

Abstract
Toxoplasma gondii is a protozoan parasite belonging to the phylum Apicomplexa. Parasites in this phylum utilize a unique process of motility termed gliding, which is dependent on parasite actin filaments. Surprisingly, 98% of parasite actin is maintained as G-actin, suggesting that filaments are rapidly assembled and turned over. Little is known about the regulated disassembly of filaments in the Apicomplexa. In higher eukaryotes, the related actin depolymerizing factor (ADF) and cofilin proteins are essential regulators of actin filament turnover. ADF is one of the few actin-binding proteins conserved in apicomplexan parasites. In this study we examined the mechanism by which T. gondii ADF (TgADF) regulates actin filament turnover. Unlike other members of the ADF/cofilin (AC) family, apicomplexan ADFs lack key F-actin binding sites. Surprisingly, this promotes their enhanced disassembly of actin filaments. Restoration of the C-terminal F-actin binding site to TgADF stabilized its interaction with filaments but reduced its net filament disassembly activity. Analysis of severing activity revealed that TgADF is a weak severing protein, requiring much higher concentrations than typical AC proteins. Investigation of TgADF interaction with T. gondii actin (TgACT) revealed that TgADF disassembled short TgACT oligomers. Kinetic and steady-state polymerization assays demonstrated that TgADF has strong monomer-sequestering activity, inhibiting TgACT polymerization at very low concentrations. Collectively these data indicate that TgADF promoted the efficient turnover of actin filaments via weak severing of filaments and strong sequestering of monomers. This suggests a dual role for TgADF in maintaining high G-actin concentrations and effecting rapid filament turnover.
Keywords: Cell/Motility, Cytoskeleton/Actin, Enzymes/ATPases, Organisms/Parasite, Protein/Protein-Protein Interactions, Protein/Structure, Subcellular Organelles/Microfilaments
Introduction
Toxoplasma gondii is an obligate intracellular protozoan parasite that belongs to the phylum Apicomplexa. In addition to being a significant cause of disease in immunocompromised individuals (1), T. gondii also provides an excellent model system for other members of the phylum (such as the medically important Plasmodium species), due to the presence of a variety of experimental tools. Transmission of T. gondii typically occurs via the ingestion of tissue cysts in undercooked meat or oocysts that are shed by cats and that can contaminate water (2). Once inside the host, the parasite utilizes a unique mode of motility, termed gliding, to move across epithelial barriers and migrate into deeper tissues (3). Gliding motility is conserved across the Apicomplexa and is responsible for the parasite's invasion of host cells (4). Both gliding and cell invasion depend on actin filaments within the parasite (5, 6).
In contrast to higher eukaryotic cells where typically 50% of the actin is filamentous (7), actin in T. gondii is almost exclusively unpolymerized (6, 8, 9). T. gondii has one actin allele (TgACT)2 that has 83% identity to vertebrate actin and is expressed throughout the life cycle of the parasite (8). Recent work has shown that parasite actins are inherently unstable (9–11), and this is likely due to key substitutions in their molecular structure (9). Actin typically undergoes self-polymerization above a critical concentration of 0.12 μm (12); however, the critical concentration for TgACT is surprisingly low at only 0.04 μm (9). Despite maintaining high cellular concentrations of G-actin, filamentous actin is essential for both gliding motility and host cell invasion by T. gondii (5). Treatment of parasites with cytochalasin D, renders parasites non-motile and unable to penetrate host cells, and genetic studies using mutants indicate that the polymerization of parasite filaments is crucial for both gliding and invasion (5). Additionally, hyperstabilizing actin filaments by treatment with jasplakinolide (13, 14) is also toxic to the parasite. Although jasplakinolide-treated parasites move with 3-fold increased speed (6) (indicating that actin polymerization is rate-limiting for motility), the prevention of filament turnover results in the inability of parasites to pursue directional movement and invade host cells (6). Collectively, these studies indicate that the control of parasite filament turnover is critical for productive gliding and invasion.
The unique actin dynamics observed in apicomplexan parasites suggests that actin polymerization and turnover are tightly regulated. However, in contrast to higher eukaryotes, which have many actin-binding proteins to regulate the microfilament system (15), only a small set of actin-binding proteins are conserved in the Apicomplexa (16–18). This subset includes formins, profilin, capping protein, actin depolymerizing factor (ADF), cyclase-associated protein (CAP), and coronin, which represent little more than the core set of actin-binding proteins required to reconstitute motility in vitro (19). Considering the high G-actin concentrations in apicomplexan parasites, it is surprising that no dedicated G-actin sequestering proteins are present, such as β-thymosin, which is found in higher eukaryotes (20).
Recent work has shed some light on how actin filaments may be assembled in apicomplexan parasites. Plasmodium formins have been shown to nucleate heterologous actin polymerization in vitro (21), and Toxoplasma profilin was shown to allow steady-state barbed-end growth while causing depolymerization at the pointed end (22). Most recently Plasmodium capping protein was shown to cap heterologous actin filaments (23). Although these studies provide insight into the actin polymerization machinery in apicomplexan parasites, little is known about the regulated disassembly of parasite actin filaments.
The ADF/cofilin (AC) family of proteins are highly conserved proteins and are essential for increasing actin filament turnover in higher eukaryotes (24). AC proteins are found ubiquitously in eukaryotes, often present in multiple isoforms, and tend to have a conserved structure and function (25). The predominant mechanism by which they are thought to increase filament turnover is by severing actin filaments (26). Electron cryomicroscopy and helical reconstructions of ADF and cofilin, bound to the side of actin filaments, reveal that they alter the twist of the filament by 4–5 °C, causing local alterations in the filament structure that are thought to lead to the fragmentation of filaments (27). AC proteins also increase the rate at which monomers are released from the pointed ends of filaments (19, 28).
Mutational analysis of yeast cofilin reveals several sites required for actin binding (29), many of which have also been confirmed in other AC proteins using mutagenesis (30–32), cross-linking (33), peptide competition, or synchrotron electron footprinting (34). The actin binding sites can be classified into two types: sites required for general actin binding and sites required exclusively for binding to filaments (29). Highly conserved residues are found at the N terminus, which includes the putative phosphorylation site serine 2 or 3 in eukaryotes or serine 6 in plants (25), in the long α3 helix, and in the turn connecting strand β6 and α4. These sites cluster together in the three-dimensional structure (35–37) and constitute a general actin-binding surface. The F-actin binding sites are less well defined but include a pair of basic residues at the beginning of the β5 strand (29, 38), which makes up part of the F-loop structure, and charged residues in the C-terminal α4 helix (29) or the C-terminal tail (32). The F-actin sites also appear to cluster together to form a binding surface (35).
Recent work has shown that mutation of the F-actin binding sites in AC proteins can uncouple their severing and depolymerizing activities (32, 38, 39). Point mutation of the critical basic residue Lys-96 in the F-loop of human cofilin, leads to a loss of severing activity and increased depolymerization activity (38). Interestingly, mutation of the homologous residue in Schizosaccharomyces pombe cofilin (Arg-78) results in a loss of nucleating activity (26). Similarly, deletion of a charged residue in the C-tail of the Caenorhabditis elegans AC homologue, Unc60B, results in a loss of severing activity and increased depolymerization activity (32, 39). Collectively, these studies highlight how a reduction in the affinity for the filament can uncouple the various activities of AC proteins, and identify specific molecular features that may also influence function in other AC proteins.
Although severing is the main activity typically associated with AC proteins, they can also interact with actin in other ways, depending on the isoform, organism, or cell type in which they are normally expressed (25). For example, S. pombe cofilin was recently shown to nucleate filaments when present at high concentrations (26). In direct contrast, the C. elegans isoform Unc60A was found to inhibit the steady-state polymerization of actin (40). Thus AC proteins can influence actin dynamics in a variety of ways depending on the specific cellular context in which they function.
AC proteins are conserved in the Apicomplexa, in which they are named ADF, and most parasites possess only a single isoform, except Plasmodium, which has two isoforms. Preliminary characterization of T. gondii ADF (TgADF) (41) demonstrates that it binds to G-actin and causes the net disassembly of rabbit actin filaments in vitro, suggesting that TgADF accelerates filament turnover (41). However, the mechanism by which it does so remains unknown. Interestingly, one of the ADF homologues, PfADF1, from the related apicomplexan parasite, Plasmodium falciparum, does not interact with F-actin or disassemble filaments (42). Instead, PfADF1 slightly enhances nucleotide exchange on G-actin, in contrast to the typical inhibition of nucleotide exchange caused by AC proteins (43, 44), suggesting an alternative role for ADF1 in these parasites. More recently, ADF from the apicomplexan parasite Eimeria tenella was reported to be transcriptionally up-regulated in the motile stages of the parasite, but little is known about the biochemical activity of this protein (45).
To determine how TgADF accelerates actin filament turnover we purified recombinant TgADF and examined its interactions with both heterologous actin and T. gondii actin in a variety of biochemical assays. We determined that the in vitro properties of TgADF are related to specific features in its molecular structure and are particularly well suited to control the unique actin dynamics found in apicomplexan parasites. Because other apicomplexan ADFs share the same molecular features, it is likely that the properties observed here are conserved in the phylum.
EXPERIMENTAL PROCEDURES
Secondary Structure-based Sequence Alignment and Homology Modeling of TgADF Structure
A multiple sequence alignment, including TgADF and other AC proteins described below, was generated using ClustalX (46) with the following parameters: gap opening = 15.00, gap extension = 0.30, delay divergent sequence = 25%. The following protein sequences (with the GenBankTM sequence ID nos.) were retrieved from NCBI and used in the alignment: AtADF1, Arabidopsis thaliana ADF1 (AAC72407); AcActophorin, Acanthamoeba castellani actophorin (AAA02909); ScCOF, Saccharomyces cerevisiae cofilin (AAA13256); SpCOF, S. pombe cofilin (CAB11258); CeUnc60A, C. elegans Unc60A (AAL02461); PfADF2, Plasmodium falciparum ADF2 (NP705497); HsADF, Homo sapiens ADF (AAB28361); PfADF1, P. falciparum ADF1 (NP703379); and TgADF, T. gondii ADF (AAC47717). The structure of TgADF was modeled onto the known crystal structure of A. castellani actophorin (Protein Data Bank code 1ahq) by submitting the sequence of TgADF to the Swiss Protein Data Bank. This structural model was used to subsequently manually adjust the sequence alignment of TgADF. Other sequences in the AC alignment were also manually adjusted based on the secondary structure alignment of Bowman et al. (47).
Proteins
Lyophilized rabbit skeletal muscle actin was obtained from Cytoskeleton (Denver, CO) and reconstituted according to manufacturer's recommendation, whereas recombinant T. gondii actin was purified as previously described (9). TgADF was amplified from RH strain T. gondii cDNA using specific primers (supplemental Table S1), and cloned into the pET16b+ vector (Novagen, Darmstadt, Germany) at the Nde1-BamH1 site, which resulted in the addition of an N-terminal His10 tag. S. pombe cofilin (SpCOF) and A. castellani actophorin were digested from separate plasmids made in the pMW172 background (48, 49) (kindly provided by Thomas Pollard, Yale University, New Haven, CT), and cloned into the NdeI-BamHI site of pET16b+. Proteins were purified using Ni-NTA-agarose according to the manufacturer's recommendation (Invitrogen). All purified proteins were stored in G buffer (5 mm Tris-HCl, pH 8.0, 0.2 mm CaCl2, 0.2 mm ATP, 0.5 mm dithiothreitol). Point mutations were introduced into pET16b+/TgADF (supplemental Table S1) using specific primers (supplemental Table S1) and the QuikChange mutagenesis kit (Stratagene), and the sequence was verified by DNA sequencing. TgADF containing the S. pombe cofilin C terminus (ADF-t) was constructed using PCR amplification with specific primers (supplemental Table S1) to add sequence encoding the last seven amino acids (LEKVTRK) of S. pombe cofilin to TgADF. ADF-t was cloned into the NdeI-BamHI site of the pET16b+ expression vector, and the protein was expressed and purified as described above.
Actin Sedimentation Assays
Rabbit actin (10 μm) was polymerized by the addition of 1/10th volume of 10× F buffer (500 mm KCl, 20 mm MgCl2, 10 mm ATP) for 30 min before the addition of AC proteins. Incubation with AC proteins (0–20 μm) was carried out for 1 h at room temperature, and filaments were separated by ultracentrifugation at 100,000 × g for 1 h (TL100 rotor, Beckman Optima TL Ultracentrifuge, Beckman Coulter, Fullerton, CA). Pellet fractions were washed once with F buffer to remove traces of the soluble fraction. Proteins were precipitated from the soluble fractions using acetone, and equivalent amounts of pellet and supernatant fractions were resolved by 15% SDS-PAGE. Quantitation of protein in the pellet and supernatant fractions was done by phosphorimaging analysis of Sypro-Ruby stained gels using a FLA-5000 phosphorimaging device (Fuji Film Medical Systems, Stamford, CT). For examining the effect of pH on activity, TgADF and actin were incubated in 1× F buffer maintained at either pH 8.2 or pH 6.8 (with the addition of 10 mm PIPES). For sedimentation assays with T. gondii actin, 5 μm actin was polymerized with the addition of 10× F buffer in the presence or absence of 5 μm phalloidin (Molecular Probes, Eugene, OR). Following incubation with 2-fold molar excess TgADF, samples were centrifuged at either 100,000 × g or 350,000 × g and processed as described above. Data are presented as the percent actin (of total) that pelleted after ultracentrifugation at the indicated speed. All average results are presented as mean ± S.E. Unpaired, equal variance, two-tailed Student's t tests were used to determine the statistical significance of differences observed between the indicated groups.
Direct Observation of Actin Filament Severing
Unlabeled and Alexa Fluor 488-labeled rabbit muscle actin (Molecular Probes) were reconstituted in G buffer, incubated on ice to depolymerize oligomers, and centrifuged at 100,000 × g to remove aggregates. Protein concentrations were determined using the Coomassie Plus (Bradford) protein assay (Pierce). Unlabeled (1.4 μm) and Alexa Fluor 488-labeled rabbit actin (0.6 μm) were copolymerized at room temperature for 2 h in ISAP buffer (50 mm KCl, 5 mm EGTA, 2 mm MgCl2, 1 mm ATP, 1 mm dithiothreitol, and 20 mm HEPES-KOH, pH 7.2). Flow chambers were assembled by mounting a 22-mm square coverslip (number 1, Corning, Corning, NY) on a 22- × 40-mm coverslip (number 0, Fisher Scientific) with two pieces of double-sided adhesive tape (Scotch), and solutions were loaded via capillary action. The following procedure was adapted from a previous study (50). Chambers were coated with 10 μg/ml N-ethylmaleimide-inactivated myosin (Cytoskeleton, inactivated as described before (51)), for 5 min, and subsequently washed with 10 mg/ml bovine serum albumin in high salt Tris-buffered saline (50 mm Tris-Cl, pH 7.6, 600 mm NaCl), followed by 10 mg/ml bovine serum albumin in low salt Tris-buffered saline (50 mm Tris-HCl, pH 7.6, 50 mm NaCl). Polymerized actin was diluted 10-fold and mixed 1:1 in 2× TIR buffer (100 mm KCl, 0.2 mm MgCl2, 2 mm EGTA, 20 mm imidazole, pH 7.0, 100 mm dithiothreitol, 0.4 mm ATP, 30 mm glucose, 2% methylcellulose, 40 μg/ml catalase, 200 μg/ml glucose oxidase) just before addition to the flow chamber. The chamber was mounted on the stage of a 1X-81 inverted microscope (Olympus America, Center Valley, PA), and filaments were allowed to settle for 5 min before washing twice with 1× TIR buffer. TgADF and S. pombe cofilin were diluted in 1× TIR buffer to 0.3–1.5 μm and loaded into the chamber. Filaments were observed by total internal reflection fluorescence (TIRF) microscopy using a 60× Plan Apo objective (oil, numerical aperture of 1.4). Images were captured on a C9100–12 charge-coupled device video camera (Hamamatsu Photonics, Bridgewater, NJ) operated and collected by Slidebook software (Intelligent Imaging Innovations, Denver, CO). Frames of 100-ms duration were collected every 5 s, and images were processed and filament lengths measured manually using ImageJ software (rsb.info.nih.gov/ij/).
Polymerization Kinetics
Actin (5 μm) was preincubated with 0–10 μm TgADF for 10 min before converting Ca-ATP-actin to Mg-ATP-actin with the addition of 10× ME (500 μm MgCl2, 2 mm EGTA) for 5 min. Polymerization was induced with 10× KMEI (500 mm KCl, 10 mm MgCl2, 10 mm EGTA, 100 mm imidazole HCl, pH 7.0), and light scattering was measured over time in a PTI Quantmaster spectrofluorometer (Photon Technology International, Santa Clara, CA). For experiments with Toxoplasma actin, 5 μm phalloidin was also added at the time polymerization was induced.
Steady-state Actin Polymerization
Rabbit actin (2.5–15 μm) was polymerized in the presence of 2.5 molar excess TgADF for 15 h at 25 °C. Steady-state levels of polymerization were measured by light scattering as above. For Toxoplasma actin, polymerization was carried out in the presence of equimolar phalloidin to actin, and 2.5 molar excess of TgADF. Light scattering was measured after 9 h at 25 °C.
Nucleotide Exchange
The effect of TgADF on the rate of nucleotide exchange by monomeric actin was assayed using rabbit or Toxoplasma actin labeled with 1,N6-ethenoadenosine 5′-triphosphate (ϵ-ATP, Molecular Probes) as described (52). Briefly, G-actin was treated with 20% volume of a 50% slurry of Dowex 1X8 Cl (200–400 mesh, Sigma-Aldrich) to remove excess nucleotide. Actin was subsequently labeled with 200 or 500 μm ϵ-ATP for 1 h at 4 °C. Following labeling, unbound ϵ-ATP was removed with Dowex treatment, and 20 μm ϵ-ATP was added back to prevent actin denaturation. To assay for the effect of TgADF on the rate of nucleotide exchange on Mg-ATP-G-actin, 1 μm actin was preincubated with 0–20 μm TgADF for 5 min, and the actin was converted to Mg-ATP-actin with the addition of 100 μm MgCl2 and 200 μm EGTA for 5 min. Unlabeled ATP (1.25 mm) was then added to displace ϵ-ATP from actin, and the corresponding loss in fluorescence was monitored over time using the PTI Quantmaster spectrofluorometer (excitation = 360 nm, emission = 410 nm). To estimate the apparent affinity of TgADF for monomeric actin, curve-fitting was conducted on the combined data from three experiments, using values that were consistently observed in two or more experiments. The data were fit using nonlinear regression analysis based on first order exponential decay kinetics using Prism (GraphPad software, San Diego, CA).
RESULTS
Comparison of Apicomplexan ADFs with Other AC Family Members
To determine the similarity of apicomplexan ADFs with other ADF/AC family members, a structure-based multiple sequence alignment was generated (Fig. 1A) using the homology model of the structure of TgADF, based on the known crystal structure of A. castellani actophorin (Fig. 1B). The apicomplexan ADF proteins are the smallest in the AC family, with TgADF encoding a protein of ∼118 amino acids that shares ∼63% homology and 39% identity with actophorin, and ∼47% homology and ∼30% identity with AC isoforms from the higher eukaryotes depicted in Fig. 1A. The predicted structure of TgADF maps onto the structure of actophorin quite well, with the conserved actin binding sites overlapping between the two structures. However, a few striking differences were apparent, and these are discussed in light of the biochemical activities described below.
FIGURE 1.

Comparison of apicomplexan ADFs with representative ADF/Cofilin proteins. A, ClustalX alignment of ADF/Cofilin family members highlighting key features. Actin binding sites previously identified in S. cerevisiae cofilin by mutagenesis (29) or synchrotron protein footprinting (34) are indicated by asterisk or circle, respectively. Residues required exclusively for F-actin binding are boxed in black. Serine 3, glycine 66, and C-terminal residues that were mutated or added back in TgADF to analyze the actin binding sites in Fig. 3, are boxed in purple. Sequences shown are: AtADF1, A. thaliana ADF1; AcActophorin, A. castellani actophorin; ScCOF, S. cerevisiae cofilin; SpCOF, S. pombe cofilin; CeUnc60A, C. elegans Unc60A; PfADF2, P. falciparum ADF2; HsADF, H. sapiens ADF; PfADF1, P. falciparum ADF1; and TgADF, T. gondii ADF. B, homology model of T. gondii ADF (TgADF, shown in yellow) based on A. castellani actophorin (Actophorin, Protein Data Bank entry 1ahq, shown in blue). Arrow points to the short F-loop in TgADF compared with actophorin.
Actin binding sites identified by site-directed mutagenesis (29) and synchrotron protein footprinting (34) have previously been mapped on yeast cofilin and are conserved across AC family members (Fig. 1A, denoted by the asterisk or circle, respectively). These sites are found primarily at the N terminus, the long α3 helix, and at the C terminus (Fig. 1A). Two regions are identified as being required for interacting with F-actin exclusively (Fig. 1A, black boxes). Charged residues at the C terminus in the α4 helix and/or in the C-tail extension are thought to pack against the side of the filament and stabilize the interaction between ADF and the filament. Additionally, the F-loop, consisting of strands β4 and β5, which typically projects out of the AC structure (Fig. 1B, arrow), is predicted to intercalate within the filament (38). These F-actin binding sites cluster together to form an interface on the opposite side of the molecule from the G-actin binding interface, which is composed of the α3 helix and the N terminus (35–37).
Apicomplexan ADFs have conserved most of the G-actin binding sites (Fig. 1A). However, the sites required exclusively for binding to F-actin are notably absent (Fig. 1A, black boxes). This is true across the apicomplexan ADFs, with the exception of PfADF2 (Fig. 1A and data not shown). Apicomplexan ADFs possess a truncated C terminus, which may not fold into the terminal α4 helix, and they lack the C-terminal charged residues predicted as necessary for F-actin interactions (Fig. 1B). The conserved basic F-loop residue (Arg-78 in S. pombe cofilin, Gly-66 in TgADF) shown to be critical for F-actin binding in human and yeast cofilin (29, 38), is also absent in apicomplexan ADFs (Fig. 1A, black boxes). The F-loop in TgADF and other apicomplexan ADFs (45), is itself much shorter compared with other AC proteins due to shorter β4 and β5 strands and a smaller transition between them, and therefore does not project out of the structure (Fig. 1B, arrow), and presumably would not interact with the filament. These striking differences in the predicted actin binding sites are likely to have important consequences for the activity and function of apicomplexan ADF proteins.
Characterization of T. gondii ADF Activity
To characterize the activity of TgADF, recombinant protein was expressed and purified in Escherichia coli and tested for its ability to disassemble actin filaments using a sedimentation assay (Fig. 2A). Because TgADF lacks many key residues required for F-actin binding, the activity of the wild-type protein was first established. The effect of TgADF on rabbit actin filaments was analyzed by quantifying the proportion of F-actin sedimenting at 100,000 × g after the addition of F buffer and 1-h incubation with different concentrations of protein. TgADF caused the net disassembly of rabbit actin filaments in a dose-dependent manner, resulting in almost complete disassembly of filaments when present at 2:1 molar excess (Fig. 2A). Interestingly, very little TgADF (∼8%), co-sedimented with actin filaments at any dose, and this proportion did not increase when higher concentrations of TgADF were used. This is in contrast to what is typically seen with AC proteins such as plant ADF and human cofilin (19, 38). These data indicated that the disassembly of filaments does not require the stable association of AC proteins with F-actin.
FIGURE 2.
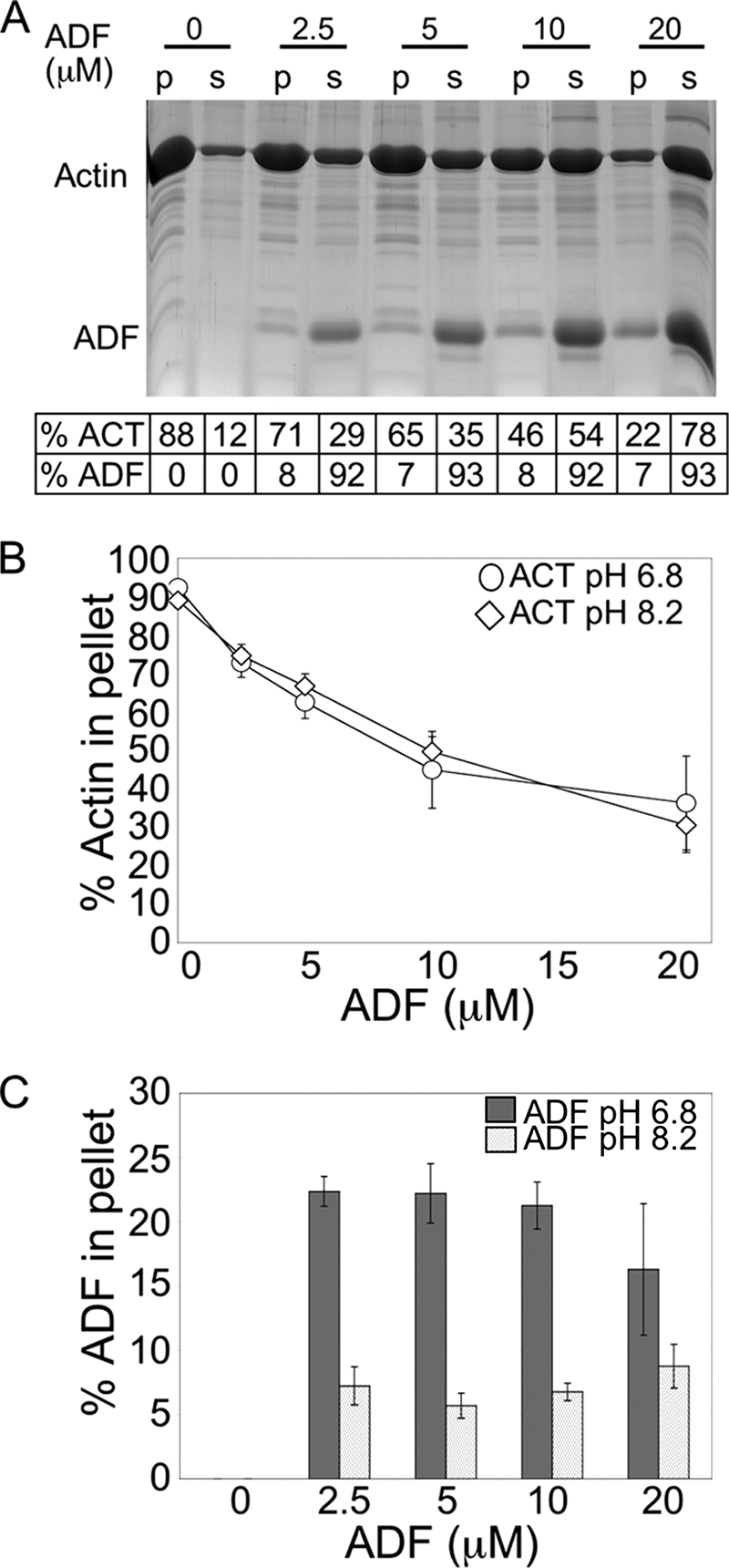
Characterization of T. gondii ADF activity. A, dose-dependent disassembly of rabbit actin filaments by TgADF. A representative Sypro-Ruby-stained gel showing the effect of increasing concentrations of TgADF on the amount of F-actin pelletting at 100,000 × g. Quantitation of the proportion of actin and TgADF in the pellet and supernatant fractions is given below. Rabbit actin (10 μm) was polymerized into filaments by the addition of F buffer, before incubation with TgADF (0–20 μm). Samples were centrifuged (100,000 × g) to sediment actin filaments, and the pellet (p) and supernatant (s) fractions were analyzed by SDS-PAGE. Bands were quantified by phosphorimaging analysis of Sypro-Ruby-stained gels. B, effect of pH on the disassembly of filaments by TgADF. Quantitation of the proportion of actin in the pellet fraction after interaction with TgADF at pH 6.8 or 8.2 is shown. Rabbit actin was polymerized under normal conditions (pH 8) and interacted with TgADF at either pH 6.8 or 8.2 (n = 3 experiments, mean ± S.E.). C, effect of pH on TgADF co-sedimentation with actin filaments. Quantitation of the proportion of TgADF in the pellet fraction after interaction with rabbit actin at pH 6.8 or 8.2 is shown (n = 3 experiments, mean ± S.E.).
Most AC proteins show pH-dependent activity, binding to filaments at neutral pH, and causing the net disassembly of filaments under slightly alkaline conditions (52–54). pH regulation has been posited as a mechanism to regulate the activity of AC proteins in vivo (55). Because our polymerization buffer is buffered at the pH permissive for activity, we tested the effect of lower pH on TgADF activity and co-sedimentation with actin filaments. TgADF was equally active at both pH 6.8 and pH 8.2 (Fig. 2, B and C). Although more TgADF co-sedimented with filaments at pH 6.8 (∼20% compared with ∼8% at pH 8.2), this did not affect its filament disassembly activity, perhaps because the fraction co-sedimenting with filaments was still relatively low compared with what has been observed with other AC proteins. Additionally, even though the absolute amount of TgADF co-sedimenting with filaments increased as more protein was added to the reaction, the proportion of TgADF that co-sedimented remained approximately the same or decreased, and this was true at both pHs, suggesting that filament binding was not saturated. Thus, despite lacking some conserved actin binding sites, TgADF was active and able to cause extensive net disassembly of actin filaments, while not stably associating with filaments. Activity was pH-independent suggesting that TgADF does not require pH activation.
Comparison of TgADF Activity with Other ADF/Cofilin Proteins, and Mutational Analysis of Actin Binding Sites
Although TgADF demonstrated potent net filament disassembly activity, we wanted to directly compare its activity to commonly studied AC proteins. S. pombe cofilin and A. castellani actophorin were chosen as representative proteins to provide a spectrum of activity. S. pombe cofilin efficiently disassembles actin filaments by severing, whereas the severing activity of actophorin is relatively weak (12). Both AC proteins were expressed and purified in the same way as TgADF and tested in parallel actin co-sedimentation assays (Fig. 3A). All three AC proteins showed negligible sedimentation (1–5% of the total protein) in the absence of actin (data not shown). As seen above, TgADF caused the efficient net disassembly of rabbit actin filaments, with very little protein co-sedimenting with filaments (Fig. 3A). In contrast, both S. pombe cofilin and actophorin showed very modest net filament disassembly, with S. pombe cofilin showing slightly better activity. A maximal effect of 20% less F-actin was observed with 2-fold molar excess S. pombe cofilin, compared with 65% less F-actin with 2-fold excess TgADF (Fig. 3A). Interestingly, ∼45% of both actophorin and S. pombe cofilin co-sedimented with actin filaments (data not shown).
FIGURE 3.
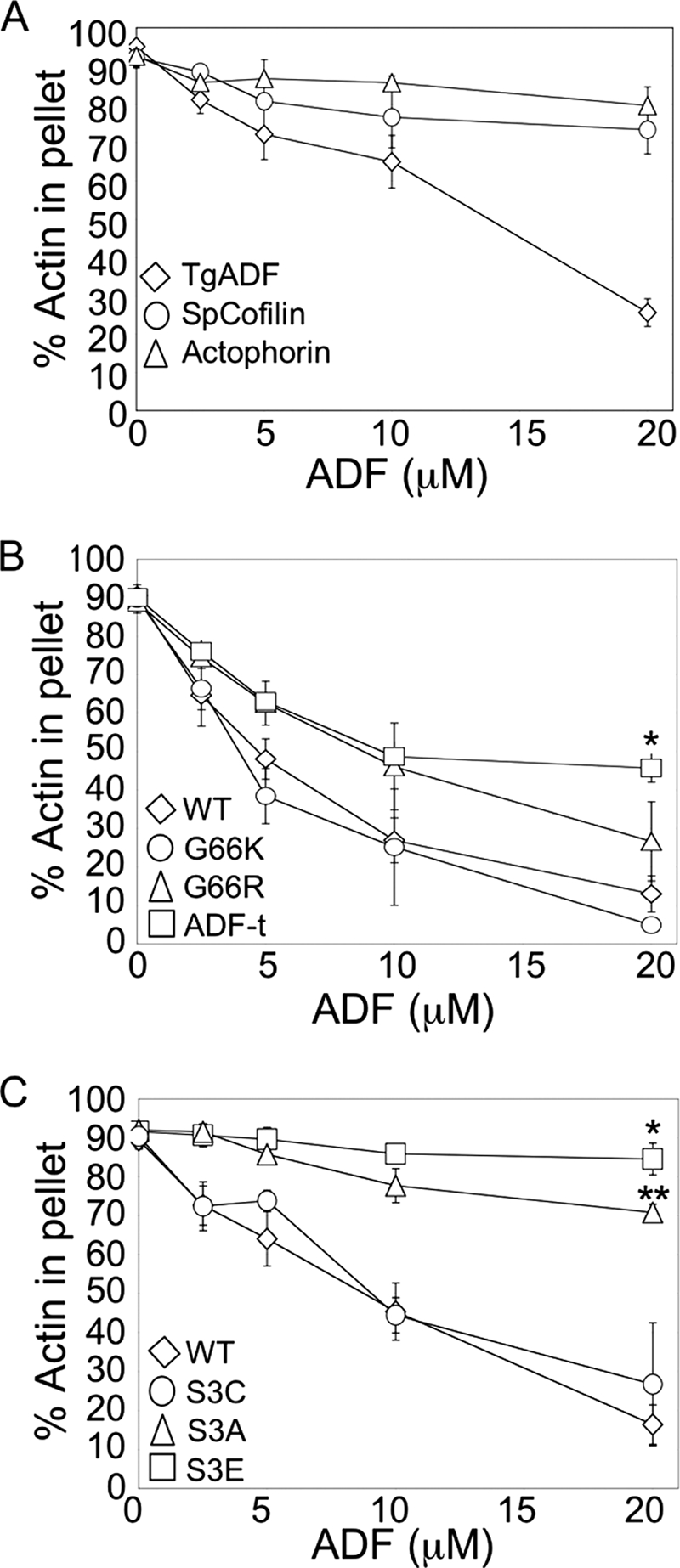
Comparison of TgADF activity with other ADF/Cofilin proteins, and mutational analysis of actin binding sites. A, comparison of TgADF activity with ADF/Cofilin proteins S. pombe cofilin and A. castellani actophorin. Quantitation of the proportion of actin sedimenting at 100,000 × g after polymerization by the addition of F buffer and incubation with TgADF, S. pombe cofilin (SpCofilin) or A. castellani actophorin (Actophorin). Experiments were done as described in Fig. 2. (n = 3 experiments, mean ± S.E.). B, effect of putative F-actin binding sites on TgADF activity. The filament disassembly activity of TgADF expressing the conserved basic F-loop residue (G66K or G66R) or the C-terminal residues of S. pombe cofilin (ADF-t) was compared with wild-type (WT) TgADF. The graph shows the relative proportion of actin sedimenting at 100,000 × g (n = 3 experiments, mean ± S.E.; *, p < 0.005 Student's t test, ADF-t versus WT). C, activity of TgADF serine 3 mutants. Actin filament disassembly activity of TgADF with mutations at the serine 3 residue to cysteine (S3C), alanine (S3A), or glutamic acid (S3E), were compared with WT TgADF (WT). The graph shows the relative proportion of actin sedimenting at 100,000 × g (n = 3 experiments, mean ± S.E.; *, p < 0.001 Student's t test, S3E versus WT; **, p < 0.05 Student's t test, S3A versus S3E).
To determine whether differences in TgADF activity could be attributed to the lack of F-actin binding sites in TgADF, mutation analysis was used to determine the relative importance of previously defined F-actin binding sites (Fig. 3, B and C). Earlier work by Pope et al., (38) demonstrated that mutating the first (Arg-96 in S. pombe cofilin) of the two basic residues in the F-loop region that are critical for F-actin binding, results in human cofilin losing its ability to bind F-actin, and instead causes it to effect the extensive depolymerization of filaments, similar to what was seen with TgADF (Fig. 3A). To determine if the lack of a basic residue at this site might play a role in the differential activity of TgADF compared with S. pombe cofilin and actophorin, the corresponding point mutations were made in TgADF (G66R and G66K). Conversion to a basic residue at this site had little effect on TgADF co-sedimentation or disassembly of actin filaments (Fig. 3B), indicating that this site alone is not sufficient to confer F-actin binding to TgADF.
A second site for F-actin binding is located at the C terminus of AC proteins, which is truncated in apicomplexan ADFs (29). To determine if a more stable interaction between TgADF and F-actin could be reconstituted, the last seven residues of S. pombe cofilin were added on to TgADF to generate TgADF-t, and the activity of this protein was assessed in the actin sedimentation assay (Fig. 3B). Addition of the cofilin tail residues to TgADF had a partial effect on activity, resulting in a 2-fold increase in the amount of TgADF-t co-sedimenting with F-actin (data not shown), and decreased disassembly of F-actin (Fig. 3B). Addition of the F-loop mutations, G66K or G66R, to TgADF-t, did not however have any additional effect on the activity or co-sedimentation of the TgADF-t protein with filaments (data not shown). These data indicate that addition of charged residues to the C terminus of TgADF was able to partially restore binding to F-actin, and this resulted in decreased net filament turnover.
The N terminus of AC proteins is highly conserved and has previously been identified by mutagenesis in yeast cofilin to be an important site for binding to both F- and G-actin (29). In particular the serine 3 residue is an important contact site for interactions with actin, and the activity of some AC proteins is negatively regulated by phosphorylation at this site (56, 57). Although apicomplexan ADFs lack key F-actin binding sites, the N terminus is very highly conserved with the AC family. To test if this site is important for TgADF activity, point mutations were made at the serine 3 residue. The S3E mutation was made to mimic potential phosphorylation at this site and has been shown to inhibit actin binding for other AC proteins (56, 57), whereas the S3A mutation was made as a control to show that any loss of activity was due to the introduction of negative charge at this site. These proteins were then compared using the actin sedimentation assay (Fig. 3C). Although the S3E mutation resulted in a complete loss of activity, as has previously been seen with the analogous point mutations in human cofilin (38), chick ADF (56), and plant ADF (58, 59), the S3A mutation also resulted in a significant loss of TgADF activity, rendering it only 25% as active as the wild-type protein (Fig. 3C). This is in contrast to plant ADF (58) and actophorin (60) where the analogous mutants demonstrate identical or 75% activity compared with the respective wild-type proteins, indicating that TgADF was particularly sensitive to mutation at this site (Fig. 3C). Because the amino acid cysteine more closely mimics serine (both small, polar amino acids), serine 3 was mutated to cysteine. This mutant showed wild-type activity (Fig. 3C) suggesting that the polar nature of this residue was particularly important for actin interactions. Thus, despite differences in the strength of its interaction with F-actin, TgADF appears to still share the same general actin binding sites, and these are critical for its activity.
Severing Activity of TgADF
S. pombe cofilin is reported to have extremely efficient severing activity (26). To examine the severing activity of TgADF, TIRF microscopy was used to directly observe filament severing. Rabbit muscle actin was co-polymerized with Alexa Fluor 488-labeled actin, and filaments were tethered to glass flow chambers with N-ethylmaleimide-inactivated myosin. TgADF or S. pombe cofilin were flowed into the chamber at time zero, and time-lapse TIRF microscopy was used to capture severing of filaments over time (Fig. 4).
FIGURE 4.

Severing activity of TgADF as observed by TIRF microscopy. A, severing of actin filaments by TgADF and S. pombe cofilin. Fluorescence time-lapse micrographs of actin filaments were taken over a period of 0–12 min after the addition of 0.3 μm S. pombe cofilin (SpCOF, top), 0.3 μm TgADF (middle), or 1.5 μm TgADF (bottom) at time zero. Rabbit actin co-polymerized with Alexa Fluor 488-labeled actin was tethered to flow chambers with N-ethylmaleimide-treated myosin. Time-lapse TIRF microscopy was used to visualize filament severing by TgADF and S. pombe cofilin over time. Scale bar, 10 μm. B, quantitation of the rate of filament disassembly by TgADF and S. pombe cofilin. The average length (mean ± S.E.) of the 15 longest filaments in the field of view was calculated at the indicated time points after TgADF or SpCOF addition and plotted for each condition (n = 3 experiments). C, detailed montage of actin filaments being severed by 1.5 μm TgADF over time. Scale bar, 10 μm.
At a concentration of 0.3 μm, S. pombe cofilin caused the rapid disassembly of actin filaments within the first 3 min (Fig. 4A, SpCOF, top). After 6 min there was little further change, with severed fragments drifting out of view, occasionally leaving behind longer stable fragments that remained attached to the surface of the chamber (Fig. 4A, SpCOF, top). In contrast, very little activity was observed with 0.3 μm TgADF (Fig. 4A, middle), except for the occasional breakage of longer filaments after 9 min. To determine if TgADF had any severing capacity, activity was examined at 5-fold higher concentrations of TgADF. At 1.5 μm TgADF (Fig. 4A, bottom), severing was clearly visible after 6 min, indicating that TgADF can sever filaments when present at higher concentrations.
To quantitate the relative rate of filament disassembly for both TgADF and SpCOF under these conditions, the average length of the longest class of filaments was calculated over time (Fig. 4B). TgADF at a concentration of 1.5 μm was found to disassemble actin filaments at a rate approximately half that of 0.3 μm S. pombe cofilin (initial rates of ∼1.1 ± 0.442 μm/min compared with 2.0 ± 0.404 μm/min, respectively). To directly visualize severing activity, single filaments in the presence of 1.5 μm TgADF were tracked over time (Fig. 4C). Severing activity was visualized as a break in the filament and the subsequent fragmentation into more pieces (Fig. 4C).
Interaction of TgADF with T. gondii Actin Filaments during Sedimentation
Studying the interaction of TgADF with a heterologous protein such as rabbit actin is useful to provide a frame of reference for identifying the salient properties of TgADF in comparison to other AC family members. However, to determine the biologically relevant functions of TgADF it was important to examine its interaction with its homologous actin substrate. T. gondii expresses one actin allele, TgACT1, which shares 83% identity with rabbit actin (8). Actin sedimentation assays were used to investigate how TgADF interacts with TgACT filaments.
As previously reported, T. gondii actin is inherently unstable, and, unlike higher eukaryotic actins, the addition of F buffer does not induce formation of long stable filaments that sediment at 100,000 × g (Fig. 5A) (9). However, stable TgACT filaments can be rescued when TgACT is polymerized in the presence of equimolar phalloidin (Fig. 5A).3 Small actin oligomers that formed in F buffer were also sedimented by centrifugation at 350,000 × g, as described previously for Toxoplasma and Plasmodium actins (9, 11). The interaction between TgADF and TgACT filaments was examined under these three conditions.
FIGURE 5.

Interaction of TgADF with T. gondii actin filaments during sedimentation. A, effect of TgADF on the sedimentation activity of T. gondii actin filaments polymerized by the addition of F buffer. A representative Sypro-Ruby-stained SDS-PAGE gel shows the proportion of T. gondii actin sedimenting at 100,000 × g or 350,000 × g in the absence and presence of phalloidin or TgADF. Quantitation of the actin bands is indicated in the table below. p = pellet, s = supernatant. B, quantitation of the percentage of actin in the pellet fraction under the conditions in (A) based on three independent experiments, mean ± S.E.; *, p < 0.005 Student's t test, TgACT alone versus TgACT plus TgADF.
In the absence of phalloidin, only 20–30% of the TgACT in F buffer sedimented at 100,000 × g, and TgADF had little effect on this behavior (Fig. 5B). In the presence of equimolar phalloidin in F buffer, there was a 50% increase in the amount of TgACT sedimenting at 100,000 × g, and a 2-fold molar excess of TgADF had only a modest effect on this population (Fig. 5B). However, when TgACT was polymerized by the addition of F buffer and centrifuged at 350,000 × g to pellet small oligomers, 70–80% of the total actin was pelleted (Fig. 5A). A 2-fold molar excess of TgADF was able to disassemble 50% of the actin that pelleted under these conditions (Fig. 5B). Interestingly, TgADF did not co-sediment with TgACT under any of the conditions tested indicating that it did not stably associate with TgACT filaments or oligomers (Fig. 5A). This is in contrast to S. pombe cofilin, which co-sedimented with TgACT at 350,000 × g (data not shown). These data indicate that TgADF can efficiently disassemble small TgACT oligomers.
Effects of TgADF on Actin Polymerization Kinetics
The data thus far suggested that TgADF had a weak affinity for actin filaments, yet it could disassemble small oligomers of TgACT1 and rabbit actin filaments. To investigate the effect of TgADF on actin polymerization, light scattering was used. Actin was incubated with TgADF for 5 min before polymerization was induced with the addition of KMEI buffer. The increase in light scattering was measured over time (Fig. 6A). At ≤ 1:1 molar ratio TgADF:rabbit actin, there was an increase in the initial rate of polymerization with TgADF (Fig. 6A). This could be due to weak severing of filaments, which would generate more seeds for elongation. Alternatively, transient binding of TgADF to filaments could cause an increase in light scattering. However, when present in 2-fold molar excess, TgADF inhibited the nucleation and polymerization of rabbit actin filaments, suggesting that TgADF was sequestering actin monomers. When TgACT was used as the substrate and polymerization induced by the addition of KMEI buffer and equimolar phalloidin, TgADF inhibited polymerization at all doses, even when extremely low TgADF concentrations were used (Fig. 6B). The more potent effect of TgADF on TgACT polymerization compared with rabbit actin could be due to higher affinity interactions between TgADF and TgACT, or because TgACT polymerization was slower and more sensitive to inhibition. These data demonstrated that TgADF strongly inhibited TgACT polymerization even at low doses, and this occurred much more efficiently than with rabbit actin.
FIGURE 6.

Effects of TgADF on actin polymerization kinetics. A, effect of TgADF on rabbit actin polymerization. Polymerization of rabbit actin (RbACT, 5 μm) was measured by light scattering in the presence of 0–10 μm TgADF. Rabbit actin was incubated with TgADF for 10 min prior to initiation of polymerization with the addition of 1/10th the volume of 10× KMEI. A representative experiment is shown (n = 3). B, effect of TgADF on T. gondii actin polymerization. Polymerization of T. gondii actin (TgACT, 5 μm) in the presence of 0–5 μm TgADF was measured over time by light scattering. Experiments were done as in A, with 5 μm phalloidin added at the time of polymerization, to stabilize T. gondii actin filaments. A representative experiment is shown (n = 2).
To test whether TgADF was able to sequester G-actin, the effect of TgADF on steady-state actin polymerization was examined (Fig. 7). Rabbit actin was polymerized in the presence of 2.5 molar excess TgADF or S. pombe cofilin, and steady-state polymerization was measured by light scattering (Fig. 7A). TgADF inhibited the steady-state polymerization of at least 15 μm rabbit actin. In contrast, S. pombe cofilin caused an increase in light scattering at actin concentrations of 10 μm and above. This increased light scattering could be due to enhanced polymerization in the presence of S. pombe cofilin, which has been shown to nucleate filaments when present at 2-fold molar excess (26), and/or from S. pombe cofilin binding to filaments and leading to increased mass. When the effect of TgADF on steady-state TgACT polymerization was examined (Fig. 7B), TgADF reduced the extent of steady-state polymerization at all actin concentrations tested. This clearly indicates that TgADF inhibits actin polymerization by sequestering actin monomers.
FIGURE 7.
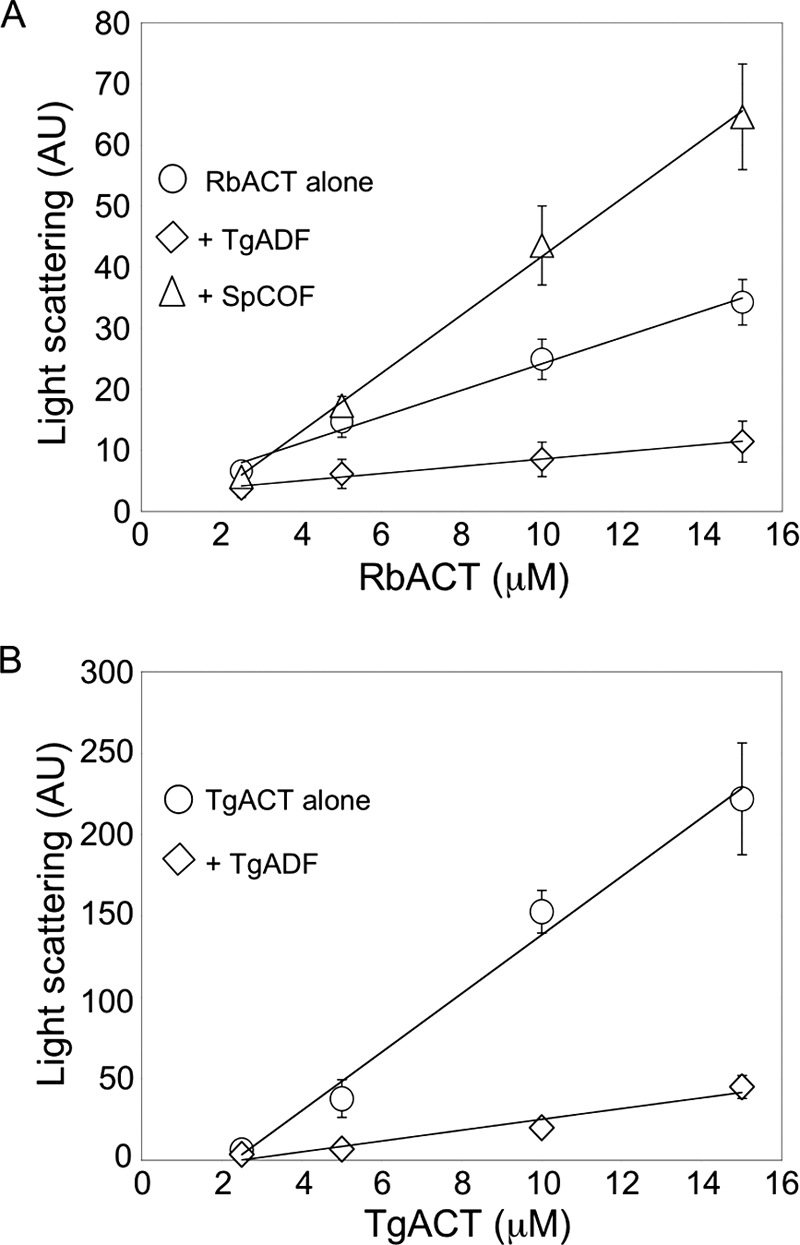
Effect of TgADF on steady-state actin polymerization. A, effect of TgADF on the steady-state polymerization of rabbit actin as measured by light scattering. Varying concentrations of rabbit actin (RbACT, 2–15 μm) were polymerized in KMEI buffer, in the presence of 2.5 molar excess TgADF or S. pombe cofilin (SpCOF) at 25 °C until steady state was achieved. The data represents the average (mean ± S.E.) of three independent experiments. B, effect of TgADF on T. gondii actin steady-state polymerization as measured by light scattering. Varying concentrations of T. gondii actin (TgACT, 2–15 μm) were polymerized in KMEI buffer, in the presence of equimolar phalloidin and 2.5 molar excess TgADF at 25 °C until steady state was achieved. The data represent the average (mean ± S.E.) of three independent experiments.
Effects of TgADF on Nucleotide Exchange of G-actin
To demonstrate a direct interaction between TgADF and G-actin, we measured the effect of TgADF on the exchange of nucleotide bound to actin monomers (Fig. 8). AC proteins typically bind to G-actin and inhibit the exchange of actin bound nucleotides (26, 40, 43, 52, 53), with the exception of Plasmodium ADF1 and Tetrahymena ADF73p, which were recently shown to stimulate nucleotide exchange (42, 61). Monomeric actin was labeled with the ATP analogue ϵ-ATP, which fluoresces when bound to actin, and the rate of nucleotide exchange was measured as a decrease in fluorescence when the actin-bound ϵ-ATP was displaced with unlabeled ATP. In the absence of TgADF, the nucleotide exchange rate of TgACT was ∼2- to 3-fold faster than RbACT (Fig. 8, A and B). However, in the presence of TgADF, nucleotide exchange was inhibited on both RbACT and TgACT monomers in a dose-dependent manner (Fig. 8, A and B). Interestingly, at low ratios (<1:1) of TgADF and TgACT, an ∼2-fold increase in the initial rate of nucleotide exchange was often observed (Fig. 8B, green curve (0.25 μm TgADF) compared with dark blue curve (0 μm)), suggesting that, at very low concentrations, TgADF may stimulate nucleotide exchange. Using the dose-dependent inhibition of nucleotide exchange in the presence of TgADF, we estimated the apparent affinity of TgADF for TgACT and RbACT monomers to be ∼0.81 and 0.64 μm, respectively. These data demonstrate a direct interaction of moderate affinity between TgADF and ATP-actin monomers, providing additional support for the sequestering activity of TgADF.
FIGURE 8.

Effect of TgADF on nucleotide exchange of G-actin. A, effect of TgADF on the nucleotide exchange rate of monomeric rabbit actin (RbACT). The nucleotide exchange rate of ϵ-ATP-labeled Mg-actin monomers (1 μm), in the presence of varying concentrations of TgADF (0–20 μm), was monitored by measuring the loss in fluorescence over time (emission = 410 nm), upon the addition of 1.25 mm unlabeled ATP at time = 0. A representative experiment is shown (n = 3). The concentrations of TgADF used, given in order of appearance, were 0, 0.1, 0.25, 1, 2, 0.5, 10, and 20 μm (represented by a shaded triangle to the right of the graph). AU = arbitrary units. B, effect of TgADF on the nucleotide exchange rate of T. gondii actin (TgACT) monomers. The rate of nucleotide exchange on TgACT monomers was measured in the presence of varying concentrations of TgADF (0–20 μm). Experiments were done as in A. A representative experiment is shown (n = 3). The concentrations of TgADF used, given in order of appearance, were 0.25, 0, 0.5, 1, 2, 10, and 20 μm (represented by a shaded triangle to the right of the graph). C, plot of the observed rate constants for nucleotide exchange on TgACT and RbACT monomers in the presence of varying concentrations of TgADF. Rate constants were derived from the initial reaction rates calculated from curves similar to those shown in A and B and plotted against TgADF concentration. The data were fit using first-order exponential decay kinetics and represent the averages of two (RbACT), or three (TgACT), independent experiments (mean ± S.E.).
DISCUSSION
To determine the mechanism by which TgADF accelerates actin filament turnover, we analyzed its biochemical interactions with both mammalian and T. gondii actin in vitro. Although severing is typically the main mechanism by which AC proteins are thought to effect filament turnover (62), TgADF was a comparatively weak severing protein. We found that the absence of key F-actin binding sites in TgADF was associated with high net filament disassembly activity. We demonstrate that the primary mechanism for the efficient net disassembly of actin filaments by TgADF is due to the strong sequestering of actin monomers. These properties identify an ADF that is adapted to function in a primarily G-actin-rich environment, where filaments are rare and rapidly assemble and turn over for very specific biological processes.
Two sites have previously been identified in AC proteins as critical for F-actin interactions: charged residues at the C terminus of the protein (29, 39), and two basic residues in the F-loop (29, 38) that extends out of the crystal structure of AC proteins. Most apicomplexan ADFs, excluding Plasmodium PfADF2, are deficient at both sites. Apicomplexan ADFs are truncated at the C terminus and therefore lack the C-terminal α4 helix and the C-tail extension. The first of the two basic residues in the F-loop is also missing in all apicomplexan ADFs. In addition to this, the apicomplexan ADFs that have been examined by homology modeling (present study and Ref. 45) appear to contain a very short F-loop, which likely does not promote tight binding to the filament. Consistent with the lack of key F-actin binding sites in its molecular structure, TgADF displayed limited co-sedimentation with actin filaments, indicating the absence of stable interactions with filaments. This property is unusual for proteins in the AC family (19, 28, 38). However, the lack of a stabilizing interaction with F-actin was associated with a greater effect on the net disassembly of actin filaments. Addition of the C-terminal F-actin binding site from S. pombe cofilin to TgADF stabilized the interaction of TgADF with F-actin, but concomitantly decreased the net filament disassembly activity, indicating that the two properties are inversely related. Restoration of the conserved basic F-loop residue to TgADF had no effect on its activity. However, this is likely due to the F-loop being considerably shorter in TgADF, such that it may not facilitate contact with the filament even when these positive charges are restored. Such high net filament disassembly activity has only been previously observed with a very small subset of AC proteins (Unc60A (40), human ADF (52), chick ADF (53), and echinoderm depactin (63)). This activity is typically pH-dependent, with human and chick ADF binding to the filaments between pH 6.8–7.1, and causing net disassembly above pH 7.5 (52, 53) (pH dependence has not been examined for depactin). In contrast, TgADF displayed potent activity at both the permissive and non-permissive pH, suggesting that wild type TgADF did not have the features to strongly interact with actin filaments.
S. pombe cofilin and actophorin are two well characterized members of the AC family, and group into the cofilin-like class of AC proteins as described by Chen et al. (64). Both proteins co-sedimented with actin filaments and showed only modest net filament disassembly, even at the pH permissive for activity. AC proteins that can bind better to F-actin may be more effective at filament severing, and there is some data to support this model (39). When the severing activity of TgADF was compared with S. pombe cofilin, TgADF was found to be a comparatively weak severing protein, with 5-fold higher concentrations of TgADF yielding a filament disassembly rate that was still 2-fold less than S. pombe cofilin. Our data suggest that TgADF requires much higher concentrations to sever filaments than is typical for AC proteins (26). The severing activity of TgADF was similar to the reported activity of the worm AC homologue Unc60A, which also requires concentrations as high as 2 μm to detect severing (40). The requirement for comparatively high concentrations of TgADF to detect severing, and the absence of stable association with actin filaments, suggested that TgADF had a low affinity for actin filaments and that severing alone could not be the primary mechanism by which TgADF caused the efficient net disassembly of actin filaments.
The activities of most other apicomplexan actin-binding proteins have been suggested based solely on their interactions with heterologous actin proteins (21–23). Because key differences exist between the molecular structure of apicomplexan actins compared with conventional eukaryotic actins (9), we wanted to confirm that TgADF interactions relevant to the apicomplexan actin system were being captured. Using recombinant TgACT we found that TgADF efficiently disassembled short TgACT oligomers that sedimented at 350,000 × g. Interestingly, TgADF failed to co-sediment with TgACT at either 100,000 × g or 350,000 × g, indicating that the failure to co-sediment with rabbit actin filaments was not due to potential structural differences between the heterologous and homologous substrates, but rather that TgADF does not interact strongly with F-actin. The disassembly of short TgACT oligomers was a specific activity of TgADF, because S. pombe cofilin did not cause the disassembly of these oligomers, and instead co-sedimented, and increased the proportion of TgACT that sedimented at 350,000 × g (data not shown). The limited interaction of TgADF with actin filaments and oligomers, while causing their efficient disassembly, suggested that TgADF had a weak affinity for polymerized actin, but a strong affinity for actin monomers.
To directly ascertain the result of TgADF interactions with actin monomers, we monitored its effect on actin polymerization. Low concentrations of TgADF efficiently inhibited the polymerization of TgACT, with an almost complete inhibition of polymerization in the presence of equimolar TgADF. A qualitatively similar result was seen with rabbit actin, although excess TgADF was required to observe this effect. This inhibition of polymerization is in contrast to most AC proteins, which typically cause increased polymerization or overshoot kinetics with increasing amounts of protein (19, 65). However, an inhibitory effect on actin polymerization has previously been observed with the C. elegans AC homologue Unc60A (65), embryonic chicken skeletal muscle ADF (66), and echinoderm depactin (63). Similar to the effect of TgADF on TgACT polymerization, Unc60A and embryonic chick ADF inhibit actin polymerization at substoichiometric concentrations in a dose-dependent manner (40, 66). Additionally, when present at a 1:1 or 2:1 molar ratio with actin, Unc60A strongly inhibits actin nucleation, similar to what is seen with TgADF (40). In contrast, echinoderm depactin causes an initial overshoot in actin polymerization before inhibiting overall polymerization levels, and this overshoot is possibly due to severing activity, making more free ends available for polymerization (63).
Because the delay in the nucleation phase of polymerization was suggestive of monomer sequestration, steady-state polymerization assays were done in the presence of TgADF to further investigate this. The steady-state polymerization of up to 15 μm actin (for both T. gondii and rabbit actin) was inhibited in the presence of TgADF. This assay conclusively demonstrated that TgADF primarily interacts with actin by sequestering actin monomers. This fairly unusual finding of strong sequestering activity by an AC protein has previously only been directly shown for Unc60A (40), which also inhibits the steady-state polymerization of 15 μm actin, and for the embryonic chicken skeletal muscle ADF (66), which inhibits the steady-state polymerization of actin at substoichiometric concentrations (as determined by reduced viscosity measurements, and higher G-actin concentrations).
To demonstrate a direct interaction between TgADF and monomeric actin, we examined the effect of TgADF on the rate of nucleotide exchange by G-actin. AC proteins typically bind to and inhibit nucleotide exchange by G-actin (43, 52, 53), and TgADF was found to similarly inhibit nucleotide exchange by both TgACT and rabbit actin in a dose-dependent manner. Based on this, the apparent affinity of TgADF for Mg-ATP-G-actin was calculated to be 0.81 and 0.64 μm for TgACT and rabbit actin, respectively. Although most AC proteins bind to ADP-G-actin with affinities of 0.5–1 μm, there is a 10- to 20-fold decrease in the affinity for Mg-ATP-G-actin (64). In contrast, the higher affinity of TgADF for Mg-ATP-G-actin is a property shared with the other monomer sequestering AC proteins. For example, Unc60A and chick ADF have affinities for Mg-ATP-G-actin of ∼1.6 μm and 1 μm, respectively (40, 64). The higher affinity of these AC proteins is consistent with the observed sequestering of Mg-ATP-G-actin, which is thought to be the predominant form of G-actin in vivo (64).
Although strong depolymerizing activity was the basis for how many ADF proteins were originally identified (63, 67–69), the mechanism for how depolymerization was occurring was not defined. Although there is no obvious signature in the amino acid sequence that distinguishes the AC proteins with strong monomer sequestering activity as being more similar, structural features that suggest the lack of, or perturbation to F-actin binding sites may be a unifying property. This is observed with TgADF, which lacks known F-actin binding sites, and Unc60A, in which the surface-exposed insertion of charged residues (65) may disrupt filament interactions. Another shared feature of this sub-type of AC isoforms, is their expression in G-actin rich environments (63, 66, 67). In the case of embryonic chicken skeletal muscle ADF, its expression has been shown to correlate with the G-actin content in the cell (66). In embryonic cells, a large proportion of the actin is present as G-actin (70). ADF is abundantly expressed in these cells, and both inhibits actin polymerization and depolymerizes F-actin (66). As the skeletal muscle develops, the proportion of F-actin in the cell increases and the amount of actin turnover decreases (66). This change in the actin milieu also corresponds to a drop in ADF expression (66), suggesting that the ADF isoform is expressed when rapid changes in the actin cytoskeleton are required.
In T. gondii, 98% of the actin is unpolymerized (6, 8, 9), yet the parasite depends on filamentous actin to achieve rapid rates of motility with speeds of ∼1–10 μm/s (71) and to productively invade host cells. This scenario is similar to the actin dynamics in the embryonic skeletal muscle described above, in that most of the actin is in the G-form, and filaments must be rapidly assembled and disassembled. We predict that TgADF plays a key role in regulating this dynamic actin equilibrium in the parasite, through its interaction with both F- and G- actin. The moderate affinity of TgADF for TgACT monomers, and the presence of equimolar concentrations of TgADF and TgACT in the parasite (∼8–10 μm, data not shown), suggest that TgADF will have an important role in sequestering actin monomers in the parasite. Additionally, profilin and CAP, the other highly conserved G-actin-binding proteins found in apicomplexan parasites (16, 17), may play important roles in maintaining the parasite actin monomer pool. TgProfilin has previously been shown to be essential for gliding motility and to interact with actin from parasite lysate (22), but the affinity of this interaction is not known. Although it has weak sequestering activity with heterologous actin, the biochemical interaction between TgProfilin and TgACT has not yet been examined (22). Similarly, little is known about the biochemical activities of TgCAP. Further studies will be necessary to assess the relative roles of all three proteins in buffering the substantial G-actin pool in the parasite.
Parasite actin must be rapidly turned over to generate productive gliding motility, because jasplakinolide-treated parasites with hyperstabilized filaments are unable to undergo directional movement (6). TgADF is an excellent candidate for controlling filament turnover based on its ability to sever actin filaments and promote their net disassembly. The relatively weak severing activity of TgADF may provide a mechanism to regulate this activity, so that TgADF only functions as a severing protein when present at high local concentrations in the parasite. Alternatively, a lower level of activity may be sufficient to efficiently sever less stable TgACT filaments. This situation shares parallels with the worm system, where two ADF isoforms (Unc60A and Unc60B) are expressed in different tissues. The Unc60A isoform shows similar properties to TgADF, with strong sequestering activity and weak severing activity (40). Unc60A is expressed in early embryos and is required for embryonic cytokinesis, an environment where actin is likely to be undergoing rapid turnover (72). Ono et al. (73) recently demonstrated that a knockdown of Unc60A is unable to be functionally complemented by Unc60B, the differentially spliced variant of Unc60A, which has strong severing activity. However, Unc60B mutants that have weak severing activity are able to rescue Unc60A knockdown cells, suggesting that the property of weak severing is functionally important in cells expressing Unc60A (73). This suggests that weak severing activity is likely to be an important property when filaments are transient and are being turned over rapidly, because strong severing activity may prevent transient filaments from being stable long enough to carry out their function. We predict that weak severing will also be an important property of T. gondii ADF for controlling actin turnover during gliding motility.
Recent work has demonstrated the essential nature of various actin-binding proteins in apicomplexan parasites (22, 23, 42), and the importance of careful regulation of the actin machinery for productive gliding motility, a process essential for host cell invasion and successful completion of the parasite life-cycle. In this study, we found that TgADF is potent at sequestering actin monomers in vitro and has weak filament-severing activity. The ability to both maintain high concentrations of G-actin and to regulate the turnover of actin filaments, positions ADF to play a critical role in regulating the unique actin dynamics found in apicomplexan parasites. In addition to this, our work points to an underappreciated role for ADF proteins as monomer-sequestering proteins in G-actin-rich environments.
Supplementary Material
Acknowledgments
We are grateful to Sho Ono for generosity with technical advice and comments on the work. We also thank John Cooper and David Sept for helpful comments, Boyd Butler for assistance and helpful suggestions with the TIRF microscopy experiments, Kristen Skillman for help with the TgACT expression and purification, and Thomas Pollard for the parental cofilin and actophorin plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grant AI073155 (to L. D. S.). This work was also supported by a Washington University Morse/Berg Fellowship (to S. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
K. M. Skillman and L. D. Sibley, unpublished data.
- TgACT
- T. gondii actin
- ADF
- actin depolymerizing factor
- AC
- ADF and cofilin
- TgADF
- T. gondii actin depolymerizing factor
- TIRF
- total internal reflection fluorescence
- CAP
- cyclase-associated protein
- SpCOF
- S. pombe cofilin
- ADF-t
- TgADF containing the S. pombe cofilin C terminus.
REFERENCES
- 1.Joynson D. H., Wreghitt T. J. (2001) Toxoplasmosis: A Comprehensive Clinical Guide, Cambridge University Press, New York [Google Scholar]
- 2.Mead P. S., Slutsker L., Dietz V., McCaig L. F., Bresee J. S., Shapiro C., Griffin P. M., Tauxe R. V. (1999) Emerg. Infect. Dis. 5, 607–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barragan A., Sibley L. D. (2003) Trends Microbiol. 11, 426–430 [DOI] [PubMed] [Google Scholar]
- 4.Sibley L. D. (2004) Science 304, 248–253 [DOI] [PubMed] [Google Scholar]
- 5.Dobrowolski J. M., Sibley L. D. (1996) Cell 84, 933–939 [DOI] [PubMed] [Google Scholar]
- 6.Wetzel D. M., Håkansson S., Hu K., Roos D., Sibley L. D. (2003) Mol. Biol. Cell 14, 396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korn E. D. (1978) Proc. Natl. Acad. Sci. U.S.A. 75, 588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobrowolski J. M., Niesman I. R., Sibley L. D. (1997) Cell Motil. Cytoskeleton 37, 253–262 [DOI] [PubMed] [Google Scholar]
- 9.Sahoo N., Beatty W., Heuser J., Sept D., Sibley L. D. (2006) Mol. Biol. Cell 17, 895–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schüler H., Mueller A. K., Matuschewski K. (2005) FEBS Lett. 579, 655–660 [DOI] [PubMed] [Google Scholar]
- 11.Schmitz S., Grainger M., Howell S., Calder L. J., Gaeb M., Pinder J. C., Holder A. A., Veigel C. (2005) J. Mol. Biol. 349, 113–125 [DOI] [PubMed] [Google Scholar]
- 12.Pollard T. D., Blanchoin L., Mullins R. D. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 545–576 [DOI] [PubMed] [Google Scholar]
- 13.Poupel O., Tardieux I. (1999) Microbes Infect. 1, 653–662 [DOI] [PubMed] [Google Scholar]
- 14.Shaw M. K., Tilney L. G. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 9095–9099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.dos Remedios C. G., Chhabra D., Kekic M., Dedova I. V., Tsubakihara M., Berry D. A., Nosworthy N. J. (2003) Physiol. Rev. 83, 433–473 [DOI] [PubMed] [Google Scholar]
- 16.Baum J., Papenfuss A. T., Baum B., Speed T. P., Cowman A. F. (2006) Nat. Rev. Microbiol. 4, 621–628 [DOI] [PubMed] [Google Scholar]
- 17.Schüler H., Matuschewski K. (2006) Traffic 7, 1433–1439 [DOI] [PubMed] [Google Scholar]
- 18.Gordon J. L., Sahoo N., Mehta S., Sibley L. D. (2007) in: Toxoplasma: Molecular and Cellular Biology (Ajioka J. W., Soldati D. S. eds) pp. 555–570, Horizon Biosciences, Norfolk, UK [Google Scholar]
- 19.Carlier M. F., Laurent V., Santolini J., Melki R., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D. (1997) J. Cell Biol. 136, 1307–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nachmias V. T. (1993) Curr. Opin. Cell Biol. 5, 56–62 [DOI] [PubMed] [Google Scholar]
- 21.Baum J., Tonkin C. J., Paul A. S., Rug M., Smith B. J., Gould S. B., Richard D., Pollard T. D., Cowman A. F. (2008) Cell Host Microbe 3, 188–198 [DOI] [PubMed] [Google Scholar]
- 22.Plattner F., Yarovinsky F., Romero S., Didry D., Carlier M. F., Sher A., Soldati-Favre D. (2008) Cell Host Microbe 3, 77–87 [DOI] [PubMed] [Google Scholar]
- 23.Ganter M., Schüler H., Matuschewski K. (2009) Mol. Microbiol. 74, 1356–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lappalainen P., Drubin D. G. (1997) Nature 388, 78–82 [DOI] [PubMed] [Google Scholar]
- 25.Bamburg J. R. (1999) Annu. Rev. Cell Dev. Biol. 15, 185–230 [DOI] [PubMed] [Google Scholar]
- 26.Andrianantoandro E., Pollard T. D. (2006) Mol. Cell 24, 13–23 [DOI] [PubMed] [Google Scholar]
- 27.McGough A., Pope B., Chiu W., Weeds A. (1997) J. Cell Biol. 138, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maciver S. K., Pope B. J., Whytock S., Weeds A. G. (1998) Eur. J. Biochem. 256, 388–397 [DOI] [PubMed] [Google Scholar]
- 29.Lappalainen P., Fedorov E. V., Fedorov A. A., Almo S. C., Drubin D. G. (1997) EMBO J. 16, 5520–5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriyama K., Yahara I. (1999) EMBO J. 18, 6752–6761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriyama K., Yonezawa N., Sakai H., Yahara I., Nishida E. (1992) J. Biol. Chem. 267, 7240–7244 [PubMed] [Google Scholar]
- 32.Ono S., Baillie D. L., Benian G. M. (1999) J. Cell Biol. 145, 491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yonezawa N., Nishida E., Iida K., Kumagai H., Yahara I., Sakai H. (1991) J. Biol. Chem. 266, 10485–10489 [PubMed] [Google Scholar]
- 34.Guan J. Q., Vorobiev S., Almo S. C., Chance M. R. (2002) Biochemistry 41, 5765–5775 [DOI] [PubMed] [Google Scholar]
- 35.Fedorov A. A., Lappalainen P., Fedorov E. V., Drubin D. G., Almo S. C. (1997) Nat. Struct. Biol. 4, 366–369 [DOI] [PubMed] [Google Scholar]
- 36.Hatanaka H., Ogura K., Moriyama K., Ichikawa S., Yahara I., Inagaki F. (1996) Cell 85, 1047–1055 [DOI] [PubMed] [Google Scholar]
- 37.Leonard S. A., Gittis A. G., Petrella E. C., Pollard T. D., Lattman E. E. (1997) Nat. Struct. Biol. 4, 369–373 [DOI] [PubMed] [Google Scholar]
- 38.Pope B. J., Gonsior S. M., Yeoh S., McGough A., Weeds A. G. (2000) J. Mol. Biol. 298, 649–661 [DOI] [PubMed] [Google Scholar]
- 39.Ono S., McGough A., Pope B. J., Tolbert V. T., Bui A., Pohl J., Benian G. M., Gernert K. M., Weeds A. G. (2001) J. Biol. Chem. 276, 5952–5958 [DOI] [PubMed] [Google Scholar]
- 40.Yamashiro S., Mohri K., Ono S. (2005) Biochemistry 44, 14238–14247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen M. L., Dobrowolski J. M., Muller H., Sibley L. D., Mansour T. E. (1997) Mol. Biochem. Parasitol. 88, 43–52 [DOI] [PubMed] [Google Scholar]
- 42.Schüler H., Mueller A. K., Matuschewski K. (2005) Mol. Biol. Cell 16, 4013–4023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishida E. (1985) Biochemistry 24, 1160–1164 [DOI] [PubMed] [Google Scholar]
- 44.Blanchoin L., Pollard T. D. (1998) J. Biol. Chem. 273, 25106–25111 [DOI] [PubMed] [Google Scholar]
- 45.Xu J. H., Qin Z. H., Liao Y. S., Xie M. Q., Li A. X., Cai J. P. (2008) Parasitol. Res. 103, 263–270 [DOI] [PubMed] [Google Scholar]
- 46.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 47.Bowman G. D., Nodelman I. M., Hong Y., Chua N. H., Lindberg U., Schutt C. E. (2000) Proteins 41, 374–384 [DOI] [PubMed] [Google Scholar]
- 48.Chan C., Beltzner C. C., Pollard T. D. (2009) Curr. Biol. 19, 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quirk S., Maciver S. K., Ampe C., Doberstein S. K., Kaiser D. A., VanDamme J., Vandekerckhove J. S., Pollard T. D. (1993) Biochemistry 32, 8525–8533 [DOI] [PubMed] [Google Scholar]
- 50.Kuhn J. R., Pollard T. D. (2005) Biophys. J. 88, 1387–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amann K. J., Pollard T. D. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 15009–15013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hawkins M., Pope B., Maciver S. K., Weeds A. G. (1993) Biochemistry 32, 9985–9993 [DOI] [PubMed] [Google Scholar]
- 53.Hayden S. M., Miller P. S., Brauweiler A., Bamburg J. R. (1993) Biochemistry 32, 9994–10004 [DOI] [PubMed] [Google Scholar]
- 54.Yonezawa N., Nishida E., Sakai H. (1985) J. Biol. Chem. 260, 14410–14412 [PubMed] [Google Scholar]
- 55.Bernstein B. W., Painter W. B., Chen H., Minamide L. S., Abe H., Bamburg J. R. (2000) Cell Motil. Cytoskeleton 47, 319–336 [DOI] [PubMed] [Google Scholar]
- 56.Agnew B. J., Minamide L. S., Bamburg J. R. (1995) J. Biol. Chem. 270, 17582–17587 [DOI] [PubMed] [Google Scholar]
- 57.Moriyama K., Iida K., Yahara I. (1996) Genes Cells 1, 73–86 [DOI] [PubMed] [Google Scholar]
- 58.Smertenko A. P., Jiang C. J., Simmons N. J., Weeds A. G., Davies D. R., Hussey P. J. (1998) Plant J. 14, 187–193 [DOI] [PubMed] [Google Scholar]
- 59.Ressad F., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D., Carlier M. F. (1998) J. Biol. Chem. 273, 20894–20902 [DOI] [PubMed] [Google Scholar]
- 60.Blanchoin L., Robinson R. C., Choe S., Pollard T. D. (2000) J. Mol. Biol. 295, 203–211 [DOI] [PubMed] [Google Scholar]
- 61.Shiozaki N., Nakano K., Takaine M., Abe H., Numata O. (2009) Biochem. Biophys. Res. Commun. 390, 54–59 [DOI] [PubMed] [Google Scholar]
- 62.Maciver S. K. (1998) Curr. Opin. Cell Biol. 10, 140–144 [DOI] [PubMed] [Google Scholar]
- 63.Mabuchi I. (1983) J. Cell Biol. 97, 1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen H., Bernstein B. W., Sneider J. M., Boyle J. A., Minamide L. S., Bamburg J. R. (2004) Biochemistry 43, 7127–7142 [DOI] [PubMed] [Google Scholar]
- 65.Ono S., Benian G. M. (1998) J. Biol. Chem. 273, 3778–3783 [DOI] [PubMed] [Google Scholar]
- 66.Abe H., Obinata T. (1989) J. Biochem. 106, 172–180 [DOI] [PubMed] [Google Scholar]
- 67.Bamburg J. R., Harris H. E., Weeds A. G. (1980) FEBS Lett. 121, 178–182 [DOI] [PubMed] [Google Scholar]
- 68.Harris H. E., Bamburg J. R., Weeds A. G. (1980) FEBS Lett. 121, 175–177 [DOI] [PubMed] [Google Scholar]
- 69.Nishida E., Muneyuki E., Maekawa S., Ohta Y., Sakai H. (1985) Biochemistry 24, 6624–6630 [DOI] [PubMed] [Google Scholar]
- 70.Shimizu N., Obinata T. (1986) J. Biochem. 99, 751–759 [DOI] [PubMed] [Google Scholar]
- 71.Håkansson S., Morisaki H., Heuser J., Sibley L. D. (1999) Mol. Biol. Cell 10, 3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ono K., Parast M., Alberico C., Benian G. M., Ono S. (2003) J. Cell Sci. 116, 2073–2085 [DOI] [PubMed] [Google Scholar]
- 73.Ono K., Yamashiro S., Ono S. (2008) J. Cell Sci. 121, 2662–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.