

Abstract
Transgenic mice with lymphoid-restricted overexpression of the double bromodomain protein bromodomain-containing 2 (Brd2) develop splenic B-cell lymphoma and, upon transplantation, B-cell leukemia with leukemic infiltrates in liver and lung. Brd2 is a nuclear-localized transcription factor kinase that is most closely related to TATA box binding protein–associated factor, 250 kDa (TAFII250) and the Drosophila developmental protein female sterile homeotic. Constitutive expression of BRD2 in the lymphoid compartment increases cyclin A transcription, “priming” transgenic B cells for proliferation. Mice stochastically develop an aggressive B-cell lymphoma with the features of B-1 cells, including CD5 and surface IgM expression. The B-cell lymphoma is monoclonal for immunoglobulin gene rearrangement and is phenotypically stable. The lymphoblasts are very large and express a transcriptome that is similar to human non-Hodgkin lymphomas. Both a wild-type BRD2 transgene and a kinase-null point mutant drive lymphomagenesis; therefore we propose that, rather than kinase activity, Brd2-mediated recruitment of E2 promoter binding factors (E2Fs) and a specific histone acetyltransferase to the cyclin A promoter by both types of transgene is a mechanistic basis for neoplasia. This report is the first to describe a transgenic mouse model for constitutive expression of a protein with more than one bromodomain.
Introduction
Proper transcriptional control of the cell cycle is necessary to avoid errors that can lead to neoplastic transformation. Bromodomain-containing proteins1-3 recently garnered attention for their roles as transcriptional regulators and their links to chromatin-modifying activities. The bromodomain motif4-6 is found in proteins with intrinsic histone acetyltransferase (HAT) activity,7-9 such as CREB (cyclic adenosine monophosphate [cAMP]–response element binding protein) binding protein (CBP), Gcn5, and TAFII250; in protein adapters that associate with HAT-containing multiprotein complexes, such as polybromo10; in certain transcription factors,11 such as suppressor of Ty 7 (Spt7); or in chromatin remodeling machines, such as switch 2/sucrose nonfermenting 2 (Swi2/Snf2) and brahma.12 Bromodomain proteins bind ∈-aminoacetyl-lysines of core histones, at least in the case of p300/CBP-associated factor (p/CAF), facilitating changes in nucleosome structure.13 The bromodomain/extraterminal domain (BET) protein family, defined by amino-terminal bromodomains and an “extraterminal domain,”14 includes (formerly15 RING3 [really interesting new gene 3]) and female sterile homeotic.16 These transcriptional regulators contain 2 mutually related bromodomains that are most similar in primary sequence to the bromodomain of CBP, yet in overall structural organization most similar to TATA box binding protein–associated factor, 250 kDa (TAFII250), which has 2 bromodomains.14 The yeast BET protein bromodomain factor 1 (Bdf1) binds acetylated nucleosomal lysines17,18 and combines with yeast TATA box binding protein-associated factor, 145 kDa (yTaf145) to perform a function similar to TAFII250 in mammals.19 Recent studies implicated bromodomain proteins in multiple transcriptional regulatory systems, including those that control the cell cycle,20-22 generating interest in their role in carcinogenesis.23,24
Human leukemic cell lines or primary leukemic blasts have high levels of bromodomain-containing 2 (Brd2) autophosphorylation and substrate-directed phosphorylation activity, unlike normal lymphocytes.25 We hypothesized a functional link between the increased kinase activity and oncogenic transformation, particularly because mitogenic signals increase autophosphorylation25-27 and induce nuclear translocation.28 Nuclear-localized Brd2 or Brd2-like proteins participate in multiprotein transcription complexes, such as murine mediator, where Brd2 associates with proteins homologous to yeast transcriptional repressors suppressor of RNA polymerase B 7 (Srb7) and required for glucose repression in yeast 1 (Rgr1), and coactivator RNA polymerase transcriptional regulation mediator subunit 7 (Med7), consistent with the contention that Brd2 provides a transcriptional end point for mitogenic signaling.29 Synergistically with oncogenic ras30 or ras effectors, overexpressed Brd2 forms foci in monolayers of NIH/3T3 cells and transactivates reporter constructs of the promoters of the E2 promoter binding factor (E2F)–regulated cell cycle genes cyclin D1, cyclin E, cyclin A, and dihydrofolate reductase.30 Endogenous Brd2 participates in E2F-containing nuclear complexes.30 A kinase-inactive point mutant of Brd2 does not transactivate synergistically with constitutively active mitogen-activated kinase kinase kinase 1 (MEKK), a ras effector, but wild-type does30; however, both forms transactivate synergistically with oncogenic ras (G.V.D., unpublished data, December 2002). These results likely mean that both kinase-dependent and independent ras pathways are involved in Brd2 transactivation. Ectopically expressed Brd2 transactivates endogenous cyclin A and accelerates the G1 → S transition (A.S., G.V.D., unpublished data, May 2003), providing a mechanism for its observed transforming properties.
A logical next step was developing a transgenic mouse model, wherein constitutive overexpression of recombinant Brd2 would be predicted to deregulate the cell cycle and lead to transformation. We used a murine immunoglobulin (Ig) heavy-chain promoter-enhancer construct pIgTE/N to express BRD2 cDNA in the lymphoid compartment (Eμ-BRD2; transgenic [Tg]), or the same vector incorporating a kinase-inactive point mutant (Eμ-BRD2K574A; Tgmut), to test the hypothesis that kinase activity is linked to oncogenesis. The Eμ system has been used successfully to express myc in the B lineage to produce B-cell31 and pre–B-cell32 lymphomas and the α subunit of casein kinase II in the T lineage to produce thymomas.33 Malignancies arise stochastically in these models and in Eμ-BRD2 mice: transgene expression is elevated in B cells but not T cells or other tissues; they sporadically develop a B-cell lymphoma. The malignancy is monoclonal, transplantable, and phenotypically uniform, bearing many of the hallmarks of B-1 cells.34 Both Tg and Tgmut mice develop the same disease but littermate controls do not. This result suggests that intrinsic kinase activity of Brd2 is not required for oncogenesis in this system and evinces that BRD2 has oncogenic properties in vivo.
Materials and methods
Molecular cloning
A consensus Kozak sequence was engineered into the 2.28-kilobase (kb) human Brd2 cDNA with Vent DNA Polymerase (New England Biolabs, Beverly, MA). The cDNA was cloned into the XhoI site of pIgTE/N and propagated in Escherichia coli STBL2 (Invitrogen Life Technologies, Carlsbad, CA). Restriction analysis and DNA sequencing verified the Tg construct. A Tg construct harboring a point mutation of catalytic lysine 574 to alanine25 was generated simultaneously (Tgmut). A 5.22-kb SalI/MluI fragment containing the Eμ enhancer and simian virus 40 (SV40) poly A was purified and injected into FVB oocytes.
Construction of transgenic mice and genotyping
Southern blotting and polymerase chain reaction (PCR) analysis of tail DNA identified founders. A primer set distinguished between endogenous and transgenic DNA (5′-CAAGAGCTGGTAGTGACCATCCCT-3′ and 5′AGGAGGGCTAGCTGGAGAACCAGGAGC-3′). Taq DNA Polymerase (Invitrogen Life Technologies) amplified the templates in 2.0 mM Mg2+ for 35 cycles at a melting temperature of 94°C (tm; 30 seconds), an annealing temperature of 68°C (ta; 30 s), and an extension temperature of 72°C (te; 1 minute). Three Tg founders and 10 Tgmut founders were obtained; breeding lines were culled to 2 Tg and 2 Tgmut lines, based on transgene passage and mRNA expression.
Expression analysis
Anti-CD45R (B220) or anti-CD3 magnetic beads (Miltenyi Biotec, Auburn, CA) positively selected B or T cells, respectively, from spleens, in phosphate-buffered saline (PBS) supplemented with 2 mM ethylenediaminetetraacetic acid (EDTA) and 0.5% bovine serum albumin. RNA was isolated from purified B and T cells or other tissues by guanidine denaturation, resolved, blotted, and detected with a BRD2 probe in ExpressHyb (BD Biosciences, Palo Alto, CA). To detect cyclin A, transgene, or Ig gene expression, RNA was reverse-transcribed with random hexamer primers using ImProm-II reverse transcriptase (RT; Promega, Madison, WI) and amplified with PCR. The cyclin A gene was amplified with Taq DNA Polymerase (Invitrogen Life Technologies) using primers 5′-TGAAGGCCGGGAACGTGCGTGGACCCGCGC-3′, 5′-CTTCTCCCACCTCAACCAGCCAGTCCACAA-3′; and 5′-CTCTGGGATTAAAGGTATGTACCACAATGCACT-3′, 5′-TGGTCGCTGGGTGGCGCCTAGGCAGGAGCG-3′ in 2.5 mM Mg2+ for 40 cycles at 95°C (tm; 1 min), 55°C (ta; 1 min), and 72°C (te; 1.5 min). The BRD2 transgene was amplified with forward primer 5′-CTCGAGGCCGCCGCCATGGCTTCGGTG-3′ and reverse primer 5′-AGTACCCATGTCCATAGGCTG-3′ in 2.0 mM Mg2+ for 35 cycles at 94°C (tm; 30 s), 68°C (ta; 30 s), and 72°C (te; 1 min). Ig gene cDNA templates were amplified with 3′ primers to constant regions of expressed Ig genes and degenerate 5′ primers to variable regions.35 Engineered EcoRI sites permitted cloning and sequencing. Primers were 3′Cμ, 5′-CCCGAATTCGCTCTCGCAGGAGAC-3′; 3′Cκ, 5′-CCCGAATTCTAACTGCTCACTGGA-3′;5′VH1, 5′-CCCGAATTCGAGGTGAAGCTGGTGGAGA/TCA/TGG-3′; 5′VH2, 5′-CCCGAATTCCAGGTCCAGTTGCAGCAGA/TCA/TGG-3′; 5′Vκ1, 5′-CCCGAATTCGATATTGTGATGACA/T/CCAG-3′; 5′Vκ2, 5′-GCGGAATTCCATATTGTTCTGACCCAGTCTCC-3′ and were used in pairs to amplify expressed Ig gene κ chains in 2.5 mM Mg2+ for 30 cycles at 95°C (tm; 1 min), 55°C (ta; 1 min), and 72°C (te; 2 min). PCR products were resolved in ethidium bromide and DNA fluorescence was measured. If the gene product was derived from a polyclonal Ig gene, a smear was expected after gel separation, but if the Ig gene was monoclonal, a single band was expected. Single bands were isolated and cloned into a plasmid for cDNA expression pcDNA(I) (Amp) (Invitrogen Life Technologies) for sequencing.
To assess B-cell chronic lymphocytic leukemia lymphoma 6 (Bcl6), B-lymphocyte–induced maturation protein-1 (Blimp-1), or β2-microglobulin expression, cDNA was amplified with a real-time PCR detection system (BioRad, Richmond, CA) using primers 5′-GCAAC ATCTACTCGCCCAAG-3′, 5′-CTTCTTCTTTGCTGGCTTTGTC-3′; and 5′-AAGAGGTTATTGGCGTGGTAAG-3′, 5′-ACTTCCTGTTGGCATTCTTGG-3′; and 5′-CCCGCCTCACATTGAAATCC-3′, 5′-GCGTATGTATCAGTCTCAGTGG-3′ correspondingly for 40 cycles at 95°C (tm; 0.5 min) and 62°C (ta/e; 1 min).
Proliferation assays
B cells were isolated with B220 magnetic beads (Miltenyi Biotec); cultured in a 96-well plate in RPMI1640 + 5% fetal bovine serum supplemented with glutamine, penicillin, and streptomycin; and stimulated for 48 hours with different concentrations of mitogens. Goat antimouse IgM F(ab')2 antibody, μ-chain specific, was from Jackson ImmunoResearch (West Grove, PA) and rat antimouse CD40 was from BD Pharmingen (San Diego, CA). Proliferation was measured in quadruplicate by incorporation of [3H] thymidine (New England Nuclear, Boston, MA).
Flow cytometry
Single cell suspensions were prepared from spleen, thymus, or lymph nodes in Hanks buffered salt solution (HBSS) after erythrocyte lysis and stained in PBS supplemented with 0.1% bovine serum albumin. Peritoneal washes were prepared with 5 mL of HBSS. Fluorescein isothiocyanate (FITC)– and phycoerythrin (PE)–coupled antibodies against mouse CD5, CD11b, CD19, CD49b, CD117, CD127, IgM, and Ly-6G for surface cytofluorimetric analyses were from eBioscience (San Diego, CA). Antibodies against CD4, CD5, CD8, CD9, CD11b, CD25, CD44, CD62L, CD69, B220, B7-1, B7-2, Syndecan-1, and rat isotype controls were from BD Pharmingen. Cell staining was performed in the presence of Fc receptor blocking antibody (clone 2.4G2; eBioscience), and cells were detected by flow cytometry with a FACSCalibur system and Cell Quest software (Becton Dickinson, San Jose, CA). Viability (> 99%) was determined by trypan blue. Cells were stained with CD19-FITC or B220-FITC antibodies and compared with PE-coupled antibodies for other antigens.
Histology
Tissue sections were fixed overnight in 10% formalin in PBS, preserved in 70% ethanol, mounted, and stained with hematoxylin and eosin (H&E) or anti-B220 antibody. Blood smears were stained with Wright-Giemsa (Fisher, Pittsburgh, PA).
Transplantation
FVB female mice (6 to 8 weeks old; Taconic Farms, Germantown, NY) were sublethally irradiated with 6 or 8 Gy from a 137Cs gamma source and maintained on acidified water. For adoptive transfer experiments, irradiated mice were each injected subcutaneously with 1 × 107 B cells positively selected from donor spleens by B220 magnetic beads. Mice were monitored for the development of lymphoma or leukemia and were killed at the first appearance of clinical symptoms, without undergoing unnecessary pain and suffering, in compliance with Federal and Institutional Animal Care and Use Committees (IACUC) requirements.
GeneChip analysis of transcriptomes
In accordance with standard Affymetrix (Santa Clara, CA) protocols, splenic B-cell RNA from lymphoma or control spleen was labeled and hybridized to a set of 3 arrays (Murine Genome MG-U74v2 arrays A, B, and C; Affymetrix). GeneChips were scanned; scans were quantified with Affymetrix Microarray Suite 5.0 software; normalization and analysis was performed in Microsoft Excel (Redmond, WA).
Results
Tg passage and expression
PCR analysis established heterozygous passage in Tg and Tgmut mice, which Southern blot confirmed (results not shown). Heterozygous male mice were mated with FVB females to propagate 4 independent lines; passage has been maintained through 6 generations. RT-PCR analysis of lymphoid and control tissues from Tg mice and littermate controls revealed greatly increased transcription of BRD2 in Tg spleen and negligible expression in thymus (Figure 1A). The underexposed image emphasizes comparison of non-Tg and Tg spleen. All tissues express an endogenous Brd2 message, apparent at longer exposures. This evidence shows that Eμ drives transgene expression exceptionally well in B cells. Eμ expression in T lineages has been reported,33,36,37 although malignancies arise more frequently in B lineages.31 To confirm B-restriction, we performed RT-PCR with B220-selected and CD3-selected splenocytes (Figure 1B). A Tg-specific message was highly expressed in B220+ Tg cells and low but detectable in CD3+ Tg cells, about one tenth that of the B-cell level. However, Tg or Tgmut mice never develop T-cell lymphomas.
Figure 1. Expression of transgene in lymphoid tissues.
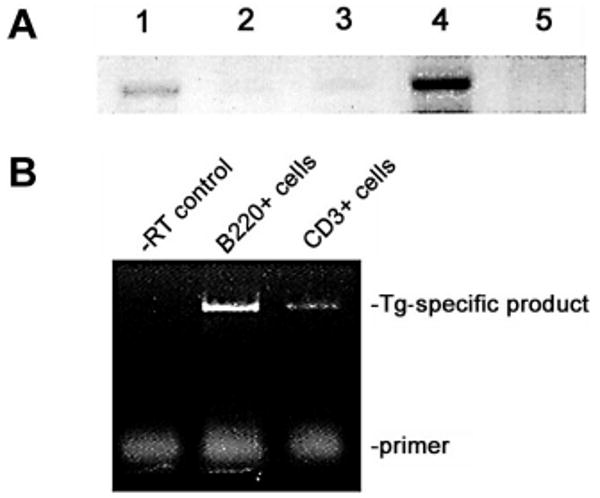
(A) RT-PCR analysis of Eμ-BRD2 expression in different Tg tissues. Lane 1, non-Tg spleen control; lane 2, Tg heart control; lane 3, Tg skeletal muscle control; lane 4, Tg spleen; lane 5, Tg thymus. Primers amplified both endogenous murine message and Tg message. Negative controls without RT verified the RT dependence of the signal (results not shown). (B) Tg expression in Tg B and T cells. The 5′ primer recognizes a Tg vector–specific sequence at the translation start site and the 3′ primer a sequence within the first bromodomain, common to both endogenous and Tg sequences. Analysis was performed with purified B-cell RNA (B220) and T-cell RNA (CD3). Control reaction without reverse transcriptase is shown (−RT control).
B-cell proliferation analysis
To explore whether Tg and Tgmut constructs confer the hypothesized proliferative advantage to B cells, we measured in vitro proliferative responses to anti-CD40 and anti-IgM stimulation. Purified splenic B cells from age-matched Tg, Tgmut, and littermate control mice (20 weeks; asymptomatic by visual inspection) responded to anti-CD40 and anti-IgM (10 μg/mL). Control cells showed a maximal burst of proliferation at 24 hours and no more through 48 hours (Figure 2). Tg and Tgmut B cells proliferated at twice control levels at 24 hours and 3 times control through 48 hours. Tg and Tgmut B cells responded similarly and both underwent apoptosis in a few days. Without mitogens, Tg and Tgmut B cells did not proliferate (results not shown).
Figure 2. Proliferation of Tg and Tgmut B cells in response to anti-CD40 and anti-IgM stimulation.
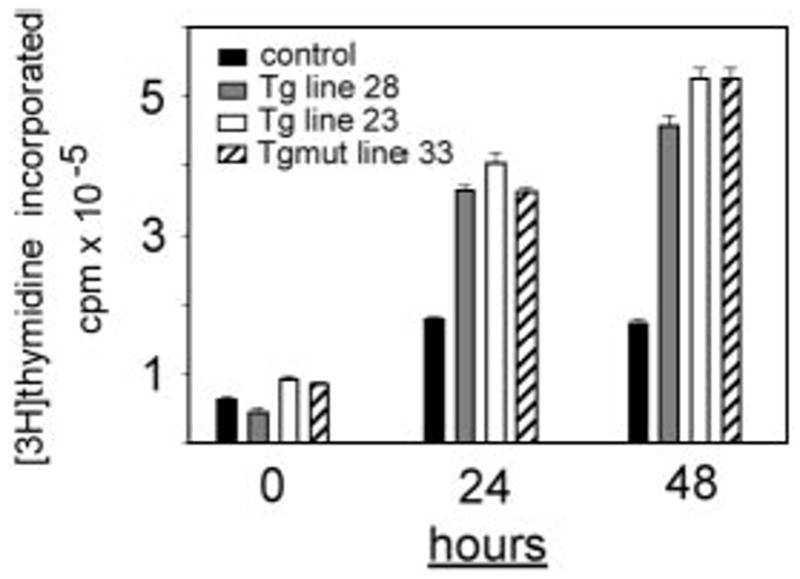
B cells from spleens of littermate control (■), Tg line 28 ( ), Tg line 23 (□), or Tgmut line 33 (▨) or were stimulated in vitro for 24 or 48 hours with anti-CD40 and anti-IgM. Incorporation of [3H] thymidine is shown with standard deviation (n = 4) for each bar.
), Tg line 23 (□), or Tgmut line 33 (▨) or were stimulated in vitro for 24 or 48 hours with anti-CD40 and anti-IgM. Incorporation of [3H] thymidine is shown with standard deviation (n = 4) for each bar.
Incidence of lymphoma
All 4 founder lines of Tg and Tgmut mice sporadically developed clinical signs (ruffled fur, failure to nest, listlessness, hunched back, loss of appetite) after 28 weeks among PCR-positive littermates at approximately 10% annual incidence. Symptomatic mice were killed; spleen, thymus, liver, mesenteric lymph node, and bone marrow were examined. Splenomegaly was always apparent accompanied by abdominal lymphadenopathy and leukemic infiltrations of liver and lung. To date, the numbers of Tg and Tgmut mice that sporadically developed lymphoma are 6 and 5, respectively, out of colonies of 38 and 59. The calculated χ2 for this dataset is 0.61, below the 3.84 value for statistical significance (α = 0.05). Therefore, Tg is not different from Tgmut if “lymphoma” is the dependent variable. Thus, kinase function per se is not important for lymphomagenesis. Another 10% of mice die annually of causes unrelated to lymphoma or leukemia.
Tg- and Tgmut-driven B-cell lymphomas exhibit B-1 characteristics
Flow cytometry was performed with unfractionated splenocytes from leukemic Tg and Tgmut mice and compared with littermate controls. Tg and Tgmut mice did not differ significantly. The vast majority of cells were B220+ CD19+, with very few CD3+, CD4+, or CD8+ cells. Note that there are very few B220− cells among unfractionated lymphoma cells. B220+ splenocytes expressed surface IgM, suggesting they are mature B cells, and CD5 (Figure 3A row 1). The proportion of CD5+ B220+ cells in the control is higher than usually reported because of the particular isotype control antibody used to define flow cytometry parameters. B220 was compared with CD19 to confirm B-cell lineage. Both were consistently expressed in independently arising lymphomas. The pattern of CD5 or B7-1 versus B220 is very similar to versus CD19 (Figure 3 rows 1-2). B-cell activation markers B7-1 and B7-2 were also elevated (Figure 3 row 2) and CD23 was low (Figure 3A row 3). CD11b (which associates with CD18 to form macrophage antigen-1 [Mac-1] and is expressed on granulocytes, natural killer [NK] cells, and macrophages) and Ly-6G (a myeloid marker) were both low (Figure 3A row 3), as was CD49b (an NK marker, results not shown), supporting the interpretation of lymphoid lineage. In addition, a number of markers for immature B cells were not detected, including CD117 (c-kit) and CD127 (interleukin-7 receptor α-chain; results not shown). Lymphoma cells were negative for CD25, syndecan-1, and CD69; positive for CD9; and showed a high IgM/low IgD ratio (Figure 3A rows 4-6). Collectively, these markers suggested that the lymphoma cells bear B-1 hallmarks.34
Figure 3. Flow cytometry of lymphoma cells.
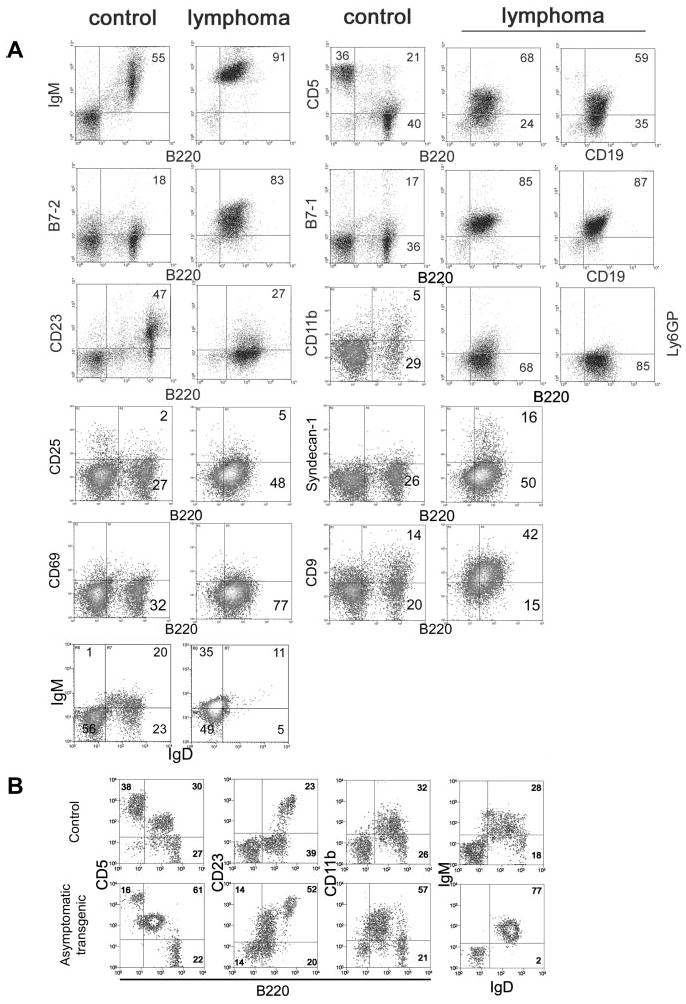
(A) Unfractionated splenocytes from mice diagnosed with splenic lymphoma and peripheral leukemia (lymphoma) were stained for various markers of B-cell differentiation and activation and compared with asymptomatic non-Tg littermate splenocytes (control). Numbers in each square refer to percentages of gated events in a particular quadrant. (B) Peritoneal lymphocytes of premalignant, asymptomatic Tg mice (asymptomatic) were compared with age-matched littermate (control) to assess expansion of the B-1 cell population in the peritoneum.
Levels of B7-1 and B7-2 on splenic B cells were comparable in asymptomatic Tg and Tgmut mice as they aged (8 through 60 weeks, not shown) and similar to those in littermate controls. Expression patterns also held constant with age for T-cell markers in asymptomatic Tg and Tgmut mice such as CD4 versus CD8 in spleen and thymus and CD4 versus CD25, CD4 versus CD44, CD4 versus CD62L, and CD4 versus CD69 in spleen (results not shown). However, we observed increased CD5 versus B220, CD23 versus B220, CD11b versus B220, and IgM versus IgD markers among peritoneal cells of premalignant mice with no clinical signs or splenomegaly, compared with age-matched control mice, suggesting that peritoneal B-1 cells had expanded in the premalignant mice and supporting the B-1 origin of the lymphoma.
Histochemistry of splenic lymphoma and peripheral leukemia
Lymphoid organs of asymptomatic, heterozygous Tg, and Tgmut mice were compared with littermate controls with B220, CD3, and H&E stains and were normal. However, spleens of leukemic mice were enlarged between × 5 and × 50, splenic architecture was disrupted (Figure 4A; Tg, panel ii; Tgmut, panel iii), and invasive lymphoid populations showed numerous mitoses (Figure 4Av; Tg). Bone marrow showed lymphocytic lymphoma (Figure 4Avi; Tg). Elevated numbers of apoptotic cells were not observed.
Figure 4. Tg-driven splenic lymphoma and peripheral leukemia.
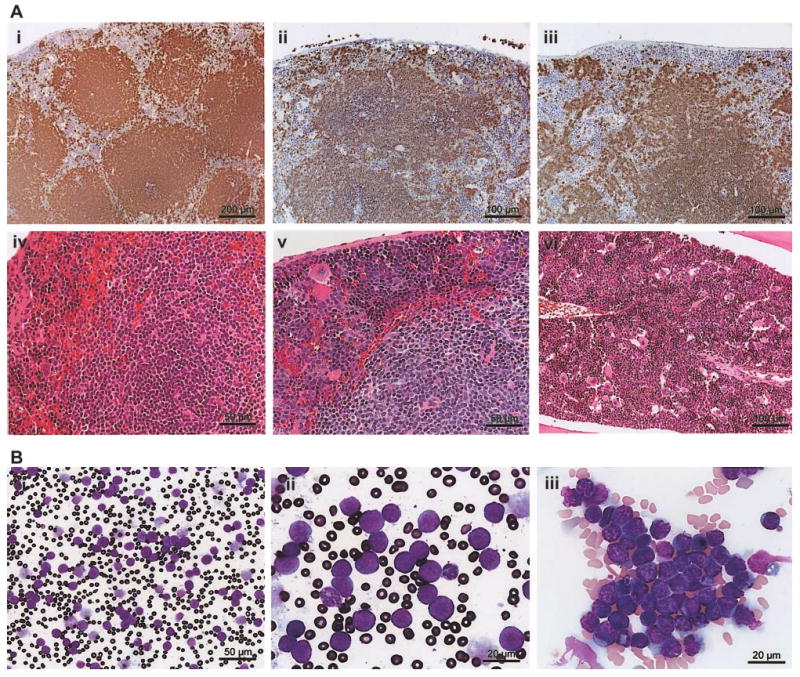
(A) A B220-stained section of asymptomatic Tg spleen (i; bar, 200 μm) is compared with a B220-stained section of a Tg mouse (ii; bar, 100 μm) or Tgmut mouse (iii; bar, 100 μm) with lymphocytic lymphoma. An H&E-stained section of asymptomatic Tg spleen (iv; bar, 50 μm) is compared with an H&E-stained section of a Tg mouse with lymphocytic lymphoma (v; bar, 50 μm). An H&E-stained section of bone marrow of Tgmut mouse with lymphocytic lymphoma is also shown (vi; bar, 100 μm). (B) A Wright-Giemsa–stained smear of peripheral blood of a leukemic mouse that received Tg B-cell lymphoma transplants shows overproliferation of lymphoblasts (i; bar, 50 μm). Blasts exhibit prominent, multiple chromocenters and scanty dark blue cytoplasm (ii; bar, 20 μm). Note the polymorphonuclear granulocyte for comparison. Additional field emphasizes homotypic adhesion and mixed chromatin states (iii; bar, 20 μm).
Normal, syngeneic female mice were irradiated and received transplanted B cells purified from splenic lymphomas. Mice that received transplants developed leukemias and blood-clotting problems; peripheral blood contained large numbers of morphologically similar, large lymphoblasts (Figure 4Bi). The white blood count was 790 (± 40) × 109/L (7.9 [± 0.4] × 105/mm3). The ratio of erythrocytes to lymphoblasts was 6.6 to 1. Leukemic blasts (Figure 4Bii) exhibited scant cytoplasm, cytoplasmic vacuolation, homotypic adhesion, and abundant chromocenters that reflect mixed heterochromatin and euchromatin states, indicating transcriptional activity (Figure 4Biii). Together, these histologic features suggest greatest similarity to the centroblastic variant of diffuse large B-cell lymphoma, following current convention for classifying murine lymphoid neoplasms.38
Monoclonal immunoglobulin gene rearrangement
To determine whether the splenic B-cell neoplasm comprised a clonal or polyclonal population, total RNA from B220-selected cells was isolated for cDNA synthesis. Whereas normal littermate control spleens weighed 0.2 g and yielded approximately 300 μg of total RNA, spleens from lymphoma mice weighed up to 10 g and yielded 30 mg total RNA. Expressed Ig sequences were amplified with 24-mer primers to the 3′ constant region (3′Cμ and 3′Cκ) and degenerate primers to the variable regions (5′VH and VK) to determine if a specific Ig gene rearrangement, or none, predominates in the expanded B-cell population.35 B220+ splenocytes of a non-Tg littermate were the control group. B-cell lymphoma RNA gave a single RT-PCR product (Figure 5A left), but polyclonal B-cell RNA gave smears with each primer pair (Figure 5A right). In this particular lymphoma, an approximately 0.5-kb product arose only from the CμVH1 primer pair. Cloning and sequencing identified a GenBank entry for Mus musculus V23-D-J-C μ mRNA, accession no. AB069913. Three independent, monoclonal IgM+, κ+ B-cell lymphomas have been sequenced to date, but the number of clones remains too small to determine if the transgene promotes the emergence of a particular clonotype. Premalignant mice without clinical signs or splenomegaly showed expansion of oligoclonal splenic B cells in the weeks before clonal malignancy emerged (results not shown). We determined Bcl6 expression using real-time PCR with a standard set of primers for cDNAs synthesized from RNA of purified splenic B-cell lymphoma cells of the first and sixth passage after transplantation (to assess stability of phenotype) and compared the result with Bcl6 expression in splenic B2 cells (Figure 5B). Bcl6 expression was low and remained unaltered over 6 months of serial transplantations, described in the next paragraph.
Figure 5. Immunoglobulin gene rearrangements are monoclonal.
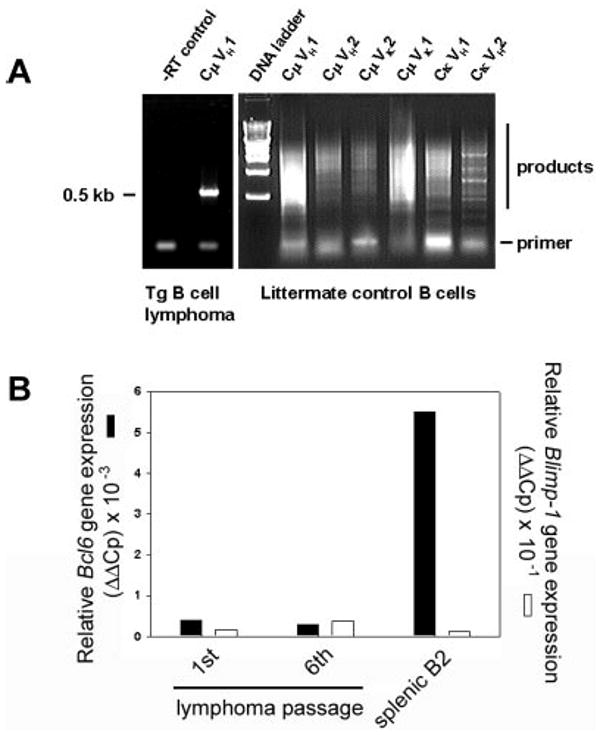
(A) Ig RNAs were isolated from purified B cells of a Tg splenic B-cell lymphoma, amplified by RT-PCR, and resolved. −RT control is shown (left). Primers to the 3′ constant region (3′Cμ and 3′Cκ) Ig sequences and degenerate primers to the variable regions (5′VH and Vk) were used in pairs as indicated. RNA of purified, polyclonal B cells from normal, non-Tg littermate control (right) is compared with Tg lymphoma (left). (B) Lymphoma RNA from first or sixth transplanted passage of malignancy was amplified with primers specific for Bcl6 (■) or Blimp-1 genes (□) by real-time PCR and compared with splenic B2 control. Relative expression levels were determined by the 2−ΔΔCt method where ΔΔCt = ΔCt, sample − ΔCt, reference with Bcl6 or Blimp-1 serving as samples and β2-microglobulin as a reference.
Transplantation
Malignant B cells were purified from Tg lymphoma and transplanted (Figure 6). Radiation dose partly determined susceptibility to establishment of the lymphoma in the transplant recipient39,40; median survival time for mice that received transplants of 107 malignant B cells (DLCL) after irradiation with 6 Gy was 28 days (dotted line), whereas median survival time after irradiation with 8 Gy was 19 days (solid line). In all cases, autopsy confirmed that mice that received transplants died of complications from lymphoma, including massive splenomegaly, peripheral leukemia, internal hemorrhage, and leukemic infiltrates in liver and lung. Irradiated controls (6 Gy) that received B cells purified from an asymptomatic Tg donor survived beyond 60 days (long dashed line). Splenocytes from Tg leukemic donors and transplants were indistinguishable by flow cytometry, Ig gene rearrangement, in vivo growth characteristics, organ sites of leukemic infiltration, or morphology of peripheral blood lymphoblasts (results not shown), suggesting the characteristics of the B-cell lymphoma were stable, even after 6 serial transplantations. The lymphoma cannot be maintained in ordinary tissue culture, as the cells appear not to have undergone an immortalizing event that permits proliferation in media without B-cell mitogens (lipopolysaccharide, interleukin-4, or anti-CD40 + anti-IgM). We have maintained individual lymphoma clones by serial transplantation into irradiated recipients.
Figure 6. Kaplan-Meier plots showing survival of mice that received Tg B-cell lymphoma transplants.
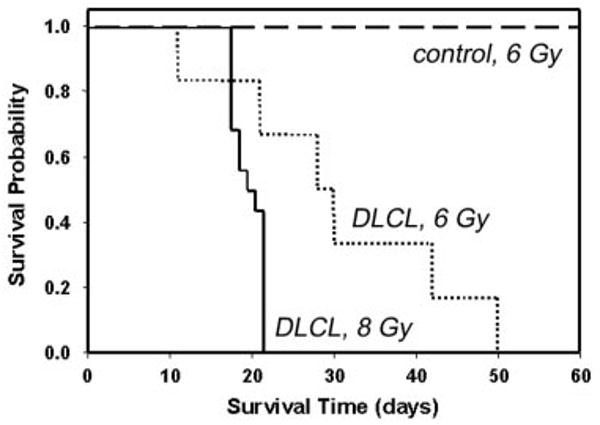
Non-Tg mice were irradiated with 6 Gy (dashed line and dotted line) or 8 Gy (solid line) and inoculated subcutaneously with 107 purified B cells from the spleen of a Tg donor with lymphoma (dotted line and solid line) or 107 purified B cells from the spleen of an asymptomatic Tg donor (dashed line). For dashed line, n = 4; for dotted line, n = 6; and for solid line, n = 15. Control indicates B cells from asymptomatic Tg donor; and DLCL, diffuse large B-cell lymphoma.
Cyclin A expression
We examined cyclin A transcription in the malignant cells, based on our published model for Brd2-dependent transcriptional activation of the cyclin A locus, and on the cell cycle acceleration that we propose arises from Eμ-BRD2 Tg expression30 (A.S., G.V.D., unpublished data, May 2003). RT-PCR analysis of several independently arising splenic B-cell lymphomas in both Tg and Tgmut mice showed that, in all cases, cyclin A transcription was elevated in the primary malignant B cells (Figure 7; lymphoma lanes 1-2) and remained stably elevated in malignant splenic B cells isolated from each passage of mice that received serial transplants that had developed B-cell lymphoma and in all individuals that received transplants, all of which developed the same lymphoma and peripheral B-cell leukemia (Figure 7; lymphoma lanes 3-4, second passage, 2 representative recipients; lymphoma 5-6, fourth passage, 2 representative recipients). Cyclin A message was slightly elevated in B cells purified from asymptomatic Tg mice or Tgmut mice (Tg B cells) but barely detectable in T cells from Tg mice (Tg T cells). Tg mice with B-cell lymphoma had very few splenic T cells (Figure 3). This pattern of cyclin A expression may represent a premalignant marker.
Figure 7. Cyclin A expression in Tg B-cell malignancy.
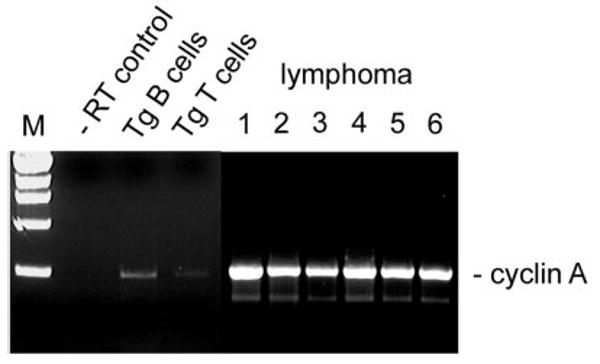
RT-PCR was performed with RNA from purified B cells (Tg B cells) or purified T cells (Tg T cells) from spleens of asymptomatic Tg mice, using primers to the cyclin A gene. Results were compared with negative control (−RT) and to RNA from B cells obtained from independently arising B-cell lymphomas in 6 different mice. Lymphoma lane 1, Tg leukemic mouse (transplant donor); lane 2, Tgmut leukemic mouse; lane 3, second serial transplantation; lane 4, second serial transplantation; lane 5, fourth serial transplantation; lane 6, fourth serial transplantation.
The annual rate of 10% of sporadic Tg lymphoma suggests that an additional “hit” is required for tumorigenesis to progress. That second hit could be engineered various ways. We chose an amphotropic retrovirus system that expresses the H-ras oncogene. Tg B cells “primed” to proliferate are expected to be susceptible to cooperation with the activated ras gene. Lymphomagenesis was accelerated from several months to 4 weeks. Seven Tg and 7 non-Tg mice were injected intraperitoneally with either H-ras– expressing amphotropic retrovirus or neo-expressing retrovirus alone. After 4 weeks, Tg mice injected with the H-ras retrovirus developed severe splenomegaly; non-Tg mice injected with H-ras did not develop illness. Analysis of histopathology, flow cytometry of cell surface antigens, transplantability, and monoclonality of Ig gene rearrangements indicated a diagnosis of B-cell lymphoma indistinguishable from sporadic cases. We then performed Affymetrix GeneChip analysis with B-cell RNA purified from both sporadic and ras-accelerated splenic lymphomas. Controls were, respectively, splenic B cells purified from normal littermate controls and from apparently normal mice infected with neo retrovirus. The 3 mouse arrays A, B, and C permitted whole-genome analysis of the transcriptome of the Brd2-induced lymphomas, resulting in molecular signatures for malignancy that we compared with published databases of human lymphomas. Selected significant markers for named genes are shown in Table 1.
Table 1. Molecular signature of Tg B-cell lymphoma.
| DLCL | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | SPOR | RAS | GC | AB | FL | AT | TC | RPB | CLL |
| Bcl2 | +1.6 | +2.7 | − | + | + | − | − | o | + |
| Bcl6 | −4.4 | −13.4 | + | − | + | o | − | − | − |
| Casp1 | +5.0 | +3.2 | − | o | o | − | − | o | o |
| Ccna1 | +2.9 | +1.6 | + | − | o | o | ++ | −− | −− |
| Ccnb1 | +2.4 | +2.4 | o | − | − | − | + | o | −− |
| Ccne2 | +3.1 | +8.7 | o | o | o | o | o | o | − |
| Cd3d | −30.1 | −2.6 | − | − | + | ++ | + | o | − |
| Cd3e | −40.8 | −2.6 | − | − | + | ++ | + | o | − |
| Cd38 | +1.6 | +3.2 | + | − | o | o | − | o | − |
| Cd44 | +10.6 | +5.9 | − | + | − | + | − | o | o |
| Cdk5 | +1.6 | +2.6 | o | o | o | o | + | o | − |
| Ches1 | −3.1 | −1.4 | + | − | o | o | o | − | o |
| Fos | +52.2 | +20.0 | o | o | + | ++ | −− | ++ | −− |
| Prkcb | +2.4 | +2.0 | o | o | o | − | − | + | ++ |
| Rara | −6.5 | −18.0 | − | + | o | o | o | o | o |
| Rb1 | +2.2 | +2.1 | o | o | o | − | o | o | − |
| Spib | +1.5 | +2.8 | o | o | o | −− | −− | o | − |
| Tcrb | −5.7 | −3.9 | − | o | + | ++ | o | o | o |
| Tubb1 | −6.1 | −1.9 | o | o | − | − | + | −− | −− |
| Vegfa | −9.5 | −6.6 | o | + | o | o | o | o | − |
Most significant changes in gene expression profile of named genes in lymphoma compared with control. About one third of the genome (12000 genes) was significantly expressed. Given the preliminary estimate of variance, about 35% of these were differentially expressed. Comparisons are made to entries in the online databases of Alizadeh et al41 and Shipp et al.42
DLCL indicates diffuse large B-cell lymphoma; SPOR, sporadic Tg B-cell lymphoma; RAS, ras-accelerated Tg B-cell lymphoma; GC, germinal center-like lymphoma41,42; AB, activated peripheral B-cell-like lymphoma41,42; FL, B-cell follicular lymphoma41,42; AT, activated/resting T cell41; TC, transformed cell lines41; RPB, resting peripheral B cell41; CLL, B-cell chronic lymphocytic leukemia41; +, small increase; ++, large increase; −, small decrease; −−, large decrease; and o, no report, no significant change, or no consistent change within a subclass.
Gene entries (mouse gene abbreviation/human homolog, name): Bcl2/BCL2 indicates B-cell leukemia/lymphoma 2; Bcl6/BCL6, B-cell leukemia/lymphoma 6; Casp1/CASP1, caspase-1; Ccna1/CCNA1, cyclin A1; CCnb1/CCNB1, cyclin B1; Ccne2/CCNE2, cyclin E2; Cd3d/CD3D, CD3 antigen, δ polypeptide; Cd3e/CD3E, CD3antigen, ε polypeptide; Cd38/CD38, CD38 antigen; Cd44/CD44, CD44 antigen; Cdk5/CDK5, cyclin-dependent kinase 5; Ches1/CHES1, checkpoint suppressor 1; Fos/FOS, FBJ osteosarcoma oncogene/v-fos FBJ murine osteosarcoma viral oncogene homolog; Prkcb/PRKCB1, protein kinase Cβ; Rara/RARA, retinoic acid receptor α, Rb1/RB1, retinoblastoma protein; Spib/SPIB, Spi-B transcription factor (Spi-1/PU.1 related); Tcrb/TCRB, T-cell receptor β chain; Tubb1/TUBB1, tubulin β1; Vegfa/VEGF, vascular endothelial growth factor A.
In the Tg B-cell lymphoma, most transcriptional changes of greatest estimated significance were 50- to 100-fold decreases in mouse immune cell markers, mostly Ig gene κ-chain sequences (results not shown). Because the B-cell lymphomas are monoclonal, the transcriptomes show a marked decrease in the abundance of diverse Ig gene transcripts when compared with the polyclonal Ig gene transcripts found in the normal littermate B-cell controls. In analyses of the most significant fold changes, we excluded Ig gene sequences because of this trivial explanation. To a first approximation, the transcriptome indicates similarity to diffuse large B-cell lymphoma (DLCL) in humans, the most common type of non-Hodgkin lymphoma.41 Histopathology supports this conclusion. The molecular signature of other B-cell phenotypes, such as resting or activated B cells in the periphery, B-cell follicular lymphoma, transformed B-cell lines, or B-cell chronic lymphocytic leukemia, are not characteristic of this B-cell lymphoma.41 The sporadically arising Tg B-cell lymphomas (Table 1; sporadically arising Tg B-cell lymphomas [SPORs]) and ras-accelerated lymphomas (RASs) shared a close but not identical molecular signature. Most fold changes of named marker genes identified in the literature as lymphoid biomarkers were changed in the same direction and often by a similar magnitude in sporadic and ras-accelerated Tg lymphomas. The Tg lymphoma transcriptional signature did not resemble signatures of human DLCL cases independently established through clinical measures to have a favorable or unfavorable prognosis42 (results not shown). We did not observe a good correlation between the Tg lymphoma transcriptional signature and signatures41,42 of either germinal center–like or activated B-cell–like human DLCLs (Table 1).
Discussion
We designed transgenic mice to test our hypothesis that constitutive expression of BRD2 in the lymphoid compartment would cause lymphoproliferative disease. We based this hypothesis on substantial in vitro evidence that ectopic overexpression of Brd2 transactivates E2F-regulated cell cycle genes such as cyclin D1, cyclin E, cyclin A, and dihydrofolate reductase in synergy with oncogenic ras30 and that Brd2 participates in transcription complexes that contain a histone H4–specific HAT activity and E2F proteins. Furthermore, Brd2 responds to mitogenic signals; when overexpressed, it transforms NIH/3T3 fibroblasts in synergy with ras and accelerates the G1 → S transition through direct action on the cyclin A promoter. Double bromodomain proteins such as Brd2 have been insufficiently studied. The occurrence of a B-cell lymphoma in these transgenic mice now establishes a link between this class of proteins and neoplasia.
Considerable effort has been expended recently to explore the possible functions of the bromodomain motif in nuclear factors associated with transcription and chromatin restructuring.4 The subject has been controversial because sometimes deletion of the motif has no phenotype.6 Virtually all of the nuclear HATs contain bromodomains, but not all bromodomain proteins are HATs.6 The solution of the bromodomain structure and interaction with nucleosomal histone acetyl-lysines3,13 has clarified the matter. Recent studies suggest that bromodomain-containing proteins provide a “scaffolding” or platform function for transcription or chromatin remodeling complexes, anchoring them to nucleosomes.1,2,23,24 Furthermore, oncogenic fusion proteins that contain bromodomains participate in neoplasia, such as the BET protein14 Brd4 (also called22 mitotic chromosome-associated protein [MCAP]), which contains 1400 amino acids and is closely related to Brd2, sharing its dual bromodomain and extraterminal (ET) domain structure.14 BRD4 is rearranged in t(15;19) translocations associated with aggressive carcinomas of the respiratory tract and its fusion partners likely encode oncoproteins.43 Also, translocations that fuse the bromodomain and HAT domain of CBP to the mixed-lineage leukemia factor44-48 (MLL; also called All-1) or to the monocytic leukemia zinc finger protein,49 or that fuse p300 to MLL, create oncoproteins50 that are associated with acute leukemias (reviewed in Filetici and Ballario23). The bromodomain is required for full transforming activity in at least one of these cases.47 Deletion of the Brd2 bromodomains destroys Brd2-dependent transcriptional activation of cyclin A (G.V.D., unpublished data, May 2003) and eliminates association of Brd2 with the multiprotein complex that contains a histone H4–specific HAT and E2F proteins.
Ectopic expression of Brd2 leads both to cell cycle destabilization and transformation in synergy with oncogenic ras or its effectors.30 We hypothesized that Eμ-directed expression of BRD2 in the lymphoid compartment would lead to leukemia or lymphoma, and we obtained this result. Malignant cells from enlarged spleens bear cell surface markers consistent with B-1 lineage.34 This apparently differentiated phenotype remains constant after 6 serial transplantations, with respect to antigen expression in flow cytometry, Ig gene rearrangement, in vivo growth characteristics, elevated cyclin A transcription, organ sites of leukemic infiltration, and morphology. From these data we speculate that the malignant cells do not have a high degree of genetic instability or flexibility in their transcriptional profile. Transcriptome analysis over time will be necessary to confirm this supposition.
The Drosophila homolog of BRD2 is female sterile homeotic,15,51,52 which encodes a homeotic protein and probable transcription regulator that is an upstream activator of the trithorax locus.53 MLL, the human homolog of trithorax, is disrupted in mixed-lineage (myeloid and lymphoid) human leukemias associated with 11q23 translocations.44,45 The functional relationships established in Drosophila (between fsh and trithorax) may be conserved in humans (between Brd2 and MLL), as we hypothesized.25 We therefore looked for a mixed-lineage leukemic phenotype in the mice that received leukemic transplants, but flow cytometry suggests that myeloid components are not present among the leukemic B cells.
We have proposed a mechanism for Brd2-driven transcriptional activation of important cell cycle regulatory genes.30 Brd2 recruits both E2F proteins and HAT activity to the cyclin A gene in particular (A.S., G.V.D., unpublished data, May 2003). These results agree with literature that has established a dual role for E2F recruitment and chromatin restructuring in cyclin A transcriptional control.54-56 Coimmunoprecipitate complexes of Brd2 contain a histone H4–specific HAT (A.S., G.V.D., unpublished data, May 2003). We speculate that a similar enzyme present in transgenic B cells provides crucial nucleosomal modification functions to Brd2 target genes. Several HATs capable of specific acetylation of histone H4 have been identified, including Tip60, implicated in leukemias.57,58 The composition of the multiprotein Brd2 complex29,30 may change across the cell cycle and contribute to cyclin A repression during G0 or to transactivation during cell cycle progression. This model could explain why Tg and Tgmut mice share a similar in vitro proliferative (Figure 2) and in vivo lymphoma phenotype (Figure 4A): apart from a point mutation in the kinase domain of the transgene of Tgmut mice, both lines overexpress the same Brd2 protein, probably involving the same HAT and E2F recruitment mechanism in both cases. This view implies that HAT and E2F recruitment is more important than kinase activity as an oncogenic mechanism in this model. The fine-structural characteristics of B-cell lymphomas may nevertheless be different between Tg and Tgmut mice. Microarray technology will elucidate the transcriptomes of the lymphomas in each breeding line. Reports of oncogenic bromodomain fusion proteins23,43,46-50 lead us to speculate that HAT recruitment to key cell cycle regulatory genes, either via intrinsic HAT domains or via bromodomain protein–mediated “scaffolding,” is important for cell cycle control.1,23,24 Oncogenic fusion proteins or deregulated bromodomain protein function could upset the histone acetylation/deacetylation balance of these genes and lead to their improper transactivation, with possible neoplastic consequences.
Ectopic overexpression of Brd2 transactivates a heterologous cyclin A promoter luciferase construct in synergy with mitogenic signals from ras or MEKK, an effector of ras,59 and increases expression of endogenous cyclin A in synchronized fibroblasts that have been stimulated to enter the cell cycle; these cells show Brd2-driven accelerated kinetics of S-phase entry and mobilization of cyclin A–associated cdk activity (A.S., G.V.D., unpublished data, May 2003). Accordingly, we examined cyclin A transcription in B220+ cells purified from several sporadically arising Tg B-cell lymphomas, compared with B220+ cells from asymptomatic Tg controls. Cyclin A is under negative transcriptional control until S phase54-56 and is frequently elevated in tumors60; it is indeed transcriptionally activated in the Tg malignancy and slightly activated in B cells from asymptomatic Tg mice but not T cells. Therefore, the transgene destabilizes cyclin A in B cells as predicted.
The morphology, surface antigen phenotype, and transcriptome signature of Eμ-BRD2–driven B-cell lymphomas suggest they are most related to human non-Hodgkin lymphomas of the DLCL type.38,41 BRD2 is not present in lymphoma databases we queried,41,42 so comparison of its level in lymphoid malignancies was not possible. We queried Cancer Genome Anatomy Project databases for evidence of transcriptional up-regulation of Brd2 message in human malignancies and found an increase of 4.2 fold (P = .04) reported for 3 cancerous or precancerous lymph nodes compared with 4 normal lymph node controls. Furthermore, the Mitelman database (http://cgap.nci.nih.gov/chromosomes/mitelman) of recurrent chromosomal abnormalities associated with cancer identifies 386 references and 546 patient hematologic malignancies involving breakpoints at 6p21, which includes the class II major histocompatibility complex wherein BRD2 is located, but only a small minority have received fine-structure mapping. Therefore, whereas some cases are likely to show breakpoints within the BRD2 locus, insufficient data exist to make generalizations about the role of BRD2 in the pathogenesis of human DLCL.
Human DLCLs exhibit a variety of genetic abnormalities. The most common is structural alteration of the BCL6 locus,61 often resulting in deregulated Bcl6 activity.62,63 We determined that Bcl6 transcript levels are low in Tg B-cell lymphoma, consistent with the view that Bcl6 activation is more commonly associated with lymphoblastic lymphoma in mice64; furthermore the Bcl6 locus is not normally mutated in mouse B-cell lineage lymphomas.65 Unaltered Bcl6 transcript levels in Tg lymphoma are also consistent with the posited function of the Tg as a transcriptional coactivator and cell cycle regulator30; no current evidence shows a role for BRD2 overexpression in genetic instability.
Human DLCL is thought to arise from germinal center B cells, or less commonly, from marginal zone B cells. However, our expression profile data indicate that the Tg B-cell malignancy does not obviously share a signature with any subclass of human DLCL, and flow cytometry analysis unambiguously identifies the Tg malignancy as composed of B-1 cells. A subclass of murine DLCL, which has neither follicular nor marginal zone origins, has been discussed38 and seems appropriate to classify this Tg lymphoma. An ongoing theme in the lymphoma literature is the extreme diversity of DLCL. Expression profile data41,42 demonstrate that even among ostensible “biomarker” genes for specific subclasses of DLCL or clinical outcomes, remarkable variance exists across cases, as though each DLCL ought to be considered a completely unique neoplasm. Nevertheless, expression profiling with DNA microarrays remains the best method so far to link specific DLCLs with prognosis and most effective course of treatment. In addition, whereas splenic lymphoma cells inoculated subcutaneously into irradiated transplant recipients (Figure 6) exhibit a stable splenic B-1 phenotype after several months of serial transplantations, intraperitoneal inoculation shifts the phenotype to peritoneal B-1 after only a few days (J.R.T., T.L.R., G.V.D., unpublished observations, June 2003), suggesting that environment partially determines the transcriptome. Previously profiled B lymphocytes were obtained from tonsils, peripheral blood, cord blood or lymph nodes,41 or lymph nodes alone,42 not from spleen as in our case, so both organ sites and species variation likely contributed to differences between databases and our profiles. Interestingly, cyclin A is elevated in several subclasses of DLCL but not in all cases41; DLCL pathogenesis could conceivably follow multiple routes from initiation to malignancy. The early elevation of cyclin A transcription in Tg B cells (Figure 7) may represent a premalignant change. Inroads are just being made into understanding the transcriptional and coactivator functions of double bromodomain proteins such as Brd2. The Tg-dependent transcriptional up-regulation of cyclin A represents a novel pathway for B-cell proliferation and may represent a useful model for lymphomagenesis.
Like the Eμ-BRD2 system, the well-known Eμ-myc system causes B-lineage–restricted malignancies that are monoclonal for Ig gene rearrangement, resemble non-Hodgkin lymphomas,66 and may exhibit a lengthy premalignant phase.31 This latent period, beginning prenatally, during which the myc transgene is active throughout the entire B-lymphoid compartment, suggests that an additional genetic change is required to effect full malignant transformation.31,67 However, unlike Eμ-myc mice,31,66,67 the B-cell lymphomas of Eμ-BRD2 mice comprise only mature B cells, despite Eμ activity early in B-cell development and despite the TAF-like properties of Brd2. We propose that Eμ-BRD2 acts at a specific, late stage of B-cell development, probably on the B-1 compartment, to promote the proliferation that gives rise to the DLCL presentation; we will investigate this system further. Weinberg and colleagues68 originated a model of oncogenesis wherein cooperation between different oncogenes such as myc and ras greatly increases the frequency of neoplastic transformation; we observe analogous behavior here with ras acceleration of Eμ-BRD2– driven lymphomagenesis. It would be interesting to determine whether myc synergizes with Brd2 by mating Eμ-myc mice with Eμ-BRD2 mice. Deregulated recruitment of HATs to susceptible promoters may provide a common mechanism for myc- and Brd2-dependent tumorigenesis, as myc has been shown to recruit transformation transcription domain-associated protein (TRRAP)– and Gcn5-associated HAT activities.69,70 TRRAP and Gcn5 also interact with E2F-1 and E2F-4 transactivation domains,71 suggesting a model of transcriptional activation dependent on both specific promoter-bound transcriptional activators and recruited HATs to account for the oncogenic activity72,73 of myc and E2Fs, recapitulating the link between HATs and E2Fs that Brd2 provides.
Given the pleiotropic nature of similar bromodomain “scaffolds,” coactivators, and TAFs, such as TAFII250, improper activation of cyclin A is unlikely to be the sole consequence of deregulated Brd2 signaling. Other transcriptional targets are likely to exist; Brd2 may not act solely in S phase. We speculate that some of these targets include virally transactivated genes associated with neoplasia, because Brd2 interacts with the latent nuclear antigen of Kaposi sarcoma–associated herpesvirus74 (HHV-8), which also targets E2F-regulated genes.75 We expect future studies will illuminate the role of bromodomain proteins as a scaffold that recruits chromatin restructuring activities and transcription factors to the cyclin A promoter, not unlike studies in TAFII105 dominant-negative transgenic mice, which have revealed interactions between transcription activators and TAFs that have consequences for nuclear factor κB (NFκB) signaling and survival in B and T cells.76 Understanding how Brd2 functions in normal cells should provide insight into the origins and nature of B-cell lymphomas that are promoted by improper Brd2 signaling in B cells.
Acknowledgments
We thank Stephan Beck for cDNA that encodes Brd2; Philip Leder for pIgTE/N vector; Katya Ravid and the Transgenic Core Facility of Boston University School of Medicine (BUSM) for production of transgenic founders; Robin MacDonald of the BUSM Laboratory Animal Science Center for help with the mouse colony; Sherrie Sharp for help with flow cytometry; Bill Joyce for help with hematology; Robert J. Munn and Judith Walls for help with histology; Joan Press for help with determining immunoglobulin gene clonality; Norman Gerry, Marc Lenburg, and Garrett Frampton of the BUSM Microarray Resource for help with Affymetrix GeneChip technology; and Mary Ballestas, Janet Buhlmann, David Seldin, and Nancy Zeleznik-Le for critical review of the manuscript.
Supported by United States Public Health Service grants RR14905 from National Institutes of Health (NIH; National Regional Resource Centers; R.D.C.); and CA84294 (R.D.C.), CA84193 (D.V.F.) and CA75107 (G.V.D.) from the National Cancer Institute.
References
- 1.Denis GV. Bromodomain motifs and “scaffolding”? Front Biosci. 2001;6:D1065–D1068. doi: 10.2741/a668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horn PJ, Peterson CL. The bromodomain: a regulator of ATP-dependent chromatin remodeling? Front Biosci. 2001;6:D1019–1023. doi: 10.2741/horn. [DOI] [PubMed] [Google Scholar]
- 3.Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 4.Haynes SR, Dollard C, Winston F, Beck S, Trowsdale J, Dawid IB. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992;20:2603. doi: 10.1093/nar/20.10.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeanmougin F, Wurtz JM, Le Douarin B, Chambon P, Losson R. The bromodomain revisited. Trends Biochem Sci. 1997;22:151–153. doi: 10.1016/s0968-0004(97)01042-6. [DOI] [PubMed] [Google Scholar]
- 6.Winston F, Allis CD. The bromodomain: a chromatin-targeting module? Nat Struct Biol. 1999;6:601–604. doi: 10.1038/10640. [DOI] [PubMed] [Google Scholar]
- 7.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 8.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 9.Dunphy EL, Johnson T, Auerbach SS, Wang EH. Requirement for TAF(II)250 acetyltransferase activity in cell cycle progression. Mol Cell Biol. 2000;20:1134–1139. doi: 10.1128/mcb.20.4.1134-1139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolas RH, Goodwin GH. Molecular cloning of polybromo, a nuclear protein containing multiple domains including five bromodomains, a truncated HMG-box, and two repeats of a novel domain. Gene. 1996;175:233–240. doi: 10.1016/0378-1119(96)82845-9. [DOI] [PubMed] [Google Scholar]
- 11.Gansheroff LJ, Dollard C, Tan P, Winston F. The Saccharomyces cerevisiae SPT7 gene encodes a very acidic protein important for transcription in vivo. Genetics. 1995;139:523–536. doi: 10.1093/genetics/139.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamkun JW, Deuring R, Scott MP, et al. brahma – a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SWI2/SNF2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. [DOI] [PubMed] [Google Scholar]
- 13.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 14.Florence B, Faller DV. You bet-cha: a novel family of transcriptional regulators. Front Biosci. 2001;6:D1008–1018. doi: 10.2741/florence. [DOI] [PubMed] [Google Scholar]
- 15.Beck S, Hanson I, Kelly A, Pappin DJC, Trowsdale J. A homologue of the Drosophila female sterile homeotic (fsh) gene in the class II region of the human MHC. DNA Seq. 1992;2:203–210. doi: 10.3109/10425179209020804. [DOI] [PubMed] [Google Scholar]
- 16.Haynes SR, Mozer BA, Bhatia-Dey N, Dawid IB. The Drosophila fsh locus, a maternal effect gene, encodes apparent transmembrane proteins. Dev Biol. 1989;134:246–257. doi: 10.1016/0012-1606(89)90094-8. [DOI] [PubMed] [Google Scholar]
- 17.Ladurner AG, Inouye C, Jain R, Tjian R. Bromodomains mediate an acetyl-histone encoded anti-silencing function at heterochromatin boundaries. Mol Cell. 2003;11:365–376. doi: 10.1016/s1097-2765(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 18.Matangkasombut O, Buratowski S. Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol Cell. 2003;11:353–363. doi: 10.1016/s1097-2765(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 19.Matangkasombut O, Buratowski RM, Swilling NW, Buratowski S. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev. 2000;14:951–962. [PMC free article] [PubMed] [Google Scholar]
- 20.Du J, Nasir I, Benton BK, Kladde MP, Laurent BC. Sth1p, a Saccharomyces cerevisiae Snf2p/Swi2p homolog, is an essential ATPase in RSC and differs from Snf/Swi in its interactions with histones and chromatin-associated proteins. Genetics. 1998;150:987–1005. doi: 10.1093/genetics/150.3.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A. 1997;94:11268–11273. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dey A, Ellenberg J, Farina A, et al. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Mol Cell Biol. 2000;20:6537–6549. doi: 10.1128/mcb.20.17.6537-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filetici P, Ballario P. The bromodomain: a chromatin browser? Front Biosci. 2001;6:D866–D876. doi: 10.2741/filetici. [DOI] [PubMed] [Google Scholar]
- 24.Dyson MH, Rose S, Mahadevan LC. Acetyllysine-binding and function of bromodomain-containing proteins in chromatin. Front Biosci. 2001;6:D853–D865. doi: 10.2741/dyson. [DOI] [PubMed] [Google Scholar]
- 25.Denis GV, Green MR. A novel, mitogen-activated nuclear kinase is related to a Drosophila developmental regulator. Genes Dev. 1996;10:261–271. doi: 10.1101/gad.10.3.261. [DOI] [PubMed] [Google Scholar]
- 26.Rachie NA, Seger R, Valentine MA, Ostrowski J, Bomsztyk K. Identification of an inducible 85-kDa nuclear protein kinase. J Biol Chem. 1993;268:22143–22149. [PubMed] [Google Scholar]
- 27.Ostrowski J, Florio SK, Denis GV, Suzuki H, Bomsztyk K. Stimulation of p85/RING3 kinase in multiple organs after systemic administration of mitogens into mice. Oncogene. 1998;16:1223–1227. doi: 10.1038/sj.onc.1201624. [DOI] [PubMed] [Google Scholar]
- 28.Guo N, Faller DV, Denis GV. Activation-induced nuclear translocation of RING3. J Cell Sci. 2000;113:3085–3091. doi: 10.1242/jcs.113.17.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang YW, Veschambre P, Erdjument-Bromage H. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc Natl Acad Sci U S A. 1998;95:8538–8543. doi: 10.1073/pnas.95.15.8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denis GV, Vaziri C, Guo N, Faller DV. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 2000;11:417–424. [PMC free article] [PubMed] [Google Scholar]
- 31.Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt EV, Pattengale PK, Weir L, Leder P. Transgenic mice bearing the human c-myc gene activated by an immunoglobulin enhancer: a pre-B-cell lymphoma model. Proc Natl Acad Sci U S A. 1988;85:6047–6051. doi: 10.1073/pnas.85.16.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seldin DC, Leder P. Casein kinase II alpha transgene-induced murine lymphoma: relation to theileriosis in cattle. Science. 1995;267:894–897. doi: 10.1126/science.7846532. [DOI] [PubMed] [Google Scholar]
- 34.Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 35.Kavaler J, Caton AJ, Staudt LM, Schwartz D, Gerhard W. A set of closely related antibodies dominates the primary antibody response to the antigenic site Cb of the A/PR/8/34 influenza virus hemagglutinin. J Immunol. 1990;145:2312–2321. [PubMed] [Google Scholar]
- 36.Kemp DJ, Harris AW, Cory S, Adams JM. Expression of the immunoglobulin Cμ gene in mouse T and B lymphoid and myeloid cell lines. Proc Natl Acad Sci U S A. 1980;77:2876–2880. doi: 10.1073/pnas.77.5.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kemp DJ, Harris AW, Adams JM. Transcripts of the immunoglobulin Cμ gene vary in structure and splicing during lymphoid development. Proc Natl Acad Sci U S A. 1980;77:7400–7404. doi: 10.1073/pnas.77.12.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morse HC, Anver MR, Fredrickson TN, et al. Hematopathology subcommittee of the Mouse Models of Human Cancers Consortium: Bethesda proposals for classification of lymphoid neoplasms in mice. Blood. 2002;100:246–258. doi: 10.1182/blood.v100.1.246. [DOI] [PubMed] [Google Scholar]
- 39.Chiu KM, Knospe WH. Inhibitor of granulocyte-macrophage colony formation in plasma of mice rendered aplastic by allogeneic lymph node cells. Exp Hematol. 1989;17:335–339. [PubMed] [Google Scholar]
- 40.Hudson WA, Liu Q, Le C, Kersey JH. Xenotransplantation of human lymphoid malignancies is optimized in mice with multiple immunologic defects. Leukemia. 1998;12:2029–2033. doi: 10.1038/sj.leu.2401236. [DOI] [PubMed] [Google Scholar]
- 41.Alizadeh AA, Eisen M, Davis RE, et al. Distinct types of diffuse large-B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 42.Shipp MA, Ross KN, Tamayo P, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- 43.French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003;63:304–307. [PubMed] [Google Scholar]
- 44.Gu Y, Nakamura T, Alder H, et al. The t(4;11) chromosomal translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- 45.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 46.Sobulo OM, Borrow J, Tomek R, et al. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3) Proc Natl Acad Sci U S A. 1997;94:8732–8737. doi: 10.1073/pnas.94.16.8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lavau C, Du C, Thirman M, Zeleznik-Le N. Chromatin-related properties of CBP fused to MLL generate a myelodysplastic-like syndrome that evolves into myeloid leukemia. EMBO J. 2000;19:4655–4664. doi: 10.1093/emboj/19.17.4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liedman D, Zeleznik-Le N. Retroviral transduction model of mixed lineage leukemia fused to CREB binding protein. Curr Opin Hematol. 2001;8:218–223. doi: 10.1097/00062752-200107000-00007. [DOI] [PubMed] [Google Scholar]
- 49.Borrow J, Stanton VP, Jr, Andresen JM. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet. 1996;14:33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- 50.Ida K, Kitabayashi I, Taki T, et al. Adenoviral E1A-associated protein p300 is involved in acute myeloid leukemia with t(11;22)(q23;q13) Blood. 1997;90:4699–4704. [PubMed] [Google Scholar]
- 51.Forquignon F. A maternal effect mutation leading to deficiencies of organs and homeotic transformations in the adults of Drosophila. Wilhelm Roux Arch Dev Biol. 1981;190:132–138. doi: 10.1007/BF00867798. [DOI] [PubMed] [Google Scholar]
- 52.Gans M, Forquignon F, Masson M. The role of dosage in the region 7D1-7D5-6 of the X chromosome in the production of homeotic transformations in Drosophila melanogaster. Genetics. 1980;96:887–902. doi: 10.1093/genetics/96.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mozer BA, Dawid IB. Cloning and molecular characterization of the trithorax locus of Drosophila melanogaster. Proc Nat'l Acad Sci U S A. 1989;86:3738–3742. doi: 10.1073/pnas.86.10.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schulze A, Zerfass K, Spitkovsky D, et al. Cell cycle regulation of the cyclin A gene is mediated by a variant E2F site. Proc Natl Acad Sci U S A. 1995;92:11264–11268. doi: 10.1073/pnas.92.24.11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stiegler P, De Luca A, Bagella L, Giordano A. The COOH-terminal region of pRb2/p130 binds to histone deacetylase 1 (HDAC1), enhancing transcriptional repression of the E2F-dependent cyclin A promoter. Cancer Res. 1998;58:5049–5052. [PubMed] [Google Scholar]
- 56.Knudsen KE, Fribourg AF, Strobeck MW, Blanchard JM, Knudsen ES. Cyclin A is a functional target of retinoblastoma tumor suppressor protein-mediated cell cycle arrest. J Biol Chem. 1999;274:27632–27641. doi: 10.1074/jbc.274.39.27632. [DOI] [PubMed] [Google Scholar]
- 57.Clarke AS, Lowell JE, Jacobson SJ, Pillus L. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol Cell Biol. 1999;19:2515–2526. doi: 10.1128/mcb.19.4.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bird AW, Yu DY, Pray-Grant MG, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–415. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 59.Lange-Carter CA, Johnson GL. Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science. 1994;265:1458–1461. doi: 10.1126/science.8073291. [DOI] [PubMed] [Google Scholar]
- 60.Yam CH, Fung TK, Poon RY. Cyclin A in cell cycle control and cancer. Cell Mol Life Sci. 2002;59:1317–1326. doi: 10.1007/s00018-002-8510-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ye BH, Lista F, Lo Coco F, et al. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science. 1993;262:747–750. doi: 10.1126/science.8235596. [DOI] [PubMed] [Google Scholar]
- 62.Ye BH, Chaganti S, Chang CC, et al. Chromosomal translocations cause deregulated BCL6 expression by promoter substitution in B cell lymphoma. EMBO J. 1995;14:6209–6217. doi: 10.1002/j.1460-2075.1995.tb00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS, Dalla-Favera R. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. 2003;101:2914–2923. doi: 10.1182/blood-2002-11-3387. [DOI] [PubMed] [Google Scholar]
- 64.Hori M, Xiang S, Qi CF, et al. Non-Hodgkin lymphomas of mice. Blood Cells Mol Dis. 2001;27:217–222. doi: 10.1006/bcmd.2000.0375. [DOI] [PubMed] [Google Scholar]
- 65.Hori M, Qi CF, Torrey TA, Huppi K, Morse HC. The Bcl6 locus is not mutated in mouse B-cell lineage lymphomas. Leukemia Res. 2002;26:739–743. doi: 10.1016/s0145-2126(01)00200-4. [DOI] [PubMed] [Google Scholar]
- 66.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eμ-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in Eμ-myc transgenic mice. Cell. 1986;47:11–8. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- 68.Land H, Parada LF, Weinberg RA. Cellular oncogenes and multistep carcinogenesis. Science. 1983;222:771–778. doi: 10.1126/science.6356358. [DOI] [PubMed] [Google Scholar]
- 69.Nikiforov MA, Chandriani S, Park J, et al. TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol Cell Biol. 2002;22:5054–5063. doi: 10.1128/MCB.22.14.5054-5063.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lang SE, McMahon SB, Cole MD, Hearing P. E2F transcriptional activation requires TRRAP and GCN5 cofactors. J Biol Chem. 2001;276:32627–32634. doi: 10.1074/jbc.M102067200. [DOI] [PubMed] [Google Scholar]
- 72.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- 73.Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001;15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Platt GM, Simpson GR, Mittnacht S, Schulz TF. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J Virol. 1999;73:9789–9795. doi: 10.1128/jvi.73.12.9789-9795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med. 2000;6:1121–1127. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 76.Silkov A, Wolstein O, Shachar I, Dikstein R. Enhanced apoptosis of B and T lymphocytes in TAFII105 dominant-negative transgenic mice is linked to nuclear factor-kappa B. J Biol Chem. 2002;277:17821–17829. doi: 10.1074/jbc.M200696200. [DOI] [PubMed] [Google Scholar]