walsh C. T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 13321–13326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y. M., Hurlbert J., White S. W., Rock C. O. (2006) J. Biol. Chem. 281, 17390–17399 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.