

Abstract
The c-Fos proto-oncogenic transcription factor defines a multigene family controlling many processes both at the cell and the whole organism level. To bind to its target AP-1/12-O-tetradecanoylphorbol-13-acetate-responsive element or cAMP-responsive element DNA sequences in gene promoters and exert its transcriptional part, c-Fos must heterodimerize with other bZip proteins, its best studied partners being the Jun proteins (c-Jun, JunB, and JunD). c-Fos expression is regulated at many transcriptional and post-transcriptional levels, yet little is known on how its localization is dynamically regulated in the cell. Here we have investigated its intranuclear mobility using fluorescence recovery after photobleaching, genetic, and biochemical approaches. Whereas monomeric c-Fos is highly mobile and distributed evenly with nucleolar exclusion in the nucleus, heterodimerization with c-Jun entails intranuclear redistribution and dramatic reduction in mobility of c-Fos caused by predominant association with the nuclear matrix independently of any binding to AP-1/12-O-tetradecanoylphorbol-13-acetate-responsive element or cAMP-responsive element sequences. In contrast to c-Jun, dimerization with JunB does not detectably affect c-Fos mobility. However, dimerization with JunB affects intranuclear distribution with significant differences in the localization of c-Fos·c-Jun and c-Fos·JunB dimers. Moreover, c-Jun and JunB exert comparable effects on another Fos family member, Fra-1. Thus, we report a novel regulation, i.e. differentially regulated intranuclear mobility and distribution of Fos proteins by their Jun partners, and suggest the existence of intranuclear storage sites for latent c-Fos·c-Jun AP-1 complexes. This may affect the numerous physiopathological functions these transcription factors control.
Keywords: Nucleus, Oncogene, Transcription/AP-1, DNA-binding protein, Intracellular Trafficking, c-Fos, c-Jun, Protein Mobility
Introduction
The AP-1 transcriptional complex comprises a large family of dimeric transcription factors involved in the control of numerous physiological and pathological processes. These include among others cell proliferation, differentiation, apoptosis, responses to environmental cues, tumorigenesis, developmental defects, and immune diseases (1–5). Its best studied components are the Fos (c-Fos, Fra-1, Fra-2, and FosB) and Jun (c-Jun, JunB, and JunD) family proteins, all of which necessitate dimerization via a leucine zipper (LZ)5 to acquire transcriptional competence. Fos proteins can only heterodimerize with other AP-1 components, whereas Jun proteins can also homodimerize, even though heterodimerization with any of the Fos is favored (6, 7). Thanks to both the LZ and the adjacent basic DNA-binding domains (DBD), Fos·Jun AP-1 dimers bind defined DNA sequences known as 12-O-tetradecanoylphorbol-13-acetate-responsive elements (TREs) and less well to cAMP-responsive elements (CREs) that are found in many gene promoters, explaining the diversity of AP-1 effects. Importantly, AP-1 can act as a positive or a negative transcriptional regulator depending on its composition, the target gene, the cell context, the extracellular environment, and which intracellular signaling cascades are activated (2, 6).
c-Fos is the first discovered and best studied member of the Fos family (8, 9). It is constitutively expressed in a limited number of tissues but is rapidly and transiently induced in many other cell types by a large variety of stimuli (9). In the latter case, c-Fos accumulation is controlled at the level of transcription, mRNA turnover, and protein stability (8, 10, 11). Moreover, c-Fos intracellular localization (see below) and transcriptional activity are tightly regulated (8). In particular, transcriptional activity can be enhanced via phosphorylation of various serines and threonines that may also participate in protein stabilization. The kinases include the MAPK p38, ERK1/2, and ERK5 (12–25), where the role of ERK5 is disputed (26), as well as the ERK1/2-activated kinases Rsk1/2 (12, 14, 16, 22) and IκB kinase (27). In contrast, c-Fos can be transcriptionally repressed by sumoylation at a specific lysine (28, 29). Interestingly, sumoylation of this lysine is antagonistic to a nearby transcription-activating phosphorylation (28).
Usually, c-Fos accumulates predominantly, if not exclusively, within the nucleus. However, it can also localize within the cytoplasm under certain conditions (17, 24, 25, 30, 31). Cytoplasmic c-Fos can even associate with the endoplasmic reticulum to activate phospholipid metabolism in a transcription activity-independent manner (32, 33). c-Fos intracellular localization is regulated by both intra- and extracellular signals with demonstrated roles for cAMP-dependent protein kinase A (34), the p38 MAPK (25), and the STAT3 transcription factor when ERK5 is inactivated (24). Consistent with this dual intracellular localization, we and others (24, 35, 36) have shown that c-Fos can shuttle between the nucleus and the cytoplasm. Entry into the nucleus is controlled by at least two nuclear localization signals: a conventional basic nuclear localization signal (35–37) that most likely utilizes the nuclear import receptor Impβ1 (35, 36) and an unconventional nuclear localization signal located in the N-terminal moiety of the protein that requires the nuclear importin Transportin 1 (35, 36). Depending on the conditions, both Crm-1 exportin-dependent (24) and -independent (36) mechanisms involving different nuclear export signals are responsible for c-Fos nuclear exit. Interestingly, this involves primarily monomeric c-Fos and is inhibited upon heterodimerization with the Jun proteins (36). This indicated that dimerization is important, not only for the formation of active AP-1 transcription complexes but also to keep them in the nucleus where they play their transcriptional part. c-Fos nuclear retention, however, varies according to its Jun partner. It is much stronger with c-Jun than with JunB or JunD. This correlates with the strength of interaction of the various c-Fos·Jun dimers (38). Here, as a first step to understand why c-Fos nucleo-cytoplasmic shuttling is differentially affected by the Juns, we used fluorescence recovery after photobleaching (FRAP) together with genetic and biochemical approaches to study the intranuclear mobility of different Fos·Jun dimers.
MATERIALS AND METHODS
Plasmids, Cloning, and Mutagenesis
Cloning and mutagenesis were performed using standard PCR-based methods. All of the constructs were entirely sequenced. Plasmids and related information are available on request. pcDNA3-based expression plasmids for wild type and mutant rat c-Fos and mouse JunB-FLAG and c-Jun-FLAG were previously described (16, 36). EGFP chimeras were obtained by insertion of either rat c-Fos or human Fra1 coding sequences into the pEGFP-C1 vector (Clontech).
Cell Culture and Transfection
Conditions for culturing and transfecting human HeLa cells by the calcium phosphate co-precipitation technique were previously described (36). 3 μg of plasmid/106 cells was routinely used, and the transfection time was limited to 16 h to avoid protein overexpression.
Immunofluorescence and Confocal Microscopy
To set up the conditions of FRAP experiments on cells expressing levels of EGFP-c-Fos or EGFP similar to that of endogenous c-Fos, we compared the expression level of transfected EGFP-c-Fos with that of endogenous c-Fos expressed upon 1 h of serum stimulation. To this aim, HeLa cells were either transfected for 16 h with the EGFP- or EGFP-c-Fos-encoding vector or stimulated with 20% serum for 1 h. The cells were fixed in 4% paraformaldehyde and permeabilized in the presence of 0.2% Triton X-100. They were then successively incubated with the H125 rabbit anti-c-Fos antibody (Santa Cruz Biotechnology), detecting equally c-Fos and EGFP-c-Fos and an Alexa 647-labeled anti-rabbit antibody (Molecular Probe). The nuclei were stained with Hoechst 33342, and coverslips were mounted in Permafluor. Cells expressing EGFP-c-Fos and emitting a fluorescence signal in the far red channel similar to that of cells expressing endogenous c-Fos were selected to set up the signal intensity range in the green channel. This allowed us to select under the microscope cells expressing EGFP or EGFP-c-Fos at a level comparable with that of endogenous c-Fos in the green channel. The far red fluorescence (Alexa 647) was monitored using a 560-nm long wavelength path filter, whereas EGFP fluorescence was monitored using a 505–550-nm wavelength band pass filter. Twelve-bit image acquisition was performed using a LSM510 meta microscope (Zeiss) equipped with a plan Apochromat 40× water immersion lens (1.2 numeric aperture) and with a confocal plane of 5 μm.
FRAP Experiments
FRAP analyses were performed at 37 °C using a Zeiss LSM 510 Meta microscope equipped with a heating chamber and a plan Apochromat 40× water immersion lens (1.2 numeric aperture). To monitor EGFP fluorescence, the cells were excited with an argon laser at a wavelength of 488 nm, and emission was collected using a 505–550-nm wavelength band pass filter. The experiment was divided in three sequences: (i) a prebleaching period, during which 15 images were acquired to define the initial level of fluorescence; (ii) photobleaching, which was carried out on a 2-μm radius circular area of the targeted nucleus using the 488-nm wavelength laser at maximal power with 100 iterations of 256 μs/pixel; and (iii) a postbleaching period, during which fluorescence recovery was monitored every 150 ms for 30 s. Fluorescence recovery was then extracted on recorded images from the bleach area and corrected for experimental fluctuations during acquisition. Correction was carried out by dividing the value of the fluorescence in the bleach area by that of another region in the nucleus far away from the bleached volume. Finally, fluorescence intensities in the bleach area were normalized against the prebleach level of fluorescence intensity. Data fitting was performed using the Levenberg-Marquadt algorithm to determine both the mean half-time of fluorescence recovery (<t½>) and the mobile fraction of the protein (Fmob) using the model in Equation 1,
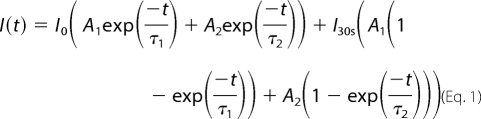 |
where I0 is the fluorescence intensity at t = 0 (immediately after bleaching) and I∞ is the fluorescence intensity at 30 s (end of recording) and with <t½> = A1τ1 + A2τ2 and with Equation 2.
 |
The choice of this model was conditioned to a c2 statistical test showing the best fit for all of the data as compared with a monoexponential or a two-dimensional diffusion under a Gaussian illumination model. This model was used without any a priori knowledge of either the geometry of the bleaching or the process of fluorescence recovery. It was therefore used as a model to compare mean half-times of fluorescence recovery. Finally, I0 was determined by bleaching under the same experimental conditions as those described above for c-Fos-EGFP-expressing cells after their fixation with 4% paraformaldehyde for 30 min at room temperature. I0 was set to 0.4.
Cell Fractionation in the Presence of Triton X-100
Cell fractionation procedures are those described in reference (39). Approximately 107 cells were scrapped in PBS (150 mm NaCl, 10 mm sodium phosphate, pH 7) on ice, harvested by low speed centrifugation, resuspended in 200 μl of Buffer A (10 mm Hepes, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, 0.34 m sucrose, 10% glycerol, 1 mm dithiothreitol, 1 Complete Mini protease inhibitor mixture tablet (Roche Applied Science)/10 ml of buffer) and let on ice for 8 min. The cells were lysed in the presence of 0.15% Triton X-100 on ice for 4 min and subjected to centrifugation (3500 rpm for 5 min). The supernatant (S) contained both the cytoplasmic and the soluble nuclear fractions. The nuclei were then washed once in 0.15% Triton X-100-containing buffer A and recentrifuged at 3500 rpm for 5 min. After resuspension in 200 μl of Buffer B (3 mm EDTA, 0.2 mm EDTA, 1 mm dithiothreitol, 1 Complete Mini protease inhibitor mixture tablet/10 ml of buffer), the nuclei suspension was left on ice for 30 min for membrane disruption and then centrifuged at 4000 rpm for 5 min. The supernatant corresponded to the wash fraction (W). The pellet (P) was subjected to a round of washing in buffer B and centrifugation and finally resuspended in Laemmli electrophoresis loading buffer. The various fractions were then submitted to immunoblotting analyses as previously described (36).
Immunoprecipitation and Immunoblotting Experiments
Immunoprecipitations were performed as described in Ref. 28. 107 cells were lysed in 600 μl of radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.02% NaN3, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 Complete Mini protease inhibitor mixture tablet/10 ml of buffer). To immunoprecipitate FLAG-tagged proteins, 200 μl of lysates were incubated for 3 h in the presence of 30 μl of anti-FLAG M2 affinity gel from Sigma. After centrifugation of cell extract-containing suspensions, the supernatants were collected, whereas the pellets were subjected to five cycles of washing in radioimmune precipitation assay buffer and centrifugation. For immunoblotting analyses, total extracts, supernatants, and immunoprecipitated fractions were electrophoresed through 12% polyacrylamide gels containing SDS and electrotransferred onto polyvinylidene difluoride membranes. Immunodetections were carried out with appropriate dilutions of the various following primary antibodies: rabbit anti-c-Fos H125 (sc-7202), rabbit anti-c-Jun H79 (sc-1694), and goat anti-JunB (sc-46G) from Santa Cruz Biotechnology. We also used rabbit antisera directed to topoisomerase I (kind gift from Dr J. Soret). The anti-Phax mouse monoclonal and the anti-lamin rabbit polyclonal antibodies were kindly provided by Drs. D. Lener, and H. Wodrich, respectively. Secondary horseradish peroxidase-coupled antibodies were either from Santa Cruz Biotechnology (sc-2313 anti-rabbit and sc-2033 anti-goat horseradish peroxidase conjugates) or from Sigma (A-9044 anti-mouse horseradish peroxidase conjugate). Chemoluminescence was detected with the Chemoluminescence reagent Plus kit from PerkinElmer Life Sciences using Biomax XAR Kodak films.
Chromatin and Nuclear Matrix Fractionation: Biochemical and Microscope Analyses
For analysis of endogenous c-Fos, HeLa cells were grown for 4 days without any change of culture medium and stimulated for 1 h by the addition of 20% fresh serum. For analysis of exogenous c-Fos, HeLa cells were transfected with a vector for EGFP-c-Fos alone or in combination with vectors for either c-Jun or JunB in a 1:2 ratio. Our fractionation procedure combined the methods described in Refs. 40 and 41. For immunoblotting analyses, 4 × 106 cells/sample were rinsed with PBS and then scraped from plates in ice-cold CSK buffer (10 mm Tris-HCl, pH 7.4, 300 mm sucrose, 100 mm NaCl, 3 mm MgCl2, 1 mm EGTA). After centrifugation, the pellets were rinsed once with 1 ml of CSK buffer and recentrifuged, and cell lysis was allowed to proceed for 10 min at 0 °C (107 cells/ml) in CSK buffer containing 0.5% Triton X-100, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin A/ml, 25 μg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, and 400 units/ml RNasin. Half of each sample was mixed with Laemmli sample buffer to constitute the total cell extract (T). The nuclei of the other half were pelleted by centrifugation at 5000 rpm for 2 min at 4 °C. The supernatants (S1), which contained solubilized cytoplasmic and nuclear proteins, were collected. The nuclei were washed once with 200 μl of the above ice-cold lysis buffer and pelleted by centrifugation. Centrifugation supernatants corresponded to the wash fractions (W). The nuclei were then treated with microccocal nuclease (MNase; Roche Applied Science) to remove chromatin- and DNA-bound proteins. To this aim, they were resuspended (107 nuclei/ml) in ice-cold CSK containing 0.5% Triton X-100, protein, and RNase inhibitors as above, 10 mm CaCl2 and 200 units/ml of MNase. After a 10-min incubation at 30 °C, they were centrifuged, and supernatants (S2) were collected. After resuspension in CSK buffer containing 2 m NaCl and protease and RNase inhibitors as above, they were let for 5 min at 0 °C to remove the remaining DNA and histones. Pellets (P) containing nuclear matrix and associated proteins were collected by centrifugation. Supernatants (S3) contained nuclear proteins soluble in 2 m NaCl. Fluorescence microscope analyses of cells were conducted in parallel. To this aim, the coverslips were placed in culture dishes before cell seeding and processed separately at the time of biochemical cell/nucleus fractionation following the same steps as those described above. After rinsing in the presence of PBS, the cells were subsequently fixed in the presence of 4% paraformaldehyde (i) before lysis, (ii) after Triton X-100 cell lysis, (iii) after MNase treatment, and (iv) after 2 m NaCl treatment. When necessary (non-Triton X-100 lysed cells), an additional permeabilization treatment with 0.2% Triton X-100 in PBS for 5 min at room temperature was added before microscopic analysis. EGFP-c-Fos was followed up using direct fluorescence, whereas endogenous c-Fos required indirect fluorescence analysis using the sc-52 (Santa Cruz Biotechnology), anti-c-Fos antibody, and an Alexa 488-labeled anti-rabbit antibody (Molecular Probes). The nuclei were stained with Hoechst 33342 at a concentration of 0.2 μg/ml. The coverslips were mounted in Permafluor.
Fluorescence Analysis of Fixed Cells
Observations were performed using a Leica DMRA microscope equipped with a 63× oil lens and with an enlargement of 1.6×, leading to a magnification of 100×. 12-Bit image acquisition was performed using Metamorph software. All of the images were acquired using the same exposure time to compare cells with equivalent c-Fos levels.
RESULTS
Monomeric c-Fos Is Highly Mobile in the Nucleus
To study c-Fos intranuclear mobility, we used the technique of FRAP (42–44) on live human HeLa cells expressing an EGFP-c-Fos fusion protein. This chimera, in which c-Fos is fused to the C terminus of EGFP, has been shown to retain the following properties of wild type c-Fos: dimerization with all Jun family members, short half-life and proteasome-dependent degradation, quantitative nuclear accumulation, and nucleo-cytoplasmic shuttling inhibited by c-Jun (16, 36). We also verified that it transactivates an AP-1-luciferase reporter gene to a similar extend as wild type c-Fos (data not shown).
When expressed alone in transient transfection assays, c-Fos and EGFP-c-Fos are essentially monomeric (36) and predominantly nuclear despite their active nucleo-cytoplasmic shuttling (36). Moreover, they show a typical diffuse distribution with nucleolar exclusion, as assayed both by indirect immunofluorescence (36) or by direct visualization of EGFP-c-Fos in living cells (Fig. 1, A and B). We used FRAP (n > 20) to evaluate nuclear mobility of EGFP-c-Fos relative to EGFP, which diffuses freely within cells (45). Because it was important to exclude any bias possibly resulting from overexpression, cells treated with fluorescence recovery after photobleaching were carefully selected for expressing levels of EGFP-c-Fos or EGFP similar to that of a physiologically expressed endogenous c-Fos. To this aim, we took advantage that c-fos is an immediate early gene rapidly and transiently induced upon growth factor stimulation (8, 9) and compared exogenous EGFP-c-Fos and EGFP abundance in transfected HeLa cells with that of endogenous c-Fos in HeLa cells stimulated for 1 h by serum using standardized fluorescence acquisition procedures, as detailed under “Materials and Methods” and shown Fig. 1A. Typical FRAP experiments are presented in Fig. 1B. Mean half-time fluorescence recovery (<t½>), as well as the mobile fractions 30 s after the bleach (Fmob30), were calculated for EGFP and EGFP-c-Fos using a two-exponential fit, as described under “Materials and Methods.” Fmob30 were 99 ± 2 and 97 ± 4% for EGFP and EGFP-c-Fos, respectively, indicating that the molecules are highly mobile in this time frame (Fig. 1C). The <t½> were 0.27 ± 0.05 s for EGFP and 0.98 ± 0.12 s for EGFP-c-Fos. In fact, EFGP was so mobile that its fluorescence recovered partially before the first post-bleach experimental measurement. This gave the impression of a less efficient photobleaching and led to an overestimation of the <t½> (compare EGFP minimal fluorescence value to that of EGFP-c-Fos in Fig. 1C). This indicated that EGFP-c-Fos, despite its high mobility, moves at least 4-fold more slowly than EGFP in the nucleus. If the mobility of the proteins is due to Brownian diffusion, this difference in mean half-time fluorescence recovery cannot be due to the increased mass of the chimeric protein as compared with EGFP because the diffusion constant varies little with mass (Da1/M3, also see Ref. 45). Instead, the reduced mobility of EGFP-c-Fos would reflect weak binding to nuclear structures.
FIGURE 1.

Monomeric c-Fos is highly mobile in the nucleus. A, comparison of transfected EGFP-c-Fos and endogenous c-Fos expressions. HeLa cells were either stimulated for 1 h with 20% serum (left panel) or transfected for 16 h with the EGFP-c-Fos-encoding plasmid (right panel). After cell fixation and permeabilization, c-Fos and EGFP-c-Fos were stained with a rabbit anti-c-Fos antiserum followed by an Alexa 647-labeled anti-rabbit antiserum. EGFP-c-Fos-expressing cells emitting a far red signal close to that of cells expressing endogenous c-Fos were selected and observed in the green channel to set up the green fluorescence intensity range usable in FRAP experiments. B, FRAP experiment. Asynchronously growing HeLa cells were transfected with expression plasmids encoding EGFP-c-Fos or EGFP, and FRAP experiments were carried out as described under “Materials and Methods.” A typical experiment with EGFP-c-Fos is presented. Arrows indicate the bleached area, before, during, and after the bleach. C, FRAP data. Typical fluorescence recovery curves are presented for both EGFP- and EFGP-c-Fos-expressing cells. <t½> and Fmob30 calculated from 20 FRAP experiments are given. They include standard deviations. D, immunoblotting analysis of cell fractions. Cells transfected as in A were lysed in the presence of Triton X-100, and soluble (S), wash (W), and nonsoluble (NS) fractions were prepared and analyzed by immunoblotting as described under “Materials and Methods.” Phax and topoisomerase I (Topo 1) were used as internal controls of soluble and nonsoluble proteins, respectively.
To complement these FRAP studies, we performed fractionation experiments on transfected HeLa cells, comparing the distribution of EGFP-c-Fos with that of c-Fos in Triton X-100-soluble and -insoluble fractions. Monomeric EGFP-c-Fos and monomeric c-Fos were principally found in the soluble fraction (S) together with the nucleoplasmic protein Phax (46) taken as a control (Fig. 1D; also see Fig. 2, D and E). Sometimes, a minor proportion of monomeric EGFP-c-Fos or c-Fos remained in the insoluble fraction (NS), which was monitored using chromatin-bound topoisomerase I (47) (Fig. 1D; also see Fig. 2, D and E). Thus, the high mobility of monomeric c-Fos in the nucleus correlates with its lack of strong interaction with intranuclear structures.
FIGURE 2.

Reduced intranuclear mobility of c-Fos in the presence of c-Jun. A, alteration of intranuclear distribution of EGFP-c-Fos in the presence of c-Jun. HeLa cells were transfected with EGFP-c-Fos in the absence or in the presence of a 2-fold excess of c-Jun expression plasmid and analyzed by confocal microscopy on living cells. B, Fmob30 of EGFP-c-Fos in the presence of varying amounts of c-Jun. HeLa cells were co-transfected with c-Jun and EGFP-c-Fos expression plasmids in the indicated ratios. FRAP experiments were performed as in Fig. 1A. Fmob30 values were calculated 30 s after the end of the bleach from 20 experiments for each condition and presented as histograms. The error bars indicate standard deviations. C, FRAP experiment. Asynchronously growing HeLa cells were transfected with expression plasmids encoding EGFP with or without a 2-fold excess of c-Jun vector. Each curve corresponds to the averages of 20 FRAP experiments. D and E, cell fractionation experiments. Fractionation experiments were carried out as in Fig. 1C using HeLa cells co-transfected with expression plasmids for (i) EGFP-c-Fos in the absence or in the presence of a 2-fold excess of c-Jun expression plasmid (D) or (ii) c-Fos in the absence or in the presence of a 2-fold excess of c-Jun expression plasmid (E). S, soluble; W, wash; NS, nonsoluble.
c-Jun Reduces c-Fos Intranuclear Mobility
We next tested whether dimerization with c-Jun altered c-Fos intranuclear mobility. Fos and Jun family members undergo dimerization/dedimerization cycles in vivo. Nevertheless, they interact for at least several minutes (48). Thus, the mobility of Fos·Jun dimers can be analyzed in short (30 s) FRAP experiments. Moreover, the formation of c-Fos·c-Jun heterodimers is strongly favored over that of c-Jun·c-Jun homodimers (6, 49), meaning that a slight excess of c-Jun will ensure quantitative c-Fos recruitment in heterodimers. Because c-Fos and c-Jun show half-lives of ∼1 h (48), this is easily accomplished by transfecting an excess of c-Jun expression vector over that for c-Fos, provided that plasmid backbones are identical.
Strikingly, co-expression of c-Jun led to a dramatic intranuclear redistribution of EGFP-c-Fos, which became less homogenous showing irregular areas of accumulation (Fig. 2A). We then performed FRAP experiments on HeLa cells, where EGFP-c-Fos was expressed together with varying amounts of c-Jun. Interestingly, the mobile fraction of EGFP-c-Fos progressively diminished as the amount of c-Jun increased. At 2- and 5-fold excesses of c-Jun expression plasmid, the Fmob30 of EGFP-c-Fos was ∼40% that of monomeric c-Fos (Fig. 2B). Moreover, the recovery of fluorescence was still not complete 10 min after bleaching (not shown). This effect was specific to the c-Fos·c-Jun interaction because co-transfection of c-Jun and EGFP did not alter the mobility of EGFP in FRAP assays (Fig. 2C). These data suggested that dimerization with c-Jun causes c-Fos to associate with nuclear components, which was a notion further supported by cell fractionation experiments, because EGFP-c-Fos was principally recovered in the Triton X-100-insoluble fraction (Fig. 2D). Importantly, a similar distribution was found for wild type c-Fos in the presence of c-Jun in parallel experiments (Fig. 2E), ruling out an artifact caused by the fusion to EGFP. Because a 2-fold excess of c-Jun expression plasmid over that for c-Fos showed sufficient to obtain maximal reduction in c-Fos mobility (Fig. 2B), this plasmid ratio was used in the above fractionation experiments as well as in our further experiments.
We noted that only 60% of c-Fos remained immobile during the time course of the experiments, even though co-immunoprecipitation experiments indicated quantitative association of c-Fos with c-Jun. Thus, 40% of dimers appeared mobile for the 30-s post-bleach. This likely reflected saturation of the nuclear binding sites for c-Fos·c-Jun dimers in the window of time analyzed possibly in association with slow exchange rate between bound and unbound dimers. Consistent with the idea of mobility for a fraction of dimers, some c-Fos and EGFP-c-Fos (albeit less than 40%) were found in the soluble fraction in our cell fractionation experiments (Fig. 2, D and E).
Reduced c-Fos Intranuclear Mobility upon Heterodimerization with c-Jun Does Not Require DNA Binding
One obvious explanation for the reduction of c-Fos intranuclear mobility by c-Jun would be binding of heterodimers to genomic AP-1 motifs. To test this possibility, we compared the behavior of different heterodimers composed of (i) wild type EGFP-c-Fos or EGFP-c-Fos mutated in either the leucine zipper or the DNA-binding domain and (ii) wild type or leucine zipper mutants of c-Jun (Fig. 3A). The dimerization mutants of EGFP-c-Fos were either deleted of the LZ (EGFP-c-FosΔLZ) or mutated at the level of three of its leucines (EGFP-c-FosVAV) (50). The DNA-binding mutants were either deleted of the DBD (EGFP-c-FosΔDBD) or mutated at two critical residues suppressing recognition of AP-1 motifs (EGFP-c-FosVV) (51). For c-Jun, we used only a dimerization mutant lacking the LZ (c-JunΔLZ), because deletion of the DBD, which contains the c-Jun nuclear localization signal, entails a dramatic intracellular redistribution of c-Jun (52).6
FIGURE 3.

Role of heterodimerization with c-Jun and binding to genomic AP-1 DNA sequences on c-Fos intranuclear dynamics. A, structures of wild type and mutant c-Fos and c-Jun proteins. B, Fmob30 of wild type and mutant EGFP-c-Fos proteins in the presence of wild type and mutant c-Jun. Co-transfections were conducted in the presence of 2-fold more c-Jun expression plasmids than vectors for either wild type or mutant EGFP-c-Fos. FRAP experiments were conducted as in Fig. 1B. Fmob30 are the averages of 6–20 experiments. The given values include standard deviations. C, localization of wild type and mutant EGFP-c-Fos in the presence of wild type c-Jun. Transfection conditions were as in B. Confocal microscopy was conducted on living cells. D, dimerization of EGFP-c-Fos mutants with c-Jun. Transfection conditions were as in B. Co-immunoprecipitations were performed with the anti-FLAG antibody, because c-Jun constructs contained a FLAG tag at the C terminus, and immunoblotting experiments were conducted with the indicated antibodies as described under “Materials and Methods.” E, cell fractionation experiments. Transfection conditions were as in B, and cell fractionations were conducted and analyzed as in Fig. 1C. T, total cell extract; SN, supernatant; S, soluble; W, wash; NS, nonsoluble; IP, immunoprecipitation.
Despite the presence of a 2-fold excess of the c-Jun expression plasmid, EGFP-c-FosVAV and EGFP-c-FosΔLZ showed a mobility similar to that of monomeric EGFP-c-Fos (Fig. 3B, left panel). Consistent with this, c-JunΔLZ did not reduce EGFP-c-Fos intranuclear mobility (Fig. 3B, left panel). In contrast, EGFP-c-FosΔDBD and EGFP-c-FosVV, which cannot bind DNA but can heterodimerize, showed the same drastic reduced mobility as wild type c-Fos in the presence of an excess of c-Jun (Fig. 3B, right panel). Accordingly, all three proteins exhibit comparable intranuclear distributions as assessed by confocal microscopy (Fig. 3C). Importantly, (i) co-immunoprecipitation experiments showed that neither point mutations within the DBD nor its deletion suppressed dimerization of c-Fos and c-Jun in HeLa cells (Fig. 3D) and (ii) heterodimers made up of c-Jun and c-Fos DBD mutants cannot bind to AP-1/TRE DNA motifs in in vitro binding assays (not shown). This supported the idea that heterodimerization and not binding to AP-1/TRE or CRE DNA sequences is the primary event responsible for this c-Jun-mediated reduction of c-Fos mobility. Furthermore, cell fractionation experiments showed that EGFP-c-FosΔDBD and EGFP-c-FosVV are redistributed in the insoluble nuclear fraction in the presence of c-Jun (Fig. 3E).
JunB Does Not Affect c-Fos Intranuclear Mobility
It seemed important to test whether another dimerization partner could have the same effect on c-Fos intranuclear mobility. JunB is also a highly documented c-Fos partner (6, 15, 48, 53). Therefore, we analyzed its effects on EGFP-c-Fos intracellular localization and mobility as we did with c-Jun. Under conditions of quantitative association with JunB, as assayed by co-immunoprecipitation (Fig. 4A), EGFP-c-Fos was redistributed within the nucleus, as seen upon dimerization with c-Jun, albeit with a reproducible significantly different distribution (Fig. 4B; compare with Fig. 2A). FRAP experiments showed both an initial recovery of fluorescence (Fig. 4C) and an Fmob30 (Fig. 4D) similar for JunB·EGFP-c-Fos and monomeric EGFP-c-Fos. Despite their high mobility, JunB·EGFP-c-Fos heterodimers associated principally with the Triton X-100-insoluble nuclear fraction (Fig. 4E), unlike monomeric c-Fos (Fig. 1D). Thus, our data indicate that c-Jun and JunB alter c-Fos intranuclear behavior with, however, different outcomes.
FIGURE 4.

Effect of JunB on c-Fos intranuclear distribution and mobility. A, heterodimerization of JunB-FLAG with EGFP-c-Fos. The expression plasmid for EGFP-c-Fos was transfected in HeLa cells in the presence of a 2-fold excess of JunB-FLAG construct. Co-immunoprecipitations were conducted with the anti-FLAG antibody as in Fig. 3D. The antibodies used in immunoblotting analyses are indicated. B, intranuclear distribution of EGFP-c-Fos in the presence of JunB. EGFP-c-Fos localization in the presence of JunB-FLAG was assessed by confocal microscopy on living cells. The JunB versus EGFP-c-Fos expression plasmid ratio was of 2. C, FRAP experiments. FRAP experiments were conducted in HeLa cells transfected with expression plasmids for either EGFP-c-Fos or EGFP-c-Fos + JunB. In the latter case, the plasmid ratio was 2. The curves correspond to the averages of 20 FRAP experiments in each case. D, mobility of EGFP-c-Fos in the presence of different amounts of JunB. HeLa cells were transfected in the presence of different ratios of JunB versus EGFP-c-Fos plasmids as indicated. Fmob30 were calculated from 15–20 FRAP experiments in each case. E, cell fractionation experiments. Cell fractionation experiments of cells transfected with plasmids encoding JunB and EGFP-c-Fos in a ratio of 2 were conducted and analyzed as in Fig. 1D. T, total cell extract; SN, supernatant; IP, immunoprecipitation; S, soluble; W, wash; NS, nonsoluble.
Fra-1 Intranuclear Distribution and Mobility Are Affected by c-Jun and JunB in a Manner Similar to c-Fos
We then tested whether c-Jun and JunB similarly influenced the behavior of the Fos family protein Fra-1, because endogenous c-Jun and JunB are known to interact with endogenous Fra-1 in a variety of situations (6, 48, 53). EGFP-Fra-1 quantitatively dimerizes with ectopic c-Jun and JunB in HeLa cells (Fig. 5A). Interestingly, monomeric EGFP-Fra-1 was weakly cytoplasmic, which was no longer seen in the presence of co-transfected c-Jun and JunB (Fig. 5B). Like for c-Fos, FRAP analyses indicated that Fra-1 intranuclear mobility was dramatically reduced when co-expressed with c-Jun but was unaffected by co-expression of JunB (Fig. 5C). The mobile fraction of Fra-1 decreased to ∼40% in the presence of c-Jun and remained unchanged in the presence of JunB (Fig. 5D). EGFP-Fra-1 associated with the Triton X-100-insoluble nuclear fraction when co-expressed with either Jun dimerization partner (Fig. 5E). Thus, c-Jun and JunB affect Fra-1 intranuclear distribution and mobility in a manner similar to c-Fos.
FIGURE 5.

Alteration of Fra-1 intranuclear distribution and mobility by c-Jun and JunB. A, dimerization of EGFP-Fra-1 with c-Jun and JunB. Heterodimerization assays were conducted as in Fig. 3D using HeLa cells transfected with plasmids for either c-Jun-FLAG + EGFP-Fra-1 or JunB-FLAG + EGFP-Fra-1 in a ratio of 2. B, intracellular localization of EGFP-Fra-1 in the presence of c-Jun and JunB in a 2-fold excess. Intracellular localization was assessed on living cells by confocal microscopy analysis of HeLa cells transfected as in A. C, FRAP experiments. FRAP experiments were conducted in HeLa cells transfected with expression plasmids for either EGFP-Fra1 or EGFP-Fra1 and a 2-fold excess of JunB- or c-Jun plasmid. The curves correspond to the averages of 10–20 FRAP experiments. D, Fmob30 of EGFP-Fra-1 in the presence of c-Jun and JunB. Transfection conditions were as in A. Fmob30 were calculated from more than 10 FRAP experiments. E, fractionation experiments. Fractionation and analyses of HeLa cells transfected as in A were conducted as in Fig. 1C. T, total cell extract; SN, supernatant; IP, immunoprecipitation; S, soluble; W, wash; NS, nonsoluble.
Differential Association of c-Fos·c-Jun and c-Fos·JunB Heterodimers with the Nuclear Matrix
In a final step, we tested whether c-Fos·c-Jun and c-Fos·JunB heterodimers could associate differentially with the chromatin and/or the nuclear matrix.
First, we conducted classical biochemical fractionation experiments (see “Materials and Methods”) followed by immunoblotting assays. In these experiments: (i) S1 corresponded to both cytoplasmic and nuclear proteins solubilized upon cell lysis in the presence of 0.5% Triton X-100, (ii) S2 corresponded to the chromatin proteins released upon subsequent extensive DNA hydrolysis by MNase, and (iii) S3 corresponded to the remnant of chromatin proteins and DNA solubilized after additional high salt (2 m NaCl) treatment, and (P) corresponded to the nuclear-matrix-containing fraction. Fractionation patterns of (i) the nucleosoluble Phax protein, (ii) the weakly chromatin-associated HCF-1 protein (54), (iii) tightly chromatin-associated histone H3, and (iv) the nuclear lamina-constituting lamin A/C proteins confirmed the efficiency of the procedure (Fig. 6). In contrast to EGFP-c-Fos expressed alone, which localized predominantly in the S1 fraction, EGFP-c-Fos·c-Jun dimers were essentially found associated with the nuclear matrix. However, a very minor fraction of EGFP-c-Fos was found in the P fraction. This may be due to dimerization of the protein with endogenously expressed AP-1 protein, although partial association of the monomer with the nuclear matrix cannot be formally excluded. Differing from EGFP-c-Fos·c-Jun dimers, EGFP-c-Fos·JunB dimers were distributed in three fractions: S1, S2, and P, with approximately half of them in the P fraction. We also analyzed the distribution of endogenous c-Fos in HeLa cells stimulated with 20% serum for 1 h. Interestingly, it behaves as EGFP-c-Fos·JunB heterodimers. This was, however, not surprising because it was previously described that 70% of c-Fos associates with JunB in comparable serum stimulation experiments (48).
FIGURE 6.

Cell and subnuclear fractionation. HeLa cells were (i) transfected with the EGFP-c-Fos vector in the absence or in the presence of an equivalent vector for either c-Jun or JunB in a 1:2 ratio or (ii) simply stimulated with 20% serum for 1 h. They were then fractionated as described under “Materials and Methods.” Equivalent amounts of each fraction were loaded in each lane for immunoblotting analysis. The names of the various proteins assayed are indicated in the figure. T, total cell extract; S1, Triton X-100-soluble fraction; W, wash; S2, soluble fraction after MNase digestion; S3, soluble fraction after 2 m NaCl treatment; P, nuclear matrix fraction.
We then complemented these biochemical experiments by microscopic analyses (Fig. 7) of cells subjected to the same succession of treatments (Triton X-100 solubilization, DNA hydrolysis by MNase, and high salt washing) as above. Hoescht 33342 staining revealed large and total elimination of DNA after MNase and high salt treatment, respectively. EGFP-c-Fos·c-Jun and EGFP-c-Fos·JunB dimers were clearly visible in nuclei after quantitative DNA elimination. Interestingly, endogenous c-Fos in serum-stimulated HeLa cells was also found associated with the nuclear matrix with a distribution similar to that of EGFP-c-Fos in the presence of c-Jun or JunB. This was consistent with intranuclear retention caused by dimerization with endogenous AP-1 proteins. Taken together, these observations were coherent with the fact that abolishing the DNA binding ability of c-Fos altered neither intranuclear mobility (Fig. 3B) nor its subcellular distribution (Fig. 3D) when dimerized with c-Jun. The fact that EGFP-c-Fos·c-Jun, EGFP-c-Fos·JunB, and endogenous c-Fos in serum-stimulated cells appeared in discrete structures resulting from MNase treatment-linked intranuclear reorganization was not investigated further. Whatever the exact nature of these structures, the combination of our FRAP experiments with the above biochemical fractionation and microscopic analyses indicates that, at variance with monomeric c-Fos, which is highly mobile within the nucleus, both c-Fos·c-Jun and c-Fos·JunB heterodimers can associate with the nuclear matrix. This association is, however, more quantitative and more stable in the case of c-Fos·c-Jun complexes.
FIGURE 7.

Microscopic analysis of c-Fos and EGFP-c-Fos intranuclear localization in fractionated cells. These experiments were carried out in parallel with those presented in Fig. 6 with HeLa cells seeded and grown on coverslips. Endogenous c-Fos was detected using sequentially the sc52 rabbit anti-c-Fos antibody and an Alexa 488-labeled anti-rabbit secondary antibody. The nuclei were stained with Hoechst 33342. All of the pictures in the EGFP channel were acquired using the same time exposure.
DISCUSSION
This study reveals a novel level of regulation operating on c-Fos, namely the differential control of its intranuclear mobility and distribution by its Jun partners. This regulation also applies to at least another member of the Fos family, Fra-1.
Our data indicate that EGFP-c-Fos does not diffuse as freely as EGFP in FRAP experiments, indicating weak interactions between monomeric c-Fos and some nuclear structures. Interestingly, heterodimerization with c-Jun results in a dramatic reduction of c-Fos intranuclear mobility. Such a mobility change is not the result of dimerization per se but is selective because JunB does not detectably alter c-Fos intranuclear dynamics (as visualized by FRAP) under conditions where c-Fos is quantitatively associated with JunB (as shown by co-immunoprecipitation). Although the FRAP data presented in Figs. 2–4 were obtained in human epithelial HeLa cells, similar differential effects of c-Jun and JunB were observed in Balb/C 3T3 mouse embryo fibroblasts (supplemental Fig. S1), indicating that immobilization of c-Fos by c-Jun is neither cell- nor species-specific.
It is most often considered that reduced intranuclear mobility of transcription factors is largely determined by binding to target DNA sequences because disabled DNA-binding domain mutants of proteins such as NF-κB p65 (55), Acep1 (56), interferon regulatory factor 8 (57), the androgen receptor (58, 59), or the glucocorticoid receptor (60) are more mobile than their wild type counterparts. However, immobilization of c-Fos by c-Jun is not primarily mediated by binding to AP-1/TRE or CRE target sequences (also see below) because EGFP-c-Fos and EGFP-c-FosΔDBD behave similarly in FRAP experiments, when quantitatively heterodimerized with c-Jun. Strengthening the idea of a DNA-independent mechanism responsible for c-Fos mobility reduction, cell fractionation experiments (Figs. 6 and 7) showed that c-Fos·c-Jun dimers predominantly associate with the nuclear matrix, which is, by definition, devoid of chromatin and DNA (61, 62).
There may be an apparent paradox between the well known strong transcriptional activity of c-Fos·c-Jun dimers, especially under the transfection conditions used in this work, and the fact that a large majority of these complexes were found in the nuclear matrix fraction during the course of this work. The observation that 40% of c-Fos·c-Jun complexes are mobile (as assayed 30 s post-bleach), however, helps resolve it with a likely scenario as follows. transcriptional activity at a given time would be due to a minor fraction of c-Fos·c-Jun dimers, whereas a large majority of these complexes would be latent and stored in intranuclear non-chromatin domains. Storage would, however, be relatively limited in time, as indicated by FRAP experiments, at least for a fraction of them. Important challenges will now be to elucidate whether these nuclear matrix storage sites are located near transcription factories or not and why and how latent c-Fos·c-Jun dimers are released from the nuclear matrix and addressed to the transcription sites. Along this line, it is of note that ∼50% of endogenous c-Fos is found associated with the nuclear matrix at the peak of expression in serum-stimulated cells, whereas the rest equally distributes between soluble and chromatin fractions (Fig. 6). Although the respective roles of the latter two populations of c-Fos molecules still require clarification, this observation is consistent with the idea of storage sites for latent AP-1 complexes under physiological conditions.
Very interestingly, Andrés and co-workers (63, 64) have recently reported that c-Fos can associate with the nuclear matrix component lamin A/C. This was proposed to constitute an AP-1 suppression mechanism. Although the authors did not address c-Fos mobility directly, their data are consistent with our cell fractionation experiments showing interaction with the nuclear matrix. From in vitro pull-down experiments, these authors hypothesized that c-Fos would interact with lamin A/C via its leucine zipper at the detriment of dimerization with its Jun partners (63). Although not questioning the principle of AP-1 inactivation by lamin A/C, our data, however, argue against LZ-mediated sequestration of monomeric c-Fos, because we clearly show that (i) monomeric c-Fos is highly mobile and quantitatively found in the soluble protein fraction and (ii) only dimeric c-Fos is found associated with the nuclear matrix.
The behavioral differences between c-Fos·c-Jun and c-Fos·JunB dimers are interesting to consider. First, in addition to partly different intranuclear distribution, as visualized by direct fluorescence analysis of transfected cells (Figs. 2A and 4B), c-Fos·JunB dimers showed more heterogeneous in our fractionation experiments than c-Fos·c-Jun ones. Whereas c-Fos·c-Jun dimers were found quantitatively associated with the nuclear matrix, c-Fos·JunB dimers were detected not only in the nuclear matrix fraction (∼50%) but also in the soluble and chromatin ones (Figs. 6 and 7). If one takes into consideration that dimerization with JunB little affects c-Fos mobility, as assayed by FRAP (Fig. 4C), it ensues that association of c-Fos·JunB dimers with the nuclear matrix must be much more dynamic than that of c-Fos·c-Jun dimers. Thus, interaction with the nuclear matrix may differentially control the abundances of the different AP-1 dimers available for gene regulation. It will next be interesting to establish (i) whether this is due to differential affinity for the same nuclear matrix sites or to interaction with different sites and (ii) whether the slowed down mobility of c-Fos is contributed by both c-Fos and c-Jun or principally by c-Jun, which would, in the later case, serve as an anchor to nuclear structures. Unfortunately, we could not answer this question in FRAP experiments, because EGFP-c-Jun chimeras behave aberrantly because they do not show the same intracellular distribution as c-Jun.6
c-Fos constitutes an heterogeneous collection of molecules at any moment in any cell because of dimerization with multiple AP-1 proteins, interaction with multiple partners, diverse post-translational modifications, widespread distribution in the nucleus, the multiple genes to which it can bind, etc. An important technical point must therefore be taken into account when considering the FRAP experiments presented in this work. Those could only address the behavior of the bulk of c-Fos and not those of the various (and possibly many) subpopulations of this protein, which may be different. In particular, it is possible that the small fraction of transcriptionally active c-Fos·c-Jun dimers may be immobilized on AP-1/TRE or CRE sequences for periods of time longer than those of association with the nuclear matrix for efficient stimulation of target gene transcription. In support of such a possibility, several other transcription factors were previously shown to display low mobility in their presumably active states. This was, for example, the case of nuclear hormone receptors, such as androgen receptor (58, 65), glucocorticoid receptor (60), estrogen receptor α and β (66, 67), liver X receptors α and β (68), peroxisome proliferator-activated receptor γy (69), and progesterone receptor (70) after ligand binding and/or association with transcriptional co-activators and/or chromatin remodeling complexes (60, 67, 68, 70). In the same vein, intranuclear diffusion of CREB (71) and liver X receptor α (68) depends on the integrity of specific trans-activation domains, the intranuclear mobility of interferon regulatory factor 8 is decreased upon association with PU.1 and interferon regulatory factor 1 (57), and that of β-catenin is reduced upon association with its transcriptional partner TCF4 (72).
Finally, by demonstrating stronger association of c-Fos·c-Jun dimers than c-Fos·JunB dimers to intranuclear structures, we provide here a straightforward explanation of our previous observation of more efficient inhibition of c-Fos nuclear export by c-Jun than by JunB (36), even though an additional reduced c-Jun-dependent recognition of c-Fos by the nuclear export machinery cannot be formally excluded. This resembles the situation of β-catenin, where interaction with TCF4 leads to reduced nucleo-cytoplasmic shuttling primarily reflecting a decrease in intranuclear mobility and not that of nuclear export efficiency per se (72). Further work will determine the role of association with intranuclear components in the transcriptional activity of c-Fos·c-Jun heterodimers.
Supplementary Material
Acknowledgments
We thank Dr. R. Hipskind for critical reading of the manuscript. All of the imaging analyses were performed on the Montpellier RIO Imaging (Infrastructures Biologie Santé et Agronomie) imaging platform.
This work was supported by grants from the ARC, Association pour la Recherche sur le Cancer, the Agence Nationale de Recherches (ANR-08-BLAN-007–01), the French Ligue Nationale Contre le Cancer, and by the CNRS.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
C. E. Malnou, F. Piechaczyk, and I. Jariel-Encontre, unpublished data.
- LZ
- leucine zipper
- FRAP
- fluorescence recovery after photobleaching
- CRE
- cAMP-responsive element
- TRE
- 12-O-tetradecanoylphorbol-13-acetate-responsive element
- DBD
- DNA-binding domain(s)
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- STAT
- signal transducers and activators of transcription
- EGFP
- enhanced green fluorescent protein
- PBS
- phosphate-buffered saline
- MNase
- microccocal nuclease.
REFERENCES
- 1.Eferl R., Wagner E. F. (2003) Nat. Rev. Cancer 3, 859–868 [DOI] [PubMed] [Google Scholar]
- 2.Hess J., Angel P., Schorpp-Kistner M. (2004) J. Cell Sci. 117, 5965–5973 [DOI] [PubMed] [Google Scholar]
- 3.Jochum W., Passegué E., Wagner E. F. (2001) Oncogene 20, 2401–2412 [DOI] [PubMed] [Google Scholar]
- 4.Shaulian E., Karin M. (2002) Nat. Cell Biol. 4, E131–E136 [DOI] [PubMed] [Google Scholar]
- 5.Zenz R., Eferl R., Scheinecker C., Redlich K., Smolen J., Schonthaler H. B., Kenner L., Tschachler E., Wagner E. F. (2008) Arthritis Res. Ther. 10, 201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinenov Y., Kerppola T. K. (2001) Oncogene 20, 2438–2452 [DOI] [PubMed] [Google Scholar]
- 7.Vinson C., Acharya A., Taparowsky E. J. (2006) Biochim. Biophys. Acta 1759, 4–12 [DOI] [PubMed] [Google Scholar]
- 8.Jariel-Encontre I., Piechaczyk M. (2008) Targeted Protein Database [22540], 10.2970/tpdb.2009.0218 [DOI] [Google Scholar]
- 9.Piechaczyk M., Blanchard J. M. (1994) Crit. Rev. Oncol. Hematol. 17, 93–131 [DOI] [PubMed] [Google Scholar]
- 10.Basbous J., Jariel-Encontre I., Gomard T., Bossis G., Piechaczyk M. (2008) Biochimie 90, 296–305 [DOI] [PubMed] [Google Scholar]
- 11.Gomard T., Jariel-Encontre I., Basbous J., Bossis G., Mocquet-Torcy G., Piechaczyk M. (2008) Biochem. Soc. Trans. 36, 858–863 [DOI] [PubMed] [Google Scholar]
- 12.Bossis G., Ferrara P., Acquaviva C., Jariel-Encontre I., Piechaczyk M. (2003) Mol. Cell Biol. 23, 7425–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chalmers C. J., Gilley R., March H. N., Balmanno K., Cook S. J. (2007) Cell Signal. 19, 695–704 [DOI] [PubMed] [Google Scholar]
- 14.Chen R. H., Abate C., Blenis J. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 10952–10956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng T., Karin M. (1994) Nature 371, 171–175 [DOI] [PubMed] [Google Scholar]
- 16.Ferrara P., Andermarcher E., Bossis G., Acquaviva C., Brockly F., Jariel-Encontre I., Piechaczyk M. (2003) Oncogene 22, 1461–1474 [DOI] [PubMed] [Google Scholar]
- 17.Higashi N., Kunimoto H., Kaneko S., Sasaki T., Ishii M., Kojima H., Nakajima K. (2004) Genes Cells 9, 233–242 [DOI] [PubMed] [Google Scholar]
- 18.Mackeigan J. P., Murphy L. O., Dimitri C. A., Blenis J. (2005) Mol. Cell Biol. 25, 4676–4682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monje P., Hernández-Losa J., Lyons R. J., Castellone M. D., Gutkind J. S. (2005) J. Biol. Chem. 280, 35081–35084 [DOI] [PubMed] [Google Scholar]
- 20.Monje P., Marinissen M. J., Gutkind J. S. (2003) Mol. Cell Biol. 23, 7030–7043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy L. O., MacKeigan J. P., Blenis J. (2004) Mol. Cell Biol. 24, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy L. O., Smith S., Chen R. H., Fingar D. C., Blenis J. (2002) Nat. Cell Biol. 4, 556–564 [DOI] [PubMed] [Google Scholar]
- 23.Okazaki K., Sagata N. (1995) EMBO J. 14, 5048–5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki T., Kojima H., Kishimoto R., Ikeda A., Kunimoto H., Nakajima K. (2006) Mol. Cell 24, 63–75 [DOI] [PubMed] [Google Scholar]
- 25.Tanos T., Marinissen M. J., Leskow F. C., Hochbaum D., Martinetto H., Gutkind J. S., Coso O. A. (2005) J. Biol. Chem. 280, 18842–18852 [DOI] [PubMed] [Google Scholar]
- 26.Gilley R., March H. N., Cook S. J. (2009) Cell Signal. 21, 969–977 [DOI] [PubMed] [Google Scholar]
- 27.Koga K., Takaesu G., Yoshida R., Nakaya M., Kobayashi T., Kinjyo I., Yoshimura A. (2009) Immunity 30, 372–383 [DOI] [PubMed] [Google Scholar]
- 28.Bossis G., Malnou C. E., Farras R., Andermarcher E., Hipskind R., Rodriguez M., Schmidt D., Muller S., Jariel-Encontre I., Piechaczyk M. (2005) Mol. Cell Biol. 25, 6964–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tempé D., Piechaczyk M., Bossis G. (2008) Biochem. Soc. Trans 36, 874–878 [DOI] [PubMed] [Google Scholar]
- 30.Roux P., Blanchard J. M., Fernandez A., Lamb N., Jeanteur P., Piechaczyk M. (1990) Cell 63, 341–351 [DOI] [PubMed] [Google Scholar]
- 31.Vriz S., Lemaitre J. M., Leibovici M., Thierry N., Méchali M. (1992) Mol. Cell Biol. 12, 3548–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bussolino D. F., Guido M. E., Gil G. A., Borioli G. A., Renner M. L., Grabois V. R., Conde C. B., Caputto B. L. (2001) FASEB J. 15, 556–558 [DOI] [PubMed] [Google Scholar]
- 33.Gil G. A., Bussolino D. F., Portal M. M., Pecchio A. A., Renner M. L., Borioli G. A., Guido M. E., Caputto B. L. (2004) Mol. Biol. Cell 15, 1881–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roux P., Carillo S., Blanchard J. M., Jeanteur P., Piechaczyk M. (1994) The c-fos and c-jun Families of Transcription Factors, pp. 87–95, CRC Press Inc. [Google Scholar]
- 35.Arnold M., Nath A., Wohlwend D., Kehlenbach R. H. (2006) J. Biol. Chem. 281, 5492–5499 [DOI] [PubMed] [Google Scholar]
- 36.Malnou C. E., Salem T., Brockly F., Wodrich H., Piechaczyk M., Jariel-Encontre I. (2007) J. Biol. Chem. 282, 31046–31059 [DOI] [PubMed] [Google Scholar]
- 37.Tratner I., Verma I. M. (1991) Oncogene 6, 2049–2053 [PubMed] [Google Scholar]
- 38.Mason J. M., Schmitz M. A., Müller K. M., Arndt K. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 8989–8994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wysocka J., Reilly P. T., Herr W. (2001) Mol. Cell Biol. 21, 3820–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burch P. M., Yuan Z., Loonen A., Heintz N. H. (2004) Mol. Cell Biol. 24, 4696–4709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spector D. L., Goldman R. D., Leinwand L. A. (1998) Cells: A Laboratory Manual, Vol. 1, pp. 44.2–44.6, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 42.Houtsmuller A. B., Vermeulen W. (2001) Histochem. Cell Biol. 115, 13–21 [DOI] [PubMed] [Google Scholar]
- 43.Reits E. A., Neefjes J. J. (2001) Nat. Cell Biol. 3, E145–E147 [DOI] [PubMed] [Google Scholar]
- 44.van Drogen F., Peter M. (2004) Methods Mol. Biol. 284, 287–306 [DOI] [PubMed] [Google Scholar]
- 45.Sprague B. L., McNally J. G. (2005) Trends Cell Biol. 15, 84–91 [DOI] [PubMed] [Google Scholar]
- 46.Ohno M., Segref A., Bachi A., Wilm M., Mattaj I. W. (2000) Cell 101, 187–198 [DOI] [PubMed] [Google Scholar]
- 47.Christensen M. O., Krokowski R. M., Barthelmes H. U., Hock R., Boege F., Mielke C. (2004) J. Biol. Chem. 279, 21873–21882 [DOI] [PubMed] [Google Scholar]
- 48.Kovary K., Bravo R. (1991) Mol. Cell Biol. 11, 2451–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vinson C., Myakishev M., Acharya A., Mir A. A., Moll J. R., Bonovich M. (2002) Mol. Cell Biol. 22, 6321–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schuermann M., Neuberg M., Hunter J. B., Jenuwein T., Ryseck R. P., Bravo R., Müller R. (1989) Cell 56, 507–516 [DOI] [PubMed] [Google Scholar]
- 51.Ransone L. J., Visvader J., Wamsley P., Verma I. M. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 3806–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waldmann I., Wälde S., Kehlenbach R. H. (2007) J. Biol. Chem. 282, 27685–27692 [DOI] [PubMed] [Google Scholar]
- 53.Kovary K., Bravo R. (1992) Mol. Cell Biol. 12, 5015–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Julien E., Herr W. (2004) Mol. Cell 14, 713–725 [DOI] [PubMed] [Google Scholar]
- 55.Schaaf M. J., Willetts L., Hayes B. P., Maschera B., Stylianou E., Farrow S. N. (2006) J. Biol. Chem. 281, 22409–22420 [DOI] [PubMed] [Google Scholar]
- 56.Karpova T. S., Chen T. Y., Sprague B. L., McNally J. G. (2004) EMBO Rep. 5, 1064–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laricchia-Robbio L., Tamura T., Karpova T., Sprague B. L., McNally J. G., Ozato K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 14368–14373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farla P., Hersmus R., Geverts B., Mari P. O., Nigg A. L., Dubbink H. J., Trapman J., Houtsmuller A. B. (2004) J. Struct. Biol. 147, 50–61 [DOI] [PubMed] [Google Scholar]
- 59.Farla P., Hersmus R., Trapman J., Houtsmuller A. B. (2005) J. Cell Sci. 118, 4187–4198 [DOI] [PubMed] [Google Scholar]
- 60.Schaaf M. J., Cidlowski J. A. (2003) Mol. Cell Biol. 23, 1922–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albrethsen J., Knol J. C., Jimenez C. R. (2009) J. Proteomics 72, 71–81 [DOI] [PubMed] [Google Scholar]
- 62.Lever E., Sheer D. (2010) J. Pathol. 220, 114–125 [DOI] [PubMed] [Google Scholar]
- 63.González J. M., Navarro-Puche A., Casar B., Crespo P., Andrés V. (2008) J. Cell Biol. 183, 653–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ivorra C., Kubicek M., González J. M., Sanz-González S. M., Alvarez-Barrientos A., O'Connor J. E., Burke B., Andrés V. (2006) Genes Dev. 20, 307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marcelli M., Stenoien D. L., Szafran A. T., Simeoni S., Agoulnik I. U., Weigel N. L., Moran T., Mikic I., Price J. H., Mancini M. A. (2006) J. Cell Biochem. 98, 770–788 [DOI] [PubMed] [Google Scholar]
- 66.Damdimopoulos A. E., Spyrou G., Gustafsson J. A. (2008) Endocrinology 149, 339–345 [DOI] [PubMed] [Google Scholar]
- 67.Maruvada P., Baumann C. T., Hager G. L., Yen P. M. (2003) J. Biol. Chem. 278, 12425–12432 [DOI] [PubMed] [Google Scholar]
- 68.Prüfer K., Hernandez C., Gilbreath M. (2008) Exp. Cell Res. 314, 2652–2660 [DOI] [PubMed] [Google Scholar]
- 69.Feige J. N., Gelman L., Tudor C., Engelborghs Y., Wahli W., Desvergne B. (2005) J. Biol. Chem. 280, 17880–17890 [DOI] [PubMed] [Google Scholar]
- 70.Rayasam G. V., Elbi C., Walker D. A., Wolford R., Fletcher T. M., Edwards D. P., Hager G. L. (2005) Mol. Cell Biol. 25, 2406–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayr B. M., Guzman E., Montminy M. (2005) J. Biol. Chem. 280, 15103–15110 [DOI] [PubMed] [Google Scholar]
- 72.Krieghoff E., Behrens J., Mayr B. (2006) J. Cell Sci. 119, 1453–1463 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.