

Abstract
Copper-transporting ATPase ATP7B is essential for human copper homeostasis and normal liver function. ATP7B has six N-terminal metal-binding domains (MBDs) that sense cytosolic copper levels and regulate ATP7B. The mechanism of copper sensing and signal integration from multiple MBDs is poorly understood. We show that MBDs communicate and that this communication determines the oxidation state and conformation of the entire N-terminal domain of ATP7B (N-ATP7B). Mutations of copper-coordinating Cys to Ala in any MBD (2, 3, 4, or 6) change the N-ATP7B conformation and have distinct functional consequences. Mutating MBD2 or MBD3 causes Cys oxidation in other MBDs and loss of copper binding. In contrast, mutation of MBD4 and MBD6 does not alter the redox status and function of other sites. Our results suggest that MBD2 and MBD3 work together to regulate access to other metal-binding sites, whereas MBD4 and MBD6 receive copper independently, downstream of MBD2 and MBD3. Unlike Ala substitutions, the Cys-to-Ser mutation in MBD2 preserves the conformation and reduced state of N-ATP7B, suggesting that hydrogen bonds contribute to interdomain communications. Tight coupling between MBDs suggests a mechanism by which small changes in individual sites (induced by copper binding or mutation) result in stabilization of distinct conformations of the entire N-ATP7B and altered exposure of sites for interactions with regulatory proteins.
Keywords: Metals, Transport, ATPases, Copper, Protein Folding, ATP7A, ATP7B, Atox1, CxxC, Copper
Introduction
Copper is an essential trace element used as a cofactor by numerous enzymes, which take advantage of its low redox potential to catalyze electron transfer reactions. Perhaps because of this high reactivity, copper is kept under strict homeostatic control. The copper-transporting P-type ATPases (Cu-ATPases), ATP7A and ATP7B, are essential for maintaining cellular copper levels. These transporters deliver copper into the secretory pathway for the biosynthesis of cuproenzymes and facilitate export of excess copper from the cells. In humans, mutations in either ATP7A or ATP7B disrupt copper homeostasis and lead to the severe disorders Menkes disease and Wilson disease, respectively (1). Copper is not only transported by Cu-ATPases; it also acts as a regulator of their activity, post-translational modification, and intracellular localization (2). The mechanism of copper-dependent regulation of Cu-ATPases is poorly understood.
ATP7A and ATP7B are highly homologous proteins and share many aspects of function and regulation. In this study, we focus on ATP7B. Cu-ATPase ATP7B is a 165-kDa protein with eight transmembrane segments and most of the soluble parts exposed to the cytosol. The large cytosolic N-terminal domain of ATP7B (N-ATP7B)2 binds copper and plays a key role in regulation of ATP7B. The intracellular copper donor metallochaperone Atox1 docks to N-ATP7B, transfers copper, and stimulates the activity of ATP7B. N-ATP7B is also phosphorylated by a kinase in response to copper binding (3), houses the sequence determinants for the copper-dependent apical targeting of ATP7B (4), and recognizes dynactin, a component of the trafficking machinery, in a copper-dependent manner (5). It remains unknown whether all of these copper-induced events are directly coupled. It is also unclear how many copper-binding sites in N-ATP7B participate in each regulatory event or how signals from multiple copper-binding events are integrated, if at all.
N-ATP7B contains six metal-binding subdomains (MBDs), each coordinating one copper between 2 Cys residues in an invariant CxxC motif (6, 7). The six MBDs are connected by flexible loops of varying lengths (Fig. 1A). The three-dimensional structure of N-ATP7B is unknown; however, the structures of individual subdomains and two pairs (MBD3/4 and MBD5/6) have been solved (8–15). These studies revealed that all MBDs have a ferridoxin-like βαββαβ-fold, with the copper-coordinating site situated in a solvent-exposed loop between the β1-strand and α1-helix (Fig. 1B). In individual recombinant MBDs, copper binding to this loop does not induce significant changes in the structure of MBDs: the loop takes on a more fixed, rigid state (9), whereas the rest of the protein shows very minor changes. However, when copper binds to N-ATP7B, significant changes in secondary structure are detected by circular dichroism and proteolysis (3, 7). Thus, very small structural alterations in individual MBDs upon copper binding are sufficient to trigger a rearrangement of the entire N-terminal domain.
FIGURE 1.

Organization of N-ATP7B. A, schematic illustrating the relative length of the loops connecting the N-terminal MBDs; copper-binding sites, tryptophan residues, and trypsin recognition sites are as indicated. B, structure of a representative MBD (MBD5; adapted from Ref. 8) (Protein Data Bank code 2ew9) The GMxCxxC metal-binding loop is shown in light gray; Cys residues are indicated.
The mechanism by which copper regulates the N-ATP7B conformation is not obvious because, in N-ATP7B, the copper-binding sites of MBDs appear fairly exposed. Specifically, all MBDs in N-ATP7B can be labeled with the Cys-directed fluorescent probe, although with different efficiency (16, 17), indicating accessibility of sites to solvent. Similarly, all six sites in N-ATP7B can be loaded with copper in the presence of Cu-Atox1 (18). Although copper exchange between individual MBDs can occur in solution (8), such interdomain exchange is not essential for Atox1-mediated metalation of N-ATP7B (16).
The above structural data showing relative exposure of metal- binding sites (MBSs) and the lack of significant structural changes upon copper binding appear at odds with the studies showing functional coupling between MBDs. In N-ATP7B, MBD2 is the preferential site for copper transfer from Atox1, and only after MBD2 is filled can other sites be loaded with copper (18). In the context of the full-length ATP7B, the CxxC-to-AxxA substitution within MBD2 completely disrupts the activation of ATP7B by Cu-Atox1 without altering protein ability to be activated by free copper (17). This observation suggests that MBD2 may act as a gateway for Atox1-mediated copper transfer to the rest of the protein and modulate access to other N-terminal MBDs.
To better understand the relationship between MBDs in the context of a fully folded N-ATP7B, we characterized the effect of mutating individual sites on copper binding by N-ATP7B. Our studies revealed the key roles of MBD2 and MBD3 in maintaining the structural and functional integrity of N- ATP7B. We observed a tight packing of MBDs within N-ATP7B that depends on the ability of the CxxC-containing loops to form hydrogen bonds and that has important functional consequences. The interdomain contacts maintain the reduced state of MBSs as well as distinct conformations of loops connecting MBDs. Our results suggest a mechanism that allows for small perturbations in structure (because of mutation or copper binding) to alter the tertiary structure of the entire N-ATP7B.
MATERIALS AND METHODS
Constructs/Mutants
Mutations in N-ATP7B were generated using the pMal-N-ATP7B plasmid as a template and the Stratagene QuikChange XLII kit with the following mutagenic primers (or combinations thereof in the case of m2/3A): M2A, GCATGACCGCCCAGTCCGCTGTCAGCTCCATTGAAGG (forward) and CTGACAGCGGACTGGGCGGTCATGCCCTCCACCC (reverse); M2S, AGGGCATGACCAGCCAGTCCAGTGTCAGCTCC (forward) and GGAGCTGACACTGGACTGGCTGGTCATGCCCT (reverse); M3A, GGAATGCATGCTAAGTCTGCCGTCTTGAATATTG (forward) and CAAGACGGCAGACTTAGCATGCATTCCATCTATTC (reverse); M4A, CCATTGCCGGCATGACCGCTGCATCCGCTGTCCATTCCATTGAAG (forward) and CTTCAATGGAATGGACAGCGGATGCAGCGGTCATGCCGGCAATGG (reverse); and M6A, GGATGACCGCCGCGTCCGCTGTCCACAACATAGAGTC (forward) and GGACAGCGGACGCGGCGGTCATCCCTGTGATTGTCAG (reverse). The correctness of the coding sequences in all constructs was verified by automatic DNA sequencing.
Expression and Purification of Recombinant Proteins
N-ATP7B (and mutants thereof) was expressed and purified as a fusion with maltose-binding protein (MBP) as described previously (6). In brief, the fusion protein was coexpressed with thioredoxin in Escherichia coli to maintain solubility and reducing state, purified by affinity chromatography over amylose resin (New England Biolabs, Ipswich, MA), and eluted into buffer containing 10 mm maltose, 25 mm NaH2PO4, and 150 mm NaCl (pH 7.5). The levels of protein expression for mutants and wild-type N-ATP7B were comparable. For copper binding in vivo, 250 μm CuCl2 was added to the growth medium prior to inducing protein expression for 3 h at 25 °C. Atox1 was expressed in E. coli as a fusion with the chitin-binding protein in the pTYB12-intein vector as described previously (18) and purified by affinity chromatography over chitin resin (New England Biolabs). Intein-mediated cleavage of chitin-binding protein and elution of Atox1 were accomplished by incubating resin-bound fusion protein with buffer containing 50 mm dithiothreitol, 25 mm NaH2PO4, and 150 mm NaCl (pH 7.5) for 36 h at room temperature. Atox1 was concentrated using Amicon ultrafiltration devices (Millipore, Billerica, MA) and then dialyzed against buffer containing 25 mm NaH2PO4 and 150 mm NaCl (pH 7.5) overnight at 4 °C to remove dithiothreitol.
Atox1-mediated Copper Transfer
Prior to copper transfer experiments, 1 mg of purified N-ATP7B (at 9 mm) or 1 mg of Atox1 (at 130 μm) was reduced with 1 mm tris(carboxyethyl)phosphine (TCEP) and then dialyzed into 1 liter of buffer A (25 mm NaH2PO4 and 150 mm NaCl (pH 7.5)) for 1 h. A 10:1 GSH/CuCl2 solution made in buffer A was added to the reduced Atox1 at a 1:1 molar ratio for 15 min at room temperature, followed by overnight dialysis to remove excess glutathione. For transfer reactions, increasing amounts of Cu-Atox1 were added to 100 ng of apo-N-ATP7B for 15 min at room temperature. Atox1 was then removed from the mixture by centrifugation through a Microcon YM-50 filter (Millipore); the concentrate containing N-ATP7B was diluted with buffer A, and centrifugations were repeated three times to completely remove Atox1. N-ATP7B was eluted with 100 μl of 25 mm NaH2PO4 (pH 7.5) containing 25 mm NaCl.
Copper-binding Stoichiometry
The concentration of purified proteins was determined with the Lowry assay (19). Copper concentration in samples was measured using atomic absorption spectroscopy (AA-6650G, Shimadzu, Columbia, MO), and a copper/protein molar ratio was averaged for each mutant in four independent experiments.
X-ray Absorption Spectroscopy
N-ATP7B mutants were loaded with copper in cells as described above, purified, and concentrated to concentrations yielding >100 μm bound copper. CuK edge (8.9 keV) extended x-ray absorption fine structures (EXAFS) were collected at the Stanford Synchrotron Radiation Laboratory operating at 3 GeV with currents between 100 and 50 mA. All samples were measured and analyzed as described previously (20). For edge comparisons, all edges were normalized to have an intensity of 0 at 8970 eV and an intensity of 1 at 9000 eV.
Fluorescent Labeling of Cys Residues
10 μg (91 pmol) of purified apo-N-ATP7B (wild-type and mutant) in elution buffer was taken for labeling of cysteines. 7-Diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM; Invitrogen, Carlsbad, CA) was added in the dark for 5 min at a concentration of 10 μm and then quenched with 100 μm glutathione. Samples were run on a 12% Laemmli gel and rinsed 2× 15 min in double-distilled H2O, and fluorescent images were taken using a FluorChem 5500 (Alpha Innotech Corp., San Leandro, CA). Gels were then fixed in 50% ethanol and 8% phosphoric acid for a minimum of 3 h, stained with colloidal Coomassie Blue G-250, and imaged again to normalize fluorescence intensity per protein present in the corresponding band. The fluorescence of fully copper-loaded N-ATP7B was defined as the background and was subtracted. Where indicated, samples were incubated in solutions containing either 100 μm TCEP for 10 min (to reduce disulfide bonds) or 6 m urea for 30 min (to expose buried Cys residues) prior to labeling with 10 μm CPM as described above.
Quantitation of Reduced Cys Residues
Further quantitation of available cysteine residues was carried out using Ellman's reagent (5,5′-dithiobis(2-nitrobenzoic acid)) as described (21). Briefly, 10 μg (91 pmol) of purified apo-N-ATP7B (wild-type ± copper and m2A) in elution buffer was used for Ellman's reaction after adjusting the protein concentration for A280 measurements. Where indicated, samples were incubated in solutions containing either 100 μm TCEP for 10 min (to reduce oxidized cysteines) or 6 m urea for 30 min (to expose buried Cys residues), with all samples suspended in final volumes of 50 μl. Samples were added to 750 μl of 1 m Tris-HCl (pH 8.0), followed by the addition of 50 μl of 2 mm 5,5′-dithiobis(2-nitrobenzoic acid (in 50 mm sodium acetate) for 15 min and reading the absorbance at 412 nm. A calibration curve of acetylcysteine ranging from 31.25 μm to 1 mm was assayed in duplicate along with each set of test samples. The assay was repeated three times using the calibration curve to estimate total cysteines available per protein in the N-ATP7B samples.
Limited Proteolysis
15 μg of eluted apo-N-ATP7B (wild-type and mutant) at 10 μm was digested with tosylphenylalanyl chloromethyl ketone-treated bovine pancreatic trypsin (Sigma) for 3 h at room temperature at a 1:3000 (w/w) protease/protein ratio in 83 mm ammonium bicarbonate (pH 8.0) containing 10 mm CaCl2. The reaction was stopped by the addition of 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride. The protein fragments were separated on a 15% Laemmli gel and then fixed and stained with colloidal Coomassie Blue G-250. In some instances, CPM labeling and quenching were performed as described above prior to proteolysis.
Native Gel Electrophoresis
20 μg of wild-type N-ATP7B was subject to limited proteolysis (see above) and then separated on a 10% Tris/glycine gel that did not contain urea, SDS, or reducing agents. The gel was fixed in 50% ethanol and 8% phosphoric acid for a minimum of 3 h and stained overnight with colloidal Coomassie Blue G-250. The lane containing the protein fragments was destained in 7% acetic acid, equilibrated in Laemmli running buffer containing 2 m urea for 1 h to redissolve proteins, layered onto a 12.5% Laemmli gel, and separated in the presence of 2 m urea and 0.1% SDS. This gel was then fixed and silver-stained as described (22).
In-gel Digests/Extractions
For protein identification by tandem mass spectrometry, Coomassie Blue-stained gels were washed in double-distilled H2O, and relevant spots were excised. Spots were incubated twice in 500 μl of 50 mm NH4HCO3 in 50% acetonitrile for 30 min and then dried by removing the wash buffer and incubating in 100% acetonitrile for 2 min. If the samples had not been previously reduced and alkylated, 100 μl of buffer containing 10 mm dithiothreitol and 100 mm NH4HCO3 was added for 45 min at 56 °C and removed, and 100 μl of buffer containing 55 mm iodoacetamide and 100 mm NH4HCO3 was added for 30 min at room temperature. This solution was removed, and 500 μl of buffer containing 50 mm NH4HCO3 and 50% acetonitrile was added for 15 min at room temperature. This solution was replaced with 200 μl of 100% acetonitrile for 2 min at room temperature. 20 μl of digestion solution (0.01 μg of trypsin, 44 mm NH4HCO3, and 4.4 mm CaCl2) was added for 15 min at 4 °C. (For large spots, this was repeated until liquid was no longer absorbed.) Excess solution was removed, and 60 μl of digestion solution (without trypsin) was added at 37 °C for 16 h. To extract peptides, 3 μl of 88% formic acid was added for 15 min at 37 °C, and all of the supernatant was then collected for tandem mass spectrometry analysis.
Fluorescence Spectroscopy
Purified N-ATP7B-MBP was dialyzed into buffer containing 25 mm NaH2PO4 and 150 mm NaCl (pH 7.5) for 1 h and diluted to 0.3 mg/ml in the same buffer using A280 to evaluate and equate final concentrations. MBP was prepared in the same manner as N-ATP7B-MBP, dialyzed, and diluted to a final concentration of 0.12 mg/ml. 400-μl samples were taken for fluorescence emission scans using a SpectraMax M2 fluorescence cuvette reader (Molecular Devices, Sunnyvale, CA). Excitation wavelengths were set at either 280 or 295 nm, and emission readings were taken between 250 and 450 nm at 1-nm intervals using 10 scans/emission wavelength. Following scans, A280 measurements were taken to normalize the data for protein concentration. The experiment was performed three times, with a representative scan presented herein.
Modeling and Molecular Dynamics Simulations
Structures for MBD1 and MBD2 were generated through homology modeling (Modeler version 9v6) using human ATP7B (NCBI Protein Database accession number P35670) and then used to generate distance constraints for ab initio modeling. Ab initio structure prediction was carried out using locally installed Rosetta ab initio software (version 2.0, licensed through the University of Washington). The fragment libraries were generated using the Web version of the Rosetta fragment server (Robetta). Briefly, fragments for residues 56–214 of ATP7B were generated using the Robetta fragment server, and 10,000 independent structures were predicted using the above-generated distance constraints for MBD1 and MBD2. The connecting loop along with 5-residue anchors from MBD1 and MBD2 (residues 121–148) were extracted using the MMTSB tool set and subjected to a clustering analysis. Loop extraction was performed to avoid biasing the clustering algorithm through constrained MBDs (which are significantly longer than the connecting loop). The centers of the three largest clusters were chosen as the best models. The MBDs were then “ligated” back to the models for the connecting loops using the 5-residue anchors. The final models were minimized using CHARMM27 force fields and validated as described previously (23).
A 10.0-ns molecular dynamics simulation of MBD2 from ATP7A in explicit solvent was conducted using the NAMD version 2.6 molecular dynamics simulation package and the CHARMM27 force field. Because the structure of MBD2 from ATP7B was not available and because one can argue that the ab initio models generated from Rosetta may not be accurate enough for molecular dynamics simulations, we used the structure of MBD2 from ATP7A (Protein Data Bank code 1s6o) (13). This structure of MBD2 (code 1s6o) was introduced in a box of TIP3P explicit water molecules that extended at least 10 Å away from the protein surface, and counterions were introduced to obtain a neutral system. The protonation status of the titratable residues corresponds to pH 7.0. The system was minimized in two steps: water minimization and entire system minimization. First, the system was subjected to 1000 steps of conjugate gradient energy minimization with the coordinates of the protein frozen to allow the solvent molecules to relax. A second 1000 steps of conjugate gradient energy minimization were then performed after unfreezing the protein to remove steric clashes found in the protein structure. The root mean square deviation (r.m.s.d.) as a function of time for the simulated structures was stable for 200 ps. The final equilibrated system that uses periodic boundary conditions has dimensions of 59.18, 64.22, and 64.92 Å. The resulting system was used in a 10.0-ns simulation that was performed in the NVT ensemble using a 2-fs time step and a Langevin thermostat with a 5 ps−1 damping parameter. The system temperature was coupled using the Berendsen algorithm at 300 K. Electrostatic interactions were calculated using the particle mesh Ewald with a real space cutoff of 0.9 nm. Cutoff for van der Waals interactions was set at 0.9 nm. A SHAKE algorithm was applied using a tolerance of 10−8Å to maintain all bonds containing hydrogen at their equilibrium length. The time step for integration was 2 fs, and coordinates and velocities were saved every 2 ps. Simulations were analyzed using NAMD routines, and the data were plotted using GraphPad version 4.0.
RESULTS
MBD2 Influences the Copper-binding Capability of Other N-terminal MBDs
To understand the relationships between different MBDs, we first generated and characterized the m2A mutant of N-ATP7B, in which the copper-binding CxxC motif of MBD2 was replaced with AxxA (Fig. 2A). Earlier experiments suggested that MBD2 may regulate access to other MBSs (see above). We hypothesized that MBD2 performs this gating role by interacting with other MBDs and changing the protein conformation of N-ATP7B (thus allowing access to other sites) in response to copper binding. Mutating MBD2 would disrupt this communication and cause insufficient exposure of other MBDs to Atox1 and inadequate copper transfer. To test this hypothesis, we compared the copper loading of wild-type N-ATP7B and the m2A mutant by Cu-Atox1. For wild-type N-ATP7B, as shown previously (18), all six MBDs could be loaded with copper (Fig. 2B). However, for the m2A mutant, instead of the expected stoichiometry of 5 copper atoms/protein, <3 copper atoms were transferred (maximum of 2.8 ± 0.5) (Fig. 2B) even using up to 60-fold excess of Cu-Atox1 over N-ATP7B.
FIGURE 2.

Atox1-mediated copper transfer is impaired in the MBD2 mutant. A, diagram of the N-ATP7B mutants used in this work. B, copper transfer from Atox1 to N-ATP7B. Points represent the amount of copper that remains bound to N-ATP7B after incubation with the indicated amounts of Cu-Atox1 and removal of Cu-Atox1 from the mixture. Error bars indicate S.D. across three independent experiments. wt, wild-type N-ATP7B.
To test whether the decreased copper-binding stoichiometry of the m2A mutant was due to structural changes, we performed limited proteolysis on wild-type and m2A N-ATP7B and compared proteolytic patterns by separating fragments on an SDS-polyacrylamide gel (Fig. 3). At the concentration used for limited proteolysis, trypsin cuts N-ATP7B at a limited number of specific sites in the loops connecting the MBDs, whereas individual 7–8-kDa MBDs and the 16-kDa MBD5/6 pair remain intact (see Fig. 1A) (3, 17). The fragmentation patterns of wild-type and mutant N-ATP7B were markedly different (Fig. 3), demonstrating that the m2A mutation caused a conformational change in N-ATP7B. The mobility of several bands was increased (i.e. the fragments became shorter), suggesting that additional trypsin recognition sites in the loops were exposed. We investigated the kinetics of the proteolysis and found that the difference in pattern remained stable for several hours before patterns converged (supplemental Fig. 1). Thus, the change in the MBS led to a new configuration of the loops connecting MBDs (see more under “Discussion”).
FIGURE 3.
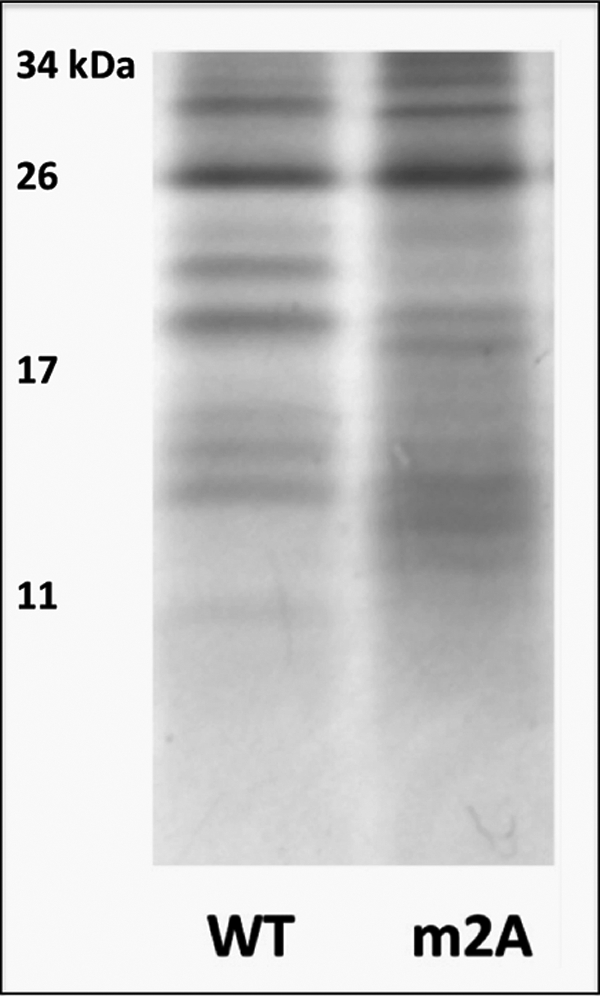
Mutation m2A induces structural changes in N-ATP7B. The Coomassie Blue-stained gel compares limited proteolytic patterns of wild-type (WT) and m2A N-ATP7B.
If the structural changes in the m2A mutant preclude appropriate exposure of the remaining MBDs to Atox1, then incubating m2A with free copper (rather than with a relatively bulky Atox1) is likely to yield higher copper-binding stoichiometry. To test this prediction, wild-type N-ATP7B and the m2A mutant were loaded with copper in cells by growing E. coli in medium supplemented with CuCl2. Although E. coli lacks Atox1 orthologs, wild-type N-ATP7B can be loaded with ∼5.5 copper atoms/protein by metal taken up by cells (6). Contrary to our expectations, the m2A mutant still showed significantly reduced copper binding (2.6 ± 0.3 copper atoms/protein compared with the control at 5.3 ± 0.1 copper atoms/protein) (Fig. 4). This result suggested that the m2A mutation impeded more than one MBD from binding (or retaining) copper and that the copper-binding status of the N-terminal MBDs depends on a functional MBD2.
FIGURE 4.

Mutations in MBDs have distinct effects on the copper-binding stoichiometry of N-ATP7B mutants. Wild-type (wt) and mutant N-ATP7B were loaded with copper in E. coli prior to purification. Light gray bars indicate the number of CxxC motifs left intact. Dark gray bars represent experimental values as determined by atomic absorption (copper) and Lowry assay (protein). Error bars indicate S.D. across three experimental repeats.
Mutations of Different MBDs Have Distinct Effects on Copper Binding by N-ATP7B
To determine whether the CxxC-to-AxxA mutations in other MBDs would have a similar effect on copper binding by N-ATP7B, we generated a series of mutants (Fig. 2A). The mutant proteins were expressed in E. coli, loaded with copper in cells, and purified, and their copper-binding stoichiometry was measured (Fig. 4). Mutating MBD3 (m3A) decreased copper binding (2.2 ± 0.4 copper atoms/molecule) to a level similar to the m2A mutation. In contrast, mutants of MBD4 (m4A) and MBD6 (m6A) bound copper with stoichiometry close to what was expected for domains containing five intact MBSs (4.6 ± 0.2 and 4.9 ± 0.1, respectively). Interestingly, when both MBD2 and MBD3 were mutated (in the m2/3A variant), the remaining sites bound copper with a stoichiometry close to the expected 4 copper atoms (3.6 ± 0.3) as if a negative effect of each individual mutation was counteracted. Altogether, these data suggest that MBD2 and MBD3 may work together to regulate access to other MBSs, whereas MBD4 and MBD6 receive copper independently, following transfer of copper to MBD2 and MBD3.
Decrease in Copper-binding Stoichiometry Is Due to Cys Oxidation
To better understand how mutation in MBD2 decreases the number of available copper-binding sites, we first examined whether the mutation altered the arrangement of the remaining intact sites, possibly causing more than one MBD to coordinate the same copper molecule. In wild-type N-ATP7B, each copper is coordinated by two sulfur groups of the CxxC motif in a linear fashion with bond lengths of 2.2 Å (24). EXAFS characterization of copper bound to m2A showed no significant deviation from this wild-type coordination (supplemental Fig. 2). Similar copper coordination was also observed for the m2/3A mutant. These results suggested that the decrease in copper-binding stoichiometry was unlikely due to changes in the copper-coordinating environment but rather was caused by the loss of copper-binding sites.
To estimate the number of available sites, Cys residues in both wild-type and mutant apo-N-ATP7B were labeled with the Cys-directed fluorescent maleimide CPM (Fig. 5A). The m2A mutant showed a marked decrease in CPM labeling (∼33% compared with wild-type N-ATP7B). To further test for specificity of the m2A mutation with respect to Cys reactivity, we performed CPM labeling of other mutants described in Fig. 2A. The only mutant besides m2A to have a significant reduction in CPM labeling was m3A (Fig. 5B), which was also the only mutant to have a sharp decrease in copper binding. To ensure that this observation was not the artifact of the purification procedure, we labeled crude E. coli lysates with CPM prior to protein purification (supplemental Fig. 3A). Lower fluorescence of m2A was again detected (supplemental Fig. 3A), demonstrating that the loss of available cysteines happened during the expression of the protein, consistent with the loss of in-cell copper binding by these mutants (Fig. 4).
FIGURE 5.

Comparison of Cys-directed fluorescent labeling for wild-type and mutant N-ATP7B. A, fluorescent (upper) and Coomassie Blue-stained gel (lower) images of wild-type (WT) and m2A N-ATP7B labeled with CPM. The tables indicate intensity of fluorescent labeling per protein compared with wild-type apo-N-ATP7B. The indicated samples were treated with TCEP prior to labeling. The lane on the right shows CPM labeling of copper-loaded wild-type N-ATP7B used as a background control. N/A, not applicable. B, fluorescent (upper) and Coomassie Blue-stained gel (lower) images of additional mutants shown in Fig. 2A, with label/protein ratios indicated in the table. Average percentages and S.D. are indicated for three experimental repeats.
To examine whether the decrease in CPM labeling was due to blocked access to the sites or oxidation of Cys residues, we repeated labeling experiments after treatment of N-ATP7B with the disulfide-reducing agent TCEP (Fig. 5A). With TCEP treatment, the m2A mutant regained about half of the lost fluorescence (to 67% of the wild-type protein). (The wild-type protein also gained ∼10% CPM labeling after TCEP treatment, which we ascribe to the recovery from normal oxidation during protein purification.) The lower free thiol reactivity of the m2A mutant was further confirmed using Ellman's reaction (supplemental Fig. 3B). Altogether, these data suggest that the m2A and m3A mutations render N-ATP7B susceptible to oxidation.
Tight Packing of MBDs Allows for Cross-communication
The marked effect of the m2A and m3A mutations on other MBDs suggested that the MBDs closely interact with each other despite being connected by long loops. To examine how tightly the MBDs interact, we subjected wild-type apo-N-ATP7B to limited proteolysis and separated the resulting fragments on a native Tris/glycine gel. Fig. 6A illustrates that, under nondenaturing conditions, most of the fragments remained associated and migrated together. The 8–12-kDa bands that did dissociate were further trypsinized in-gel, extracted, and analyzed by mass spectroscopy. They were identified as containing peptides from MBD1 and MBD2 (supplemental Fig. 4). The band containing associated fragments was excised and separated on a regular Laemmli gel. The reducing and denaturing conditions restored the banding pattern (Fig. 6B). The 8–12-kDa bands were also detected in the second dimension, indicating that these fragments interact with the rest of the bundle, but perhaps less tightly.
FIGURE 6.

MBDs in N-ATP7B are tightly packed. A, wild-type N-ATP7B was subjected to limited proteolysis as described in the legend to Fig. 3 and separated on a native Tris/glycine gel. Under these conditions, most of the fragments stayed together. The gel was stained with Coomassie Blue. B, the gel lane containing fragments run on a native gel was then cut and placed on top of a Laemmli gel and separated in the second dimension under reducing and denaturing conditions. The fragments no longer migrated together (silver staining).
The interdependence of MBDs in N-ATP7B was further confirmed by performing limited proteolysis of other mutants described in Fig. 2A. Distinct proteolytic patterns for these mutants (supplemental Fig. 5) indicated that MBD packing controls the exposure/orientation of connecting loops in a very precise manner.
Flexibility of the Copper-binding Loop Allows for Changing Exposure of Sulfhydryl Groups
Having established interaction between MBDs, we investigated the mechanism of interdomain communication. The copper-binding CxxC site is situated at the surface of MBD2 and must be sufficiently exposed for Atox1 to dock and transfer copper (Fig. 1B). Therefore, it was not apparent how mutating Cys residues to Ala in the exposed loop would affect the conformation of the entire N-ATP7B. We considered that the sulfhydryl group of Cys, unlike the side chain of Ala, is capable of forming a hydrogen bond. Others have demonstrated differential dynamics and flexibility of the metal-binding loops of various MBDs using molecular dynamics simulations (25). We hypothesized that, in the apo form, the metal-binding loop of MBD2 may be flexible enough to reach and interact with other MBDs and that the ability of sulfhydryl groups to hydrogen bond would be important for the interdomain contacts.
To test this hypothesis, we first examined the flexibility of MBD2 using molecular dynamics simulations. The structure of MBD2 of ATP7B is not available; rather than using a model, we characterized the dynamics of MBD2 of ATP7A, for which structures have been solved (13) and which has 60% identity and 77% similarity to MBS2 of ATP7B. supplemental Fig. 8 compares the structure of MBD2 (Protein Data Bank code 1s6o) with the TIP3P explicit solvent-equilibrated structure used for the molecular dynamics simulation. The results show that the initial structure and the equilibrated structure used for this simulation display a r.m.s.d. of <1.0 Å. Following a 10,000-ps simulation of dynamic movement in solution, we calculated r.m.s.d. values for the backbone carbons, as well as distances between the α-carbons and sulfurs of the metal-binding Cys residues (Fig. 7A). Overall, the structure was quite stable, with a maximum backbone r.m.s.d. of 2.12 Å. The distance between the α-carbons of the Cys residues showed higher flexibility (between 5.0 and 8.4 Å) but overall remained fairly constant. The R-groups of the Cys residues have larger rotational freedom (particularly the N-terminal Cys), as evidenced by S–S distances that ranged from 3.2 to 12.5 Å. This flexibility allowed the CxxC-containing loop to adopt conformations in which the metal-coordinating sulfurs faced either toward or away from each other (Fig. 7B). In the former conformation, the cysteines are well positioned to bind copper. In the latter conformation, the N-terminal Cys is angled away from the rest of the MBD and could potentially be involved in inter-MBD interactions.
FIGURE 7.

Molecular dynamics simulation of MBD2 of ATP7A illustrates the mobility and exposure of cysteines. A, relevant distances calculated for each frame of a 10,000-ps simulation. Black dots (RMSD) indicate overall backbone r.m.s.d. for the protein. Green dots (Cα–Cα) indicate the distance between α-carbons of metal-binding cysteines. Blue dots (S–S) indicate the distance between sulfur atoms of metal-binding cysteines. B, different orientation of the metal-binding loop from molecular dynamics simulation. Upper, loop with metal-binding cysteines facing away from each other; lower, loop with metal-binding cysteines facing toward each other.
Hydrogen Bonding May Play a Role in the N-ATP7B Folding and Redox State
To examine the role of hydrogen bonding, we generated an N-ATP7B mutant with the CxxC motif of MBD2 mutated to SxxS (m2S). We hypothesized that this mutant would lose copper binding at MBD2 but would retain the hydrogen-bonding characteristics of the wild-type protein; thus, the reduced state of other MBDs and their ability to bind copper would be preserved. Indeed, in contrast to the m2A mutant, the m2S mutant bound close to the expected 5 copper atoms (4.7 ± 0.3) (Fig. 8A). Similarly to m4A and m6A, which bound close to the expected amount of copper, the m2S mutant did not show any significant decrease in CPM labeling (Fig. 5B). In addition, when subjected to limited proteolysis, the m2S mutant displayed a fragmentation pattern that was more similar to the wild-type than to the m2A pattern (Fig. 8B).
FIGURE 8.

Mutations m2A and m2S have different effects on the conformation and oxidation state of N-ATP7B. A, copper-binding stoichiometries of wild-type N-ATP7B (wt) and the m2A and m2S mutants. B, Coomassie Blue-stained gel comparing limited proteolysis of wild-type and m2S N-ATP7B. C, fluorescence emission spectra of apo-N-ATP7B when excited with 280 nm UV light. Representative curves are shown for wild-type (blue), m2A (red), and m2S (green) N-ATP7B, with average peak emissions (342 nm) for three experiments shown in the inset. D, proteolytic patterns of wild-type apo-N-ATP7B and N-ATP7B with one or two Cu+ atoms transferred from Atox1.
These results strongly suggest that the hydrogen-bonding ability of the Cys residues is critical for the overall folding of N-ATP7B. To further test this conclusion, we compared intrinsic Trp fluorescence in wild-type N-ATP7B and the m2 mutants (Fig. 8C). N-ATP7B contains 2 Trp residues: at the very N terminus before MBD1 (residue 29) and in the loop between MBD1 and MBD2 (Fig. 1A). Compared with apo-N-ATP7B, the m2A mutant had a higher fluorescence (110 ± 4% of the wild-type protein), suggesting a greater exposure of Trp to solution. Unlike m2A, the m2S mutant did not show an increase in fluorescence (96 ± 2% compared with the wild-type protein), a result consistent with its behavior in other tests. Similar results were obtained when fluorescence was measured using an excitation wavelength of 295 nm (data not shown).
Finally, if the metal-binding Cys residues in MBD2 play a role in interdomain communication, then the transfer of copper from Atox1 to N-ATP7B is expected to have an effect on N-ATP7B conformation. Fig. 8D illustrates that the proteolytic pattern of ATP7B indeed changed significantly with each copper transferred. The changes were not identical to those observed in m2A (perhaps reflecting different functional consequences of copper binding and mutation), although the downward shift of bands resembling the m2A pattern was detected after 2 copper atoms were bound (Fig. 8D).
Ab Initio Modeling of MBD1 and MBD2 Suggests Potential Orientations for These Domains
To visualize how MBD2 might interact with neighboring MBDs, we performed ab initio folding simulations on the Val57–Lys213 sequence, corresponding to MBD1-loop-MBD2. As a control of accuracy of our predictions, we subjected sequence Gly169–Gln241 of ATP7A (corresponding to MBD2) to the same folding prediction protocol and compared the obtained model with the available structure. supplemental Fig. 6 illustrates that the most common solution cluster for MBD2 is similar to the published structure (r.m.s.d. of 1.928). For MBD1-loop-MBD2, the minimized centers of the three largest solution clusters are shown in Fig. 9. In each cluster, MBDs are folded into the expected ferridoxin-like structures. The inter-MBD loop shows a tendency to form an α-helix, giving the two-domain construct a clamshell-like structure. In this structure, the MBDs and their respective MBSs can be brought together when the loop takes on a more structured conformation, which in turns alters the exposure of proteolytic sites located within the loop.
FIGURE 9.

Structures of pairs of neighboring MBDs. A, structure of MBD5 and MBD6 of ATP7B (adapted from Achila et al. (8); Protein Data Bank code 2ew9). B–D, ab initio structures for MBD1 and MBD2. MBD1 is shown in green, the inter-MBD loop in cyan, and MBD2 in blue. The GMxCxxC loop is shown in red, and copper-binding Cys residues are shown in yellow.
DISCUSSION
We have demonstrated that the regulatory N-terminal domain of ATP7B is arranged with the MBDs in close contact and communication with each other. The tight packing of MBDs fixes the connecting loops in distinct conformations and allows for small structural changes in individual MBDs to be transmitted to other MBDs and modify the exposure of the connecting loops. The interdomain communication appears to require hydrogen bonding, which also contributes to the maintenance of the redox status of metal-binding Cys residues. MBD2, the site of primary copper transfer from Atox1 (when the fully folded N-ATP7B is used), along with MBD3 plays a central role in maintaining the conformation and reduced state of the entire N-terminal domain.
NMR and crystal structures of individual MBDs show only minor differences between the copper-bound and apo forms (8–15). However, the conformation and secondary structure of apo-N-ATP7B and copper-loaded N-ATP7B differ significantly (3, 7). Our data show that, even in the apo form, MBDs are packed tightly, albeit not rigidly, together. Copper binding to the CxxC site, which causes the inward movement of the loop, making it rigid and less exposed (13), appears to alter the contacts between MBDs (as do the Cys-to-Ala mutations) and induces changes in the structure of loops connecting MBDs. The change in loop structures/orientations is likely to be an important event in regulating ATP7B because it may expose sites for kinase-mediated phosphorylation located within the loops (3, 26).
Conformational changes in the loops may also provide docking sites for trafficking machinery (5) while burying residues that interact with the ATP-binding domain and disrupting interdomain interactions (27). It is interesting that the conformational effects of mutating different MBDs (or sequential copper binding) are not identical as evidenced by distinct and stable proteolytic patterns. This diversity of consequences may be necessary for a fined-tuned response to various concentrations of copper and may explain the poorly understood need for multiple copper-binding sites in N-ATP7B.
The effect of m2A and m3A mutations on the oxidation state and copper-binding ability of other MBDs was unexpected. The drastic difference between m2A and m2S was particularly striking. These results suggest that hydrogen bonding may be required for both the precise packing and maintenance of the reduced state of N-ATP7B. The flexibility of the metal-binding loop in MBD2 could be critical for allowing communications between MBDs (and between MBD and Atox1) (see Ref. 25). It is interesting to consider that the stabilization of a Cys residue in a more extended conformation through hydrogen bonding is likely to decrease the probability of forming disulfide bonds and thus preserve the reduced state of the MBDs. This view is supported by our observation that, in wild-type N-ATP7B, MBD2 is reduced and available for Cys labeling, whereas following proteolysis (when precise contacts are disrupted), MBD2 rapidly oxidizes and cannot be labeled with CPM (supplemental Fig. 7). Our data also suggest that some Cys residues could be oxidized irreversibly (supplemental Fig. 3B), possibly to sulfinic or sulfonic acid, as has been observed for other Cys-containing metalloproteins (29). Although this may be a by-product of our expression system, these in vitro data raise an interesting question as to how the reduced state of N-ATP7B is maintained in a cell. A previous report on a copper-dependent interaction between N-ATP7B and glutaredoxin suggests one possible reduction mechanism (30).
It is also intriguing that the m2A and m3A mutants have a similar increase in oxidation and loss of copper binding, whereas mutating both MBDs in tandem relieves this effect (Figs. 4 and 5B). This observation suggests that, although both MBDs play an important role in regulating the status of other MBDs, they each do not have individual downstream partners. The proteolytic pattern of the m2/3A mutant is distinct from that of either m2A or m3A (supplemental Fig. 5B). It is possible that this distinct conformation favors the “proper” orientation of the remaining MBDs, although we did not explore this further in this study. It is clear that not every change in conformation is associated with increased susceptibility to oxidation. The m6A mutant had a similar proteolysis pattern to the m2A mutant (supplemental Fig. 5A), even though it bound copper and labeled with CPM at predicted levels. This suggests that the conformational changes in loops and redox sensitivity of MBSs are related but not dependent on one another and further demonstrates the unique role MBD2 plays in regulating access to other MBSs in N-ATP7B.
We attribute the changes in the proteolytic pattern of the m2A mutant to be predominantly due to the dissociation of MBD3 and MBD1 from MBD2 and exposure of additional trypsin sites that are inaccessible when the domains are packed tightly together (Fig. 1A). This is supported by the increase in tryptophan fluorescence in the mutant, which we think is likely due to the exposure of the Trp residue in the loop connecting MBD1 and MBD2. We also observed some dissociation of MBD1 and MBD2 when the digested domain was separated on a native gel (Fig. 6A). Our ab initio structures (Fig. 9) provide a clue to the potential interaction between MBD1 and MBD2. The loop connecting the two domains seems to have some inherent α-helical tendencies, which could bring the two MBDs close together, but can also unfold and increase the distance between the MBDs. Molecular dynamics simulations of apo- and Cu-Atox1, which is a structural homolog of MBDs, suggest that copper binding may increase the flexibility of the portion of the MBD opposite from the metal-binding loop, thus influencing pairing of other MBDs (28). This change could be transmitted through the loop region, imparting more structure to the loop in the copper-bound state and bringing the MBDs closer together. Previous EXAFS analysis of copper-loaded N-ATP7B consistently showed a Cu–Cu distance of 2.6 Å (24), indicative of close proximity between pairs of MBSs.
In summary, we have presented a working model in which close packing of N-ATP7B allows cross-talk among MBSs. This interdomain communication is necessary for maintenance of reduced cysteines and proper copper acquisition. The MBD interactions may involve a hydrogen-bonding network that allows copper binding to be translated into larger structural rearrangements via the loops between the MBDs. These rearrangements are likely to play a major role in regulating ATPase activity as well as mediating interactions with cellular trafficking machinery. Maintaining the precise interdomain contacts could also be essential for success of high resolution structural studies of N-ATP7B, which, so far, have represented a significant challenge.
Supplementary Material
Acknowledgments
We thank the members of the Lutsenko and Hubbard laboratories for critical reading of this manuscript and helpful comments.
This work was supported by National Institutes of Health Grant R01 DK071865 (to S. L.) and Training Grant 5-T32-HL007781 (to E. S. L.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–8.
- N-ATP7B
- N-terminal domain of ATP7B
- MBD
- metal-binding domain
- MBS
- metal-binding site
- MBP
- maltose-binding protein
- TCEP
- tris(carboxyethyl)phosphine
- EXAFS
- extended x-ray absorption fine structure
- CPM
- 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1.Bertini I., Rosato A. (2008) Cell. Mol. Life Sci. 65, 89–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lutsenko S., LeShane E. S., Shinde U. (2007) Arch. Biochem. Biophys. 463, 134–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartee M. Y., Ralle M., Lutsenko S. (2009) Biochemistry 48, 5573–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo Y., Nyasae L., Braiterman L. T., Hubbard A. L. (2005) Am. J. Physiol. Gastrointest. Liver Physiol. 289, G904–G916 [DOI] [PubMed] [Google Scholar]
- 5.Lim C. M., Cater M. A., Mercer J. F., La Fontaine S. (2006) J. Biol. Chem. 281, 14006–14014 [DOI] [PubMed] [Google Scholar]
- 6.Lutsenko S., Petrukhin K., Cooper M. J., Gilliam C. T., Kaplan J. H. (1997) J. Biol. Chem. 272, 18939–18944 [DOI] [PubMed] [Google Scholar]
- 7.DiDonato M., Hsu H. F., Narindrasorasak S., Que L., Jr., Sarkar B. (2000) Biochemistry 39, 1890–1896 [DOI] [PubMed] [Google Scholar]
- 8.Achila D., Banci L., Bertini I., Bunce J., Ciofi-Baffoni S., Huffman D. L. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 5729–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banci L., Bertini I., Cantini F., Rosenzweig A. C., Yatsunyk L. A. (2008) Biochemistry 47, 7423–7429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banci L., Bertini I., Cantini F., DellaMalva N., Herrmann T., Rosato A., Wüthrich K. (2006) J. Biol. Chem. 281, 29141–29147 [DOI] [PubMed] [Google Scholar]
- 11.Banci L., Bertini I., Cantini F., Migliardi M., Rosato A., Wang S. (2005) J. Mol. Biol. 352, 409–417 [DOI] [PubMed] [Google Scholar]
- 12.Banci L., Bertini I., Ciofi-Baffoni S., Chasapis C. T., Hadjiliadis N., Rosato A. (2005) FEBS J. 272, 865–871 [DOI] [PubMed] [Google Scholar]
- 13.Banci L., Bertini I., Del Conte R., D'Onofrio M., Rosato A. (2004) Biochemistry 43, 3396–3403 [DOI] [PubMed] [Google Scholar]
- 14.DeSilva T. M., Veglia G., Opella S. J. (2005) Proteins 61, 1038–1049 [DOI] [PubMed] [Google Scholar]
- 15.Gitschier J., Moffat B., Reilly D., Wood W. I., Fairbrother W. J. (1998) Nat. Struct. Biol. 5, 47–54 [DOI] [PubMed] [Google Scholar]
- 16.Yatsunyk L. A., Rosenzweig A. C. (2007) J. Biol. Chem. 282, 8622–8631 [DOI] [PubMed] [Google Scholar]
- 17.Walker J. M., Huster D., Ralle M., Morgan C. T., Blackburn N. J., Lutsenko S. (2004) J. Biol. Chem. 279, 15376–15384 [DOI] [PubMed] [Google Scholar]
- 18.Walker J. M., Tsivkovskii R., Lutsenko S. (2002) J. Biol. Chem. 277, 27953–27959 [DOI] [PubMed] [Google Scholar]
- 19.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 20.Barry A. N., Blackburn N. J. (2008) Biochemistry 47, 4916–4928 [DOI] [PubMed] [Google Scholar]
- 21.Ellman G. L. (1958) Arch. Biochem. Biophys. 74, 443–450 [DOI] [PubMed] [Google Scholar]
- 22.Shevchenko A., Wilm M., Vorm O., Mann M. (1996) Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 23.Subbian E., Yabuta Y., Shinde U. (2004) Biochemistry 43, 14348–14360 [DOI] [PubMed] [Google Scholar]
- 24.Ralle M., Lutsenko S., Blackburn N. J. (2004) J. Inorg. Biochem. 98, 765–774 [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez-Granillo A., Crespo A., Wittung-Stafshede P. (2009) Biochemistry 48, 5849–5863 [DOI] [PubMed] [Google Scholar]
- 26.Pilankatta R., Lewis D., Adams C. M., Inesi G. (2009) J. Biol. Chem. 284, 21307–21316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsivkovskii R., MacArthur B. C., Lutsenko S. (2001) J. Biol. Chem. 276, 2234–2242 [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Granillo A., Wittung-Stafshede P. (2008) J. Phys. Chem. B 112, 4583–4593 [DOI] [PubMed] [Google Scholar]
- 29.Fujiwara N., Nakano M., Kato S., Yoshihara D., Ookawara T., Eguchi H., Taniguchi N., Suzuki K. (2007) J. Biol. Chem. 282, 35933–35944 [DOI] [PubMed] [Google Scholar]
- 30.Lim C. M., Cater M. A., Mercer J. F., La Fontaine S. (2006) Biochem. Biophys. Res. Commun. 348, 428–436 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.